

Abstract
Benzylisoquinoline alkaloids (BIAs) are produced in a wide variety of plants and include many common analgesic, antitussive, and anticancer compounds. Several members of a distinct family of S-adenosylmethionine (SAM)-dependent N-methyltransferases (NMTs) play critical roles in BIA biosynthesis, but the molecular basis of substrate recognition and catalysis is not known for NMTs involved in BIA metabolism. To address this issue, the crystal structure of pavine NMT from Thalictrum flavum was solved using selenomethionine-substituted protein (dmin = 2.8 Å). Additional structures were determined for the native protein (dmin = 2.0 Å) as well as binary complexes with SAM (dmin = 2.3 Å) or the reaction product S-adenosylhomocysteine (dmin = 1.6 Å). The structure of a complex with S-adenosylhomocysteine and two molecules of tetrahydropapaverine (THP; one as the S conformer and a second in the R configuration) (dmin = 1.8 Å) revealed key features of substrate recognition. Pavine NMT converted racemic THP to laudanosine, but the enzyme showed a preference for (±)-pavine and (S)-reticuline as substrates. These structures suggest the involvement of highly conserved residues at the active site. Mutagenesis of three residues near the methyl group of SAM and the nitrogen atom of the alkaloid acceptor decreased enzyme activity without disrupting the structure of the protein. The binding site for THP provides a framework for understanding substrate specificity among numerous NMTs involved in the biosynthesis of BIAs and other specialized metabolites. This information will facilitate metabolic engineering efforts aimed at producing medicinally important compounds in heterologous systems, such as yeast.
Keywords: enzyme kinetics, enzyme mechanism, protein structure, secondary metabolism, x-ray crystallography, Benzylisoquinoline alkaloid biosynthesis
Introduction
Benzylisoquinoline alkaloids (BIAs)3 are a structurally diverse class of plant specialized metabolites that display an impressive range of pharmacological properties, including the widely used analgesics morphine and codeine (1). Although a large body of knowledge has been acquired on numerous enzymes involved in the biosynthesis of BIAs, many basic biochemical questions remain unanswered. The molecular structural basis of substrate recognition and catalysis among BIA biosynthetic enzymes is still poorly understood for several key conversions. This information is not only of fundamental importance in plant metabolic biochemistry, but it also establishes a framework for manipulating natural BIA biosynthetic pathways as an approach toward obtaining rare or novel compounds with desirable pharmacological properties. In particular, several efforts are under way to transfer entire BIA biosynthetic pathways found exclusively in plants to heterologous systems, such as yeast (2, 3). The ability to control and modify the substrate specificities and catalytic activities of enzymes obtained from the biosynthetic machinery found in plants will be central to the success of these metabolic engineering objectives. An ultimate goal of this ambitious work is to generate novel compounds with modifications that may improve on the pharmacological properties of naturally accessible BIAs. In addition, naturally occurring compounds that are difficult to obtain from natural sources may be produced more economically through heterologous systems.
The methylation of secondary and tertiary amines occurs at several key points in BIA biosynthesis (4), with each conversion typically catalyzed by specific members of a unique N-methyltransferase (NMT) subclade. Three related NMTs with relatively distinct substrate specificities have been reported and include a coclaurine NMT (CNMT) involved in the formation of the branch-point intermediate (S)-reticuline (5); tetrahydroprotoberberine NMT (TNMT) (6), which participates in the formation of several BIAs, such as noscapine and sanguinarine; and pavine NMT (PavNMT) (7, 8) (Fig. 1). CNMT is mainly responsible for the N-methylation of the central 1-benzylisoquinonline intermediate (S)-coclaurine, whereas TNMT primarily accepts various protoberberine alkaloids, such as (S)-canadine and (S)-stylopine. PavNMT is known to accept substrates with a pavine scaffold. Although a number of NMT variants have been isolated and characterized, the molecular structural basis of catalysis and substrate specificity has not yet been determined for any NMT involved in BIA metabolism. Sequence analysis of NMTs from BIA biosynthesis indicates <20% sequence identity with the closest structurally characterized homologues. To provide insight into the structure and function of NMTs involved with BIA biosynthesis, we report a series of structural and functional studies of PavNMT from the common meadow rue (Thalictrum flavum).
FIGURE 1.

Reactions catalyzed by NMTs in BIA biosynthesis. CNMT is mainly responsible for the N-methylation of the 1-benzylisoquinonline (S)-coclaurine, generating the tertiary amine in the central branch point intermediate (S)-reticuline. In contrast, TNMT primarily accepts a variety of protoberberine alkaloids, such as (S)-canadine, yielding quaternary derivatives. PavNMT is known to prefer substrates with a pavine scaffold compared with those containing 1-benzylisoquinonline or protoberberine backbones.
Results
Substrate Specificity of PavNMT
Previously, reported substrate range for PavNMT was determined by detection of radioactively labeled reaction products separated by thin layer chromatography (21). Using reverse-phase liquid chromatography coupled to mass spectrometry detection and quantification, the enzymatic activity of PavNMT was reevaluated with a more sensitive and quantitative assay. Enzyme assays performed using potential substrates representing several major BIA subgroups showed that PavNMT exhibited a preference for (±)-pavine and the 1-benzylisoquinoline (S)-reticuline but also accepted the protoberberines scoulerine and stylopine and, to a lesser extent, tetrahydropapaverine (THP) (Table 1). Interestingly, (R)-reticuline was not accepted as a substrate, revealing a strong stereoselectivity for (S)-reticuline. Unfortunately, most of the other substrates evaluated were only available in racemic form, and the stereospecificity of PavNMT for other alkaloid substrates cannot be determined at this time. As our crystallographic results indicate below, however, the presence of two enantiomers in the racemic mixtures of most substrates could affect the apparent kinetics in a somewhat complex manner. A racemic mixture of THP was converted by PavNMT to laudanosine with a significant amount of apparent substrate inhibition seen at higher concentrations of substrate: Km = 16 μm, Kis = 120 μm, and kcat = 0.00079 s−1 (Fig. 2B). As described below, our crystallographic results suggest that the apparent substrate inhibition (i.e. a decrease in activity at higher concentrations of substrate) probably arises from the binding of the (R)-isomer in a non-productive mode that may inhibit the enzyme at higher total concentrations of the racemic substrate. Consistent with this structural information, substrate inhibition was not detected when enantiomerically pure (S)-reticuline was used as the substrate (Fig. 2C).
TABLE 1.
Substrate specificity profile of PavNMT
| Alkaloid class | Alkaloid | Substrate consumeda |
|---|---|---|
| % | ||
| Pavine | (±)-Pavine | 7.9 ± 1.9 |
| 1-Benzylisoquinoline | (R,S)-Tetrahydropapaverine | 0.10 ± 0.01 |
| Papaverine | NDb | |
| (S)-Reticuline | 9.7 ± 2.6c | |
| (R)-Reticuline | ND | |
| Protoberberine | (R,S)-Scoulerine | 0.8 ± 0.1 |
| (R,S)-Stylopine | 2.7 ± 0.3 | |
| Protopine | Cryptopine | ND |
| Aporphine | Glaucine | ND |
| Morphinan | Codeine | ND |
| Secoberberine | Canadaline | ND |
| Pthalideisoquinoline | Noscapine | ND |
| Bisbenzylisoquinoline | Berbamine | ND |
a Under standard assay conditions: 20 μm alkaloid substrate, 20 μm SAM, 100 mm Tris-HCl, pH 7.5, in a 40-μl reaction containing 2 μg of PavNMT protein and incubated for 1 h.
b ND, not detected.
c Specific activity: 10.6 picokatals mg protein−1.
FIGURE 2.

Kinetic analysis of PavNMT using racemic THP as the enzyme substrate. A, separation of R and S enantiomers of THP using chiral chromatography. B, plot of initial reaction velocity of PavNMT versus the concentration of a racemic mixture of THP as the enzyme substrate. Values represent the mean ± S.D. (error bars) of three independent assays, and the data were fit to a rate equation derived using rapid equilibrium assumptions for the formation of productive E·S and non-productive E·S·S complexes, as expected for substrate inhibition. C, plot of initial reaction velocity of PavNMT versus the concentration of (S)-reticuline as the enzyme substrate.
Overall Structure of PavNMT
Selenomethionine-substituted PavNMT was expressed in Escherichia coli and crystallized at pH 6.0 in a monoclinic crystal form containing two protomers in the asymmetric unit (Vm = 2.01 Å3/Da, 39% solvent). Anomalous differences and dispersive differences from data measured at three wavelengths were used to locate 29 well ordered selenium atoms from the 36 selenomethionine residues predicted based on amino acid sequence. The experimental electron density map extended to 2.8 Å resolution and allowed the autobuild module in PHENIX to construct an initial model for 588 of 794 residues (74%). Higher resolution data from native crystals extending to 2.0 Å were used to build and refine a model for 341 residues in each of the two polypeptide chains found in the asymmetric unit (Table 2). The native C terminus of each protomer is visible in each of the two copies in the asymmetric unit, but roughly 50 residues (51 in chain A and 48 in chain B) at the N terminus of each protomer are not well ordered. Of these disordered residues, 41 are derived from a non-natural sequence containing an N-terminal hexahistidine tag and the polylinker of the expression vector. As a result, only 10 and 7 residues from the native N termini of the two polypeptide chains are not sufficiently ordered to model. In the apoenzyme structure, two loops (residues 70–79 and 91–103 in chain A and residues 71–79 and 91–107 in chain B) are not sufficiently well ordered to model the protein completely. In the SAM and SAH complexes, however, only residues 72–78 in chain A and residues 73–77 in chain B are missing, in addition to the disordered residues at the N terminus. In the SAH·(S)-THP·(R)-THP complex, the 70s loop is completely ordered in both protomers, and only the N-terminal residues are disordered. Because all of these structures are determined in essentially the same crystal form with two copies in the asymmetric unit, the presence of order and disorder in specific loops does not appear to be related to crystal packing effects. Instead, it appears that the binding of each substrate induces a stepwise change in conformation of the protein that is consistent with an ordered sequence for substrate binding.
TABLE 2.
Crystallographic statistics
| Crystal | SeMet | Apoenzyme | SAM complex | SAH complex | SAH·THP2 complex | H206A SAM complex | ||
|---|---|---|---|---|---|---|---|---|
| PDB code | 5KN4 | 5KOC | 5KPG | 5KOK | 5KPC | |||
| Data collection | ||||||||
| Unit cell dimensions | ||||||||
| a, b, c (Å) | 53.2, 72.3, 96.7 | 53.7, 72.3, 97.1 | 54.6, 72.0, 96.2 | 53.9, 72.0, 95.1 | 53.8, 71.6, 95.2 | 54.2, 71.7, 96.3 | ||
| β (degrees) | 99.6 | 99.8 | 99.0 | 99.25 | 99.2 | 99.0 | ||
| Wavelength (Å) | 0.97915 | 0.97932 | 0.95369 | 0.984 | 0.980 | 0.980 | 0.980 | 0.980 |
| Resolution (Å)a | 40-2.8 (2.87-2.80) | 40-2.9 (2.97-2.90) | 40-2.7 (2.77-2.70) | 40-2.0 (2.05-2.00) | 40-2.3 (2.36-2.30) | 40-1.6 (1.64-1.60) | 40-1.8 (1.84-1.80) | 40-2.5 (2.57-2.50) |
| Rsymb | 0.118 (0.611) | 0.130 (0.743) | 0.104 (0.815) | 0.074 (0.794) | 0.087 (0.819) | 0.040 (0.626) | 0.037 (0.169) | 0.129 (0.820) |
| I/σI | 13.6 (3.2) | 13.8 (2.8) | 16.2 (2.5) | 11.9 (1.5) | 12.2 (1.8) | 23.7 (2.4) | 24.7 (7.57) | 8.8 (1.9) |
| Completeness (%) | 99.5 (96.5) | 99.6 (97.0) | 99.7 (99.3) | 94.1 (78.5) | 97.0 (84.6) | 99.6 (99.1) | 97.4 (92.0) | 99.6 (99.9) |
| Redundancy | 7.5 (7.5) | 7.6 (7.5) | 7.6 (7.5) | 5.3 (4.0) | 3.8 (3.6) | 4.2 (4.2) | 4.5 (4.4) | 3.8 (3.8) |
| Refinement | ||||||||
| Resolution (Å) | 40-2.0 | 40-2.3 | 40-1.6 | 40-1.8 | 40-2.5 | |||
| Unique reflections | 47,298 | 32,206 | 94,288 | 65,359 | 25,334 | |||
| Rworkc/Rfreed | 0.200/0.254 | 0.186/0.247 | 0.197/0.242 | 0.169/0.198 | 0.231/0.315 | |||
| Total no. of atoms | 5439 | 5701 | 6297 | 6261 | 5602 | |||
| Protein atoms | 5302 | 5591 | 5561 | 5734 | 5534 | |||
| SAM/SAH atoms | 54 | 52 | 102 | 54 | ||||
| Water atoms | 137 | 83 | 682 | 425 | 36 | |||
| Average B-factors (protein) | 44.1 | 47.2 | 25.8 | 27.5 | 43.9 | |||
| Average B-factors (water) | 40.7 | 44.0 | 31.9 | 22.8 | 36.8 | |||
| Average B-factors (SAM/SAH) | 55.7 | 22.1 | 20.9 | |||||
| Average B-factors (THP) | 26.7 | |||||||
| Root mean square deviation from ideal geometry | ||||||||
| Bond lengths (Å) | 0.010 | 0.011 | 0.007 | 0.008 | 0.013 | |||
| Bond angles (degrees) | 1.07 | 1.30 | 1.05 | 1.11 | 1.47 | |||
a Values from the outermost resolution shell are given in parentheses.
b Rsym = Σi|Ii − 〈I〉|/Σi Ii, where Ii is the ith integrated intensity of a given reflection and 〈I〉 is the weighted mean of all measurements of I.
c Rwork = Σ‖Fo| − |Fc‖/Σ|Fo for 95% of reflection data used in refinement.
d Rfree = Σ‖Fo| − |Fc‖/Σ|Fo| for 5% of reflection data excluded from refinement.
The overall structure of PavNMT reveals the presence of an α/β SAM-binding domain with a canonical Rossmann fold that is shared among most SAM-dependent methyltransferases (5–7) (Fig. 3). In addition to this domain, a primarily α-helical BIA-binding domain forms the majority of the putative binding site for the methyl group acceptor. The sequence, structure, and topology of the lid domain appear most similar to the Plasmodium phosphoethanolamine NMTs (PfPMT) (8, 9) and the mycolic acid cyclopropane synthases (10) from Mycobacterium tuberculosis (supplemental Movies 1 and 2). Although the sequence identity is <20% and the root mean square deviation for C-α atoms is over 3–4 Å after filtering away the most different parts of the structures, the fairly high DALI Z-scores (∼28–29) for these structures indicate substantial similarity in the arrangement of secondary structure elements (11). Three N-terminal helices that encircle and cap off the BIA-binding domain appear to be unique to PavNMT and related NMTs from plants. The functional significance of this unique structural feature is not clear at present, although some of the residues at the N terminus of helix α3 form part of the dimer interface described below.
FIGURE 3.
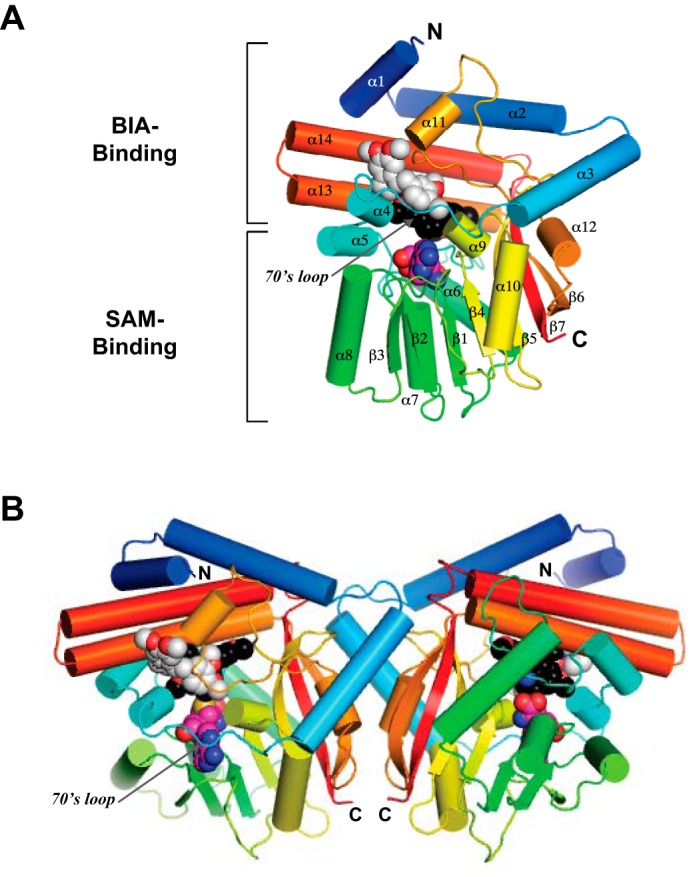
A, overall structure of the PavNMT·SAH·(S)-THP·(R)-THP complex in which the protein is drawn in a ribbon representation, and SAH (carbon atoms colored magenta), (S)-THP (carbon atoms colored black), and (R)-THP (carbon atoms colored light gray) are drawn in a space-filling representation. The polypeptide chain is colored as a gradient from the N terminus (blue) to the C terminus (red). B, dimer structure of the PavNMT·SAH·(S)-THP·(R)-THP complex.
PavNMT forms a dimer with 2-fold rotational symmetry in the asymmetric unit that is not seen in homologous NMTs (Fig. 3B). Gel filtration analysis confirms that PavNMT forms a dimer in solution. Nineteen residues from each protomer form an extensive network of interactions, resulting in the formation of 12 hydrogen bonds and two ionic interactions between subunits as well as the burial of ∼1020 Å2 of accessible surface area, which is 6.5% of the total accessible surface area per subunit (Fig. 4). Although PISA (12) predicts that this putative dimer interface is only marginally stable and may not be significant in solution, most of the interfacial residues involved with hydrogen bonding, ionic interactions, and hydrophobic interactions are conserved among eight characterized NMTs with roles in BIA biosynthesis. This observation supports the functional significance of the dimerization interface (Fig. 3). The majority of the dimer interface involves residues on the “back” side of the SAM-binding domain away from the loops that cover the “front” side and thus provide access to the active site. As a result, the loops covering the active site are located at the distal ends of the dimer. It is unclear whether the dimer structure may mediate cooperative effects with respect to substrate binding or catalysis. However, the nature of the dimer interface suggests that hinge movements between the BIA-binding domain and the SAM-binding domain of one protomer would affect the hinge movements in the second protomer. Comparisons between the structures of apo-PavNMT, the PavNMT·SAM complex, and the PavNMT·SAM·THP complex do suggest the presence of a small hinge-opening movement between the SAM-binding and BIA-binding domains that could lead to cooperative effects on substrate binding or catalysis.
FIGURE 4.

Sequence alignment. Residues highlighted in yellow indicate positions where the side chain forms either direct or water-mediated hydrogen bonds with SAM in PavNMT. Residues highlighted in red indicate positions where the side chain is either observed to form an interaction with the (S)-THP substrate in PavNMT or predicted to form an interaction with a BIA substrate in PavNMT, CNMT, or TNMT, whereas residues highlighted in blue indicate positions where the side chain is predicted to form an interaction with a BIA substrate in TNMT. Residues forming part of the dimer interface are shaded in light gray. The secondary structure assigned for PavNMT by DSSP (28) was slightly changed after manual inspection of the structure and is provided above the sequence of PavNMT, wherein H represents α-helical conformation and E represents β-strand conformation. Abbreviations and accession numbers are as follows: ECATNMT, Eschscholzia californica TNMT, EU882977 (15); PBRTNMT2, Papaver bracteatum TNMT2, EU882993 (15); PBRTNMT1, P. bracteatum TNMT1, EU882994 (15); PSOTNMT, Papaver somniferum TNMT, DQ028579 (29); PSONMT1, P. somniferum NMT1, uncharacterized N-methyltransferase, AFK73712 (30); TFLPAVNMT, Thalictrum flavum PAVNMT, EU883010 (15); PSOCNMT, P. somniferum CNMT, AY217336 (31); CJACNMT, Coptis japonica CNMT, AB061863 (32); TFLCNMT, T. flavum CNMT, EU883009 (15).
Structural Determinants of Substrate Binding
NMTs employ a surprisingly large variety of mechanisms to bind to SAM, although the overall architecture of the SAM-binding domain (i.e. the Rossmann fold) is highly conserved (6, 7). Structures of PavNMT in complex with SAM and SAH reveal a mode of cofactor binding that is most similar to Class Ia SAM-dependent NMTs (7). The side chains of several conserved polar residues form hydrogen bonds or water-mediated hydrogen bonds with the amino group (Ser140), carboxylate group (Lys95 and Ser97), vicinal hydroxyl groups of the ribose moiety (Asn159 and Gln163), and the adenine base (Asp185), which all help position the SAM methyl donor for reaction with the BIA acceptor (Fig. 5). In addition, the highly conserved GXGXG sequence motif I runs directly underneath the ribose moiety, forming the central platform for the SAM-binding site. Sequence alignments of NMTs involved with BIA biosynthesis indicate a high level of conservation in most of the residues forming direct or water-mediated hydrogen bonds, indicating a common mechanism of substrate recognition (Figs. 4 and 5). It is notable that the specific interactions between SAM and PavNMT differ from the specific SAM-binding interactions seen in other NMTs, including the most closely related enzymes with known three-dimensional structures (i.e. phosphoethanolamine NMT from Plasmodium species and the mycolic acid cyclopropane synthases from M. tuberculosis). The diversity of binding modes for SAM that is seen among different NMTs has been extensively analyzed previously (5, 7).
FIGURE 5.
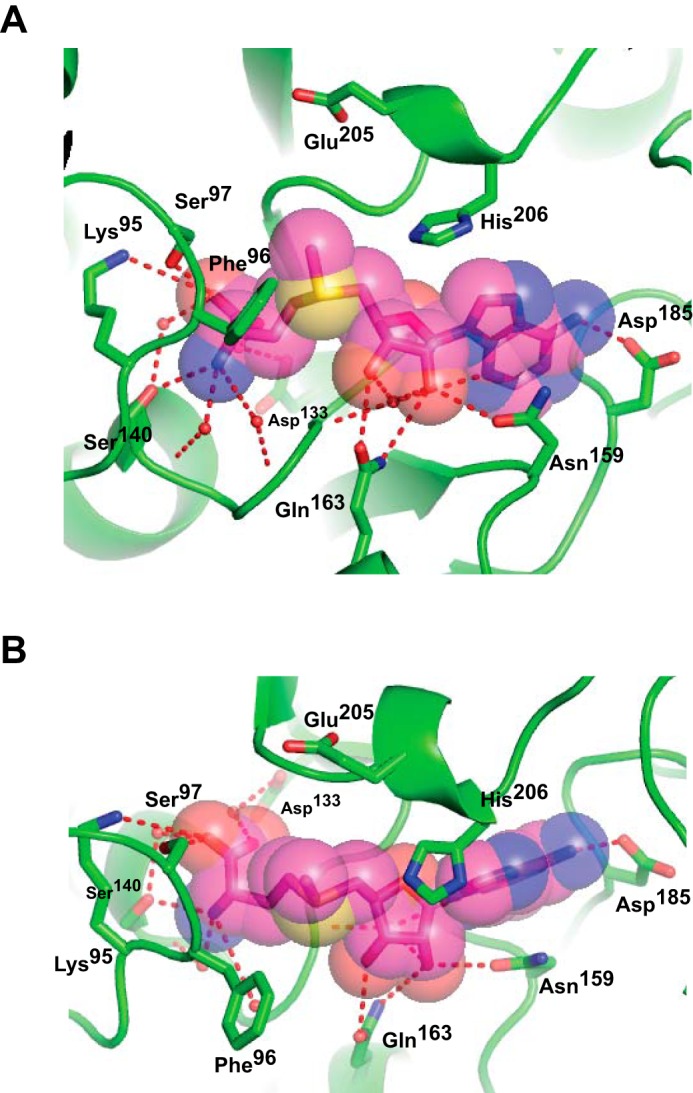
“Front” (A) and “top” (B) views of interactions between PavNMT and SAM. Hydrogen bonds (red dashes) between SAM (carbon atoms colored magenta), key water molecules (red spheres), and the protein (carbon atoms colored green) are drawn. Oxygen atoms are colored red, and nitrogen atoms are colored blue.
The structure of PavNMT was also determined in the presence of the product SAH and a racemic mixture of THP. One molecule of (S)-THP lies in a binding pocket lined primarily with aromatic residues and where the secondary amino group is positioned within ∼3 Å from the position expected for the methyl donor (Fig. 6 and supplemental Movie 3). The benzyl group of (S)-THP is most deeply buried in the substrate-binding pocket, whereas the isoquinoline moiety lies in a more exposed position where the plane of the six-carbon ring is stacked in a parallel arrangement against the benzyl group of a molecule of (R)-THP. The molecule of (R)-THP appears to be bound more loosely, with fewer direct contacts with the protein than seen for the molecule of (S)-THP. The loop formed by residues 75–91 covers both THP molecules, and the structure of this loop appears to be dependent on the binding of both THP molecules, because it is disordered when THP is not bound (e.g. in the structures of the apoenzyme as well as the complexes containing SAH or SAM). The steady-state kinetic parameters for THP appear similar to those for pavine (22), except for a significant amount of apparent substrate inhibition at higher concentrations of THP. The binding of (R)-THP in a non-productive binding mode next to (S)-THP, which is bound in a productive mode, suggests that the (R)-THP may inhibit the release of products from the enzyme when higher concentrations of the racemic substrate are added to the reaction. The biological significance of the binding of (R)-THP is unclear, but the similarity in structure of (R)-THP to the physiologically important intermediate (R)-reticuline suggests that the mode of inhibition seen here may also occur under cellular conditions. The structure of the (S)-THP·(R)-THP complex also suggests that PavNMT or perhaps other NMTs may also bind to larger, more complex alkaloid intermediates like the bisbenzylisoquinoline alkaloids. Several hundred structures are known for these compounds, including many that have been isolated from Thalictrum species (13). The role of NMTs in the biosynthesis of bisbenzylisoquinoline alkaloids is poorly understood, but the structure of the (S)-THP·(R)-THP complex raises the intriguing possibility that some of these compounds may be effective substrates for NMTs.
FIGURE 6.
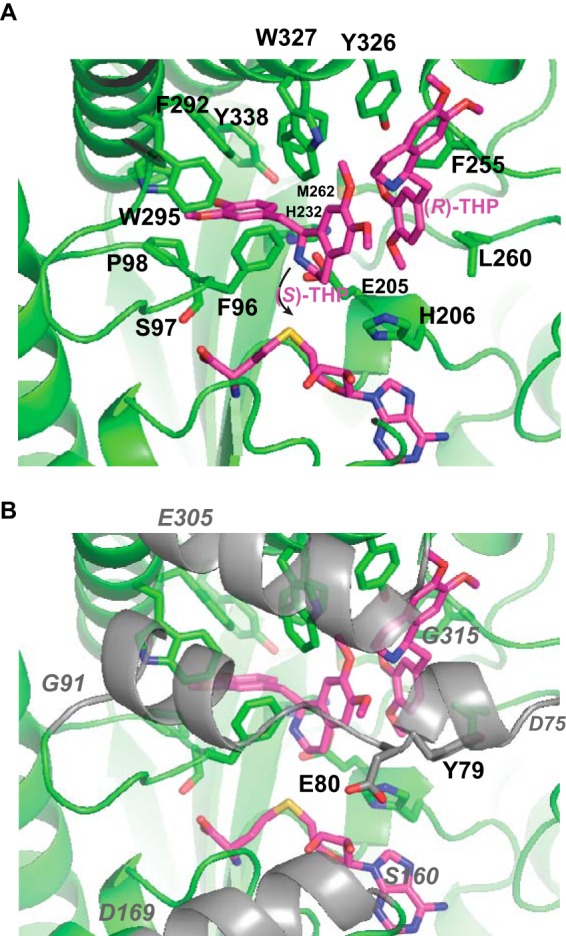
Detailed view of interactions between PavNMT and THP. Residues with side chain atoms within 4 Å of an atom of THP are labeled. In A, the segments from three surface loops (residues 75–91, 160–169, and 305–315) are removed for clarity. In B, these segments are drawn in a semitransparent representation and colored gray; the starting and ending residues of each segment are labeled in italic type. The curved arrow indicates the approximate path required for nucleophilic attack by the secondary amino group of THP on the methyl donor group of SAM. The methyl group is not shown here, because this is the experimentally determined structure of THP in complex with the product SAH. Atom coloring follows the scheme described in the legend to Fig. 4.
Although crystals of PavNMT were grown in the presence of pavine under a wide range of conditions, including conditions identical to those used for crystallizing the PavNMT·SAH·THP2 complex, pavine was not bound at the active site of PavNMT in any crystals analyzed to date. Co-crystallization of PavNMT with (S)-reticuline, which has a structure very similar to that of (S)-THP (Fig. 1), has also failed to yield the structure of an enzyme·substrate complex, although (S)-reticuline has kinetic parameters similar to those of THP and thus appears to be an equally effective substrate.
Patterns of sequence conservation at the 13 positions that line the binding pocket for (S)-THP suggest how sequence variation may determine substrate specificity in PavNMT and the other NMTs involved with BIA biosynthesis (Fig. 6). Nine positions (His232, Phe255, Leu260, Pro288, Trp295, Tyr326, Trp327, Phe330, and Tyr338) almost always contain hydrophobic or aromatic residues, but only two of these positions (Phe255 and Trp295) show a high level of sequence conservation across all NMTs. At four other positions (Phe96, Pro98, Gly291, and Phe292), there are very high levels of sequence variation. When considered in the context of substrate specificity and biological function, it is likely that these patterns of sequence variation relate to functionally important differences in substrate specificity among the broad range of BIA substrates for NMT enzymes found in these pathways. It is interesting to note that there are no direct hydrogen bonds between (S)-THP and PavNMT. Binding specificity appears to arise primarily through steric effects, van der Waals interactions, and π-stacking effects.
Putative Active Site Residues Involved in Catalysis
Two highly conserved residues at the active site (Glu205 and His206) are positioned near the activated methyl group of SAM, but these residues do not appear to have specific roles in substrate binding (Figs. 4–6). These residues are conserved in all of the NMTs involved with BIA biosynthesis as well as most other less closely related NMTs. Replacement of each residue with Ala results in a small decrease in protein stability as observed through a slight reduction in melting temperature determined using differential scanning fluorimetry (DSF) and the dye 8-anilinonaphthalene-1-sulfonate (Table 3). Replacement of both residues with alanine leads to a larger decrease in Tm, but the overall structure appears intact when monitored by intrinsic tryptophan fluorescence. Moreover, the crystal structure of the H206A mutant confirms that the mutation does not affect the structure of the protein, apart from slight changes in the immediate region around the side chain at the active site. As expected, the addition of THP and SAH either one at a time or together stabilize the wild-type protein and each mutant in a roughly additive manner, confirming that mutations at these positions affect catalysis more than substrate binding.
TABLE 3.
Melting transition temperatures as determined by differential scanning fluorimetry using 8-anilinonaphthalene-1-sulfonate
| Enzyme/mutant |
Tma (difference of Tm from the apo-state) |
|||
|---|---|---|---|---|
| Apo | +THP (1 mm) | +SAH (1 mm) | +1 mm SAH + 1 mm THP | |
| °C | ||||
| PavNMT | 69.8 | 71.4 (+1.6) | 72.7 (+2.9) | 75.8 (+6.0) |
| E205A | 67.5 | 69.0 (+1.5) | 70.5 (+3.0) | 73.0 (+5.5) |
| H206A | 68.7 | 70.1 (+1.4) | 70.3 (+1.6) | 72.4 (+3.7) |
| E205A/H206A | 66.6 | 68.3 (+1.7) | 68.8 (+2.2) | 70.4 (+3.8) |
| Y79A | 70.0 | 71.4 (+1.1) | 72.7 (+2.7) | 74.8 (+4.8) |
| E80A | 69.9 | 71.1 (+1.2) | 72.5 (+2.6) | 74.2 (+4.1) |
a Values are determined from the average of duplicate measurements, which typically differ by <0.2 °C.
Replacement of Glu205 or His206 with Ala led to large decreases in activity for three different substrates, especially the best substrate identified so far, (S)-reticuline (Fig. 7). The simultaneous replacement of both residues with Ala further decreased activity to <10% of wild-type activity at pH 7.5. These mutational studies thus confirm the involvement of these residues in catalysis, although further studies are clearly required to deduce specific roles in the enzyme mechanism.
FIGURE 7.
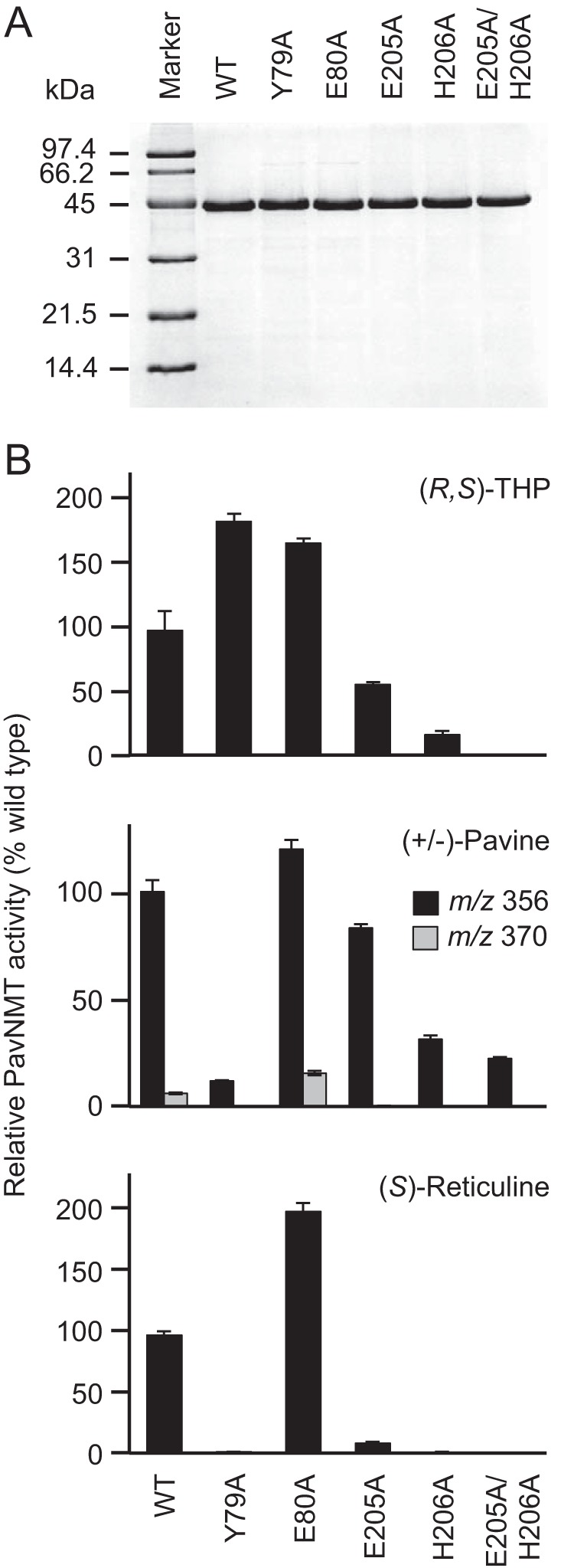
Enzymatic activities of alanine mutants for putative catalytic residues. A, SDS-PAGE analysis reveals high levels of purity for wild-type and mutant versions of PavNMT. B, black bars indicate the amounts of the monomethylated product observed from each mutant relative to the wild-type enzyme for each of the three best substrates available for PavNMT: racemic THP, racemic pavine, and (S)-reticuline. Measurements were conducted in triplicate (error bars show S.D.) in the presence of 0.2 mm SAM and either 0.1 mm (S)-reticuline, 0.2 mm racemic THP, or 0.2 mm racemic pavine at pH 7.5. Gray bars indicate the relative amounts of the dimethylated product observed with pavine.
Induced Fit Effects of Substrate Binding on Conformations of Loops
An important aspect of substrate recognition and catalysis may also depend on the structure and dynamics of two loops that are poorly defined in the electron density maps of the PavNMT apoenzyme when compared with the binary complexes formed with SAM or SAH as well as the SAH·(S)-THP·(R)-THP complex. A long loop between helices α4 and α5 (residues 91–107) shows a dramatic increase in ordered structure between the apoenzyme and the complexes containing either SAM or SAH. This loop is poorly defined in both copies of the asymmetric unit of the apoenzyme, but both copies are well defined in the complexes containing SAM or SAH. A major part of this loop forms the binding pocket for the amino and carboxylate groups of SAM and SAH. Changes in the ordering of the 90s loop, resulting from binding of the methyl donor, suggest that flexibility in this loop may help with the entry of SAM and the exit of SAH during the enzymatic cycle.
The loop and helix (α4) following helix α3, especially residues 70–79, are also poorly defined in the electron density maps of the apoenzyme as well as in the complexes with either SAM or SAH bound. However, the loop and helix α4 are well ordered in both copies of the asymmetric unit when THP is bound. This loop bridges the N-terminal region that is unique to the NMTs involved with BIA biosynthesis with the first helix of the SAM-binding domain. The change in structure accompanying the binding of THP suggests that the flexibility in this loop assists with the entry and exit of the BIA substrate.
In combination with patterns of sequence conservation, the structure and positioning of the 70s loop and α4 also suggest possible roles for residues in catalysis or substrate binding (Figs. 3 and 4). For example, Tyr79 and Glu80 are highly conserved residues that are located within 6–10 Å of the amino group of THP and the methyl donor group seen in the SAM complex. Although the side chain hydroxyl and carboxylate groups in these residues are too far away to interact directly with the substrates in the conformation seen in existing crystal structures, these functional groups are located in a region of the active site that may form a gate near the outer part of the BIA-binding pocket. Replacement of Tyr79 with Ala shows a dramatic loss of catalytic activity with (S)-reticuline and racemic pavine as substrates but surprisingly resulted in a slight increase in catalytic activity with racemic THP as the substrate. Moreover, the replacement of Glu80 with Ala shows an even more substantial increase of activity with all three tested substrates (Fig. 7). DSF measurements confirm that protein stability is not significantly affected by either the Y79A or the E80A mutations. Moreover, the significant increases in stability observed in the presence of SAH, THP, and SAH + THP are very similar to the increases in stability observed for the wild-type protein and the E205A and H206A mutants (Table 3). Altogether, these findings confirm roles for Tyr79 and Glu80 primarily in catalysis and possibly also substrate recognition. Clearly, additional work is required to define more specific functions in catalysis, which may differ depending on the specific substrate. It is notable that these two residues are found in a loop that is similar to that seen in PfPMT, where Asp128 was recently proposed to assist with general base catalysis (14). The moderate levels of sequence conservation seen for several other residues in the 70s loop and α4 of PavNMT suggest that other positions may also be of functional importance for the NMTs involved with BIA biosynthesis. Leu74, for example, shows perfect conservation among the CNMT enzymes but is replaced by Met and Phe in the TNMTs. This pattern of sequence conservation for an exposed hydrophobic residue near the active site suggests a possible role in determining substrate specificity.
Discussion
The structure of PavNMT establishes an essential structural framework for interpreting how the sequences of NMTs relate to their catalytic functions in BIA biosynthesis. Comparing the structure with sequence alignments reveals that nearly all of the key hydrophobic residues and glycine residues important for stabilizing the overall fold of PavNMT are highly conserved among other NMTs from plants, especially eight NMTs that have been shown to act on BIA substrates. In addition, the residues involved with forming the non-crystallographic dimer in PavNMT and the residues interacting with SAM are highly conserved among plant NMTs with known sequences. Altogether, these observations strongly suggest that the structure of PavNMT will provide an extremely useful template for constructing accurate homology models to better predict the molecular determinants of substrate specificity and catalysis in a wide range of plant NMTs. This is especially true for the most closely related NMTs involved with BIA biosynthesis, in which the overall level of sequence identity is ∼45–65%.
Homology models for TNMT and CNMT are particularly informative when examined in light of the multiple sequence alignment of the BIA-binding domain (Fig. 4). As discussed above, 13 positions appear poised to interact with BIA substrates based on the structure of PavNMT, and the patterns of sequence conservation in these positions appear to follow the substrate specificities of the TNMT and CNMT enzymes that have been characterized so far. For example, the TNMT enzymes that have been shown previously to accept stylopine and related compounds with a protoberberine backbone as substrates contain Ala (or Val), Val, His, and Ile at positions 260, 292, 326, and 327, respectively. In contrast, the three CNMT enzymes that prefer coclaurine and other compounds with a 1-benzylisoquinoline skeleton as substrates contain Leu (or Met), Asn (or His), Tyr (or Phe), and Trp at the same positions, respectively (Fig. 4). The strong preference for amino acids with larger side chains in the CNMT enzymes may reflect their preference for less rigid 1-benzylisoquinoline substrates as opposed to the TNMT enzymes, which prefer protoberberine compounds with a fixed benzyl group, relative to the isoquinoline moiety, due to the occurrence of a methylene bridge (15). Moreover, CNMT substrates contain a secondary amine, whereas TNMT substrates inherently possess a tertiary amine.
In addition to the 13 residues predicted to form the binding pocket for BIA compounds, there are probably other features in the structure of the BIA-binding domain that determine BIA binding specificity. For example, the side chain of Glu252 in PavNMT points away from the BIA-binding pocket, but this residue and adjacent residues contact Phe255 and Pro-256, which are part of the BIA-binding pocket. The residue equivalent to Glu252 in the TNMTs is always Gly, whereas the residue is either Glu or Asn in the CNMTs. The strict conservation of Gly at this position among the TNMTs suggests that the lack of a side chain at this position may have functional significance, even if the residue most likely does not directly contact the BIA substrate. Perhaps Gly allows a slight expansion of the BIA-binding pocket relative to the enzymes with Glu or Asn at this position. The significance of such hypothetical indirect effects on the structure of the BIA-binding pocket will become more clear when the structures of additional members of the plant NMT family are determined in complex with substrates.
Understanding the specific roles of the highly conserved Tyr79, Glu80, Glu205, and His206 residues near the donor methyl group also awaits further analysis. Recent studies of PfPMT from P. falciparum proposed that Tyr19, His132, Asp128, and a bound water molecule combine to provide general base catalysis and electrostatic positioning effects to increase the reactivity of the amino group of the methyl acceptor phosphoethanolamine (8, 14). Asp128 appears to be particularly important for the primary amine substrate but does not appear to play the same role for secondary or tertiary amine substrates. For the primary amine substrate in PfPMT, quantum mechanics and molecular modeling calculations suggest that Asp128 probably accepts a proton from a bridging water molecule that in turn activates the amino group through base catalysis (14).
Mutagenesis studies have also implicated His132 and Tyr19 in PfPMT for a possible role in catalysis, but quantum mechanics/molecular modeling calculations do not support a role in base catalysis as initially proposed (8, 14). Recent work examining changes in the kinetic and binding isotope effects of mutations to the Tyr21 residue of glycine N-methyltransferase (GNMT) suggests that the bulky aromatic side chain of the tyrosine residue may play a role in activating the donor methyl group in SAM (16). His206 in PavNMT is located in nearly the exact same location as His132 in PfPMT and His142 in GNMT, but the structure of PavNMT around the region of Tyr19 in PfPMT and Tyr21 in GNMT is not conserved. It is interesting to note, however, that Tyr79 in PavNMT is located at the end of the 70s loop (between helices α3 and α4), which becomes ordered upon binding of THP. Although the aromatic side chain of Tyr79 in the crystal structure of the PavNMT SAH·(S)-THP·(R)-THP complex adopts a rotamer that moves it farther from the donor methyl group than seen in both GNMT and PfPMT, the 70s loop appears to be quite flexible (e.g. no electron density is seen in the apoenzyme or the SAM and SAH complexes) Dissociation of the non-productive and probably inhibitory molecule of (R)-THP would allow for a modest conformational change in the 70s loop and change in the rotamer adopted by Tyr79 that would allow the aromatic side chain to adopt a position similar to that seen for the equivalent Tyr residues in PfPMT and GNMT. Some of the results from the mutagenesis of Tyr79 (Fig. 7) confirm a role in catalysis with at least some substrates, but a more definite role awaits further mechanistic and structural studies.
The range of structures seen in NMT active sites may reflect differences in catalytic strategies. The architectures of the active sites in more distantly related NMTs appear even more divergent than between PfPMT and PavNMT. A variety of catalytic mechanisms have been proposed for rat liver glycine NMT (17), rat liver guanidinoacetate NMT (18), and human phenylethanolamine NMT (19, 20). In these other NMTs, a variety of functional groups have been proposed for general base catalysis and for the stabilization of the transition state. The work presented in this study provides a foundation for further exploring how the enzymatic mechanisms and substrate specificities of NMTs relate to their functions in BIA biosynthesis. In turn, a deeper understanding of the molecular functions of NMTs in BIA biosynthesis will provide many new opportunities to facilitate synthetic biology efforts to reconstitute BIA biosynthetic pathways in heterologous systems.
Experimental Procedures
Expression and Purification of PavNMT
PavNMT was produced in E. coli and purified as described previously (21). Briefly, the PavNMT open reading frame encoding residues 1–386 was amplified and cloned into pRSETC (Invitrogen), digested with PvuII and KpnI. The resulting plasmid allowed for the expression of full-length PavNMT fused to an N-terminal MRGS sequence, followed by a hexahistidine tag and a 31-residue linker sequence (GMASMTGGQQMGRDLYDDDDKDRWIRPRDLQ). PavNMT was expressed and purified as described previously (21) but with some modifications. Clarified lysate (40 ml) from 0.5 liters of bacterial culture was loaded onto a HisTrap column (1 ml; GE Healthcare) and eluted with a linear 0–0.5 m imidazole gradient. The protein was dialyzed overnight against buffer A (20 mm Tris-Cl, pH 8.0, 30 mm sodium chloride, 0.25 mm EDTA, 1 mm DTT) and further purified using anion exchange chromatography (HiTrap Q HP column, GE Healthcare). Protein was loaded at 1 ml/min onto a column (1 ml) previously equilibrated in buffer A. After washing with buffer A, the bound protein was eluted with a linear gradient (20 mm to 1 m sodium chloride dissolved in buffer A, 30-ml total volume). Peak fractions were pooled and dialyzed against 20 mm Tris-Cl, pH 7.5, 30 mm potassium chloride, 10 g/liter glycerol, 1 mm DTT before ultrafiltration with 10,000 kDa molecular weight cut-off centrifugal filters (Millipore) to increase protein concentration to 5 g/liter, as measured by absorbance at 280 nm using the extinction coefficient (1.495 liters/g/cm or 69,330 m−1 cm−1) calculated from the amino acid sequence using the ProtParam web server (ExPASy). Aliquots (0.1 ml) of PavNMT in 0.2-ml PCR tubes were flash-frozen in liquid nitrogen and stored at −80 °C.
Selenomethionine-substituted PavNMT was expressed from cells grown in M9 minimal medium that was supplemented with 50 mg/liter each of the l-enantiomers of selenomethionine, leucine, isoleucine, and valine and 100 mg/liter of the l-enantiomers of lysine, threonine, and phenylalanine after the culture reached an optical density of 1.0. After further growth at 25 °C for 30 min, 1 mm isopropyl-β-d-galactopyranoside was added, and the culture was allowed to grow for an additional 18 h with vigorous shaking at 25 °C before the cells were harvested by centrifugation. PavNMT containing selenomethionine was purified in the same manner as the native enzyme.
Site-directed Mutagenesis
Synthetic genes (Genscript) encoding three PavNMT mutants (E205A, H206A, and E205A/H206A) were individually ligated into the PstI and KpnI restriction sites of the pRSETC vector for expression in E. coli. For E205A, the original GAG codon for Glu205 was changed to GCG, which encodes Ala. For H206A, the original CAC codon for His206 was changed to GCC, which also encodes Ala. For E205A/H206A, the original GAG codon for Glu205 and the CAC codon for His206 were changed to GCG and GCC, respectively.
Synthetic genes (Genscript) encoding two additional PavNMT mutants (Y79A and E80A) were prepared using wild-type pRSET-TfPavNMT plasmid as a template. A region containing the desired codons for mutagenesis was removed by restriction digestion with NdeI and SalI. Site-directed mutagenesis was performed on the excised fragment to create the desired products. Mutagenized sequences were then ligated back into the same restriction sites, and the resulting plasmids were sequenced to verify that the correct mutation was introduced. For Y79A, the original TAC codon for Tyr79 was replaced with GCT, which encodes for Ala. For E80A, the original GAG codon for Glu80 was also replaced with GCT.
Purification of Pavine and Tetrahydropapaverine (THP)
An ∼1:1 mixture of pavine and THP was purchased from Sigma-Aldrich. Pavine and THP were separated using a 1260 Infinity HPLC (Agilent Technologies) equipped with a semipreparative Luna 5-μm C18 100-Å column (dimensions, 250 × 10 mm; Phenomenex). Chromatographic separation was performed at a flow rate of 5 ml/min using solvent A (0.08% acetic acid in 5% acetonitrile (ACN)) and solvent B (100% ACN) under the following gradient conditions: 0–67% solvent B from 0 to 10 min, 67–99% solvent B from 10 to 10.1 min, isocratic 99% solvent B from 10.1 to 11.1 min, 99–0% solvent B from 11.1 to 11.2 min, and 0% solvent B from 11.2 to 16.2 min. Elution of compounds from the column was monitored by UV detection at 284 nm. Pavine and THP were dissolved in methanol at 30 mm before adding to the protein for crystallizations or into enzyme assay reactions. Isolated THP was identified as a 1:1 racemic mixture of enantiomers using chiral chromatography (Fig. 2A), following a previously published protocol (22).
Crystallization
Native PavNMT was mixed with 0.75 mm THP before crystallization trials using commercially prepared crystallization screens (IndexHT and CrystalHT from Hampton Research). A Hydra-II-Plus-One pipetting robot was used to mix 0.4 μl of protein with 0.4 μl of well solution in a 96-well sitting drop plate (Corning Inc.) before the crystallizations were equilibrated through vapor diffusion.
For native and selenomethionine-substituted crystals of apo-PavNMT (PDB entry 5KN4), the initial crystallization condition identified from the IndexHT screen was optimized to form well diffracting crystals using a reservoir solution of 230 g/liter pentaerythritol ethoxylate (15:4 EO/OH) (Hampton Research), 50 mm BisTris-Cl, pH 6.0, 70 mm ammonium sulfate, 120 g/liter glycerol. Reservoir solution (1.5 μl) was mixed with PavNMT (1.5 μl) and suspended as a hanging drop that was equilibrated against 0.45 ml of reservoir solution at 22 °C. Crystals appeared within a week and grew to full size (∼0.3 × 0.15 × 0.03 mm) after 2–3 weeks. Although 1.5 mm pavine was added to PavNMT before crystallization, no signs of bound pavine were seen in the electron density map.
Crystals of the PavNMT complex formed with SAM (PDB entry 5KOC) were grown in a similar manner, but starting with protein containing 1 mm SAM and 0.75 mm THP. The optimized reservoir solution contained 340 g/liter pentaerythritol ethoxylate (15:4 EO/OH), 30 mm ammonium sulfate, 50 mm BisTris-Cl, pH 7.0, and 160 g/liter glycerol. Although THP was present in the crystallization, no signs of bound THP were seen in the electron density map.
Crystals of the PavNMT complex formed with SAH alone (PDB entry 5KPG) were grown using PavNMT containing 1 mm SAH and 2.0 mm reticuline. The optimized reservoir solution contained 250 g/liter polyethylene glycol 3350, 75 mm potassium chloride, 100 mm sodium HEPES, pH 7.0, and 40 g/liter glycerol. Although reticuline was present in the crystallization, no signs of bound pavine were seen in the electron density map.
Crystals of the PavNMT complex (PDB entry 5KOK) containing both SAH and THP were grown using PavNMT containing 2 mm SAH and 1 mm THP. The optimized reservoir solution contained 210 g/liter polyethylene glycol 3350, 75 mm potassium chloride, 100 mm sodium HEPES, pH 7.25, and 120 g/liter glycerol. Crystals appeared after 1 month and grew to full size (0.15 × 0.05 × 0.02 mm) after 3 weeks.
Crystals of the H206A mutant (PDB entry 5KPC) were grown using protein containing 1 mm SAM and 1.5 mm THP and a reservoir solution of 210 g/liter pentaerythritol ethoxylate (15:4 EO/OH), 50 mm BisTris-Cl, pH 6.0, 60 mm ammonium sulfate, 140 g/liter glycerol. As for crystals of the wild-type protein, the electron density maps indicated the presence of SAM, but there was no indication of bound THP.
Crystal Structure Determination
A single crystal (0.2 × 0.1 × 0.05 mm) was quickly mounted in a polymer film Microgripper (MiTeGen) and flash-cooled in a nitrogen gas stream at ∼100 K. The crystal was stored in liquid nitrogen and transferred to the Canadian Light Source beamline 08B1-1 or Stanford Synchrotron Radiation Laboratory beamlines 12-2 or 14-1 for data collection. Data were processed and scaled using XDS (23). Solution and refinement of the selenium atom substructure as well as all phasing and electron density map calculations were performed using PHENIX (24). COOT (25) was used for model building, and PHENIX was used for refinement. Model quality was assessed using Molprobity (26).
Enzyme Assay
Individual reactions (40 μl) contained 100 mm Tris-Cl, pH 7.5, 0.5 mm SAM, 30 g/liter methanol, 0–0.5 mm alkaloid substrate, and 0.16 μm PavNMT. All reaction components except for the enzyme were mixed thoroughly before PavNMT (2 μl) was added and rapidly mixed. Reactions were incubated in a 30 °C water bath for 30 min before the addition of 100 μl of acetonitrile to stop the reaction. Assays were performed in triplicate, and 5 μl of each reaction was subjected to LC-MS/MS analysis.
LC-MS/MS Analysis
Enzyme assay samples were analyzed using a 1200 Infinity liquid chromatography system coupled with a 6410 triple-quadrupole mass spectrometer (Agilent Technologies). Chromatographic separation was achieved using a Poroshell 120 SB-C18 HPLC column at a solvent flow rate of 0.6 ml/min using solvent A (10 mm ammonium acetate, pH 5.5, 5% ACN) and solvent B (100% ACN) under the following gradient conditions: 0–80% solvent B from 0 to 9 min, 80–99% solvent B from 9 to 10 min, isocratic 99% solvent B from 10 to 11 min, 99–0% solvent B from 11 to 11.1 min, and 0% solvent B from 11.1 to 14.1 min. Chromatographic separation for THP was achieved using a Zorbax SB-C18 HPLC column at a solvent flow rate of 0.6 ml/min using solvent A (0.1% aqueous formic acid) and solvent B (100% methanol) under the following gradient conditions: 0–50% solvent B from 0 to 10 min, 50–100% solvent B from 10 to 12 min, isocratic 100% solvent B from 12 to 13 min, 100–0% solvent B from 13 to 13.1 min, and 0% solvent B from 13.1 to 17 min. Electrospray ionization, full scan mass analyses (m/z 300–400), and collisional MS/MS experiments were performed as described previously (27). Reaction product identification was based on the previously reported collision-induced dissociation spectrum for laudanosine (11). In the absence of authentic laudanosine, reaction product quantification was determined as equivalents of (S)-reticuline as a surrogate standard.
DSF
Reactions containing 8.6 μm PavNMT, 100 μm 8-anilinonaphthalene-1-sulfonate, 16 mm K2HPO4, 4 mm NaH2PO4, pH 7.5, 100 mm KCl, and 50 g/liter glycerol were incubated at room temperature for 30 min. Duplicate samples (18 μl) were transferred to 0.2-ml PCR tubes and subjected to a high resolution melt program (40–85 °C, 3.5 °C/min) in the Rotor-Gene Q (Qiagen), with fluorescence excitation at 365 ± 20 nm and emission monitored at 460 ± 20 nm. The temperature at the midpoint of the melting transition was determined by finding the maximum of the first derivative of the plot of fluorescence versus temperature.
Author Contributions
M. A. T., P. J. F., and K. K. S. N. conceived and coordinated the study. M. A. T., E. H., L. E., J. S., X. C., and J. M. designed, performed, and analyzed the experiments. M. A. T., P. J. F., and K. K. S. N. wrote the paper. All authors reviewed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
Some of the X-ray diffraction data were measured at beamline 08ID-1 of the Canadian Light Source (CLS), which is supported by the Natural Sciences and Engineering Research Council of Canada, the National Research Council Canada, the Canadian Institutes of Health Research, the Province of Saskatchewan, Western Economic Diversification Canada, and the University of Saskatchewan. Some of the diffraction data were also measured at the Stanford Synchrotron Radiation Lightsource, a Directorate of SLAC National Accelerator Laboratory and an Office of Science User Facility operated for the United States Department of Energy Office of Science by Stanford University. The SSRL Structural Molecular Biology Program is supported by the Department of Energy Office of Biological and Environmental Research and by the National Institutes of Health, NIGMS (including P41GM103393) and the National Center for Research Resources (P41RR001209). We thank Dr. Jillian Hagel for assistance with the chiral analysis of THP.
Note Added in Proof
Another article published in this issue (33) reports the characterization of reticuline N-methyltransferase, a related enzyme that shares some functional properties and substantial amino acid sequence identity with pavine N-methyltransferase.
This work was supported by NSERC Discovery Grant 05728 (to K. N.) and NSERC Discovery Grant 183573 (to P. J. F.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The atomic coordinates and structure factors (codes 5KN4, 5KOC, 5KPG, 5KOK, and 5KPC) have been deposited in the Protein Data Bank (http://wwpdb.org/).

This article contains supplemental Movies 1–3.
- BIA
- benzylisoquinoline alkaloid
- NMT
- N-methyltransferase
- CNMT
- coclaurine NMT
- DSF
- differential scanning fluorimetry
- GNMT
- glycine NMT
- PavNMT
- pavine NMT
- PfPMT
- Plasmodium falciparum phosphoethanolamine NMT
- SAH
- S-adenosylhomocysteine
- SAM
- S-adenosylmethionine
- THP
- tetrahydropapaverine
- TNMT
- tetrahydroprotoberberine NMT
- ACN
- acetonitrile
- PDB
- Protein Data Bank
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol.
References
- 1. Hagel J. M., and Facchini P. J. (2013) Benzylisoquinoline alkaloid metabolism: a century of discovery and a brave new world. Plant Cell Physiol. 54, 647–672 [DOI] [PubMed] [Google Scholar]
- 2. Fossati E., Ekins A., Narcross L., Zhu Y., Falgueyret J. P., Beaudoin G. A., Facchini P. J., and Martin V. J. (2014) Reconstitution of a 10-gene pathway for synthesis of the plant alkaloid dihydrosanguinarine in Saccharomyces cerevisiae. Nat. Commun. 5, 3283. [DOI] [PubMed] [Google Scholar]
- 3. Thodey K., Galanie S., and Smolke C. D. (2014) A microbial biomanufacturing platform for natural and semisynthetic opioids. Nat. Chem. Biol. 10, 837–844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ziegler J., and Facchini P. J. (2008) Alkaloid biosynthesis: metabolism and trafficking. Annu. Rev. Plant Biol. 59, 735–769 [DOI] [PubMed] [Google Scholar]
- 5. Kozbial P. Z., and Mushegian A. R. (2005) Natural history of S-adenosylmethionine-binding proteins. BMC Struct. Biol. 5, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Schubert H. L., Blumenthal R. M., and Cheng X. (2003) Many paths to methyltransfer: a chronicle of convergence. Trends Biochem. Sci. 28, 329–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gana R., Rao S., Huang H., Wu C., and Vasudevan S. (2013) Structural and functional studies of S-adenosyl-l-methionine binding proteins: a ligand-centric approach. BMC Struct. Biol. 13, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lee S. G., Kim Y., Alpert T. D., Nagata A., and Jez J. M. (2012) Structure and reaction mechanism of phosphoethanolamine methyltransferase from the malaria parasite Plasmodium falciparum: an antiparasitic drug target. J. Biol. Chem. 287, 1426–1434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Garg A., Lukk T., Kumar V., Choi J. Y., Augagneur Y., Voelker D. R., Nair S., and Ben Mamoun C. (2015) Structure, function and inhibition of the phosphoethanolamine methyltransferases of the human malaria parasites Plasmodium vivax and Plasmodium knowlesi. Sci. Rep. 5, 9064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Huang C. C., Smith C. V., Glickman M. S., Jacobs W. R. Jr, and Sacchettini J. C. (2002) Crystal structures of mycolic acid cyclopropane synthases from Mycobacterium tuberculosis. J. Biol. Chem. 277, 11559–11569 [DOI] [PubMed] [Google Scholar]
- 11. Holm L., and Rosenström P. (2010) Dali server: conservation mapping in 3D. Nucleic Acids Res. 38, W545–W549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Krissinel E., and Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 [DOI] [PubMed] [Google Scholar]
- 13. Ropivia J., Derbré S., Rouger C., Pagniez F., Le Pape P., and Richomme P. (2010) Isoquinolines from the roots of Thalictrum flavum L., and their evaluation as antiparasitic compounds. Molecules 15, 6476–6484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Saen-Oon S., Lee S. G., Jez J. M., and Guallar V. (2014) An alternative mechanism for the methylation of phosphoethanolamine catalyzed by Plasmodium falciparum phosphoethanolamine methyltransferase. J. Biol. Chem. 289, 33815–33825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Liscombe D. K., Ziegler J., Schmidt J., Ammer C., and Facchini P. J. (2009) Targeted metabolite and transcript profiling for elucidating enzyme function: isolation of novel N-methyltransferases from three benzylisoquinoline alkaloid-producing species. Plant J. 60, 729–743 [DOI] [PubMed] [Google Scholar]
- 16. Zhang J., and Klinman J. P. (2016) Convergent mechanistic features between the structurally diverse N- and O-methyltransferases: glycine N-methyltransferase and catechol O-methyltransferase. J. Am. Chem. Soc. 138, 9158–9165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Takata Y., Huang Y., Komoto J., Yamada T., Konishi K., Ogawa H., Gomi T., Fujioka M., and Takusagawa F. (2003) Catalytic mechanism of glycine N-methyltransferase. Biochemistry 42, 8394–8402 [DOI] [PubMed] [Google Scholar]
- 18. Komoto J., Yamada T., Takata Y., Konishi K., Ogawa H., Gomi T., Fujioka M., and Takusagawa F. (2004) Catalytic mechanism of guanidinoacetate methyltransferase: crystal structures of guanidinoacetate methyltransferase ternary complexes. Biochemistry 43, 14385–14394 [DOI] [PubMed] [Google Scholar]
- 19. Drinkwater N., Gee C. L., Puri M., Criscione K. R., McLeish M. J., Grunewald G. L., and Martin J. L. (2009) Molecular recognition of physiological substrate noradrenaline by the adrenaline-synthesizing enzyme PNMT and factors influencing its methyltransferase activity. Biochem. J. 422, 463–471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wu Q., and McLeish M. J. (2013) Kinetic and pH studies on human phenylethanolamine N-methyltransferase. Arch. Biochem. Biophys. 539, 1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jain A., Ziegler J., Liscombe D. K., Facchini P. J., Tucker P. A., and Panjikar S. (2008) Purification, crystallization and X-ray diffraction analysis of pavine N-methyltransferase from Thalictrum flavum. Acta Crystallogr. F Struct. Biol. Cryst. Commun. 64, 1066–1069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Farrow S. C., Hagel J. M., Beaudoin G. A., Burns D. C., and Facchini P. J. (2015) Stereochemical inversion of (S)-reticuline by a cytochrome P450 fusion in opium poppy. Nat. Chem. Biol. 11, 728–732 [DOI] [PubMed] [Google Scholar]
- 23. Kabsch W. (1993) Automatic indexing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J. Appl. Cryst. 26, 795–800 [Google Scholar]
- 24. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Emsley P., and Cowtan K. (2004) COOT: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 26. Davis I. W., Leaver-Fay A., Chen V. B., Block J. N., Kapral G. J., Wang X., Murray L. W., Arendall W. B. 3rd, Snoeyink J., Richardson J. S., and Richardson D. C. (2007) MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 35, W375–W383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Farrow S. C., Hagel J. M., and Facchini P. J. (2012) Transcript and metabolite profiling in cell cultures of 18 plant species that produce benzylisoquinoline alkaloids. Phytochemistry 77, 79–88 [DOI] [PubMed] [Google Scholar]
- 28. Kabsch W., and Sander C. (1983) Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22, 2577–2637 [DOI] [PubMed] [Google Scholar]
- 29. Liscombe D. K., and Facchini P. J. (2007) Molecular cloning and characterization of tetrahydroprotoberberine cis-N-methyltransferase, an enzyme involved in alkaloid biosynthesis in opium poppy. J. Biol. Chem. 282, 14741–14751 [DOI] [PubMed] [Google Scholar]
- 30. Desgagné-Penix I., Farrow S. C., Cram D., Nowak J., and Facchini P. J. (2012) Integration of deep transcript and targeted metabolite profiles for eight cultivars of opium poppy. Plant Mol. Biol. 79, 295–313 [DOI] [PubMed] [Google Scholar]
- 31. Facchini P. J., and Park S. U. (2003) Developmental and inducible accumulation of gene transcripts involved in alkaloid biosynthesis in opium poppy. Phytochemistry 64, 177–186 [DOI] [PubMed] [Google Scholar]
- 32. Choi K. B., Morishige T., Shitan N., Yazaki K., and Sato F. (2002) Molecular cloning and characterization of coclaurine N-methyltransferase from cultured cells of Coptis japonica. J. Biol. Chem. 277, 830–835 [DOI] [PubMed] [Google Scholar]
- 33. Morris J. S., and Facchini P. J. (2016) Isolation and characterization of reticuline N-methyltransferase involved in biosynthesis of the aporphine alkaloid magnoflorine in opium poppy. J. Biol. Chem. 291, 23416–23427 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.