

Abstract
Mutations of the AMP-activated kinase gamma 2 subunit (AMPKγ2), N488I (AMPKγ2NI) and R531G (AMPKγ2RG), are associated with Wolff-Parkinson-White (WPW) syndrome, a cardiac disorder characterized by ventricular pre-excitation in humans. Cardiac-specific transgenic overexpression of human AMPKγ2NI or AMPKγ2RG leads to constitutive AMPK activation and the WPW phenotype in mice. However, overexpression of these mutant proteins also caused profound, non-physiological increase in cardiac glycogen, which might abnormally alter the true phenotype. To investigate whether physiological levels of AMPKγ2NI or AMPKγ2RG mutation cause WPW syndrome and metabolic changes in other organs, we generated two knock-in mouse lines on the C57BL/6N background harboring mutations of human AMPKγ2NI and AMPKγ2RG, respectively. Similar to the reported phenotypes of mice overexpressing AMPKγ2NI or AMPKγ2RG in the heart, both lines developed WPW syndrome and cardiac hypertrophy; however, these effects were independent of cardiac glycogen accumulation. Compared with AMPKγ2WT mice, AMPKγ2NI and AMPKγ2RG mice exhibited reduced body weight, fat mass, and liver steatosis when fed with a high fat diet (HFD). Surprisingly, AMPKγ2RG but not AMPKγ2NI mice fed with an HFD exhibited severe kidney injury characterized by glycogen accumulation, inflammation, apoptosis, cyst formation, and impaired renal function. These results demonstrate that expression of AMPKγ2NI and AMPKγ2RG mutations at physiological levels can induce beneficial metabolic effects but that this is accompanied by WPW syndrome. Our data also reveal an unexpected effect of AMPKγ2RG in the kidney, linking lifelong constitutive activation of AMPK to a potential risk for kidney dysfunction in the context of an HFD.
Keywords: AMP-activated kinase (AMPK), diabetes, glycogen storage disease, insulin resistance, kidney metabolism, liver metabolism
Introduction
AMPK2 is an energy sensor that functions to maintain energy homeostasis in response to changes in the ratio of AMP to ATP in the cell (1). AMPK is a heterotrimeric complex composed of catalytic α subunits (α1 and α2), regulatory β (β1 and β2), and γ-subunits (γ1, γ2, and γ3) (2). The AMPK γ-subunit has three AMP binding sites; two sites exhibit reversible and one site irreversible binding (3, 4). AMPK is activated by its upstream kinase LKB1 via phosphorylation of Thr-172 of the α-subunit (5–7). Upon binding of AMP to AMPK γ-subunit, AMPK can be further allosterically activated (8, 9). AMPK is reported to regulate multiple metabolic functions in different organs, such as protein synthesis, lipid metabolism, insulin receptor signaling pathway, hepatic gluconeogenesis, glucose transport in skeletal muscle, cardiac function, kidney development, etc. (8, 10).
To date, AMPKγ2 (Prkag 2) is the only subunit recognized to be associated with disease-causing mutations. Human genetic studies reported association of AMPKγ2 mutations and Wolff-Parkinson-White (WPW) syndrome, a cardiac defect characterized by intermittent or persistent ventricular pre-excitation in sinus rhythm and arrhythmia and sometimes accompanied by paroxysmal tachycardia and cardiac hypertrophy (11, 12). In total, 11 mutations of and one insertion in the AMPKγ2 subunit have been identified that are correlated to WPW syndrome (13). Of these mutations/insertion, AMPKγ2NI (Asn → Ile mutation at amino acid 488) and AMPKγ2RG (Arg → Gly mutation at amino acid 531) have been intensively studied (14–17). Both AMPKγ2NI and AMPKγ2RG mutations are believed to generate constitutively active AMPK complexes. AMP can further enhance AMPK activity upon binding to AMPKγ2NI (16, 17). In contrast, AMPKγ2RG, a mutation within the irreversible AMP binding region, lacks responsiveness to AMP because it has lost its ability to bind AMP (3, 18, 19). Transgenic mice with cardiac-specific overexpression of human AMPKγ2NI and AMPKγ2RG at >20-fold over endogenous AMPKγ2 levels exhibit dramatic glycogen accumulation, cardiac hypertrophy, and ventricular pre-excitation compared with mice overexpressing human AMPKγ2WT at similar levels (16–18). The effect of the AMPKγ2RG mutation in the regulation of cardiac-specific ion channels and on early onset of pre-excitation and atrial fibrillation in the WPW patients suggested that AMPKγ2 mutations can cause abnormalities of the cardiac conduction system during development of the heart (20). In a recent report, Kim et al. (21) reported that eliminating glycogen storage in AMPKγ2NI transgenic mice by genetic inhibition of glucose 6-phosphate-stimulated glycogen synthase activity prevented pre-excitation in mice but did not affect cardiac hypertrophy, indicating a glycogen-induced abnormality in cardiac electrical conductance but a glycogen-independent effect on cardiac hypertrophy in WPW syndrome.
To investigate how AMPKγ2NI and AMPKγ2RG mutations affect cardiac function and metabolism, we generated two knock-in mice harboring single mutation of AMPKγ2NI or AMPKγ2RG. The current study was undertaken to understand the causality of AMPKγ2NI and AMPKγ2RG mutations expressed at physiological levels on the WPW syndrome and to explore whether they affect whole-body metabolism and function of other organs. We hypothesize that these mutations affect not only cardiac function but also whole-body metabolism, especially major metabolic organs, such as liver, skeletal muscle, and kidney. We describe our initial findings in this paper.
Results
Increased AMPK Activity in AMPKγ2NI and AMPKγ2RG Knock-in Mice
N488I and R531G mutations in human AMPKγ2 correspond to the N485I and R528G mutations, respectively, in mouse AMPKγ2 due to the absence of three amino acids, Pro-207, Ala-253, and Val-254, in mouse AMPKγ2 (Fig. 1A). Both AMPKγ2NI and AMPKγ2RG knock-in mice were generated on a C57BL/6N background. The genotype of all mice was confirmed by sequencing the mutated regions (Fig. 1B).
FIGURE 1.

Characterization of AMPKγ2NI and AMPKγ2RG mice. A, schematic diagram of human and mouse AMPKγ2 protein. Mouse AMPKγ2 has three amino acid deletions, Pro-207, Ala-253, and Val-254, versus the human protein; thus, N485I and R528G correspond to N488I and R531G in human AMPKγ2. B, modified sequence of AMPKγ2WT, AMPKγ2NI, and AMPKγ2RG mice. C and D, phosphorylated AMPKα (Thr-172), total AMPKα, and phospho-AMPKα (Thr-172)/total AMPKα ratio in liver and skeletal muscle of AMPKγ2WT, AMPKγ2NI, and AMPKγ2RG mice at 26 weeks of age (n = 8). E and F, phosphorylated ACC in liver and skeletal muscle of AMPKγ2WT, AMPKγ2NI, and AMPKγ2RG mice at 26 weeks of age (n = 8). RU, relative units. *, p ≤ 0.05; **, p ≤ 0.01. Error bars, S.E.
We next measured AMPKα and phospho-AMPKα protein levels in liver and skeletal muscle of the AMPKγ2WT, AMPKγ2NI, and AMPKγ2RG mice using antibodies recognizing both the α1 and α2 subunits. Knock-in of AMPKγ2NI and AMPKγ2RG mutations did not affect the protein levels of AMPKα and phospho-AMPKα in liver and skeletal muscle as compared with AMPKγ2WT controls (Fig. 1, C and D). mRNA levels of AMPK α-, β-, and γ-subunits in liver and skeletal muscle were not different in AMPKγ2NI and AMPKγ2RG mice as compared with AMPKγ2WT mice (data not shown). Taken together, these data demonstrate that the AMPKγ2NI and AMPKγ2RG knock-in mutations had no effect on expression of γ2 subunit and other AMPK subunits.
Two main isoforms of acetyl-CoA carboxylase (ACC), ACC1 and ACC2, are downstream targets of activated AMPK. Phosphorylation of ACC inhibits its enzymatic activity, and this effect is widely employed to reflect the activity of AMPK (22). We therefore used phosphorylated ACC (pACC) (both ACC1 and ACC2) to monitor AMPK activity. In skeletal muscle, pACC was significantly increased in AMPKγ2NI and AMPKγ2RGcompared with AMPKγ2WT mice (Fig. 1, E and F). Liver levels of pACC in AMPKγ2RG mice were significantly higher than in AMPKγ2NI and AMPKγ2WT mice. The pACC level in skeletal muscle was significantly lower than in the liver in AMPKγ2WT mice and showed larger differences among AMPKγ2WT, AMPKγ2NI, and AMPKγ2RG mice. Based on the level of pACC in different groups, it appeared that both AMPKγ2NI and AMPKγ2RG are intrinsically activated and that AMPKγ2RG has the highest activity in skeletal muscle as compared with AMPKγ2WT and AMPKγ2NI (Fig. 1F).
AMPKγ2NI and AMPKγ2RG Mice Develop WPW Syndrome
To investigate the effect of AMPKγ2NI and AMPKγ2RGmutations on cardiac electrical conductance, we recorded a surface electrocardiogram (ECG) by using electrocardiography in isoflurane-anesthetized, 8-week-old mice fed with a chow diet. Continuous ECG recording indicated that 50% of AMPKγ2NI and 70% of AMPKγ2RG mice had bradycardia with significantly shortened PR intervals compared with AMPKγ2WT mice (Fig. 2A and Table 1). The QRS complex in AMPKγ2NI and AMPKγ2RG mice appeared similar to that of AMPKγ2WT mice (Fig. 2A). In particular, compared with AMPKγ2WT mice, no δ wave was observed in AMPKγ2NI and AMPKγ2RG mice (Fig. 2A). Sometimes we noticed intermittent pre-excitation due to a probable retrograde conduction in the accessory pathway (Fig. 2A, bottom). Further ECG analysis demonstrated that AMPKγ2NI and AMPKγ2RG mice had intermittent pre-excitation, shortened PR interval, bradycardia, sinus arrhythmia, and occasional premature ventricular contraction (supplemental Table 1). Cardiac pACC in AMPKγ2NI and AMPKγ2RG mice was significantly increased as compared with AMPKγ2WT mice (Fig. 2B), indicating increased AMPK activity in the heart of AMPKγ2NI and AMPKγ2RG mice.
FIGURE 2.
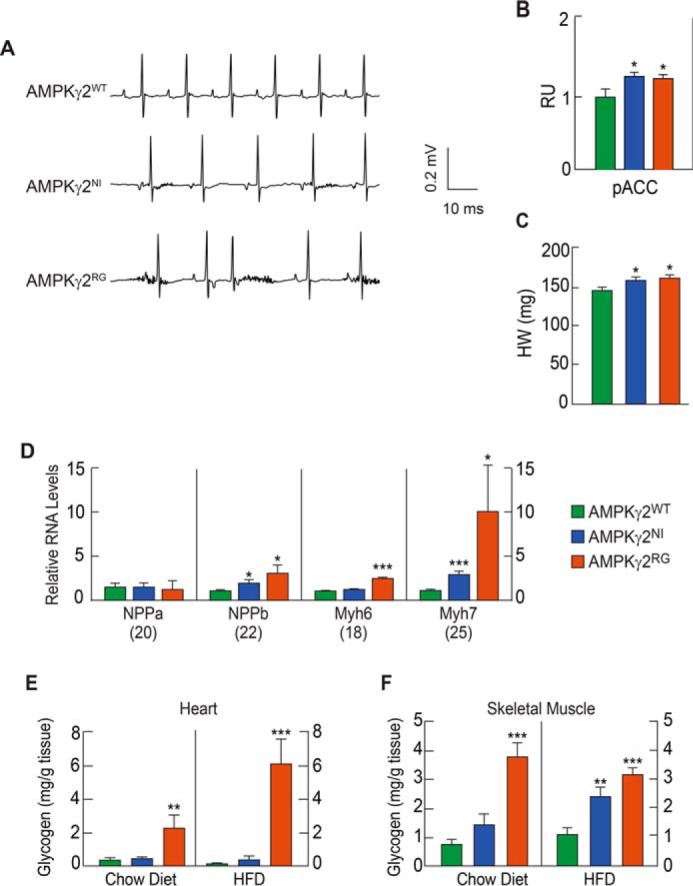
AMPKγ2NI and AMPKγ2RG mice exhibit WPW syndrome. A, ECG of AMPKγ2WT, AMPKγ2NI, and AMPKγ2RG mice under anesthetized conditions at 22 weeks of age. AMPKγ2NI and AMPKγ2RG mice have a shorter PQ interval, a feature of pre-excitation in human WPW syndrome in humans. B, pACC of the heart of AMPKγ2WT, AMPKγ2NI, and AMPKγ2RG mice fed with HFD at 26 weeks of age (n = 8). RU, relative units. C, heart weight of AMPKγ2WT, AMPKγ2NI, and AMPKγ2RG mice on chow diet (n = 8). D, relative mRNA levels of cardiac hypertrophy biomarker genes in AMPKγ2WT, AMPKγ2NI, and AMPKγ2RG mice. All mice were age-matched, maintained on a chow diet, and euthanized at 26 weeks of age. Numbers in parenthesis are the CT values of each gene in AMPKγ2WT mice (n = 8). E and F, glycogen levels in the heart and skeletal muscle of AMPKγ2WT, AMPKγ2NI, and AMPKγ2RG mice fed with chow diet and HFD at 26 weeks of age (n = 8). *, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001. Error bars, S.E.
TABLE 1.
Relative mRNA levels of AMPK isoforms in heart and kidney of AMPKγ2WT, AMPKγ2NI, and AMPKγ2RG mice fed with HFD at 26 weeks of age
Prkaa1, AMPK α1; Prgaa2, AMPK α2; Prgab1, AMPK β1; Prkab2, AMPK β2; Prkag1, AMPK γ1; Prkag2, AMPK γ2; Prkag3, AMPK γ3. *, p ≤ 0.05.
| mRNA | AMPKγ2WT (n = 4), average CT | AMPKγ2NI (n = 4), change | AMPKγ2RG (n = 4), change |
|---|---|---|---|
| -fold | -fold | ||
| Heart | |||
| Prkaa1 | 28.0 | 1.13 | 1.11 |
| Prkaa2 | 24.6 | 1.07 | 1.09 |
| Prkab1 | 26.7 | 1.12 | 1.07 |
| Prkab2 | 26.7 | 1.19 | 1.23* |
| Prkag1 | 26.8 | 0.78* | 1.00 |
| Prkag2 | 26.3 | 0.82 | 0.70 |
| Prkag3 | 29.9 | 0.73 | 0.68* |
| Kidney | |||
| Prkaa1 | 27.9 | 0.94 | 0.93 |
| Prkaa2 | 24.7 | 1.01 | 0.93* |
| Prkab1 | 26.8 | 1.10 | 1.23 |
| Prkab2 | 29.8 | 0.94 | 1.17 |
| Prkag1 | 28.5 | 1.24 | 1.26 |
| Prkag2 | 26.3 | 1.16 | 1.01 |
| Prkag3 | 30.0 | 0.76 | 0.92 |
To further understand the effect of AMPKγ2NI and AMPKγ2RGmutations on cardiac function, we measured heart weight and quantitated relative mRNA levels of genes associated with cardiac hypertrophy in AMPKγ2WT, AMPKγ2NI, and AMPKγ2RG chow diet-fed mice at 8 weeks of age by using echocardiography. AMPKγ2NI and AMPKγ2RG mice fed with a chow diet developed cardiac hypertrophy at 24 weeks of age (Fig. 2C). Feeding the mice with an HFD did not worsen cardiac function based on echocardiograms finding obtained at 24 weeks of age as compared with mice fed with chow (data not shown). Relative mRNA levels of AMPK subunits were quantitated in the heart of AMPKγ2WT, AMPKγ2NI, and AMPKγ2RG mice fed an HFD (Fig. 2D). Overall, there was no significant difference between AMPKγ2NI and AMPKγ2RG mice in all AMPK subunits except for slightly reduced γ1 and γ3 in AMPKγ2NI and AMPKγ2RG mice, respectively, and a slightly induced β2 in AMPKγ2RG mice (Table 1). Biomarkers of cardiac hypertrophy, NPPa, NPPb, Myh6, and Myh7 (23), were measured in the heart of AMPKγ2WT, AMPKγ2NI, and AMPKγ2RG mice fed with chow diet at 26 weeks of age. mRNA levels of NPPb and Myh7 were significantly increased in AMPKγ2NI and AMPKγ2RG mice, and Myh6 was significantly increased in AMPKγ2RG mice compared with AMPKγ2WT mice (Fig. 2D).
Cardiac glycogen was measured in AMPKγ2WT, AMPKγ2NI, and AMPKγ2RG mice fed with a chow diet or HFD at 26 weeks of age. Cardiac glycogen levels were not changed in AMPKγ2NI mice compared with AMPKγ2WT mice fed with either chow diet or HFD but were significantly increased by 5.6- and 46.9-fold in AMPKγ2RG mice fed with chow diet and HFD, respectively (Fig. 2E). Although the effect was less pronounced, glycogen content in skeletal muscle was significantly increased by 4- and 1.9-fold in AMPKγ2RG mice fed with the chow diet and HFD, respectively (Fig. 2F). Glycogen contents in skeletal muscle of AMPKγ2NI mice were significantly increased on HFD and showed a trend of increase on chow diet compared with that of AMPKγ2WT mice (Fig. 2F).
Improved Metabolic Parameters in AMPKγ2NI and AMPKγ2RGMice Fed with HFD
Exercise, a well known weight loss measure, is known to activate AMPK (24), and AMPK activation by AMPK activators could induce fatty acid oxidation (25). To examine these potential effects in our models, we examined the effects of AMPKγ2WT, AMPKγ2NI, and AMPKγ2RG mutations in mice fed with a chow diet or HFD on body weight and composition and other metabolic parameters. On a chow diet, body weight and body weight gain of AMPKγ2NI and AMPKγ2RG mice were similar to AMPKγ2WT controls (Fig. 3, A, B, F, and G). On the HFD, both AMPKγ2NI and AMPKγ2RG mice gained less weight as compared with AMPKγ2WT controls (Fig. 3, B and G). Body composition analysis demonstrated that the major differences in body weight between AMPKγ2WT and AMPKγ2NI and AMPKγ2RG were due to fat mass (Fig. 3, C, E, and H–J). At 20 weeks of age, the fat mass of AMPKγ2NI and AMPKγ2RG mice was lower than that of AMPKγ2WT mice by 44 and 32.5%, respectively (Fig. 3, D and I). Food intake of AMPKγ2RG mice was significantly decreased versus AMPKγ2WTmice on both diets and trended lower for AMPKγ2NI versus AMPKγ2WT mice (Fig. 3K).
FIGURE 3.
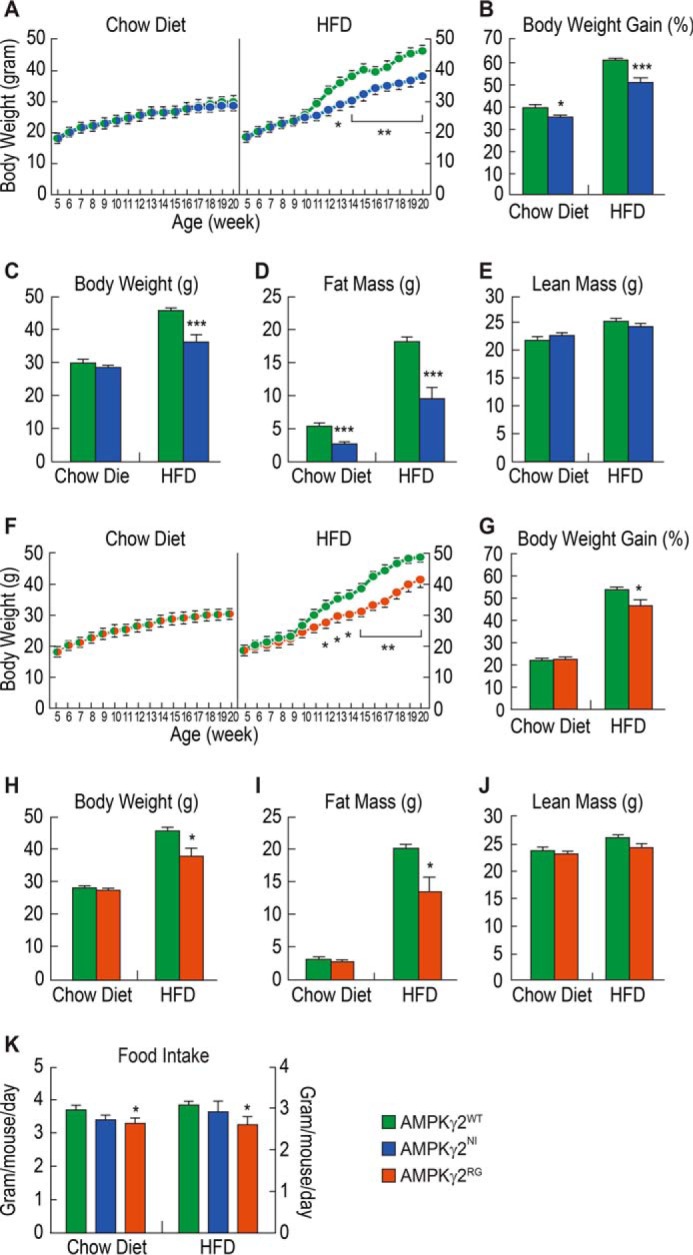
AMPKγ2NI and AMPKγ2RG mice are resistant to HFD. A, time curve of body weight and body weight gain of AMPKγ2WT and AMPKγ2NI mice on chow diet and HFD (n = 9). B–E, body weight gain, body weight, and body composition of AMPKγ2WT and AMPKγ2NI mice on chow diet and HFD at 20 weeks of age. F, time curve of body weight and body weight gain of AMPKγ2WT and AMPKγ2RG mice on chow diet and HFD. G–J, body weight gain, body weight, and body composition of AMPKγ2WT and AMPKγ2RG mice on chow diet and HFD at 20 weeks of age. K, food intake of AMPKγ2WT, AMPKγ2NI, and AMPKγ2RG mice on chow diet and HFD. *, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001. Error bars, S.E.
There were no significant differences in glucose and insulin tolerance in AMPKγ2NI and AMPKγ2RG mice fed with a chow diet compared with AMPKγ2WT mice, consistent with the lack of difference in body weight. By contrast, on an HFD, both AMPKγ2NI and AMPKγ2RG mice showed improved glucose tolerance and insulin tolerance (Fig. 4, A–F). Plasma levels of TG, cholesterol, and free fatty acid were significantly lower in AMPKγ2NI and AMPKγ2RG than in AMPKγ2WT mice on the HFD under fed conditions (Fig. 4, G and H). The plasma ketone levels were significantly increased in AMPKγ2RG mice on the HFD (Fig. 4H).
FIGURE 4.

Improved glucose homeostasis and dyslipidemia in AMPKγ2NI and AMPKγ2RG mice. A–C, GTT, AUC of GTT, and ITT of AMPKγ2WT and AMPKγ2NI mice on chow diet and HFD (n = 9). The diet in the HFD group was switched from chow diet to HFD at 9 weeks of age. GTT and ITT were performed at 20 and 21 weeks of age. D–F, GTT, AUC of GTT, and ITT of AMPKγ2WT and AMPKγ2RG mice on chow diet and HFD. G and H, fed plasma levels of TG, cholesterol, free fatty acid, and β-hydroxybutyrate of AMPKγ2WT, AMPKγ2NI, and AMPKγ2RG mice on chow diet at 26 weeks of age. I, H&E histology of livers of AMPKγ2WT, AMPKγ2NI, and AMPKγ2RG mice on HFD at 26 weeks of age. *, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001. Error bars, S.E.
At study termination, metabolic organs, including liver, heart, skeletal muscle, kidney, and pancreas of AMPKγ2WT, AMPKγ2NI, and AMPKγ2RG mice, were investigated for histological changes by H&E staining. No obvious histological changes of heart, skeletal muscle, and pancreas were observed in AMPKγ2NI and AMPKγ2RG mice compared with AMPKγ2WTmice fed with either chow diet or HFD (data not shown). AMPKγ2WT mice developed liver steatosis after being fed with HFD for 17 weeks, whereas AMPKγ2NI and AMPKγ2RG appear to be protected with no obvious accumulation of lipid droplets in the liver (Fig. 4I). Altogether, AMPKγ2NI and AMPKγ2RG mice are resistant to HFD-induced body weight gain and show improved metabolic parameters compared with AMPKγ2WT mice.
AMPKγ2RG Induces Kidney Pathology and Renal Impairment
During histopathological evaluation, we observed dramatic morphological changes in kidneys of AMPKγ2RG mice. Upon chow diet feeding, kidneys of AMPKγ2WT and AMPKγ2NI mice appeared to be normal, whereas moderate to severe cyst dilatation (occasionally with proteinosis) of distal and collector tubules was evident in the medullar rays of AMPKγ2RG mice kidneys at 26 weeks of age (Fig. 5A). Kidneys of AMPKγ2WT and AMPKγ2NI mice fed an HFD remained normal. In contrast, HFD-fed AMPKγ2RG mice developed severe kidney injury that manifests gross discoloration of the kidney (Fig. 5B). There was also dramatic tubular degeneration, as indicated by papilla dilatation and severe cystic changes (with proteinosis) in the collecting ducts (Fig. 5B). Many foci of necrosis, lymphocyte infiltration, and regeneration were observed in the tubulointerstitial compartment in HFD-fed AMPKγ2RG mice, indicating severe inflammation in the kidney (Fig. 5B). Foci of amyloid deposits and areas of collagen fibers were also observed in AMPKγ2RG mouse kidneys (Fig. 5B and supplemental Fig. 1A). Kidney glycogen content was not changed in AMPKγ2NI mice compared with AMPKγ2WTmice fed with either chow diet or HFD. By contrast, it was significantly increased by 4.3- and 6.8-fold in AMPKγ2RG mice fed with a chow diet and HFD, respectively (Fig. 5C).
FIGURE 5.

AMPKγ2RG mutation induces kidney disease and impaired renal function. A, H&E histology of kidneys of AMPKγ2WT, AMPKγ2NI, and AMPKγ2RG mice on chow diet at 26 weeks of age (×0.27 for gross kidney on the left, ×5 in the middle, and ×20 on the right). B, gross pictures and H&E histology of kidneys of AMPKγ2WT, AMPKγ2NI, and AMPKγ2RG mice on HFD at 26 weeks of age (×0.27 for gross kidney on the left, ×5 in the middle, and ×20 on the right). C, kidney glycogen content of AMPKγ2WT, AMPKγ2NI, and AMPKγ2RG mice fed with chow diet and HFD at 26 weeks of age (n = 8). D–G, concentrations of urine albumin (D), NGAL (E), KIM1 (F), and creatinine (G) of AMPKγ2WT, AMPKγ2NI, and AMPKγ2RG mice on chow and HFD at 20 weeks of age (n = 7–10). A 24-h collection of urine was used for measurement of albumin, creatinine, NGAL, and KIM1. H–J, ratios of urine albumin/creatinine (H), NGAL/creatinine (I), and KIM1/creatinine (J) of AMPKγ2WT, AMPKγ2NI, and AMPKγ2RG mice on chow and HFD at 20 weeks of age (n = 7–10). **, p ≤ 0.01; ***, p ≤ 0.001. Error bars, S.E.
To further investigate kidney function, AMPKγ2WT, AMPKγ2NI, and AMPKγ2RG mice were maintained on either the chow diet or HFD until 26 weeks of age. A 24-h urine collection sample was used to measure albumin, creatinine, NGAL, and KIM1, and these parameters were normalized to urinary creatinine concentration (Fig. 5, D–J). Creatinine in AMPKγ2RG mice on HFD showed a trend of slight reduction, which might be associated with a trend of decrease in lean mass (Fig. 3J). The urinary albumin/creatinine ratio (ACR) has been used as a marker reflecting change of renal function, and KIM-1 and NGAL levels have been used as renal tubular injury biomarkers (26). On the chow diet, all parameters in AMPKγ2NI mice were similar to those of AMPKγ2WT mice, whereas they were significantly higher in AMPKγ2RG mice (Fig. 5, H–J). On the chow diet in AMPKγ2RG mice, urinary ACR and excretion of NGAL and KIM1 were increased by 87%, 57%, and 3.4-fold compared with AMPKγ2WT mice. This situation was exacerbated on an HFD, the urinary ACR and excretion of NGAL and KIM1 in AMPKγ2RG mice were increased by 2-, 8.5-, and 27.2-fold compared with AMPKγ2WT mice, indicating impaired renal function and tubular injury in AMPKγ2RG mice (Fig. 5, H–J). Urinary ACR and excretion of NGAL and KIM1 were comparable between AMPKγ2WT and AMPKγ2NI mice on HFD (Fig. 5, H–J). Consistent with proteinurea, protein casts in kidney tubules were prominent in AMPKγ2RG mice fed with a chow diet and HFD as shown by periodic acid-Schiff (PAS) staining (supplemental Fig. 1B). There was no significant difference in kidney lipid contents among AMPKγ2WT, AMPKγ2NI, and AMPKγ2RG mice on either the chow diet or HFD, as indicated by oil red O staining and lipid content measurement (supplemental Fig. 1, C and D).
AMPKγ2RG Mutation Increases Apoptosis and Inflammation in the Kidney
To investigate the mechanism of kidney injury in AMPKγ2RG mice, we first measured mRNA levels of the seven AMPK subunits and calculated the -fold change of each subunit in AMPKγ2NI and AMPKγ2RG mice over AMPKγ2WT controls. Overall, there was no significant difference between AMPKγ2NI and AMPKγ2RG mice in all AMPK subunits except for a slightly reduced AMPKα2 in AMPKγ2RG mice (Table 1). The differences in AMPK activity in WT versus mutant mice were confirmed by measuring kidney pACC as a function of diet. When fed with a chow diet, pACC in the kidney of AMPKγ2NI and AMPKγ2RG mice was significantly higher than in AMPKγ2WT mice. When the diet was switched to HFD, pACC was significantly higher in the kidney of AMPKγ2RG mice compared with AMPKγ2WT and AMPKγ2NI mice (Fig. 6, A and B).
FIGURE 6.

AMPKγ2RG mutation induces apoptosis in the kidney. A and B, phosphorylated ACC in liver and skeletal muscle of AMPKγ2WT, AMPKγ2NI, and AMPKγ2RG mice on chow and HFD at 26 weeks of age (n = 8). C, TUNEL immunostaining of kidney of AMPKγ2WT and AMPKγ2RG mice on HFD at 26 weeks of age. D–G, phospho-Akt (Thr-308)/total Akt ratio, phospho-Akt (Ser-473)/total Akt, phospho-FOXO3a (Thr-32), phospho-mTOR (Ser-2448)/total mTOR in the kidneys of AMPKγ2WT, AMPKγ2NI, and AMPKγ2RG mice on chow diet or HFD at 26 weeks of age (n = 8). RU, relative units. *, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001. Error bars, S.E.
To investigate the cause of kidney injury in AMPKγ2RGmice on HFD, we performed TUNEL staining of kidneys. AMPKγ2RG mouse kidneys were positive for TUNEL staining, indicating active apoptosis (Fig. 6C). It has been reported that AMPK activation inhibits phosphorylation of Akt, which in turn activates FOXO3a, and the latter induces apoptosis in cancer cells (27). The insulin-independent AMPK-dependent pathway regulates cell survival under the low energy state (28). To investigate the mechanism of apoptosis in AMPKγ2RG mice on HFD, we measured pAkt (Thr-308)/Akt and pAkt (Ser-473)/Akt in kidney lysates of 26-week-old AMPKγ2WT, AMPKγ2NI, and AMPKγ2RG mice fed with the chow diet or HFD. In mice fed with the chow diet, there was no significant difference compared with AMPKγ2WT mice in the ratios of pAkt (Thr-308)/Akt and pAkt (Ser-473)/Akt in the kidney of AMPKγ2NI and AMPKγ2RG mice. On the HFD, AMPKγ2RG mice exhibited a significant decrease in the ratio of pAkt (Ser-473)/Akt and a trend toward a lower ratio of pAkt (Thr-308)/Akt (Fig. 6, D and E). FOXO3a is downstream target of Akt (29). Consistent with decreased pAkt/Akt in AMPKγ2RG mice, pFOXO3a was significantly increased in AMPKγ2RG mice on the chow diet and HFD (Fig. 6F). AMPK activation inhibits mTOR phosphorylation by phosphorylating the upstream mTOR regulator TSC2 on its Raptor subunit (30, 31). As expected, pmTOR (Ser-2448)/mTOR was significantly decreased by AMPKγ2RG on the chow diet and HFD (Fig. 6G).
To investigate the inflammatory response revealed by kidney histology in AMPKγ2RG mice fed with HFD, we performed molecular profiling of kidneys from AMPKγ2WT, AMPKγ2NI, and AMPKγ2RG mice, focusing on apoptosis, inflammatory response, and autoimmunity pathways. Caspases, positive regulators of apoptosis, death domain receptors, cytokines and chemokine receptors, and inflammatory and immune response gene expression levels were compared in AMPKγ2NI and AMPKγ2RG mice with those of AMPKγ2WT mice. AMPKγ2RG mice showed significant and dramatic induction for many of these genes in these pathways compared with AMPKγ2NI mice (Table 2), consistent with the inflammatory response by histology (Fig. 5B).
TABLE 2.
Relative mRNA levels of genes involved in apoptosis and inflammation in the kidney of AMPKγ2NI and AMPKγ2RG mice fed with HFD (n = 4)
*, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001.
| AMPKγ2NI | AMPKγ2RG | |
|---|---|---|
| Caspases | ||
| Casp1 | 1.01 | 2.73** |
| Casp2 | 1.05 | 1.50** |
| Casp3 | 1.08 | 1.18 |
| Casp4 | 1.06 | 2.75** |
| Casp6 | 1.01 | 1.25* |
| Casp7 | 1.20** | 1.29* |
| Casp8 | 1.00 | 0.50 |
| Casp12 | 0.99 | 2.34*** |
| Casp14 | 1.19* | 1.36** |
| Casp9 | 1.11 | 0.95 |
| Cflar | 1.10 | 0.91 |
| Cradd | 1.17 | 0.99 |
| Pycard | 1.09 | 1.91*** |
| Positive regulators of apoptosis | ||
| Tnfsf12 | 1.03 | 1.33* |
| Trp53 | 1.07 | 1.24** |
| Trp53bp2 | 1.18 | 1.34** |
| Traf1 | 1.73* | 2.52** |
| Traf2 | 1.23* | 1.45** |
| Death domain receptors | ||
| Nfkb1 | 1.11 | 1.59** |
| Tnfrsf11b | 0.93 | 1.85** |
| Cytokine and chemokine receptors | ||
| Il1r1 | 1.19 | 1.66** |
| Il1rap | 1.27* | 1.14* |
| Il6ra | 1.05 | 1.55* |
| Ccr2 | 0.84 | 1.96** |
| Ccr3 | 0.73 | 2.17*** |
| Cxcr4 | 1.17 | 1.60** |
| Inflammatory and immune response | ||
| C3 | 0.37* | 2.96** |
| C3ar1 | 0.76 | 2.23 |
| C4b | 1.03 | 3.86** |
| Ccl19 | 0.82* | 1.22 |
| Ccl5 | 1.06 | 2.17* |
| Ccl8 | 0.67 | 3.82* |
| Ccr2 | 0.84 | 1.96** |
| Ccr3 | 0.73 | 2.17*** |
| Cd14 | 1.24 | 2.67*** |
| Cxcr4 | 1.17 | 1.60** |
| Itgb2 | 0.85 | 1.73** |
| Il1b | 1.09 | 1.50** |
| Myd88 | 1.28* | 1.16* |
| Nfkb1 | 1.05 | 1.30** |
| Tlr2 | 1.11 | 2.20*** |
| Tlr4 | 1.08 | 1.51* |
Discussion
Human genetic studies linked AMPKγ2 mutations to WPW syndrome, and the cardiac phenotype has been recapitulated in mice by overexpressing human AMPKγ2NI and AMPKγ2RG mutations in the heart (16–18). The protein levels of human AMPKγ2NI and AMPKγ2RG in transgenic mice were ∼20-fold higher than the endogenous mouse γ2 subunit. Overexpression of human AMPKγ2WT to a similar level did not show this phenotype. In this work, we studied AMPKγ2NI and AMPKγ2RG knock-in mice, and AMPKγ2NI and AMPKγ2RG mutations expressed at physiological levels caused similar phenotypes of the WPW syndrome. AMPKγ2NI and AMPKγ2RG mutations increase the basal activity of AMPK and yield cardiac phenotype, including pre-excitation and cardiac hypertrophy. We also demonstrated the novel result that both AMPKγ2NI and AMPKγ2RG mice were resistant to HFD-induced body weight gain, leading to an improved metabolic phenotype, including glucose and insulin tolerance, plasma dyslipidemia, and liver steatosis. Unexpectedly, additional studies revealed that a constitutively active mutation, AMPKγ2RG, led to impaired kidney function as a result of apoptosis and inflammation in the kidney when fed with an HFD.
As an energy sensor, AMPK is activated by cellular energy stress, resulting in an increased concentration of AMP and the ratio of AMP/ATP. AMPK activation by AMP is a result of binding of AMP to the CBS domains of the regulatory γ-subunit, phosphorylation of residue Thr-172 on the catalytic loop of the α-subunit, and inhibition of dephosphorylation of the α-subunit Thr-172 by protein phosphatases (32). In response to increases in cellular AMP levels, activated AMPK acts to restore energy homeostasis by inhibiting ATP consumption and stimulating ATP production pathways, leading to ATP synthesis (8). Our data demonstrate that both the AMPKγ2NI and AMPKγ2RG mutations increase baseline AMPK activity; thus, pACC in mouse tissues of both AMPKγ2NI and AMPKγ2RG mice was increased compared with AMPKγ2WT mice. ACC (ACC1 and ACC2) is the rate-limiting enzyme in de novo fatty acid synthesis. ACC phosphorylation decreases its enzymatic activity, leading to inhibition of fatty acid synthesis (33). Reduction in ACC activity also decreases cellular malonyl-CoA, which in turn disinhibits CPT1, facilitates fatty acid transport into mitochondria, and increases fatty acid oxidation (25). In principle, inhibition of fatty acid synthesis should result in decreased ATP utilization, whereas activation of fatty acid oxidation should result in increased ATP production. Taken together, these effects of AMPK activation can restore cellular ATP levels under conditions of extreme energy stress (2). Consistent with predictions, AMPKγ2NI and AMPKγ2RG mice demonstrated less body weight gain, less fat mass, lower plasma lipids, and improved liver steatosis when fed with an HFD.
Cellular glycogen levels are controlled by the balance between glycogen synthesis and glycogenolysis. AMPK induces glucose uptake via increasing Glut4 translocation, but it allosterically inhibits glycogen synthase (34, 35). Gain-of-function mutations in the AMPKγ3 subunit, R225W in humans and R225Q in pigs, increase skeletal muscle glycogen content (36, 37). Overexpressing high levels of AMPKγ2NI and AMPKγ2RG led to 30- and 40-fold higher cardiac glycogen content versus non-transgenic littermates. These changes in glycogen are caused by an aberrant increase of AMPK activity (17). This was accompanied by pre-excitation arrhythmia and cardiac hypertrophy. By contrast, cardiac glycogen content was not changed in AMPKγ2NI mice irrespective of diet, whereas it was increased by 5.6- and 46.9-fold in AMPKγ2RG mice fed with chow diet and HFD, respectively. However, both AMPKγ2NI and AMPKγ2RG mice developed WPW syndrome, suggesting the existence of a glycogen-independent mechanism for WPW syndrome, at least in AMPKγ2NI mice. This is consistent with the observation that not all WPW patients carrying AMPKγ2 mutations manifest increased glycogen storage in the heart (38). The pro-arrhythmogenic phenotype in patients with AMPKγ2 mutations might be related to alterations of voltage-gated sodium channels that cause defects in conduction pathways in cardiomyocytes (39).
By rescuing the glycogen storage phenotype in AMPKγ2NI transgenic mice, Kim et al. (21) demonstrated that ablation of glycogen storage eliminated ventricular pre-excitation. Excessive cardiac growth and cardiomyopathy, however, persisted in AMPKγ2NI transgenic mice, indicating a glycogen storage-independent and AMPK intrinsic activation mechanism on cell growth of cardiac myocytes. This effect was proposed to be mediated by enhanced insulin sensitivity and activation of the Akt-mTOR-FOXO3A pathway in cardiac myocytes (21). Interestingly, another mutation of AMPKγ2, R302Q, causes a reduction in AMPK activity yet exhibits a glycogen storage hypertrophy and a shortened PQ interval as opposed to what has been observed in humans carrying the N485I mutation (38). Indeed, cardiac hypertrophy does not seem to exist in all AMPKγ2 mutations; the R531G mutation causes childhood onset WPW syndrome without cardiac hypertrophy (20). Altogether, studies from both humans and mice demonstrate that glycogen storage, ventricular pre-excitation, and cardiac hypertrophy are dissociable events in AMPKγ2 mutations.
The cellular mechanisms of AMPKγ2-induced arrhythmia have also been linked to voltage-gated sodium channels. By overexpressing constitutively active AMPKα1 (T172D), Light et al. (39) demonstrated that sodium channels are a target of activated AMPK and that phosphorylation slowed the open-state inactivation of sodium channel and shifted the voltage-activation curve in a hyperpolarizing direction (20). Whether the voltage-gated sodium channel is affected in AMPKγ2NI and AMPKγ2RG mice is unknown. Reduction of the physiological delay between atrial and ventricular electrical activation in AMPKγ2NI and AMPKγ2RG mice is similar to that in humans with Lown-Ganong-Levine syndrome, a class of the WPW syndrome (40). These knock-in mice will be valuable tools for studying the mechanism by which physiological levels of AMPKγ2NI and AMPKγ2RG affect electrophysiology of cardiac myocytes.
The observation of severe kidney injury in the AMPKγ2RG but not in AMPKγ2NI mice fed with an HFD was unexpected. AMPK subunits are abundantly expressed in the kidney (41, 42) and play critical roles in physiological processes, such as sodium transport and podocyte function, and pathological processes like diabetic renal hypertrophy, inflammation, and polycystic kidney disease (10). We are unaware of any reports of kidney injury or disease in humans or mice with the AMPKγ2RG mutation, and it is not known whether our findings are specific to C57BL/6N mice. Genome-wide association studies have identified an intronic variant, rs7805747, in Prkag2 for chronic kidney disease (43). Kidney injury in AMPKγ2RG mice is unlikely to be caused by WPW syndrome or cardiomyopathy because AMPKγ2NI mice did not have this phenotype. Notably, kidney glycogen content of AMPKγ2RG mice was dramatically increased in both diets compared with AMPKγ2WT and AMPKγ2NI mice, which may be one of the reasons for the kidney pathology observed in AMPKγ2RG mice.
It had been reported that AMPK activation suppresses Akt and induces FOXO3a in cervical cancer cells (27). This signaling pathway appears to be conserved in the kidney of AMPKγ2RG mice fed with an HFD. Consistent with a suppression of the Akt pathway by AMPK, apoptosis and inflammation were increased, as indicated by dramatic induction of apoptotic and inflammatory genes in the kidney of AMPKγ2NI and AMPKγ2RG mice. This pathway may have interactions with dyslipidemia or metabolic stress because kidney injury and functional impairment were only observed in AMPKγ2RG mice fed with an HFD. This effect does not seem to be solely caused by increased AMPK activity but specific to AMPKγ2RG mutation. It had been reported that AMPK and autophagy were activated in renal cells under stress conditions, which can cause programmed cell death (44). On the other hand, supraphysiologically activating AMPK by AICAR and metformin was reported to exert a beneficial effect in kidney ischemia-reperfusion (45). Although the underlying mechanism for why AMPKγ2RG, but not AMPKγ2NI, mice showed kidney injury is not fully understood, we speculate that the kidney injury might be associated with the higher baseline activity of AMPKγ2RG and the “loss of AMP response” of AMPKγ2RG, in which cellular AMP cannot bind to one of the binding domains in AMPK γ-subunit (3).
In summary, studies of knock-in mice carrying AMPKγ2NI and AMPKγ2RG mutations at physiological expression levels confirmed the causality of these mutations for WPW syndrome. Further understanding of the mechanisms by which AMPKγ2NI and AMPKγ2RG mutations cause the WPW phenotype can be pursued by using these arguably more relevant mouse models. Due to the elevated baseline AMPK activity, AMPKγ2NI and AMPKγ2RG mice are resistant to the metabolic phenotype characteristically induced by an HFD and exhibit an improved metabolic phenotype, including increased insulin sensitivity, improved dyslipidemia, and liver steatosis. However, the constitutively active mutation of AMPKγ2RG, but not that of AMPKγ2NI, induced kidney injury and functional impairment in mice fed with an HFD, which was accompanied by renal glycogen accumulation, apoptosis, and inflammation (Fig. 7). These AMPK-engineered mouse models provide new tools for investigating the mechanisms leading to WPW syndrome, the roles of AMPK in kidney function and disease, and the effects of physiological AMPK activation on mammalian metabolism and physiology.
FIGURE 7.

Comparison of phenotypes between AMPKγ2NI and AMPKγ2RG mice and the mechanism underlying the kidney injury in AMPKγ2RG mice. Both AMPKγ2NI and AMPKγ2RG mutations increase AMPK activity and cause WPW syndrome in mice, which are independent of glycogen accumulation in the heart. Comparing with AMPKγ2NI, AMPKγ2RG caused glycogen accumulation in the kidney, inhibited pAkt, and increased pFOXO3a, which led to apoptosis, inflammation, cyst formation, and ultimately kidney injury and impaired renal function.
Experimental Procedures
Experimental Animals
All mice used in this study were generated and maintained on C57BL/6N background at Taconic (Germantown, NY) until 8 weeks of age. AMPKγ2NI and AMPKγ2RG knock-in mice were generated by using the Flp recombination method, in which AAT (corresponding amino acid Asn-485) was replaced by ATT (corresponding amino acid Ile-485) in exon 14, and CGG (corresponding amino acid Arg-528) was replaced by GGC (corresponding amino acid Gly-528) in exon 15, respectively. Novel AMPKγ2 new splice variants were not detected by RT-PCR in AMPKγ2NI and AMPKγ2RG hearts. Age-matched AMPKγ2WT, AMPKγ2NI, and AMPKγ2RGmice were genotyped before use. Animals were maintained in a 12-h/12-h light/dark cycle with free access to food and water in an environment with the temperature maintained at 22 °C. Four mice were housed per cage and maintained on regular rodent chow diets or an HFD (20% kcal% carbohydrate and 60% kcal% fat) (Research Diet, catalog no. RD12492, New Brunswick, NJ). Two regular chow diets were used in different studies; one was 5% dietary fat (3.03 kcal/g) (PicoLab® Rodent Diet 20, catalog no. 5053, LabDiet, St. Louis, MO), and the other was 6.4% dietary fat (3.32 kcal/g) (Complete breeding diet for rats, mice, and hamsters, catalog no. D03, Scientific Animal Food and Engineering, SAFE Corporate, Augy, France). All protocols reported in this paper were reviewed and approved by the MRL Institutional Animal Care and Use Committee (Kenilworth, NJ). Animals received appropriate veterinary care throughout the studies. The Guide for the Care and Use of Laboratory Animals was followed in the conduct of the animal studies. Finally, the ARRIVE guidelines published by NC3Rs for reporting the in vivo experiments in animal research were followed.
Metabolic Assays
Two cohorts of age-matched male AMPKγ2WT, AMPKγ2NI, AMPKγ2RG mice were used. One cohort was fed with a regular rodent chow diet from 5 weeks of age until takedown at 26 weeks of age. The second cohort was fed with the regular rodent chow diet until 9 weeks of age and then switched to HFD. Body weight was recorded weekly from 5 to 20 weeks of age. Body composition was evaluated in conscious mice by quantitative NMR using the Minispec+ analyzer (Bruker) at 7 and 19 weeks of age. A glucose tolerance test was performed in mice at 20 weeks of age. Mice were fasted for 4 h starting at 8:00 a.m. followed by a bolus glucose (2 g/kg) administration by oral gavage. Blood glucose was measured at different time points by using a glucometer (Accu-Chek, Roche Diagnostics). An insulin tolerance test was performed in mice at 21 weeks of age. Mice were fasted for 2 h in the morning followed by intraperitoneal administration of insulin (0.5 units/kg). Blood glucose was measured at different time points by a glucometer (Accu-Chek). Plasma levels of TG and cholesterol were measured using the Thermo ScientificTM triglycerides and cholesterol reagent (Fisher, catalog nos. TR22421/2780-250 and TR13421). Plasma levels of free fatty acid and β-hydroxybutyrate were measured using the free fatty acids half-micro test kit (Roche Diagnostics, catalog no. 11383175001) and β-hydroxybutyrate LiquiColor® kit (StanBio, catalog no. 2440-058), respectively.
Electrocardiography and Echocardiography
The in vivo electrophysiology study was performed in chow diet-fed mice at 8 weeks of age and HFD-fed mice at 24 weeks of age. Before the study, mice were anesthetized by using 1–2% isoflurane. Surface electrodes were placed in the right arm and left hind paw (DII configuration), and an ECG was recorded by an electrocardiography ISO DAM8 amplifier (World Precision Instruments) and analogic-numeric conversion box (ITF16A/D converter, EMKA Technologies).
ELISA by MSD and Cisbio HTRF® Assay Kits
Tissues weighed at around 100 mg were homogenized in 1 ml of Tris lysis buffer containing protease and phosphatase inhibitor mixtures (Meso Scale Discovery). Total amounts of protein used for each assay point were 10 μg for liver and kidney (0.4 mg/ml) and 20 μg for skeletal muscle (0.8 mg/ml). The assay was performed according to the manufacturer's recommendations. Determination of pACC was based on the interaction between streptavidin and biotin because ACC is a highly biotinylated protein. MSD GOLD 96-well streptavidin SECTOR plates (MSD, catalog no. L15SA-5) were blocked by blocker buffer (1× Tris wash buffer + BSA (30 mg/ml)) for 1 h, the blocker was removed, and wells were washed three times by wash buffer. Twenty-five μl of samples were added to the plate and incubated at room temperature with shaking for 1 h. After washing three times with 1× Tris wash buffer, primary antibody of rabbit anti-pACC (EMD Millipore, catalog no. 07-303) was diluted in antibody dilution buffer (1 ml of blocker buffer and 2 ml of 1× Tris wash buffer) at 1:250 and 1:500, respectively, and incubated at room temperature with shaking for 1 h. After washing three times with 1× Tris wash buffer, goat anti-rabbit sulfo-tag secondary antibody (MSD, catalog no. R32AB5), diluted at 1:250 in antibody dilution buffer, was added to 25 μl of secondary antibody and incubated at room temperature with shaking for 1 h. Assays of phosphorylated FOXO3a (Thr-32) (MSD, catalog no. K150KHD) and phosphorylated mTOR (Ser-2448)/total mTOR (MSD, catalog no. K15170D) were performed according to the manufacturer's instructions of MSD. pAkt (Thr-308) (catalog no. 64AKTPEG), pAkt (Ser-473) (catalog no. 64AKSPEG), and total Akt (catalog no. 64NKTPEG) were determined by using Cisbio HTRF® assay kits (Cisbio, CA). The relative levels of proteins and ratios of WT mice were arbitrarily set as 1.
Expression Analysis by Real-time RT-PCR
Mouse tissues were collected, placed in RNAlater solution (Qiagen, Hilden, Germany), and stored at 4 °C until processing. Tissues were homogenized and total RNA was isolated by using the RNA Easy kit and QIACube instrument (Qiagen). Two μg of total RNA from each sample were reverse transcribed with a cDNA kit (Life Technologies, Inc.), and mRNA levels for the genes of interest were measured by RT-PCR with SYBR Green Mastermix reagents and RT2 ProfilerTM PCR array for apoptosis and inflammation mouse (Qiagen, catalog nos. PAMM-012Z and PAMM-077Z) on a ViiATM 7 real-time PCR system (Thermo Fisher Scientific) (46). The relative amounts of specific target amplicons for each gene were estimated by a cycle threshold (CT) value and normalized to the copy number of housekeeping genes (β-actin and GAPDH), with all genes in WT mice arbitrarily set at 1 (46). The p values were determined by two-tailed equal variance Student's t test, comparing the 2−ΔCT values of WT versus transgenic mice.
Tissue Glycogen Assay
Approximately 50–100 mg of tissue was weighed and incubated with 300 μl of 30% KOH at 70 °C for 2 h. Sixty μl of 1 m Na2SO4 and 1 ml of ethanol were added, and then the samples were vortexed well followed by centrifugation at 12,000 rpm at room temperature for 20 min. Pellets were washed twice by resuspending in 300 μl of water followed by the addition of 700 μl of ethanol and centrifuged at 12,000 rpm for 20 min. The pellet was then washed with 300 μl of 100% methanol, air-dried at room temperature for 1 h, and then dissolved in 1 ml of water. Glycogen was determined by using the BioVision glycogen assay kit (catalog no. K646-100). Briefly, glycogen samples were diluted 30× in water, and then 35 μl of hydrolysis buffer were added to 10 μl of glycogen sample. Glycogen was hydrolyzed by adding 5 μl of hydrolysis enzyme mix followed by shaking at room temperature for 30 min. Development buffer mix (50 μl) was added to the solution, followed by shaking at room temperature for 30 min. Fluorescence was read at 535-nm excitation/587-nm emission on a SpectraMax M2e reader (Molecular Devices). Tissue glycogen content was quantitated based on the glycogen standard curve and calculated as μg/mg of tissue weight.
Histology of Liver and Kidney
H&E, PAS, and Masson's trichrome stainings were performed on paraffin-embedded sections of liver and kidney tissues using standard protocols (47). Briefly, liver and kidney samples were fixed in 4% formalin for 24 h before being paraffin-embedded and sectioned into 5-μm-thick sections. For standard histology, H&E staining was used to contrast nuclei from cytoplasm. Oil red O staining was performed in cryosectioned slides by using snap frozen kidneys (American MasterTech Inc.). TUNEL staining was performed on paraffin-embedded sections of kidney using the ApopTag® Plus peroxidase in situ apoptosis detection kit (Millipore SAS, catalog no. S7101). Slides were digitized using a Nanozoomer 2.0 HT digital slide scanner (Hamamatsu).
Statistical Analyses
All data are shown as the means ± S.E. Statistical significance was calculated by one-way analysis of variance and multiple comparisons. Asterisks denote statistical significance of the AMPKγ2NI and AMPKγ2RG groups compared with the AMPKγ2WT group: *, p ≤ 0.05; **, p ≤ 0.01; ***, p ≤ 0.001.
Author Contributions
C. L. and R. A. designed the knock-in strategy of AMPKγ2NI and AMPKγ2RG mice. H. P. G. and J. M. designed the experiments. H. P. G., X. Y., and K. L. performed the initial in vivo experiments, including dosing, surgery, blood/tissues/urine collection, and biochemical assays and ELISAs. H. P. G. analyzed and interpreted the data. G. B.-A., M.-F. C., H. J., L. M., G. P., T. S., Y. H., and B. P.-D. did the follow-up metabolic phenotype and cardiac function analysis, including GTT, ITT, histology, electrocardiography, and echocardiography. W. F. and R. W. M. developed and performed the glycogen assay. H. W. supported on bioinformatic modeling of AMPK. L.-J. M. provided insights on renal function and histology analysis. H. P. G. wrote the manuscript. D. E. K., C. L., R. W. M., and M. D. E. revised the manuscript.
Supplementary Material
Acknowledgments
We acknowledge Dr. Grahame Hardie (Cell Signaling and Immunology, College of Life Sciences, University of Dundee) for suggesting that we generate knock-in mice harboring AMPK mutations associated with WPW syndrome, which helped us to understand the causality of WPW syndrome and led to unexpected findings in the kidney. Dr. Lan Yi assisted with cryosection and oil red O staining of kidneys. Dr. Zhu Chen provided suggestions on manuscript revision.
This work was supported by the Merck Research Laboratories. The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental Table 1 and Fig. 1.
- AMPK
- AMP-activated protein kinase
- WPW
- Wolff-Parkinson-White
- ACC
- acetyl-CoA carboxylase
- pACC and pAkt
- phosphorylated ACC and Akt, respectively
- ECG
- electrocardiogram
- TG
- triglycerides
- ACR
- albumin/creatinine ratio
- PAS
- periodic acid-Schiff
- HFD
- high fat diet
- AICAR
- 5-aminoimidazole-4-carboxamide ribonucleotide
- GTT
- glucose tolerance test
- ITT
- insulin tolerance test
- AUC
- area under the curve.
References
- 1. Hardie D. G. (2011) AMP-activated protein kinase: an energy sensor that regulates all aspects of cell function. Genes Dev. 25, 1895–1908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hardie D. G., Ross F. A., and Hawley S. A. (2012) AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 13, 251–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Xiao B., Heath R., Saiu P., Leiper F. C., Leone P., Jing C., Walker P. A., Haire L., Eccleston J. F., Davis C. T., Martin S. R., Carling D., and Gamblin S. J. (2007) Structural basis for AMP binding to mammalian AMP-activated protein kinase. Nature 449, 496–500 [DOI] [PubMed] [Google Scholar]
- 4. Cheung P. C., Salt I. P., Davies S. P., Hardie D. G., and Carling D. (2000) Characterization of AMP-activated protein kinase γ-subunit isoforms and their role in AMP binding. Biochem. J. 346, 659–669 [PMC free article] [PubMed] [Google Scholar]
- 5. Hawley S. A., Boudeau J., Reid J. L., Mustard K. J., Udd L., Mäkelä T. P., Alessi D. R., and Hardie D. G. (2003) Complexes between the LKB1 tumor suppressor, STRAD α/β and MO25 α/β are upstream kinases in the AMP-activated protein kinase cascade. J. Biol. 2, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Woods A., Johnstone S. R., Dickerson K., Leiper F. C., Fryer L. G., Neumann D., Schlattner U., Wallimann T., Carlson M., and Carling D. (2003) LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr. Biol. 13, 2004–2008 [DOI] [PubMed] [Google Scholar]
- 7. Shaw R. J., Kosmatka M., Bardeesy N., Hurley R. L., Witters L. A., DePinho R. A., and Cantley L. C. (2004) The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc. Natl. Acad. Sci. U.S.A. 101, 3329–3335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hardie D. G. (2015) AMPK: positive and negative regulation, and its role in whole-body energy homeostasis. Curr. Opin. Cell Biol. 33, 1–7 [DOI] [PubMed] [Google Scholar]
- 9. Carling D., Zammit V. A., and Hardie D. G. (1987) A common bicyclic protein kinase cascade inactivates the regulatory enzymes of fatty acid and cholesterol biosynthesis. FEBS Lett. 223, 217–222 [DOI] [PubMed] [Google Scholar]
- 10. Hallows K. R., Mount P. F., Pastor-Soler N. M., and Power D. A. (2010) Role of the energy sensor AMP-activated protein kinase in renal physiology and disease. Am. J. Physiol. Renal Physiol. 298, F1067–F1077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gollob M. H., Green M. S., Tang A. S., Gollob T., Karibe A., Ali Hassan A. S., Ahmad F., Lozado R., Shah G., Fananapazir L., Bachinski L. L., Roberts R., and Hassan A. S. (2001) Identification of a gene responsible for familial Wolff-Parkinson-White syndrome. N. Engl. J. Med. 344, 1823–1831 [DOI] [PubMed] [Google Scholar]
- 12. Morita H., Rehm H. L., Menesses A., McDonough B., Roberts A. E., Kucherlapati R., Towbin J. A., Seidman J. G., and Seidman C. E. (2008) Shared genetic causes of cardiac hypertrophy in children and adults. N. Engl. J. Med. 358, 1899–1908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zaha V. G., and Young L. H. (2012) AMP-activated protein kinase regulation and biological actions in the heart. Circ. Res. 111, 800–814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Burwinkel B., Scott J. W., Bührer C., van Landeghem F. K., Cox G. F., Wilson C. J., Grahame Hardie D., and Kilimann M. W. (2005) Fatal congenital heart glycogenosis caused by a recurrent activating R531Q mutation in the γ2-subunit of AMP-activated protein kinase (PRKAG2), not by phosphorylase kinase deficiency. Am. J. Hum. Genet. 76, 1034–1049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Arad M., Benson D. W., Perez-Atayde A. R., McKenna W. J., Sparks E. A., Kanter R. J., McGarry K., Seidman J. G., and Seidman C. E. (2002) Constitutively active AMP kinase mutations cause glycogen storage disease mimicking hypertrophic cardiomyopathy. J. Clin. Invest. 109, 357–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Arad M., Moskowitz I. P., Patel V. V., Ahmad F., Perez-Atayde A. R., Sawyer D. B., Walter M., Li G. H., Burgon P. G., Maguire C. T., Stapleton D., Schmitt J. P., Guo X. X., Pizard A., Kupershmidt S., Roden D. M., Berul C. I., Seidman C. E., and Seidman J. G. (2003) Transgenic mice overexpressing mutant PRKAG2 define the cause of Wolff-Parkinson-White syndrome in glycogen storage cardiomyopathy. Circulation 107, 2850–2856 [DOI] [PubMed] [Google Scholar]
- 17. Luptak I., Shen M., He H., Hirshman M. F., Musi N., Goodyear L. J., Yan J., Wakimoto H., Morita H., Arad M., Seidman C. E., Seidman J. G., Ingwall J. S., Balschi J. A., and Tian R. (2007) Aberrant activation of AMP-activated protein kinase remodels metabolic network in favor of cardiac glycogen storage. J. Clin. Invest. 117, 1432–1439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Davies J. K., Wells D. J., Liu K., Whitrow H. R., Daniel T. D., Grignani R., Lygate C. A., Schneider J. E., Noël G., Watkins H., and Carling D. (2006) Characterization of the role of gamma2 R531G mutation in AMP-activated protein kinase in cardiac hypertrophy and Wolff-Parkinson-White syndrome. Am. J. Physiol. Heart. Circ. Physiol. 290, H1942–H1951 [DOI] [PubMed] [Google Scholar]
- 19. Hawley S. A., Ross F. A., Chevtzoff C., Green K. A., Evans A., Fogarty S., Towler M. C., Brown L. J., Ogunbayo O. A., Evans A. M., and Hardie D. G. (2010) Use of cells expressing γ subunit variants to identify diverse mechanisms of AMPK activation. Cell Metab. 11, 554–565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gollob M. H., Seger J. J., Gollob T. N., Tapscott T., Gonzales O., Bachinski L., and Roberts R. (2001) Novel PRKAG2 mutation responsible for the genetic syndrome of ventricular preexcitation and conduction system disease with childhood onset and absence of cardiac hypertrophy. Circulation 104, 3030–3033 [DOI] [PubMed] [Google Scholar]
- 21. Kim M., Hunter R. W., Garcia-Menendez L., Gong G., Yang Y. Y., Kolwicz S. C. Jr., Xu J., Sakamoto K., Wang W., and Tian R. (2014) Mutation in the γ2-subunit of AMP-activated protein kinase stimulates cardiomyocyte proliferation and hypertrophy independent of glycogen storage. Circ. Res. 114, 966–975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Davies S. P., Sim A. T., and Hardie D. G. (1990) Location and function of three sites phosphorylated on rat acetyl-CoA carboxylase by the AMP-activated protein kinase. Eur. J. Biochem. 187, 183–190 [DOI] [PubMed] [Google Scholar]
- 23. Accornero F., van Berlo J. H., Benard M. J., Lorenz J. N., Carmeliet P., and Molkentin J. D. (2011) Placental growth factor regulates cardiac adaptation and hypertrophy through a paracrine mechanism. Circ. Res. 109, 272–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Birk J. B., and Wojtaszewski J. F. (2006) Predominant α2/β2/γ3 AMPK activation during exercise in human skeletal muscle. J. Physiol. 577, 1021–1032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Merrill G. F., Kurth E. J., Hardie D. G., and Winder W. W. (1997) AICA riboside increases AMP-activated protein kinase, fatty acid oxidation, and glucose uptake in rat muscle. Am. J. Physiol. 273, E1107–E1112 [DOI] [PubMed] [Google Scholar]
- 26. de Geus H. R., Betjes M. G., and Bakker J. (2012) Biomarkers for the prediction of acute kidney injury: a narrative review on current status and future challenges. Clin. Kidney J. 5, 102–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yung M. M., Chan D. W., Liu V. W., Yao K. M., and Ngan H. Y. (2013) Activation of AMPK inhibits cervical cancer cell growth through AKT/FOXO3a/FOXM1 signaling cascade. BMC Cancer 13, 327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chopra I., Li H., Bishopric N. H., and Webster K. A. (2008) An insulin-independent AMPK-dependent survival pathway is activated in glucose-starved cardiac myocytes through IRS-1/PI3-kinase and rictor-mediated activation of PDK1 and PDK2. Circulation 118, S_486 [Google Scholar]
- 29. Brunet A., Bonni A., Zigmond M. J., Lin M. Z., Juo P., Hu L. S., Anderson M. J., Arden K. C., Blenis J., and Greenberg M. E. (1999) Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 96, 857–868 [DOI] [PubMed] [Google Scholar]
- 30. Inoki K., Zhu T., and Guan K. L. (2003) TSC2 mediates cellular energy response to control cell growth and survival. Cell 115, 577–590 [DOI] [PubMed] [Google Scholar]
- 31. Gwinn D. M., Shackelford D. B., Egan D. F., Mihaylova M. M., Mery A., Vasquez D. S., Turk B. E., and Shaw R. J. (2008) AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 30, 214–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kahn B. B., Alquier T., Carling D., and Hardie D. G. (2005) AMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab. 1, 15–25 [DOI] [PubMed] [Google Scholar]
- 33. Corton J. M., Gillespie J. G., Hawley S. A., and Hardie D. G. (1995) 5-aminoimidazole-4-carboxamide ribonucleoside: a specific method for activating AMP-activated protein kinase in intact cells? Eur. J. Biochem. 229, 558–565 [DOI] [PubMed] [Google Scholar]
- 34. Hunter R. W., Treebak J. T., Wojtaszewski J. F., and Sakamoto K. (2011) Molecular mechanism by which AMP-activated protein kinase activation promotes glycogen accumulation in muscle. Diabetes 60, 766–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Halse R., Fryer L. G., McCormack J. G., Carling D., and Yeaman S. J. (2003) Regulation of glycogen synthase by glucose and glycogen: a possible role for AMP-activated protein kinase. Diabetes 52, 9–15 [DOI] [PubMed] [Google Scholar]
- 36. Costford S. R., Kavaslar N., Ahituv N., Chaudhry S. N., Schackwitz W. S., Dent R., Pennacchio L. A., McPherson R., and Harper M. E. (2007) Gain-of-function R225W mutation in human AMPKγ(3) causing increased glycogen and decreased triglyceride in skeletal muscle. PLoS One 2, e903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Milan D., Jeon J. T., Looft C., Amarger V., Robic A., Thelander M., Rogel-Gaillard C., Paul S., Iannuccelli N., Rask L., Ronne H., Lundström K., Reinsch N., Gellin J., Kalm E., et al. (2000) A mutation in PRKAG3 associated with excess glycogen content in pig skeletal muscle. Science 288, 1248–1251 [DOI] [PubMed] [Google Scholar]
- 38. Light P. E. (2006) Familial Wolff-Parkinson-White syndrome: a disease of glycogen storage or ion channel dysfunction? J. Cardiovasc. Electrophysiol. 17, S158–S161 [DOI] [PubMed] [Google Scholar]
- 39. Light P. E., Wallace C. H., and Dyck J. R. (2003) Constitutively active adenosine monophosphate-activated protein kinase regulates voltage-gated sodium channels in ventricular myocytes. Circulation 107, 1962–1965 [DOI] [PubMed] [Google Scholar]
- 40. Benditt D. G., Pritchett L. C., Smith W. M., Wallace A. G., and Gallagher J. J. (1978) Characteristics of atrioventricular conduction and the spectrum of arrhythmias in Lown-Ganong-Levine syndrome. Circulation 57, 454–465 [DOI] [PubMed] [Google Scholar]
- 41. Cammisotto P. G., Londono I., Gingras D., and Bendayan M. (2008) Control of glycogen synthase through ADIPOR1-AMPK pathway in renal distal tubules of normal and diabetic rats. Am. J. Physiol. Renal. Physiol. 294, F881–F889 [DOI] [PubMed] [Google Scholar]
- 42. Fraser S., Mount P., Hill R., Levidiotis V., Katsis F., Stapleton D., Kemp B. E., and Power D. A. (2005) Regulation of the energy sensor AMP-activated protein kinase in the kidney by dietary salt intake and osmolality. Am. J. Physiol. Renal. Physiol. 288, F578–F586 [DOI] [PubMed] [Google Scholar]
- 43. Köttgen A., Pattaro C., Böger C. A., Fuchsberger C., Olden M., Glazer N. L., Parsa A., Gao X., Yang Q., Smith A. V., O'Connell J. R., Li M., Schmidt H., Tanaka T., Isaacs A., et al. (2010) New loci associated with kidney function and chronic kidney disease. Nat. Genet. 42, 376–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kume S., Thomas M. C., and Koya D. (2012) Nutrient sensing, autophagy, and diabetic nephropathy. Diabetes 61, 23–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Decleves A. E., Sharma K., and Satriano J. (2014) Beneficial effects of AMP-activated protein kinase agonists in kidney ischemia-reperfusion: autophagy and cellular stress markers. Nephron Exp. Nephrol. 10.1159/000368932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Jensen K. K., Previs S. F., Zhu L., Herath K., Wang S. P., Bhat G., Hu G., Miller P. L., McLaren D. G., Shin M. K., Vogt T. F., Wang L., Wong K. K., Roddy T. P., Johns D. G., and Hubbard B. K. (2012) Demonstration of diet-induced decoupling of fatty acid and cholesterol synthesis by combining gene expression array and 2H2O quantification. Am. J. Physiol. Endocrinol. Metab 302, E209–E217 [DOI] [PubMed] [Google Scholar]
- 47. Cardiff R. D., Miller C. H., and Munn R. J. (2014) Manual hematoxylin and eosin staining of mouse tissue sections. Cold Spring Harb. Protoc. 2014, 655–658 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.