

Abstract
Background: Sevoflurane postconditioning (SPostC) can exert myocardial protective effects similar to ischemic preconditioning. However, the exact myocardial protection mechanism by SPostC is unclear. Studies indicate that hypoxia-inducible factor-1 (HIF-1) maintains cellular respiration homeostasis by regulating mitochondrial respiratory chain enzyme activity under hypoxic conditions. This study investigated whether SPostC could regulate the expression of myocardial HIF-1α and to improve mitochondrial respiratory function, thereby relieving myocardial ischemia-reperfusion injury in rats. Methods: The myocardial ischemia-reperfusion rat model was established using the Langendorff isolated heart perfusion apparatus. Additionally, postconditioning was performed using sevoflurane alone or in combination with the HIF-1α inhibitor 2-methoxyestradiol (2ME2). The changes in hemodynamic parameters, HIF-1α protein expression levels, mitochondrial respiratory function and enzyme activity, mitochondrial reactive oxygen species (ROS) production rates, and mitochondrial ultrastructure were measured or observed. Results: Compared to the ischemia-reperfusion (I/R) group, HIF-1α expression in the SPostC group was significantly up-regulated. Additionally, cardiac function indicators, mitochondrial state 3 respiratory rate, respiratory control ratio (RCR), cytochrome C oxidase (CcO), NADH oxidase (NADHO), and succinate oxidase (SUCO) activities, mitochondrial ROS production rate, and mitochondrial ultrastructure were significantly better than those in the I/R group. However, these advantages were completely reversed by the HIF-1α specific inhibitor 2ME2 (P<0.05). Conclusion: The myocardial protective function of SPostC might be associated with the improvement of mitochondrial respiratory function after up-regulation of HIF-1α expression.
Keywords: Sevoflurane postconditioning, myocardial protective effect, hypoxia-inducible factor-1, mitochondrial respiratory function, respiratory enzyme activity
Introduction
Ischemic heart disease is a major cause of mortality due to human cardiovascular diseases [1]. Restoration of the blood supply to the ischemic myocardia (i.e., reperfusion treatment) is considered the most effective treatment for myocardial ischemia. However, myocardial reperfusion can aggravate or even cause an irreversible injury [2]. Therefore, development of methods that effectively reduce perioperative myocardial reperfusion injury is a current research topic.
A large number of studies have indicated that inhalational anesthetics such as sevoflurane and isoflurane can relieve ischemia-reperfusion injury [3,4]. Sevoflurane has been applied extensively in clinical practice and basic research due to its stable induction and rapid recovery pharmacological properties. Sevoflurane postconditioning (SPostC) can produce myocardial protective functions similar to ischemic preconditioning and is a promising treatment against post-ischemic reperfusion injury during perioperative period [5]. Currently, the anti-reperfusion injury effect of SPostC is thought to be achieved through protection of mitochondrial functions [6]; however, the exact mechanism has not been elucidated. As an important site for ATP production via oxidative phosphorylation, mitochondria play important roles in myocardial ischemia and reperfusion injury [7-9]. The structural integrity and normal functions of mitochondria are the bases for the maintenance of the physiological activities of the heart. Mitochondrial respiratory function and respiratory enzyme activity are the major components that reflect the oxidative phosphorylation process and structural integrity of the mitochondria.
Hypoxia-inducible factor-1 (HIF-1) regulates the expression of a series of hypoxia-sensitive genes under hypoxic conditions to maintain the survival of tissues and cells. Studies have confirmed that HIF-1 is central to cardioprotection against ischemia-reperfusion injury [10]. Recent studies showed that HIF-1 maintained normal cellular respiration by regulating mitochondrial respiratory chain activity. Furthermore, HIF-1 can reduce the production of mitochondrial ROS to avoid cell injury [11]. Thus, we investigated whether the SpostC myocardial protective function was associated with the regulation of myocardial mitochondrial respiratory function.
We used an in vitro rat myocardial ischemia-reperfusion injury model and studied the mechanism underlying the association between anti-myocardial ischemia-perfusion by SPostC and HIF-1 by investigating mitochondrial respiratory function.
Materials and methods
Animals and experimental groupings
A total of 88 healthy adult male Sprague-Dawley (SD) rats with a body weight of 250-300 g were provided by the experimental animal center of the Third Military Medical University (permission number SCXK2012-0005). All SD rats were raised according to the Guide for the Care and Use of Laboratory Animals released by the National Institute of Health of the USA (1996 revision).
These rats were randomly divided into 4 groups (n=22 rats/group) as follows: normal control (C) group, ischemia-reperfusion (I/R) group, SPostC group, and HIF-1α inhibitor (2-methoxyestradiol, 2ME2) + SPostC (MSP) group. The C group received persistent perfusion of Krebs-Henseleit (K-H) solution for 180 min. The I/R group was equilibrated for 20 min, followed by perfusion of 4°C St. Thomas cardioplegia; afterwards, the rats were perfused with K-H solution for 120 min, and then, whole heart ischemia was performed at 32°C for 40 min. The SPostC group was equilibrated for 20 min, followed by perfusion of 4°C St. Thomas cardioplegia. Afterwards, the rats were perfused with 1.0 MAC (minimum alveolar concentration) of sevoflurane-saturated K-H solution for 15 min, and then, whole heart ischemia was performed for 40 min at 32°C, followed by continuous perfusion of K-H solution for 105 min. The MSP group was perfused with 2ME2 (2 µM) + 1.0 MAC of sevoflurane-saturated K-H solution for 15 min after 40 min of whole heart ischemia followed by continuous perfusion of K-H solution for 105 min (Figure 1). The preparation of 1.0 MAC of sevoflurane-saturated K-H was previously described [12,13]. The sevoflurane concentration was monitored using a ULT-Svi-22-07 gas detector (Division, Finland) and an infrared gas analyzer (Datex-Ohmeda, GE Healthcare) to ensure that the sevoflurane concentration in the K-H solution was maintained at 1.0 MAC.
Figure 1.
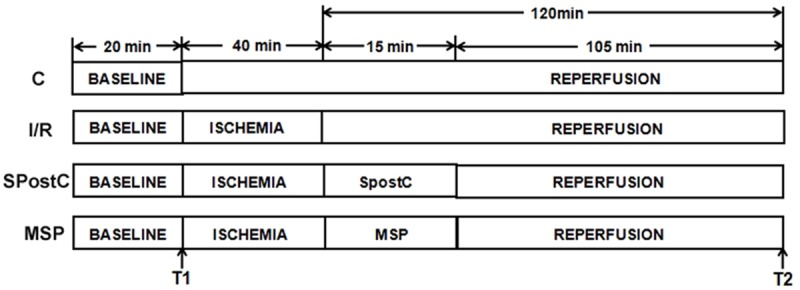
The schematic diagram of the isolated rat heart experimental procedures. With the exception of the C group, all hearts were equilibrated for 20 min, followed by whole heart ischemia for 40 min and reperfusion for 120 min. The SpostC received 1.0 MAC sevoflurane treatment for 15 min, followed by reperfusion for 105 min. T1: the end of equilibration; T2: the end of reperfusion.
Establishment of the Langendorff model [14]
The rats were intraperitoneally injected with sodium pentobarbital (40 mg/kg) and heparin (250 U/kg). After anesthetization, the heart was rapidly removed (3-4 mm of the aorta was retained) and placed in K-H buffer pre-cooled to 4°C to discharge all blood in the heart cavities. The K-H buffer solution (mmol/L) was prepared with NaCl (118), KCl (4.7), MgSO4•7H2O (1.2), KH2PO (1.2), NaHCO3 (25), glucose (11), and CaCl2 (2.5) at pH 7.45. The heart was immobilized with a Langendorff perfusion needle using a No. 4 surgical thread. Retrograde perfusion of the aorta was performed at 37°C using K-H solution equilibrated in 95% O2-5% CO2 mixed gas under 5.8 kPa perfusion pressure. The pulmonary artery and left atrial appendage were cut open; then, a pressure measuring tube with a rubber balloon was inserted into the left ventricle through the mitral valve opening and connected with a biological function experimental pressure transducer system. The perfusion pressure was maintained at approximately 60-70 mmHg. The size and position of the balloon was adjusted to maintain the left ventricular end-diastolic pressure (LVEDP) at 0-10 mmHg. The above steps were completed within 2 min. The inclusion criteria were a heart rate (HR) >250 beats/min and a left ventricular developed pressure (LVDP) >80 mmHg after the isolated heart was equilibrated for 20 min.
Monitoring of hemodynamics
The heart rate (HR, beats/min), LVDP (mmHg), LVEDP (mmHg), and maximum rate of increase of LV pressure (+dp/dtmax, mmHg/s) at the end of equilibration (T1) and the end of reperfusion (T2) were collected using the Powerlab/8SP data collection system.
Measurement of mitochondrial respiratory function
Myocardial mitochondria were extracted, and mitochondrial proteins were quantitated. Mitochondrial respiratory function was measured using a Clark oxygen electrode [15,16]. The total volume of the reaction solution was 1 mL, and the temperature was 37°C. First, 900 μL of the mitochondrial respiration measurement medium was added; after equilibration for 15 min, 50 μL of the mitochondrial suspension at a final concentration of 1 mg/mL was added, and the 20-30 sec oxygen consumption curve was recorded. After the curve was stabilized, 10 μL of succinic acid (final concentration 5 mM) and 1 μL of rotenone (final concentration 2 μM) were added. After state 4 respiration was entered, the results was recorded for 1 min. Next, 9 μL of ADP (final concentration 100 μM) was added to induce state 3 respiration. The changes in the oxygen consumption curve were recorded; when all of the ADP was completely phosphorylated to ATP, the mitochondria entered state 4 respiration. Mitochondrial respiratory control rate (RCR) = the oxygen consumption rate in state 3 respiration/the oxygen consumption rate in state 4 respiration.
Measurement of mitochondrial respiratory enzyme activity [15,17]
Extracted and quantitated mitochondria were repeatedly frozen and thawed at -80°C and 20°C to prepare the mitochondrial subunits. The mitochondrial respiratory chain enzyme activities, including that of cytochrome C oxidase (CcO), NADH oxidase (NADHO), and succinate oxidase (SUCO), were measured. The total volume of the reaction solution was 1 mL, and the temperature was 37°C. The CcO measurement medium, NADHO measurement medium, and SUCO measurement medium were added (900 μL each) and equilibrated for 15 min. The mitochondrial subunit suspension preparation (50 μL) was added, and the 5-10 min oxygen consumption curves were recorded.
Measurement of the mitochondrial ROS production rate
The myocardial mitochondrial ROS production rate was measured using fluorescence spectrometry [18]. The basic principle is that dichlorodihydrofluorescein diacetate (DCFH-DA) can diffuse through the mitochondrial membrane and form reduced dichlorodihydrofluorescein (DCFH), which does not exhibit fluorescence activity because of hydrolysis by esterases in the mitochondria. DCFH is rapidly oxidized by ROS into the highly fluorescent active substance dichlorofluorescein (DCF). The speed of the oxidization of DCFH into DCF positively correlates with the ROS production rate. In this experiment, we used the 3 mL cuvette reaction system with 2.9 mL of mitochondrial ROS measurement medium and 0.5 mg of mitochondria; we also included one cuvette that did not contain mitochondria. Succinic acid at a final concentration of 3.3 mmol/L was added and used as the substrate; then, 3 μL of DCFH-DA at a final concentration of 5 mmol/L was added and incubated at 37°C for 15 min. The fluorescence intensity of the substrate without mitochondria (F1) and the fluorescence intensity of the sample with mitochondria (F) were measured. The ROS production rate was the result of subtracting F1 from F.
Myocardial ultrastructure
After reperfusion, myocardial tissues with sizes of 1 mm×1 mm×1 mm were collected from each group. The ultrastructure of the myocardial mitochondria were observed using a transmission electron microscope after fixation, washing, dehydration, embedding, sectioning, and staining.
Western blot analysis
After the repeated reperfusion, the left ventricular myocardial tissues were cut (n=6) and immediately placed in liquid nitrogen for storage. Total proteins were extracted from the myocardial tissues and lysed in tissue lysis buffer. Samples containing 30 µg of protein were subjected to SDS-PAGE and then transferred onto a membrane. After the membrane was blocked at 37°C for 2 h, an HIF-1α primary antibody was added at a 1:1000 dilution and incubated at 4°C overnight. The membrane was washed with TBST and then incubated with an HRP-labeled secondary antibody at a 1:5000 dilution at room temperature for 1 h. The images were developed using electrochemiluminescence (ECL). Analysis of the gray density values of the target protein bands was performed using the Quantity One imaging analysis system.
Statistical analysis
SPSS 17.0 statistical software was used for the data analysis. Measurement data are presented as the mean ± standard error of the mean (SEM). The within group comparisons were performed using the analysis of variance of repeated measurement design. The comparison between groups was performed using univariate analysis of variance. P<0.05 indicated that the difference had statistical significance.
Results
Cardiac function indicators
No significant differences were detected in the HR, LVDP, LVEDP, and +dp/dtmax between groups at the end of equilibration (P>0.05). The HR, LVDP, and +dp/dtmax in all groups were significantly decreased at the end of reperfusion compared to the end of equilibration, whereas LVEDP was significantly increased (P<0.05) (Figure 2A-D). The comparison of all groups at the end of reperfusion showed that LVEDP was significantly decreased and HR, LVDP, and +dp/dtmax were significantly increased in the SPostC group compared to the I/R group (P<0.05). However, none of the cardiac function indicators between the SPostC and I/R groups were significantly different after the application of the HIF-1α inhibitor (P>0.05) (Figure 2A-D).
Figure 2.
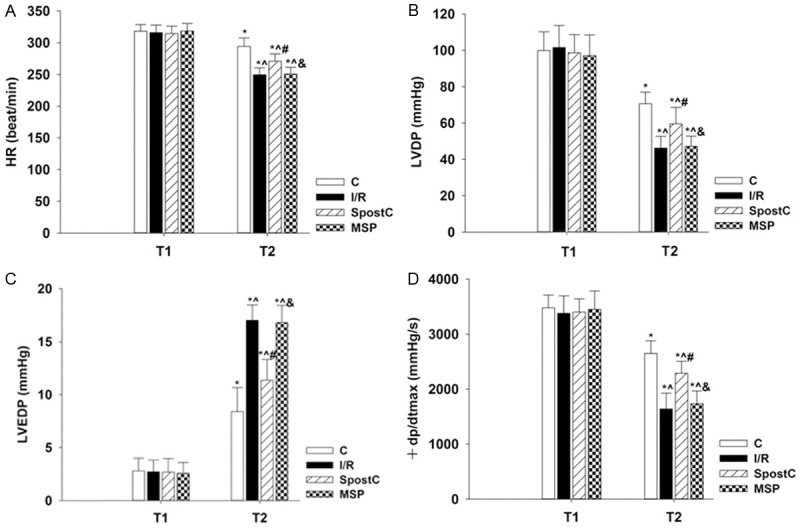
The cardiac function indicators at the end of equilibration (T1) and the end of reperfusion (T2). A: HR; B: LVDP; C: LVEDP; D: +dp/dtmax. Data are presented as the mean ± SEM (n=22). *P<0.05 vs T1; T2 time point: ^P<0.05 vs C, #P<0.05 vs I/R, &P<0.05 vs SPostC.
Expression of the HIF-1α protein
Compared to the I/R group, myocardial HIF-1α expression in the SPostC group at the end of reperfusion was significantly up-regulated. Compared to the SPostC group, HIF-1α protein expression in the MSP group at the end of reperfusion was significantly down-regulated (P<0.05); however, no significant difference was detected in HIF-1α protein expression between the MSP and I/R groups (P>0.05) (Figure 3).
Figure 3.
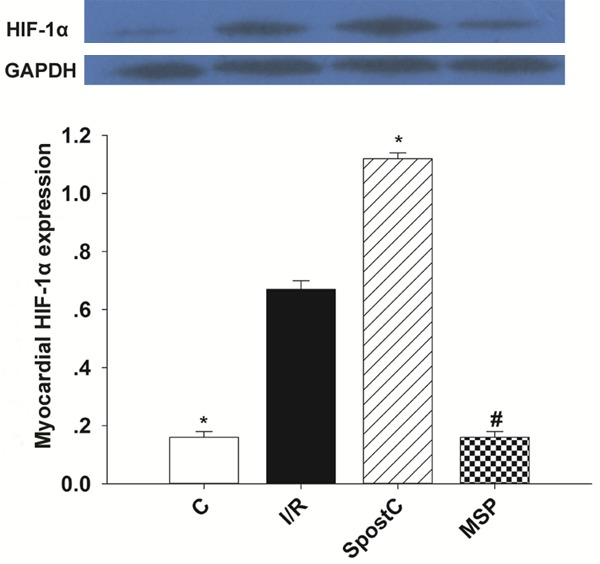
The changes in the HIF-1α protein levels at the end of reperfusion. Data are presented as themean ± SEM (n=6). *P<0.05 vs I/R; #P<0.05 vs SPostC.
Mitochondrial respiratory function
The mitochondrial state 3 respiration and RCR did not significantly differ at the end of equilibration among all groups (P>0.05). The mitochondrial state 3 respiration and RCR at the end of reperfusion were significantly decreased in all groups compared to the end of equilibration (P<0.05). The comparison of all groups at the end of reperfusion showed that the mitochondrial state 3 respiration and RCR in the SPostC group were significantly increased compared to those in the I/R group (P<0.05); no significant difference in respiration function was observed between the MSP and I/R groups (P>0.05) (Table 1).
Table 1.
The changes in mitochondrial state 3 respiration and RCR at the end of equilibration (T1) and at the end of reperfusion (T2). Data are presented as the mean ± SEM (n=6)
| T1 | T2 | |||
|---|---|---|---|---|
|
|
|
|||
| State 3 | RCR | State 3 | RCR | |
| Control | 147.31±5.42 | 2.43±0.09 | 145.23±7.59 | 2.24±0.14* |
| I/R | 145.05±8.33 | 2.35±0.17 | 70.16±5.99*,^ | 1.11±0.09*,^ |
| SpostC | 146.46±5.11 | 2.34±0.19 | 121.94±6.24*,^,# | 1.87±0.07*,^,# |
| MSP | 147.47±7.91 | 2.36±0.14 | 71.02±5.29*,^,& | 1.14±0.08*,^,& |
P<0.05 vs T1;
T2 time point:
P<0.05 vs C;
P<0.05 vs I/R;
P<0.05 vs SPostC.
Mitochondrial respiratory enzyme activity
The CcO, NADHO, and SUCO activities at the end of equilibration did not significantly differ among groups (P>0.05). The CcO, NADHO, and SUCO activities at the end of reperfusion were significantly decreased in all groups compared to the activities at the end of equilibration (P<0.05). The comparison of all groups at the end of reperfusion showed that the CcO, NADHO, and SUCO activities in the SPostC group were significantly increased compared to those in the I/R group; however, the CcO, NADHO, and SUCO activities did not significantly differ between the MSP and I/R groups (P>0.05) (Table 2).
Table 2.
The changes in mitochondrial CcO, NADHO, and SUCO activities at the end of equilibration (T1) and at the end of reperfusion (T2). Data are presented as the mean ± SEM (n=6)
| T1 | T2 | |||||
|---|---|---|---|---|---|---|
|
|
|
|||||
| NADHO | CcO | SUCO | NADHO | CcO | SUCO | |
| C | 316.56±13.93 | 97.39±5.75 | 101.85±5.73 | 287.19±12.84* | 81.10±7.53* | 80.88±6.12* |
| I/R | 313.20±12.62 | 94.27±4.31 | 99.57±6.36 | 205.11±11.05*,^ | 47.73±3.97*,^ | 46.90±3.40*,^ |
| SpostC | 311.07±14.12 | 97.18±6.66 | 99.10±6.84 | 252.82±12.34*,^,# | 69.07±4.45*,^,# | 66.95±5.41*,^,# |
| MSP | 310.60±11.35 | 96.85±5.44 | 100.43±6.24 | 209.27±9.65*,^,& | 48.95±3.39*,^,& | 48.95±3.39*,^,& |
P<0.05 vs T1;
T2 time point:
P<0.05 vs C;
P<0.05 vs I/R;
P<0.05 vs SPostC.
Mitochondrial ROS production rates
The mitochondrial ROS production rates in all groups at the end of reperfusion were significantly increased compared to the rates at the end of equilibration. The mitochondrial ROS production rate at the end of reperfusion in the SPostC group was significantly lower than the rate in the I/R group. After the application of the HIF-1α inhibitor, the ROS production rates were not significantly different between the MSP and I/R groups (P>0.05) (Table 3).
Table 3.
The mitochondrial ROS production rates (RPR) at the end of equilibration (T1) and at the end of reperfusion (T2). Data are presented as the mean ± SEM (n=6)
| T1 | T2 | |
|---|---|---|
|
|
|
|
| RPR (u.s-1.mg-1) | RPR (u.s-1.mg-1) | |
| Control | 1.97±0.13 | 6.89±0.54* |
| I/R | 2.01±0.08 | 12.96±0.82*,^ |
| SpostC | 1.99±0.11 | 9.12±0.61*,^,# |
| MSP | 2.00±0.09 | 13.01±0.69*,^,& |
<0.05 vs T1;
T2 time point:
P<0.05 vs C;
P<0.05 vs I/R;
P<0.05 vs SPostC.
Ultrastructure of myocardial mitochondria
In the C group, the myofilaments typically exhibited an ordered arrangement with no obvious dissolution or fractures; the morphology of the mitochondria was intact with a round or oval shape and an orderly arrangement, and the cristae were closely connected. In the I/R group, the myocardial structure was severely damaged, with the myofilaments dissolved or even fractured; the mitochondria were significantly swollen, the crista space was widened and broken, and the sarcoplasmic reticulum was highly expanded. In the SPostC group, the myofilaments exhibited an ordered arrangement; some myofilaments and sarcomere spaces were widened and dissolved, but the majority of the mitochondria had an intact morphology with clear and visible cristae. Conversely, some mitochondria were slightly swollen but did not exhibit dissolutions and ruptures. In the MSP group, the myocardial structure was damaged, the myofilaments were dissolved, and the mitochondria were significantly swollen (Figure 4).
Figure 4.

The changes in the myocardial ultrastructure in the left ventricle at the end of reperfusion (n=4). The magnification is 10,000×.
Discussion
Our study showed that SPostC significantly up-regulated HIF-1α protein expression when compared to the I/R group, improved mitochondrial respiratory function and mitochondrial respiratory enzyme activity, and relieved ischemia-reperfusion injury. However, the myocardial protection effect of SPostC completely disappeared after the application of the HIF-1α inhibitor. This suggested that the myocardial protection effect of SPostC might be associated with the improvement of mitochondrial respiratory function after the up-regulation of HIF-1α expression.
As a representative inhalational anesthetic, sevoflurane has been applied extensively in clinical practice. Compared to other volatile anesthetics, sevoflurane not only produces excellent anesthetic effects but also plays important protective roles in the perioperative myocardial ischemia-reperfusion process; therefore, it is considered as the choice of selection when defending the perioperative myocardial ischemia-reperfusion injury. Our results showed that LVDP, +dp/dtmax, and HR were significantly increased at the end of reperfusion in the SPostC group compared to the I/R group, which was consistent with the results of Inamura et al [19]. Currently, the function of SPostC in the improvement of cardiac functions in the ischemic heart is thought to be associated with its maintenance of normal mitochondrial structure and function [20,21].
The mitochondrion is the key organelle that provides energy to the myocardia. During ischemia-reperfusion, mitochondrial structures and mitochondrial respiratory functions are damaged, causing a reduction in ATP synthesis that further aggravates myocardial injury [22,23]. Previous studies confirmed that HIF-1 plays a critical role in the defense against myocardial ischemia-reperfusion injury [10]. Upon treatment with the HIF-1α-specific inhibitor 2ME2, HIF-1α levels after ischemia-reperfusion in isolated hearts were significantly lower than those in the ischemia-reperfusion only group and were comparable to the levels in the non-ischemic heart group [24]. Under hypoxic conditions, up-regulation of HIF-1α expression not only regulates energy production by the mitochondrial respiratory chain and maintains CcO activities but also reduces ROS production from the mitochondria to avoid myocardial injury [11].
HIF-1 is the most important transcription factor involved in the transcriptional regulation of hypoxic responses. HIF-1 is a heterodimer that is primarily composed of the HIF-1α and HIF-1β subunits. Under normal oxygen conditions, prolyl hydroxylase (PHD) and factor inhibiting HIF (FIH) in normal cells use oxygen as the substrate to hydroxylate the HIF-1α protein in the cytoplasm. Then, the hydroxylated HIF-1α is ubiquitinated by the E3 ubiquitin ligase complex von Hippel-Lindau (VHL) and degraded through the ubiquitin-proteasome degradation pathway. Under hypoxic conditions, PHD and FIH are suppressed and HIF-1α degradation is blocked. A large number of HIF-1α proteins accumulate and are translocated into the nucleus to form heterodimers with HIF-1β to produce active HIF-1. HIF-1 interacts with hypoxia response elements to regulate downstream target gene expression, which is conducive to cell survival under hypoxic conditions [25].
In this study, to confirm whether the myocardial protective function of SPostC was associated with the maintenance of mitochondrial respiratory function after HIF-1 up-regulation, we measured the HIF-1α protein expression level. The results showed that HIF-1α protein expression was significantly increased in the SPostC group and the mitochondrial state 3 respiration, RCR, and mitochondrial respiratory chain enzyme activities (CcO, NADHO, and SUCO) were significantly better than those in the I/R group. Moreover, the mitochondrial ultrastructure in the SPostC was more intact and the ROS production rate was significantly lower than that in the I/R group. To validate the experimental results, we applied the HIF-1α inhibitor 2ME2 and found that the above advantages of SPostC disappeared completely. Therefore, our results suggested that the myocardial protective function of SPostC might be due to the up-regulation of HIF-1α expression by improving mitochondrial respiratory function, thereby relieving myocardial ischemia-reperfusion injury.
The study conducted by Sirvinskas et al also found that the application of sevoflurane during coronary artery bypass grafting surgery protected the myocardial mitochondrial outer membrane from ischemia-reperfusion injury and avoided the loss of CcO [26]. However, the researchers did not identify the pathway used by sevoflurane to protect the integrity of mitochondrial structure and function. In this study, we hypothesized that SPostC could reduce ROS release by the mitochondria to avoid injury to the mitochondrial membrane system through the up-regulation of HIF-1α expression [27,28] thereby maintaining normal mitochondrial respiratory chain operations to provide energy for the restoration of cardiac function after myocardial ischemia-reperfusion; alternatively, SPostC could increase the nuclear translocation and expression of HIF-1α to promote the binding between HIF-1α and HIF-1β to form active HIF-1, which in turn could promote the expression of downstream target genes, promote mitochondrial respiration, and maintain mitochondrial enzyme activity, thereby relieving myocardial ischemia-reperfusion injury.
Conclusion
Our study showed that SPostC up-regulated HIF-1α expression in the rat myocardia, improved mitochondrial respiratory function during myocardial ischemia-reperfusion, and maintained normal mitochondrial electron transport chain function, thereby relieving myocardial ischemia-reperfusion injury in rats. This study will provide theoretical support for the mechanism underlying the myocardial protection by SPostC.
Acknowledgements
This work was supported by the National Natural Science Foundation (Grant No. U1403223) of China. We wish to thank Prof. Tian Yu, Haiying Wang and Dr. Lin Zhang, Guizhou Key Laboratory of Anesthesia and Organ Protection, Department of Anesthesiology, Zunyi Medical College, Zunyi, Guizhou, China, People’s Republic, who provided technical support to us.
Disclosure of conflict of interest
None.
References
- 1.Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, Franco S, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Huffman MD, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Magid D, Marcus GM, Marelli A, Matchar DB, McGuire DK, Mohler ER, Moy CS, Mussolino ME, Nichol G, Paynter NP, Schreiner PJ, Sorlie PD, Stein J, Turan TN, Virani SS, Wong ND, Woo D, Turner MB American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Executive summary: heart disease and stroke statistics--2013 update: a report from the American Heart Association. Circulation. 2013;127:143–152. doi: 10.1161/CIR.0b013e318282ab8f. [DOI] [PubMed] [Google Scholar]
- 2.Zhang Y, Ren J. Targeting autophagy for the therapeutic application of histone deacetylase inhibitors in ischemia/reperfusion heart injury. Circulation. 2014;129:1088–1091. doi: 10.1161/CIRCULATIONAHA.113.008115. [DOI] [PubMed] [Google Scholar]
- 3.Yao YT, Fang NX, Shi CX, Li LH. Sevoflurane postconditioning protects isolated rat hearts against ischemia-reperfusion injury. Chin Med J (Engl) 2010;123:1320–1328. [PubMed] [Google Scholar]
- 4.Hu ZY, Abbott GW, Fang YD, Huang YS, Liu J. Emulsified isoflurane postconditioning produces cardioprotection against myocardial ischemia-reperfusion injury in rats. J Physiol Sci. 2013;63:251–261. doi: 10.1007/s12576-013-0261-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gong JS, Yao YT, Fang NX, Li LH. Sevoflurane postconditioning attenuates reperfusion-induced ventricular arrhythmias in isolated rat hearts exposed to ischemia/reperfusion injury. Mol Biol Rep. 2012;39:6417–6425. doi: 10.1007/s11033-012-1447-9. [DOI] [PubMed] [Google Scholar]
- 6.Yu P, Zhang J, Yu S, Luo Z, Hua F, Yuan L, Zhou Z, Liu Q, Du X, Chen S, Zhang L, Xu G. Protective Effect of Sevoflurane Postconditioning against Cardiac Ischemia/Reperfusion Injury via Ameliorating Mitochondrial Impairment, Oxidative Stress and Rescuing Autophagic Clearance. PLoS One. 2015;10:e0134666. doi: 10.1371/journal.pone.0134666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Escobales N, Nunez RE, Jang S, Parodi-Rullan R, Ayala-Pena S, Sacher JR, Skoda EM, Wipf P, Frontera W, Javadov S. Mitochondria-targeted ROS scavenger improves post-ischemic recovery of cardiac function and attenuates mitochondrial abnormalities in aged rats. J Mol Cell Cardiol. 2014;77:136–146. doi: 10.1016/j.yjmcc.2014.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shintani-Ishida K, Yoshida K. Mitochondrial m-calpain opens the mitochondrial permeability transition pore in ischemia-reperfusion. Int J Cardiol. 2015;197:26–32. doi: 10.1016/j.ijcard.2015.06.010. [DOI] [PubMed] [Google Scholar]
- 9.Szczepanek K, Xu A, Hu Y, Thompson J, He J, Larner AC, Salloum FN, Chen Q, Lesnefsky EJ. Cardioprotective function of mitochondrial-targeted and transcriptionally inactive STAT3 against ischemia and reperfusion injury. Basic Res Cardiol. 2015;110:53. doi: 10.1007/s00395-015-0509-2. [DOI] [PubMed] [Google Scholar]
- 10.Eckle T, Kohler D, Lehmann R, El Kasmi K, Eltzschig HK. Hypoxia-inducible factor-1 is central to cardioprotection: a new paradigm for ischemic preconditioning. Circulation. 2008;118:166–175. doi: 10.1161/CIRCULATIONAHA.107.758516. [DOI] [PubMed] [Google Scholar]
- 11.Hwang HJ, Lynn SG, Vengellur A, Saini Y, Grier EA, Ferguson-Miller SM, LaPres JJ. Hypoxia Inducible Factors Modulate Mitochondrial Oxygen Consumption and Transcriptional Regulation of Nuclear-Encoded Electron Transport Chain Genes. Biochemistry. 2015;54:3739–3748. doi: 10.1021/bi5012892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Obal D, Preckel B, Scharbatke H, Mullenheim J, Hoterkes F, Thamer V, Schlack W. One MAC of sevoflurane provides protection against reperfusion injury in the rat heart in vivo. Br J Anaesth. 2001;87:905–911. doi: 10.1093/bja/87.6.905. [DOI] [PubMed] [Google Scholar]
- 13.Zhang J, Wang C, Yu S, Luo Z, Chen Y, Liu Q, Hua F, Xu G, Yu P. Sevoflurane postconditioning protects rat hearts against ischemia-reperfusion injury via the activation of PI3K/AKT/mTOR signaling. Sci Rep. 2014;4:7317. doi: 10.1038/srep07317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bell RM, Mocanu MM, Yellon DM. Retrograde heart perfusion: the Langendorff technique of isolated heart perfusion. J Mol Cell Cardiol. 2011;50:940–950. doi: 10.1016/j.yjmcc.2011.02.018. [DOI] [PubMed] [Google Scholar]
- 15.Adlam VJ, Harrison JC, Porteous CM, James AM, Smith RA, Murphy MP, Sammut IA. Targeting an antioxidant to mitochondria decreases cardiac ischemia-reperfusion injury. FASEB J. 2005;19:1088–1095. doi: 10.1096/fj.05-3718com. [DOI] [PubMed] [Google Scholar]
- 16.Frezza C, Cipolat S, Scorrano L. Organelle isolation: functional mitochondria from mouse liver, muscle and cultured fibroblasts. Nat Protoc. 2007;2:287–295. doi: 10.1038/nprot.2006.478. [DOI] [PubMed] [Google Scholar]
- 17.Schneider H, Lemasters JJ, Hochli M, Hackenbrock CR. Liposome-mitochondrial inner membrane fusion. Lateral diffusion of integral electron transfer components. J Biol Chem. 1980;255:3748–3756. [PubMed] [Google Scholar]
- 18.Bejma J, Ji LL. Aging and acute exercise enhance free radical generation in rat skeletal muscle. J Appl Physiol (1985) 1999;87:465–470. doi: 10.1152/jappl.1999.87.1.465. [DOI] [PubMed] [Google Scholar]
- 19.Inamura Y, Miyamae M, Sugioka S, Domae N, Kotani J. Sevoflurane postconditioning prevents activation of caspase 3 and 9 through antiapoptotic signaling after myocardial ischemia-reperfusion. J Anesth. 2010;24:215–224. doi: 10.1007/s00540-010-0877-6. [DOI] [PubMed] [Google Scholar]
- 20.Li J, Loukili N, Rosenblatt-Velin N, Pacher P, Feihl F, Waeber B, Liaudet L. Peroxynitrite is a key mediator of the cardioprotection afforded by ischemic postconditioning in vivo. PLoS One. 2013;8:e70331. doi: 10.1371/journal.pone.0070331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Soltysinska E, Bentzen BH, Barthmes M, Hattel H, Thrush AB, Harper ME, Qvortrup K, Larsen FJ, Schiffer TA, Losa-Reyna J, Straubinger J, Kniess A, Thomsen MB, Bruggemann A, Fenske S, Biel M, Ruth P, Wahl-Schott C, Boushel RC, Olesen SP, Lukowski R. KCNMA1 encoded cardiac BK channels afford protection against ischemia-reperfusion injury. PLoS One. 2014;9:e103402. doi: 10.1371/journal.pone.0103402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ferdinandy P, Schulz R, Baxter GF. Interaction of cardiovascular risk factors with myocardial ischemia/reperfusion injury, preconditioning, and postconditioning. Pharmacol Rev. 2007;59:418–458. doi: 10.1124/pr.107.06002. [DOI] [PubMed] [Google Scholar]
- 23.Symons JA, Myles PS. Myocardial protection with volatile anaesthetic agents during coronary artery bypass surgery: a meta-analysis. Br J Anaesth. 2006;97:127–136. doi: 10.1093/bja/ael149. [DOI] [PubMed] [Google Scholar]
- 24.Si J, Wang N, Wang H, Xie J, Yang J, Yi H, Shi Z, Ma J, Wang W, Yang L, Yu S, Li J. HIF-1alpha signaling activation by post-ischemia treatment with astragaloside IV attenuates myocardial ischemia-reperfusion injury. PLoS One. 2014;9:e107832. doi: 10.1371/journal.pone.0107832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhao HX, Wang XL, Wang YH, Wu Y, Li XY, Lv XP, Zhao ZQ, Zhao RR, Liu HR. Attenuation of myocardial injury by postconditioning: role of hypoxia inducible factor-1alpha. Basic Res Cardiol. 2010;105:109–118. doi: 10.1007/s00395-009-0044-0. [DOI] [PubMed] [Google Scholar]
- 26.Sirvinskas E, Kinderyte A, Trumbeckaite S, Lenkutis T, Raliene L, Giedraitis S, Macas A, Borutaite V. Effects of sevoflurane vs. propofol on mitochondrial functional activity after ischemia-reperfusion injury and the influence on clinical parameters in patients undergoing CABG surgery with cardiopulmonary bypass. Perfusion. 2015;30:590–595. doi: 10.1177/0267659115571174. [DOI] [PubMed] [Google Scholar]
- 27.Fukuda R, Zhang H, Kim JW, Shimoda L, Dang CV, Semenza GL. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell. 2007;129:111–122. doi: 10.1016/j.cell.2007.01.047. [DOI] [PubMed] [Google Scholar]
- 28.Tello D, Balsa E, Acosta-Iborra B, Fuertes-Yebra E, Elorza A, Ordonez A, Corral-Escariz M, Soro I, Lopez-Bernardo E, Perales-Clemente E, Martinez-Ruiz A, Enriquez JA, Aragones J, Cadenas S, Landazuri MO. Induction of the mitochondrial NDUFA4L2 protein by HIF-1alpha decreases oxygen consumption by inhibiting Complex I activity. Cell Metab. 2011;14:768–779. doi: 10.1016/j.cmet.2011.10.008. [DOI] [PubMed] [Google Scholar]