

Abstract
The aggregation of insulin into amyloid fibers has been a limiting factor in the development of fast acting insulin analogues, creating a demand for excipients that limit aggregation. Despite the potential demand, inhibitors specifically targeting insulin have been few in number. Here we report a non-toxic and serum stable-designed heptapeptide, KR7 (KPWWPRR-NH2), that differs significantly from the primarily hydrophobic sequences that have been previously used to interfere with insulin amyloid fibrillation. Thioflavin T fluorescence assays, circular dichroism spectroscopy, and one-dimensional proton NMR experiments suggest KR7 primarily targets the fiber elongation step with little effect on the early oligomerization steps in the lag time period. From confocal fluorescence and atomic force microscopy experiments, the net result appears to be the arrest of aggregation in an early, non-fibrillar aggregation stage. This mechanism is noticeably different from previous peptide-based inhibitors, which have primarily shifted the lag time with little effect on later stages of aggregation. As insulin is an important model system for understanding protein aggregation, the new peptide may be an important tool for understanding peptide-based inhibition of amyloid formation.
Keywords: amyloid, atomic force microscopy (AFM), fluorescence anisotropy, inhibitor, insulin
Introduction
Protein misfolding results in the formation of insoluble aggregates that are often in the form of elongated fibers with a characteristic cross-β-sheet structure called amyloid fibers. Excessive accumulation of these aggregates can compromise the activity of the affected tissue or organ. Although the ultimate involvement of amyloidogenic proteins in disease is a subject of considerable controversy (1), this process is often correlated with the development of severe pathological conditions such as Alzheimer's and Parkinson's disease, type II diabetes, and Huntington's disease (2, 3). The process of amyloid fibril formation initiates under certain conditions from both unstructured peptides and structured globular proteins and traverses through a series of structural modifications and metastable oligomers before ultimately forming insoluble amyloid fibers (4). Despite intensive research, the exact mechanism for amyloid fibrillation has yet to be fully understood.
One of the proteins where amyloidogenesis is of potential concern is insulin. Insulin plays a major role in blood glucose homeostasis and pharmaceutical preparations of insulin are commonly used to treat diabetes. Amyloidogenesis is a major concern during insulin manufacture, as insulin in the amyloid form is not bioavailable, and any degree of amyloid formation leads to reduced efficacy of insulin administration (5). In continuous delivery systems such as insulin pumps, amyloid formation may cause fouling of the system and reduced or erratic delivery rates (6). Insulin amyloidosis at the site of injection may also lead to poor penetration of the injected insulin and a variable insulin response (7, 8). Because insulin delivery must be tightly calibrated, any of these situations can result in diabetic ketoacidosis due to insulin deficiency, a life-threatening complication (6).
To prevent these occurrences, commercial insulin preparations are formulated with excipients to limit aggregation, typically by promoting the zinc bound hexameric form over the monomeric state, which is more prone to fibrillation (9–11). In its zinc-bound hexameric form, insulin is a helical protein with each subunit consisting of two disulfide-linked polypeptide chains of 21 and 30 amino acid residues each: an A-chain consisting of a helical hairpin (A2-A8 and A13-A19) and a B-chain with a single helix (B9-B19) (PDB code 2ZP6). Insulin is more conformationally unstable as a monomer than as a hexamer, and partial unfolding of insulin structure is believed to expose hydrophobic regions of the peptide that trigger eventual aggregation (12–14). Stabilizing the hexameric form has disadvantages, as insulin is most active in the monomeric form (15–18). Shifting the equilibrium to the hexameric form therefore delays the onset of insulin activity. Faster acting insulin analogues such as lispro that remain in the monomeric state have been created to address this problem, but these analogues suffer from more severe aggregation problems (5). An excipient that would allow monomeric insulin's advantage of fast-action but eliminate the corresponding disadvantage of aggregation would have obvious clinical utility (19).
Beyond its immediate clinical application, insulin is an important model system for the study of amyloid fibrillation. Insulin can be triggered to form amyloid fibrils rapidly in vitro upon exposure to specific conditions such as low pH, high temperature, and higher ionic strength (17). Insulin is cheap, soluble at high concentrations, and available in large amounts, factors that often limit the type of experiments that can be done on other amyloid systems. For this reason, insulin has been used extensively to understand the generic physical chemistry of aggregating systems (20–29).
Despite these advantages, known inhibitors for insulin aggregation are comparatively few, at least in comparison to other amyloid proteins such as Aβ5 and α-synuclein. A few small molecules such as epigallocatechin-3-gallate (30), quercetin (31), quinones (32) have been explored as potential inhibitors for insulin aggregation. In this work we report a non-toxic and serum stable heptapeptide named KR7 (KPWWPRR-NH2) derived from the antimicrobial peptide indolicidin that inhibits insulin amyloid fibrillation. In contrast to other insulin peptide inhibitors (19, 33, 34), KR7 does not contain a self-recognition sequence based on the insulin peptide and, therefore, represents a new point of attack. Using a variety of biophysical techniques, we show KR7 decreases the elongation rate of fibers without affecting the lag time, a different mechanism than current peptide-based inhibitors (19, 33).
Results and Discussion
Peptide Design
The designed peptide, KR7 (KPWWPRR-NH2), is derived with a slight modification from the known antimicrobial peptide indolicidin. It harbors a PWWP motif at the center flanked by the positively charged residues Lys and Arg at the N and C termini, respectively. Recently it was reported that tripeptides WWW, WPW, WWP, and PWW bind to amyloid fibrils of Aβ9–40 at the hydrophobic groove with very high affinity. Among the tripeptides, PWW exhibited the highest binding affinity toward amyloid fibers (35). Based on the hypothesis that amyloid fibers of different proteins often share similar structural properties, we were interested in testing the activity of KR7 with insulin, as the central region of the KR7 has overlapping PWW and WWP motifs. Insulin is also rich in hydrophobic amino acid residues, similar to the Aβ peptide. Interestingly, the insulin chain B is mostly conserved among mammals, whereas chain A exhibits sequence variability as shown is supplemental Fig. S1A. Insulin has a high proportion of hydrophobic residues, i.e. 5.9% Phe, 5.9% Ala, 7.8% Gly, 9.8% Val, and 11.8% Leu for the bovine variant used in this study. As per the Protscale server (36), hydrophobicity reaches a maximum at Leu-15 of chain B (supplemental Fig. S1B).
Fibrillation Kinetics of Insulin
ThT Fluorescence Assay
We first used thioflavin T (ThT) fluorescence to determine the effects of KR7 on the kinetics of insulin amyloid fibrillation at low pH and high temperature (335 K, pH 2.6), conditions in which insulin is primarily dimeric and forms fibers relatively rapidly (17). ThT binds specifically to the cross-β sheet structure of amyloid fibers and fluoresces more intensely once bound. Provided that the binding is specific, ThT fluorescence gives a measure of the fibrillar population of the amyloid protein in amyloidogenic conditions (37). The fibrillation kinetics of insulin shows the characteristic three stages typical of a nucleation dependent fibrillation process (Fig. 1A) (38) i.e. a quiescent lag phase, an elongation phase where fiber formation increases rapidly, and a saturation phase as the concentration of free peptide decreases.
FIGURE 1.
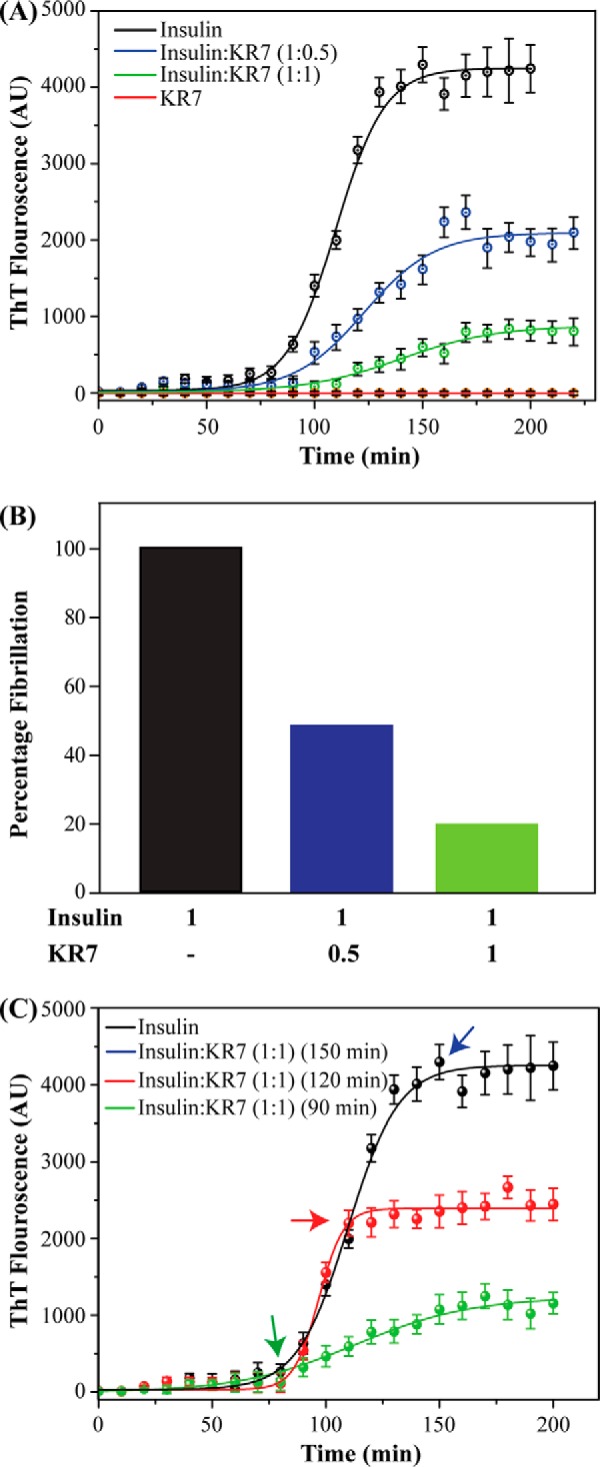
Kinetics of insulin amyloid fibrillation in the presence of KR7 by ThT assay. All experiments were performed using 2 mg/ml insulin at 335 K in 50 mm citrate phosphate buffer with 100 mm NaCl at pH 2.6. A, kinetics of insulin fibrillation in the presence of varying ratios of KR7 peptide. KR7 decreases the rate of fiber formation with little effect on the lag time before fiber formation. AU, atomic units, B, bar plot of the normalized final ThT fluorescence from A. KR7 reduces the total amount of amyloid formed after the reaction has reached completion. C, effect of KR7 addition at different time points. KR7 arrests fiber formation immediately after the addition, but breakdown of fibers is minor. This pattern is consistent with KR7 stopping the fiber elongation step.
The KR7 peptide inhibits insulin fibrillation in a concentration-dependent manner (Fig. 1A). As a control experiment, KR7 by itself was tested at a concentration of 350 μm (similar to the concentration used at the highest KR7:insulin molar ratio) and did not show any sign of fibrillation (Fig. 1A, red). Insulin to KR7 molar ratio of 1:0.5 reduces the total amount of fiber formation by 50%, whereas a 1:1 molar ratio of KR7 to insulin reduces insulin fibrillation by 80% (Fig. 1B). KR7 has little effect on the early lag phase but reduces both the rate and total amount of amyloid formed (supplemental Table S1). This specific kinetic profile suggests KR7 acts at a late stage in the aggregation process, possibly by blocking fiber elongation. To test this hypothesis (39), KR7 was added at different points along the aggregation pathway in a 1:1 ratio (Fig. 1C). Adding KR7 at the midpoint (Fig. 1C, red) arrested aggregation completely but did not decrease ThT fluorescence. Similarly, adding KR7 near the end (Fig. 1C, blue) resulted in a partial decrease of ThT fluorescence consistent with arrest of fibrillation and incomplete breakdown of the fiber product. Adding KR7 at the end of the lag phase results in normal fiber formation at a reduced rate (Fig. 1C, green). Overall these results are consistent with KR7 primarily targeting the fiber elongation step.
Insulin Fibrillation Kinetics with NMR Spectroscopy
ThT kinetic assays can be deceptive because only the final amyloid product is measured. In a complicated system in which only one species is detected many models can be consistent with the data. To gain further insight into the inhibition process at the molecular level, we turned to other biophysical techniques to directly measure kinetic events before fiber elongation. One-dimensional proton NMR is one of the most powerful of these techniques because of its high sensitivity to small changes in conformation and association (40, 41). To observe possible changes that ThT cannot detect, we took one-dimensional NMR spectra periodically under conditions similar to those used for the ThT experiments. In the absence of KR7, the intensity in the sensitive aromatic and amide region was reduced by almost 5-fold after 14 h (Fig. 2A). This decrease is an indication of the formation of aggregates too large to detect by NMR (42, 43), which can be directly seen in the inset photo of the NMR tube (Fig. 2A, inset). On the other hand, in the presence of KR7 in a 1:1 ratio, peaks could be detected even after 14 h (Fig. 2B), and no such precipitation was observed (Fig. 2B, inset). Qualitatively, the NMR results are in overall agreement with the ThT data, which suggest that KR7 suppresses the formation of large aggregates.
FIGURE 2.
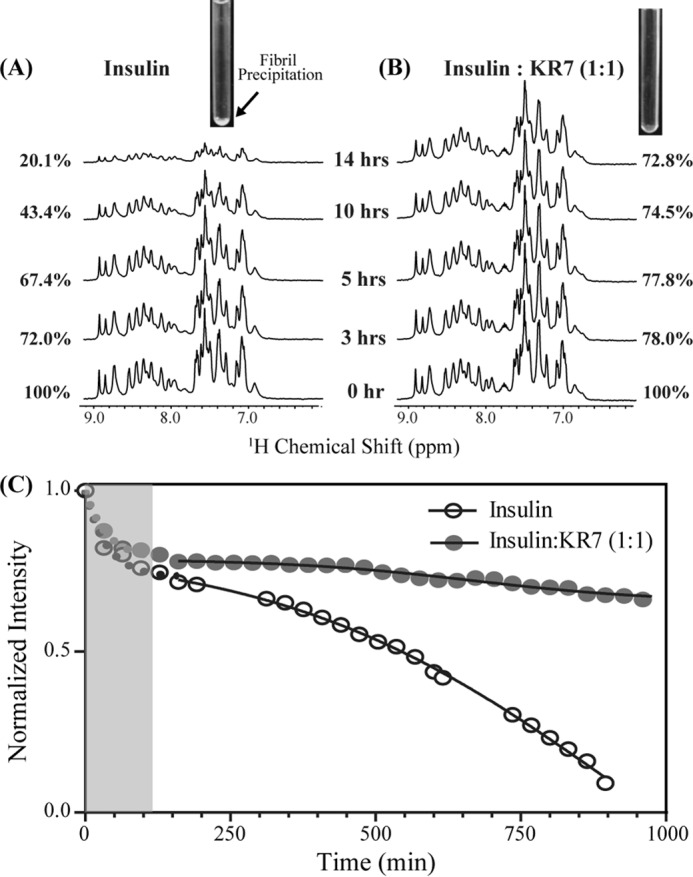
One-dimensional 1H NMR time-course of insulin at 335 K in the presence and absence of KR7 peptide. Conditions were similar to those used in the ThT experiment: 2 mg/ml insulin in 50 mm citrate phosphate buffer with 100 mm NaCl (pH 2.6). A, one-dimensional 1H NMR of the aromatic and amide region of insulin alone. Inset, image of the NMR tube at the end of the experiment. A precipitate is visible at the bottom of the tube. B, one-dimensional 1H NMR of aromatic and amide region of insulin in the presence of KR7 at 1:1 molar ratio. Inset, image of the NMR tube at the end of the experiment. No precipitate is visible at the bottom of the tube. C, time course of the integrated intensity. The data were fit to a two part model consisting of an exponential first stage (shaded region) and a sigmoidal second stage (solid line). KR7 has little effect on the initial phase in the lag phase of the ThT data but a strong effect on the latter phase.
A closer look at the one-dimensional NMR time-course allows a more precise determination of the point of KR7 inhibition. In both cases, the decay of the signal can be divided into two distinct phases (Fig. 2C): a significant decrease occurring immediately after dissolution that plateaus at around 160 min and a slower sigmoidal type transition reminiscent of the ThT kinetic profile. The absence of a corresponding decrease in the lag phase of the ThT data suggests the first phase is the formation of non-amyloid aggregates not detected by ThT. The absence of amyloid fibers in the first phase can be confirmed by circular dichroism (CD) experiments which show that insulin remains in an α-helical conformation for the first 120 min before slowly transitioning to the β-sheet structure of amyloid fibers (Fig. 3).
FIGURE 3.
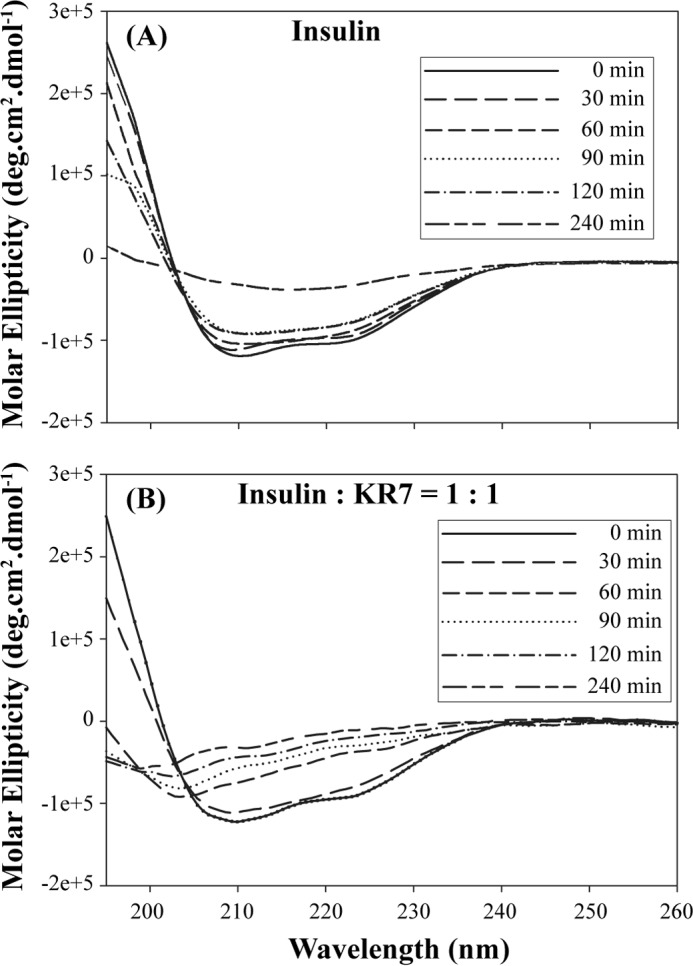
Relative changes in secondary structure of insulin during the course of fibrillation in the presence and absence of KR7 by CD spectroscopy. A, far UV CD spectra of insulin under fibrillating condition recorded at indicated time points. B, far UV CD spectra of insulin+KR7 (1:1) under fibrillating condition recorded at the indicated time points.
The two-phase profile, therefore, confirms at least two separate kinetic steps are involved in aggregation: nucleation from a non-amyloid intermediate followed by amyloid formation by fiber elongation. KR7 does not have a statistically measurable effect on the first phase linked to nucleation (τ = 37.4 m versus 37.8 m for insulin alone) but does significantly inhibit the second phase linked to fiber elongation (τ = 666 ± 21 m versus 161 ± 24 m for insulin alone), in agreement with the ThT data.
Microscopic Observation of Insulin Fibrillation with and without KR7
Confocal Microscopy
The ThT fibrillation assay showed that the KR7 peptide effectively inhibits insulin fibrillation. Because many of the biological properties of aggregates are sensitive to not only the microscopic structure but also their physical dimensions (44–47), we turned to microscopy to understand how KR7 affects the higher order morphology of insulin aggregates. First, images of insulin amyloid formation were acquired using confocal microscopy using ThT fluorescence to measure the effect of KR7 on the morphology of the amyloid fibers specifically. Representative images of selected time points are displayed in Fig. 4; representative images of the entire time course are shown in supplemental Fig. S2. In both samples with KR7 and those without, the density and size of ThT positive aggregates gradually increases with time and follows the same two-step process; amorphous aggregates are formed first followed by the dense accumulation of short clusters of curly fibers.
FIGURE 4.
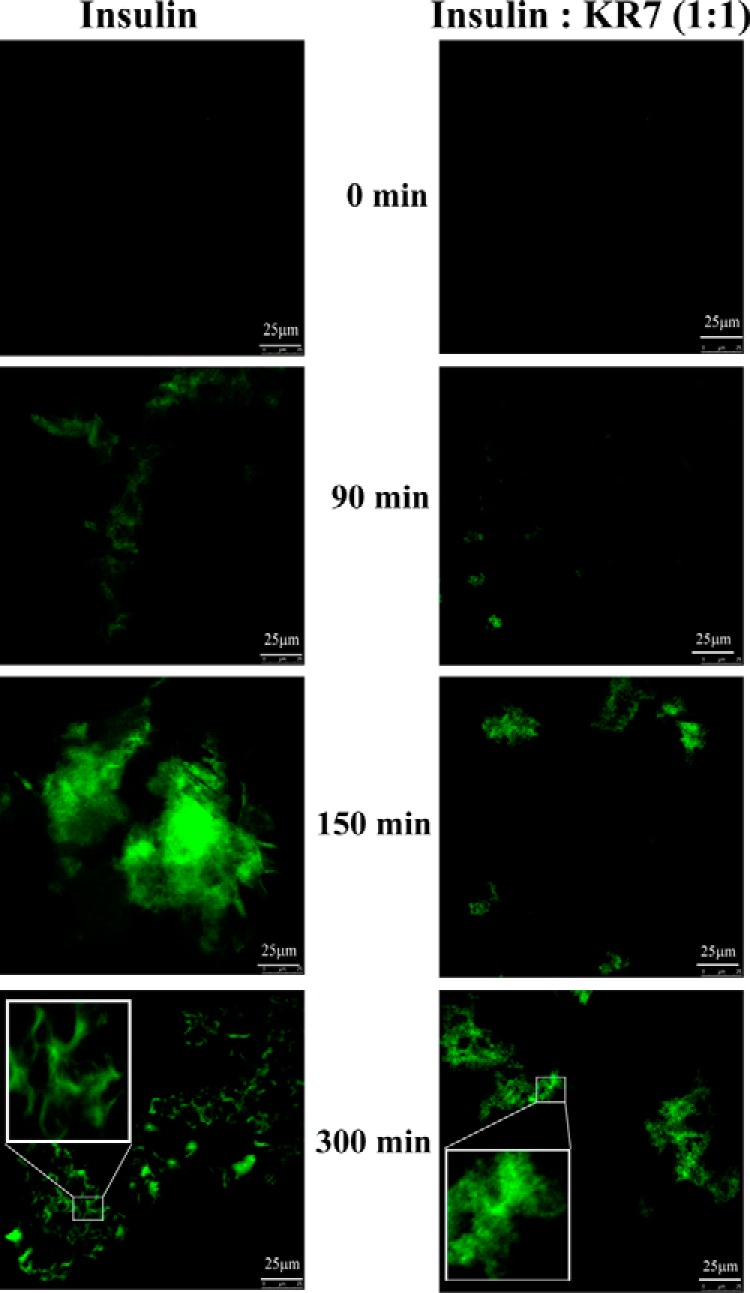
Confocal images of insulin fibrillation by ThT fluorescence. Fluorescence confocal images of insulin amyloids recorded at time points indicated during the course of insulin fibrillation in the presence and absence of KR7 peptide. 10-μl aliquots of the reaction mixture from (2 mg/ml) stock samples were placed on clean glass slides and dried inside laminar airflow in dark at each time point. The inset displays the zoomed view of indicated region in square box. Insulin alone displays a curly fibrillar mass at 300 min in the presence of KR7 (1:1) exhibits amorphous aggregates after the same 300 min of aggregation time.
Confocal ThT imaging can only detect aggregates with the cross β-sheet structure characteristic of amyloid fibers. It also lacks the resolution to distinguish distinct fibrillar species. To resolve the finer details of we employed time-lapse AFM to visualize the progression of insulin fibrillation. The results are similar to those obtained by confocal ThT imaging and consistent with the both the NMR and ThT results. Small oligomers form first within the lag time of the ThT experiment (Fig. 1) in both the insulin and equimolar insulin+KR7 samples (Fig. 5A). At around 150 min the fate of the two samples diverges. In the absence of KR7, insulin proceeds to form large amorphous aggregates (Fig. 5A) followed by dense fiber-like structures (Fig. 5A), similar to the sequence of events observed by confocal ThT imaging. With KR7, progression is halted on the amorphous aggregation stage. These results were confirmed by TEM (Fig. 5B). As described earlier (48), the amyloid products were incubated for few days at room temperature before applying sample on TEM grids. In KR7's absence, the TEM micrograph of insulin (Fig. 5B) showed densely packed branched fibers, but in the presence of KR7 peptide at a molar ratio of 1:1 no such fibrillar species were observed. Because dissolution of peptide from amyloid fibers is known to be a slow process, the system does not likely reach true equilibrium in the time frame of the ThT experiment. To see if the amyloid fibers are actually broken down at longer time points, KR7 was added at an equimolar concentration to a sample of insulin aggregated overnight, and the resulting sample was imaged by TEM (Fig. 5C). Small spherical aggregates were visible, whereas the larger fibers seen in Fig. 5B are noticeably absent in Fig. 5C.
FIGURE 5.

High resolution microscopic images of insulin fibrillation in presence and absence of KR7. A, time-lapse atomic force microscopy images of insulin fibrillation. AFM samples were prepared on mica sheets using 10-μl aliquots taken from (2 mg/ml) stock samples with or without KR7 peptide in an equimolar (1:1) ratio and dried for overnight at room temperature. Height of the sample is indicated in pseudo color. B, TEM images of mature insulin fibril in the absence (left panel) and presence (right panel) of KR7 at the end of the reaction. C, high resolution microscopic images of insulin fiber dissolution with KR7. Shown is a TEM image of 10-μl aliquots taken from (2 mg/ml) stock fibril incubated overnight with equimolar concentration of KR7 peptide.
KR7 Binds Both Free Insulin and Insulin Fibers in the Low Micromolar Range
The kinetic studies suggest KR7 inhibits the fiber elongation process but is ambiguous as to the actual target of KR7. To establish the actual molecular target, we used fluorescence anisotropy binding assays to measure the interaction of KR7 with insulin at different stages in the aggregation process. Binding of a ligand to a receptor results in a marked reduction in the rotational diffusion of the ligand, which in turn increases the value of fluorescence anisotropy. Interpretation of the fluorescence anisotropy values is simplified by the fact that KR7 has two tryptophan residues, whereas bovine insulin lacks tryptophan. Using freshly dissolved insulin, the fluorescence anisotropy increased in a dose-dependent manner with increasing insulin (Fig. 6). Fitting the data, an apparent KD value of 2.6 ± 1.1 μm was found for the initial insulin-KR7 interaction (assuming a 1:1 binding model); similar but slightly higher KD values were found at later time points (see below). Similar results were obtained at pH 7.4 (supplemental Fig. S3). The high affinity of this interaction might suggest KR7 blocks fiber extension by binding either monomeric or dimeric insulin and forming a complex incapable of supporting fiber extension (43). However, KR7 has little effect on the lag time, which suggests it binds in such a way that it does not prevent the transition to the early large oligomers seen in confocal imaging. Therefore, it is also possible that soluble oligomers are present at the intermediate time points and contribute to the anisotropy increase.
FIGURE 6.
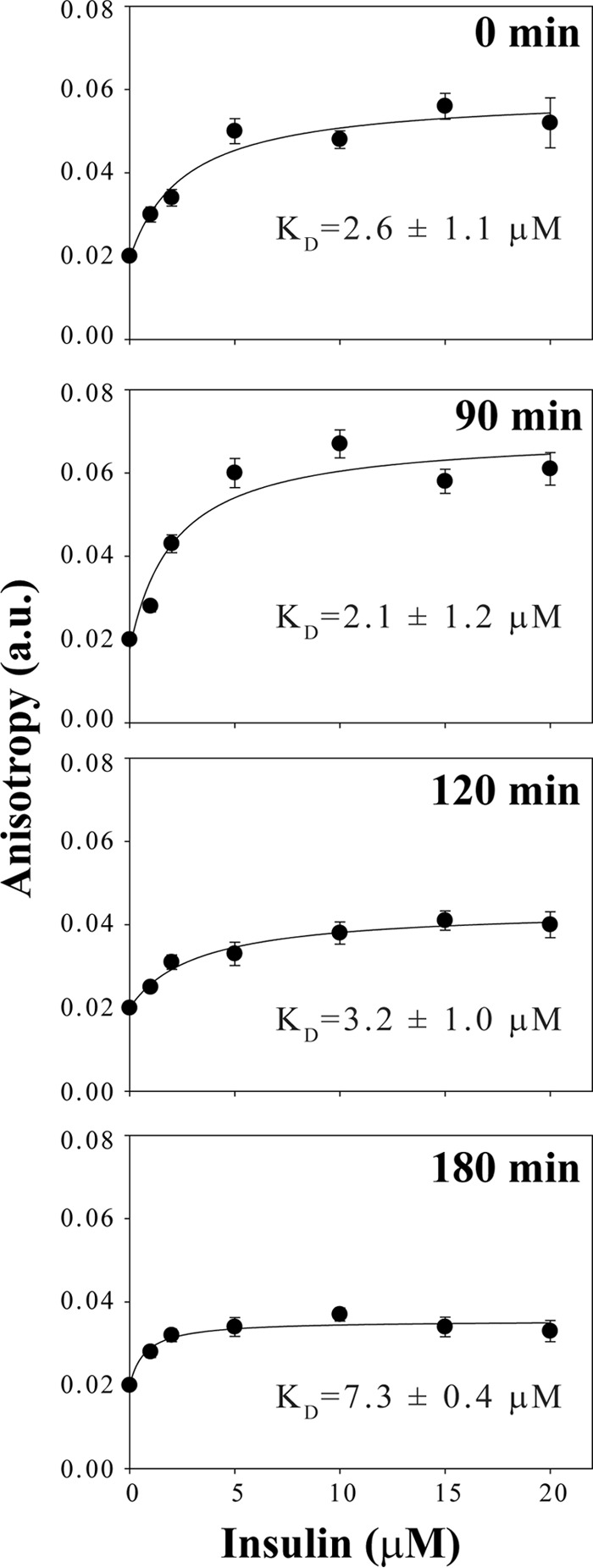
Fluorescence anisotropy measurements of the binding of KR7 to insulin at different time points. Anisotropy of the tryptophan fluorescence of 5 μm KR7 peptide in 50 mm citrate phosphate buffer, 100 mm NaCl (pH 2.6) with gradual addition of insulin from 1 μm to 20 μm concentration from a 2 mg/ml stock solution in 50 mm citrate phosphate, 100 mm NaCl (pH 2.6) aggregated at 335 K for the indicated time period.
The binding constant of interaction of KR7 was also determined at later time points, with presumably different levels of the intermediates of insulin fibrillation. The KD value for interaction at 90, 120, and 180 min were 2.1 ± 1.2 μm, 3.2 ± 1.0 μm, and 7.3 ± 0.4 μm, respectively (Fig. 6), reflecting a pattern in which the affinity of KR7 first increases then decreases as time progresses, although caution should be used in interpreting these values in a quantitative manner due to the uncertainty in the binding stoichiometry and the exact species present at each time point. Under similar experimental conditions the anisotropy of the insulin control did not show any significant change at each mentioned time points (data not shown), eliminating scattered light from insulin aggregates as a likely reason for the anisotropy increase. The fluorescence anisotropy suggests KR7 has affinity for free, oligomeric, and fibrillar insulin.
Fluorescence anisotropy cannot be used to determine the sites on insulin at which KR7 interacts. To investigate the vital residues for insulin binding with KR7, we performed a two-dimensional 1H,1H nuclear Overhauser effect spectroscopy (NOESY) experiment of insulin in the presence of 20% acetic acid-d4, a condition in which insulin is completely monomeric due to the influence of acetic acid (49, 50). The NOESY spectra is well resolved with an ample number of NOE cross-peaks (Fig. 7), indicating a folded conformation in its monomeric state. Although we could assign ∼42 of 51 amino acids of insulin, tracing of intermolecular NOE cross-peaks between insulin and KR7 was not possible due to the presence of severe signal overlap. For this reason, experimental determination of the structure of the bound complex was not possible. Determination of the structure of the insulin-bound conformations of KR7 by a two-dimensional 1H,1H-transferred NOESY (trNOESY) NMR experiment was also not feasible due to a lack of sufficient trNOEs (supplemental Figs. S4 and S5).
FIGURE 7.
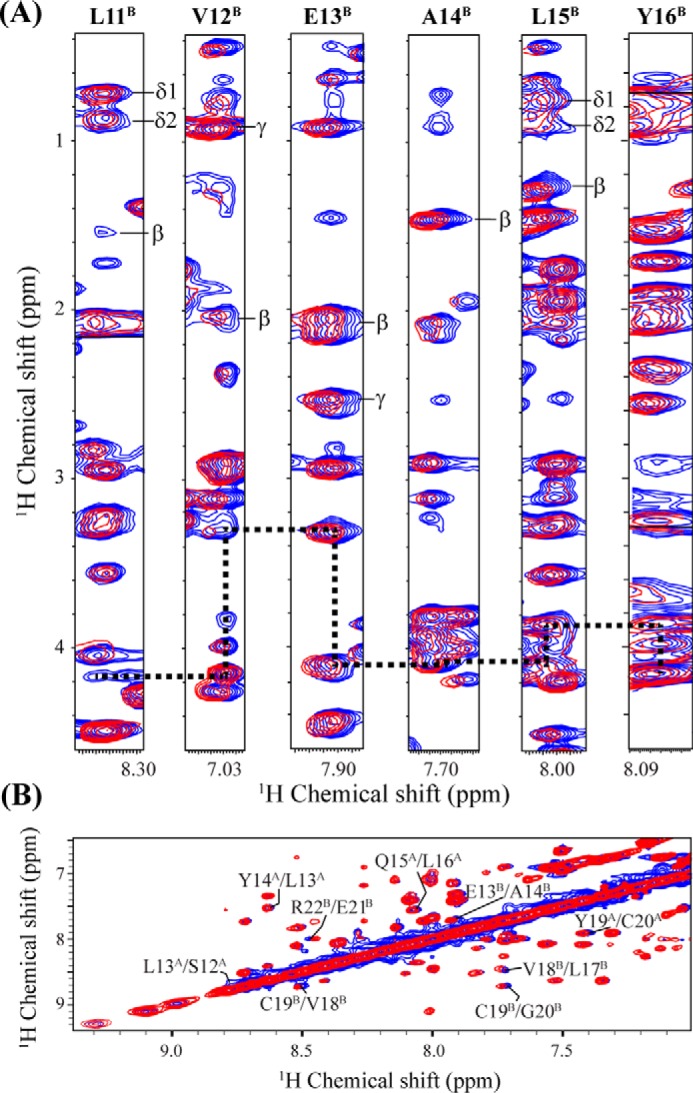
NOESY spectra of insulin in 20% acetic acid. A, strip plot displaying the chemical shift perturbation as well as sequential connectivity (dotted black line) of insulin (chain B Leu-11 to Tyr-16) from the selected region of two-dimensional 1H,1H NOESY spectra of insulin in 20% acetic acid-d4, 10% D2O and at pH 1.9 in the presence (red contour) and absence (blue contour) of a equimolar ratio (1:1) of KR7 at 298 K using a Bruker Avance III 700 MHz spectrometer with a NOESY mixing time of 200 ms. B, amide region of the NOESY spectra of insulin showing the CSP (insulin residues and chain ID are indicated with number and in superscript, respectively) of insulin in the presence of KR7.
Despite the lack of a full structure of the KR7-insulin complex, binding can still be localized from the NOESY spectra to distinct regions of KR7 and insulin. For insulin, binding mostly manifests as chemical shift perturbations (CSPs) (Fig. 7 and supplemental Table S2) along seven consecutive residues in the insulin B chain (11LVEALYL17). This aggregation prone sequence is believed to be one of the driving forces behind insulin fibrillization as it forms a energetically favorable steric zipper in the insulin amyloid fiber (51, 52). Several neighboring residues such as His-10, Cys-19, Gly-20, Glu-21, Gly-23, Phe-24, and Thr-27 in the B chain as well as a few residues in the A chain, namely Ser-12, Leu-13, Leu-16, Tyr-19, and Cys-20, also display substantial CSPs (supplemental Table S2).
On the KR7 side, the PWWP motif appears to be critical for binding. KR7 in the absence of insulin shows four indole ring proton peaks in one-dimensional spectrum for the tryptophan residues due to the presence of cis-trans conformational changes at the PWWP motif (see Fig. 8A). With the addition of insulin to KR7 at a molar ratio of 1:1, we see a slight shift (∼9.0 Hz) and significant broadening for both the major and minor indole ring protons of tryptophan residues of KR7. The line broadening effect could be due to apo-holo exchange between bound and unbound states in the intermediate kinetic regime of the NMR timescale. Taken together, CSPs in the NOESY spectra of insulin as well as broadening in the indole proton region of KR7 suggests portions of insulin B chain (11LVEALYL17) and two tryptophan residues of KR7 are actively involved in the course of binding.
FIGURE 8.

Binding of KR7 to insulin at an atomic resolution. A, selected region of the one-dimensional 1H NMR spectra of KR7 in 20% acetic acid-d4, 10% D2O and at pH 1.9 in the absence (black) and presence (red) of equimolar concentration of insulin (25 °C, Bruker Avance III 700 MHz NMR, with cryo-probe). The indole ring protons of Trp residue of KR7 are perturbed by ∼9 Hz in the presence of insulin, and 2 of 4 indole peaks of Trp disappeared (highlighted with green circle). B, STD experiments probing binding of KR7 to insulin amyloid fibers. The decrease in the STD signal of KR7 in the presence of preformed insulin fibril at 430:1 molar ratio (red) compared with the reference spectrum without insulin (black) confirms binding of KR7 to fibrillar insulin. The STD NMR experiments were carried out in aqueous solution on a Bruker Avance III 500 MHz spectrometer at 288 K. C, scatter plot showing the docked conformations obtained from HADDOCK server, choosing the lowest energy conformation (marked by a red circle) from interface r.m.s.d. versus HADDOCK score plot. D, surface representation of the proposed molecular model (docked complex) of KR7 oriented over bovine insulin; KR7 is represented by a blue stick model, and the protons which are in close proximity to insulin (<5 Å) are shown by purple spheres. E, proposed molecular model of the insulin-KR7 complex; KR7 is shown by its silver color, and insulin is shown by the olive green color for chain A and green color for chain B, respectively. The dotted line shows either H-bond or electrostatic interaction between insulin and KR7.
Epitope Mapping
Saturation Transfer Difference (STD) NMR
The tryptophan anisotropy measurements show that the dissociation constant of freshly prepared insulin and KR7 is on the micromolar scale. Moreover, the line width broadening in the amide and aromatic region of KR7 in one-dimensional 1H NMR upon the addition of insulin signifies that the protein and ligand undergo fast to intermediate exchange between the free and bound state (supplemental Fig. S4). Unfortunately, a lack of sufficient NOEs and spectral congestion prevented easy identification of binding sites. On the other hand, the fast exchange rate between KR7 and insulin fibers does make the ligand protein pair suitable for study with STD NMR, a technique that detects ligand binding to high molecular weight species indirectly by the transfer of magnetization to the unbound state (53). The efficiency of magnetization transfer is dependent on the size of the binding partner; binding to large species like amyloid fibers results in a much larger STD signal than binding to smaller species like the insulin monomer. The STD control spectrum of KR7 without insulin did not show any detectable STD signals (data not shown), whereas peaks are detectable in the aliphatic and aromatic region in the presence of preformed insulin fibers (Fig. 8B). A 1:430 molar ratio of insulin fibers to KR7 was chosen to efficiently transfer magnetization from the protein to the ligand in the bound state. The strongest STD signal was observed at 1.85 ppm corresponding to CβH2 of Arg of KR7. Other peaks such as the methylene (CH2) protons of Lys and Pro and the aromatic protons of Trp also exhibited moderate STD signals. From the STD NMR data, it can be said that the side chains of Arg, Lys, Pro, and Trp are close to insulin in the bound form.
Interaction Analysis of the KR7 Insulin Binding Pose
Molecular modeling is a useful computational approach to aid in the understanding for the probable interaction mode between KR7 and the insulin monomer, the conformational state presumed to be responsible for the elongation of the amyloid fiber. Because the structure of KR7 in the bound complex is not available, simulated annealing in implicit generalized born conditions was used to explore the energy barrier required for proper peptide folding upon binding and also to fetch a wide range of KR7 conformational possibilities. From this set, the k-means hierarchical clustering technique was employed to select a single pose representative of the KR7 population for docking.
The HADDOCK server was used for docking of insulin with KR7 using the CSPs in the NOESY and one-dimensional 1H NMR spectrum to define active residues for insulin and KR7, respectively. The minimum energy conformation of the docked complex was chosen as the model of interaction (Fig. 8C). This model is represented in Fig. 8D as a surface, where the yellow surface of the protein represents the residues within 5 Å distance of KR7. The docking result indicates KR7 is likely in close proximity to the C terminus and α-helical region of the B-chain of insulin, which is also supported by NMR analysis. Specifically, KR7 makes hydrophobic and electrostatic interactions with Glu-13, Tyr-16, Leu-17, Gly-20, Glu-21, Phe-24, and Tyr-26 of insulin chain B (Fig. 8E), consistent with the CSP values from the NOESY experiments. These residues lie along the interface of the insulin dimer, suggesting KR7 may favor the monomeric state of insulin in the monomer/dimer equilibrium, although direct evidence is lacking at this point. The absence of contacts in the N-terminal region in the docked structure is consistent with KR7's inability to delay the fibrillization lag time; the first aggregation prone intermediate is believed to result from partial unfolding of the insulin monomer at the N terminus of the A-chain (14). Further details in terms of residues involved, nature of interaction, and interatomic distance are shown in supplemental Table S3.
Serum Stability of KR7 and Cytotoxicity of Amyloid Insulin Intermediates
KR7 Peptide Is Non-cytotoxic and Stable in Serum
In vitro activity is not a guarantee of in vivo efficacy. As a first step toward this goal, we performed preliminary experiments to test the in vivo suitability of the KR7 peptide. We first confirmed that KR7 itself is not cytotoxic to cells by testing the effect of KR7 on the viability of HEK293T cell line by the 3,(4,5-dimethylthiazol-2-yl)2,5-diphenyltetrazolium bromide (MTT) assay. The viability of cells treated with KR7 for 24 h was statistically indistinguishable from the controls (Fig. 9A). Because KR7 is less effective at suppressing the formation of early insulin aggregates compared with later insulin fibers, we also tested the cytotoxic potential of early insulin fibrillation intermediates. Insulin fibrillation intermediates were prepared by heating 3.5 mg/ml insulin (in HCl pH 2.0 with 100 mm NaCl) at 335 K, and aliquots were collected from this reaction mixture at different time points for the cytotoxicity study. At relatively high final concentrations (60 μm), intermediate species of insulin fibrillation (allowed to aggregate at 335 K for 5, 7, 10, and 20 min before treatment) are moderately toxic and kill a statistically significant proportion (12–15%) of cells (Fig. 9B). This toxic effect of insulin was not observed when we treated insulin at the aforementioned condition along with our synthetic peptide KR7. This result suggests that KR7 peptide minimizes the cytotoxicity of intermediate insulin oligomers.
FIGURE 9.
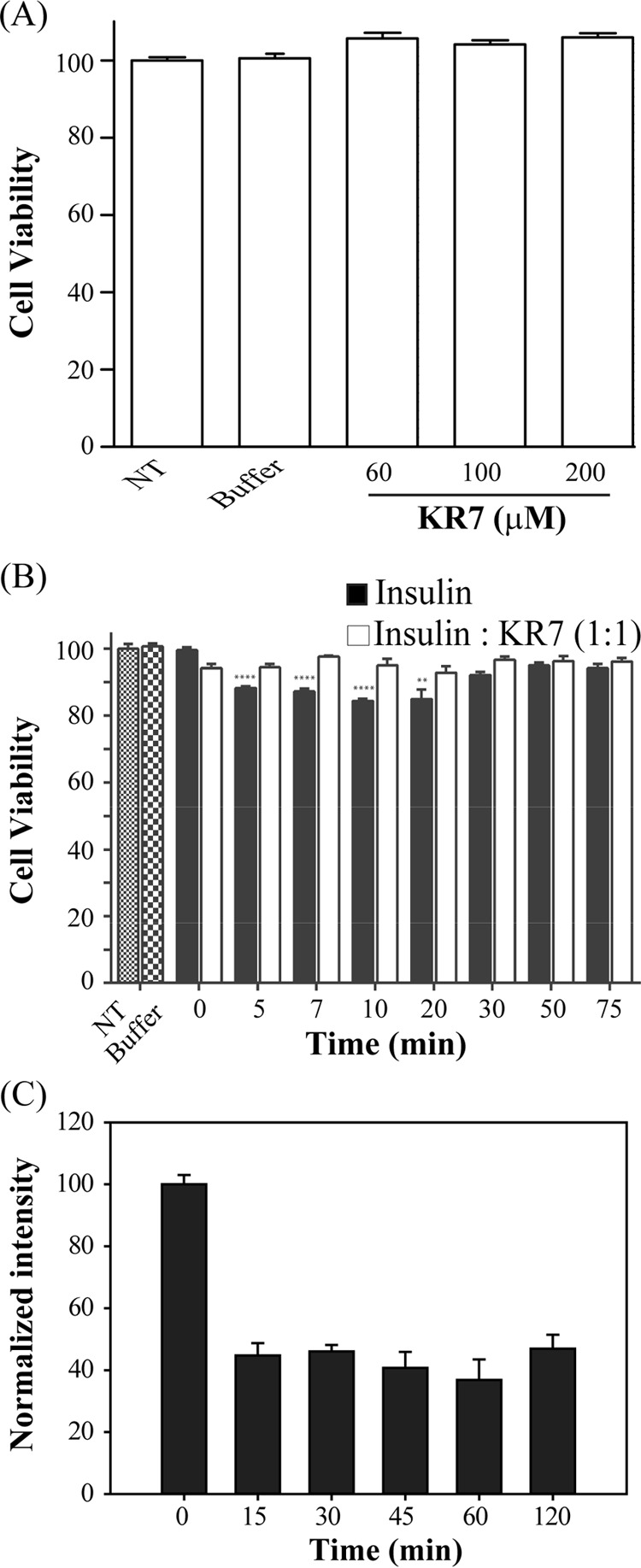
Cytotoxicity and serum stability study. A, effect of different concentration of KR7 peptide on HEK293T cells, studied by MTT reduction assay (NT stands for non-treated control cells, and buffer was used as negative control). B, comparison of cytotoxic effect of insulin amyloid intermediates (black bar) and insulin-KR7 complex (1:1 molar ratio) (white bar) on HEK293T cell at different time points. C, stability of KR7 peptide in serum was checked by RP-HPLC at different time intervals.
Peptide KR7 Is Serum Stable
Serum contains various kinds of proteases, which cause hydrolysis of peptide bonds that can inactivate peptide therapeutics. To test the potential stability of KR7 in vivo, we tested the proteolytic stability of our designed peptide KR7 in fetal bovine serum (FBS). From the proteolytic assay it could be seen that after 2 h of incubation ∼35% of the peptide remain intact (Fig. 9C). KR7 thus shows significant stability in the presence of serum proteins.
Conclusion
KR7 is an effective inhibitor of insulin aggregation with a novel inhibition mode; it appears to arrest insulin aggregation in the later stages of fiber assembly without disassembly by binding both to monomeric insulin and to insulin fibers. This inhibition mode is different from other peptide-based inhibitors, even those based on a similar Trp repeat motif (34). Understanding why such subtle differences in sequence can yield large differences in behavior may improve the design of future peptide based inhibitors against insulin and other amyloid forming peptides.
Experimental Procedures
Chemicals
Bovine insulin from bovine pancreas and the peptide KR7 were purchased from Sigma and GL Biochem (Sanghai, China), respectively. The purity of KR7 was >95%, as evidenced by HPLC and mass spectral (electrospray ionization-MS) analysis. A MTT assay was performed with phenol red free RPMI media, obtained from HiMedia Laboratories Pvt. Ltd. (Mumbai, India). All other chemicals used in this study were of highest analytical grade.
Insulin Fibrillation Kinetics by ThT Assay
Bovine insulin was dissolved in 50 mm citrate phosphate buffer (pH 2.6) with 100 mm NaCl (54). The concentration of the insulin in buffer solution was determined by observing the UV absorbance at 276 nm. The concentration of insulin was calculated by using the known extinction coefficient of 0.91 mg/ml for insulin (33). All the buffers used in the experiment were filtered through a 0.22-μm membrane filter followed by degassing with vigorous stirring. ThT was dissolved in Milli-Q water, and its concentration was determined by UV absorbance at 412 nm. The concentration of ThT was determined by using a molar extinction coefficient of 36,000 m−1 cm−1 (33). For recording ThT fluorescence, a 2 mg/ml insulin sample was incubated at 335 K in a circulating water bath, and aliquots were drawn from it at regular intervals. As described earlier (54, 55), the aliquot along with 10 μm ThT was added to 10 mm phosphate buffer (pH 7.4) with 100 mm NaCl. ThT fluorescence was measured in a quartz cuvette; Jasco spectrofluorometer (FX-8500) set at excitation wavelength of 450 nm and emission wavelength set at 485 nm.
The data collected from ThT fluorescence were fitted by Boltzmann equation (a sigmoidal function), i.e. given by Equation 1,
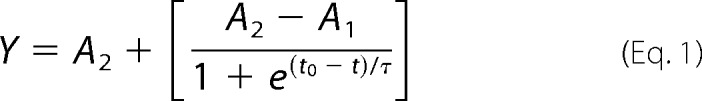 |
Here A1 is the initial fluorescence, A2 is the maximum fluorescence, t0 stands for the time where the fluorescence has reached to half that of the maximum value, and 1/τ is the apparent rate constant of fibril growth and lag time approximated to t0−2τ.
Circular Dichroism (CD) Spectroscopy
The secondary structure of insulin during fibrillation in the presence and absence of KR7 peptide was studied with Jasco J-815 spectrophotometer. 10 mm Na2HPO4 buffer containing 100 mm NaCl (pH 7.4) was used throughout the study. A quartz cuvette with a path length of 0.2 cm was used to record CD spectra at temperature 298 K for insulin in the presence and absence of KR7 peptide. Spectra were recorded over a range of 200–260 nm at 1-nm data interval with a scanning speed of 100 nm/min. Each CD spectrum represents an accumulation of four subsequent scans. To eliminate contributions of KR7 peptide in the CD spectrum, the same concentration of protein CD spectrum (without insulin) was subtracted from the resultant CD spectra. Collected spectral raw data in millidegrees were subtracted from the blank buffer data and transformed to molar ellipticity using Equation 2,
where m0 is millidegrees, Mr is the molecular weight (g mol−1), L is the path length of quartz cuvette used (cm), and C is the concentration (Molarity).
Fluorescence Anisotropy
Tryptophan fluorescence of 5 μm KR7 peptide in 50 mm citrate phosphate buffer (pH 2.6) with 100 mm NaCl was detected in a Hitachi F-7000 FL spectrometer with a 0.1-cm path length quartz cuvette at 298 K with the gradual addition of insulin from 1 μm to 30 μm concentration. The excitation wavelength was set at 295 nm. Along with the emission wavelength measurements, fluorescence anisotropy was also measured. Fluorescence anisotropy was determined by the single site ligand binding equation. The anisotropy values so obtained were plotted against the insulin concentration and fitted by Equation 3 to determine the apparent dissociation constant (KD) of the peptide-protein interaction assuming a 1:1 binding stoichiometry or equivalent and independent binding sites.
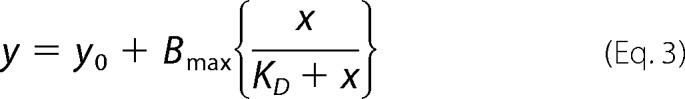 |
where, y is the measured anisotropy value, y0 is the anisotropy value determined for KR7 alone (without insulin), x is the concentration of insulin (μm) added, Bmax is the maximum value of anisotropy during the binding process, and KD is the apparent dissociation constant of insulin-KR7 complex.
Microscopy Study of Insulin Fibrils
Confocal Study
The fibrillation conditions were similar to that of the kinetics of fibrillation insulin experiments. The samples were prepared as described for ThT assay, and 10-μl aliquots of the reaction mixture were placed on clean glass slides and dried inside laminar airflow at room temperature in the dark followed by mounting with DPX mounting media (HiMedia Laboratories). A 405-nm band pass filter was used to observe the ThT fluorescence in a confocal microscope (Leica TCS SP8).
TEM Study
2 mg/ml insulin samples were subjected to fibrillation using similar conditions as the kinetics experiment in the presence or absence of KR7 peptide. After saturation of the ThT fluorescence of insulin fibrillation, the sample was incubated at room temperature for few days, then an aliquot was placed on the TEM grid, and images were taken with CM 12 PHILIPS TEM instrument.
AFM Microscopy
2 mg/ml insulin was incubated at 335 K in 50 mm citrate phosphate buffer in the presence of 100 mm NaCl with and without KR7 (1:1) peptide, and at different time points 10 μl of the sample was collected and placed onto an even-layered mica sheet for 5 min. The mica sheet was then washed with Milli-Q pure water three times followed by air-drying in a closed chamber for overnight at room temperature. Protein aggregates were imaged with an AFM (AFM workshop, Signal Hill, CA). The AFM imaging was performed in the vibrating mode. The resonance frequencies of the cantilever was 140–210 kHz and attached with a single crystal silicon tip with a nominal radius of curvature <10 nm. Typical scan size was 10 × 10 μm (512 × 512 points), and scan rate was kept below 1 Hz.
NMR Experiments
The NMR experiments were carried out at 298 K or 335 K using either on Bruker AVANCE III 500 MHz (equipped with SMART probe) or 700 MHz (equipped with QCI Cryoprobe). Topspin v3.1 software (Bruker) was used for NMR data acquisition and processing. The one-dimensional 1H NMR spectra for insulin fibrillation kinetic experiment was performed in 50 mm citrate-phosphate buffer (pH 2.6) with 100 mm NaCl, 10% D2O, and at 335 K temperature using Bruker Avance III 500 MHz NMR spectrometer.
A series of one-dimensional proton NMR spectrum of KR7 was recorded in the presence of insulin fibril to observe the line broadening effect of KR7 (without effecting the chemical shift change) in the context of insulin fibril. The trNOESY experiments of KR7 in the presence of insulin fibril at a molar ratio of 1:20 (insulin:KR7) were recorded using a Bruker Avance III 500 MHz. The 96 scans were recorded for trNOESY spectra per t1 increment. 16 dummy scans were performed in each case, and a spectral width of 12 ppm was maintained for both dimensions. 512 increments in t1 and 2048 data points in t2 dimension along with States TPPI for quadrature detection in t1 dimension and WATERGATE for water suppression were used. The total correlation spectroscopy (TOCSY) and trNOESY spectra were processed using 4 K (t2) × 1 K (t1) data matrices after zero filling.
Bovine pancreatic insulin was made zinc-free by the addition of EDTA followed by extensive dialysis and then lyophilization. The sample were then dissolved in 20% acetic acid-d4, 70% water, and 10% D2O (pH 1.9) or 20% acetic acid-d4 and 80% D2O (pD 1.9) at a final concentration of 350 μm. Two-dimensional homonuclear TOCSY (with 40 ms mixing time) and NOESY (with 200 ms mixing time) were recorded at 298 K in the absence and presence of KR7 peptide at pH 1.9 using Bruker Avance III 700 MHz spectrometer (equipped with cryo-probe), keeping same pulse program and parameter files (number of scans, water suppression technique etc.) described above. A fresh set of lyophilized sample was used for each experiment. The insulin TOCSY, NOESY, and KR7 trNOESY spectra were analyzed with Sparky software (66).
For saturation transfer difference (56, 57) NMR experiments, standard STD pulse sequences with the WATERGATE 3-9-19 water suppression method were used. 1 mm of KR7 peptide in water (pH adjusted to 2.6 with dilute HCl) was lyophilized and resuspended in an equal volume of D2O, and 0.6 μm insulin fibril was added. A train of 40-selective Gaussian-shaped pulses (49 ms each) with an interval period of 1 ms was used to saturate protein resonance for a total of 2 s saturation time. During the STD NMR experiment, on- and off-resonance frequencies were set at −1 ppm and 40 ppm, respectively. Subtraction of off-resonance spectrum from the on-resonance spectra to yield signals appearing due to saturation transfer from protein to ligand was achieved using phase cycling. The one-dimensional STD and reference spectra were acquired with 1024 and 512 scans, respectively, with 16 dummy scans using a sweep width of 12 ppm. An exponential line broadening function of 5 Hz was applied to the spectral data before Fourier transformation (58). Similar STD experiments were performed on the peptide KR7 in the absence of insulin fibril as a control.
Molecular Modeling
KR7 Structure Prediction
The structural calculation of KR7 using NMR spectroscopy was restricted due to lack of NOEs in the trNOESY spectrum. The conformational prediction of KR7 was employed using simulated annealing (SA) process, similar to our previous study (59). In particular, the linear sequence of KR7 was built in tleap of AMBER14 using f99SB-ILDN forcefield. The temperature scale used for SA procedure includes incremental steps of 50 K, where each step was followed with constant temperature of 5 ps each. The temperature window used for the procedure was from 0 K to 325 K and then cooling to 300 K with integration time step of 2 fs. The end-point conformation of the SA process was then used for a production run of 5ns, with a trajectory recording interval of 5 ps in implicit solvent conditions. Cluster analyses of structures were performed using perl scripts from the MMTSB tool-set (60). Hierarchical k-means cluster analysis procedure was used with a r.m.s.d. cut-off criteria of 2 Å distances to separate various populations of KR7 (61). Finally, the representative structure from the top-ranked cluster was used for docking studies.
Docking Calculation
High ambiguity-driven protein-protein docking (HADDOCK) was used for the docking study that helps in predicting the probable binding mode of KR7 to insulin with default server parameters. The coordinate of insulin was adopted from PDB code 2ZP6, which was used as receptor and the predicted KR7 structure as ligand with a rigid backbone and flexible side chains. There are similar reports of molecular modeling that uses insulin monomer to correlate with the experimental results. This prompts us to make use of monomeric insulin conformation for our docking studies (34, 62). Active residues of the receptor and ligand were chosen on the basis of chemical shift perturbation in two-dimensional homonuclear NOESY NMR and signals from STD NMR, respectively. The results obtained from HADDOCK server were collected as clusters based on the internal standards of the HADDOCK server. Further analysis of the docked complexes carried out based on HADDOCK score and i-I-r.m.s.d. values for the best cluster (63, 64).
Cell Culture and MTT Assay
Cell Culture and Insulin Fibrillation
HEK293T cell line was grown and maintained in complete DMEM media (HiMedia Laboratories) supplemented with 10% FBS, 150 μg/ml penicillin/streptomycin, 50 μg/ml gentamycin, and 2.5 μg/ml amphotericin B at 310 K in a humidified 5% CO2 containing incubator. 1.5 × 104 cells were seeded in 96-well plates, and insulin fibril plus KR7 peptide were treated after 48 h of seeding. Insulin fibrillation for cytotoxicity assay was carried out in the presence (1:1 molar ratio) and absence of KR7 by incubating 3.5 mg/ml insulin in HCl, 100 mm NaCl, pH 2.0, followed by incubating at 335 K (54).
MTT Reduction Assay
The viability of HEK293T cell after insulin and KR7 peptide treatment was estimated using the metabolic dye MTT reduction assay. MTT was reduced by only the viable cells by forming blue-colored formazan crystals, which was then dissolved in DMSO, and the value was measured at 570 nm. Insulin was dissolved in HCl (pH 2.0) with 100 mm NaCl, and the stock concentration of insulin was 3.5 mg/ml. The cell was incubated at 310 K in a humidified 5% CO2-containing incubator. After 24 h of treatment, MTT was added to the wells at a final concentration of 0.5 mg/ml by dissolving the MTT in phenol red-free RPMI media (HiMedia Laboratories) and again incubated for 3 h. MTT solution was discarded by decanting the 96-well plates on a tissue paper, and 100 ml of DMSO was added to dissolve the formazan crystals and incubate for 15–30 min with gentle shaking on a rocker. The absorbance at 570 nm was taken using a microplate reader (MultiscanGo, Thermo Scientific), and cell viability was calculated accordingly.
Serum Stability Assay
Serum stability was performed by a method described by Jenssen and Aspmo (65) with slight modification. In brief, 1 ml of RPMI 1640 supplemented with 25% (v/v) fetal bovine serum was equilibrated to 37 ± 1 °C for 15 min. 100 μg/ml final concentration of KR7 was added, and at the respective time points 100 μl of the reaction mixture was taken out and mixed with 400 μl of 96% ethanol for precipitation of serum proteases. The cloudy reaction mixture was cooled at 4 °C for 15 min and then centrifuged at 18,000 × g for 2 min to pellet down the precipitated serum protein.
The reaction supernatant was then analyzed using reverse phase HPLC on a 250 × 4.6-mm C18 column. Gradient elution from 0.1% TFA in 100% acetonitrile to 0.1% TFA in water was used over 35 min. A flow rate of 1 ml/min was used, and the quantity of complete peptide remaining was integrated at 220 nm. The entire analysis was done at room temperature. The area under the peak was analyzed using LCsolution software (Shimadzu). Water was used as the blank, and a reaction mixture lacking peptide served as negative control. % of peptide remaining after each time point was calculated by using Equation 4.
 |
Author Contributions
A. B. designed, performed, and analyzed NMR data. B. N. R. conducted most of the biophysical experiments, analyzed the results, and wrote the manuscript. N. G. conducted the cell biology experiments under the supervision of A. K. M. A. G. performed all the NMR experiments and analyzed the NOESY spectra of insulin. H. I. conducted the serum stability experiments. C. N. helped B. N. R. in performing the AFM experiments in the laboratory of Dr. Kanchan Garai, TIFR Centre for Interdisciplinary Sciences, Hyderabad, India. K. P. D. provided helpful discussions and reviewed the manuscript. J. R. B. analyzed the data and edited the manuscript with A. B. All authors reviewed the manuscript.
Supplementary Material
Acknowledgments
A. B. acknowledges DBT, Government of India, for infrastructure development Fund BT/PR3106/INF/22/138/2011 to the Bose Institute for procuring a 700-MHz NMR spectrometer with cryoprobe. Central Instrument Facility (CIF) of Bose Institute is greatly acknowledged.
This work was supported by Institutional Plan Project-II (to A. B.), Bose Institute, Kolkata, India. The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental Tables S1–S3 and Figs. S1–S5.
- Aβ
- amyloid beta
- AFM
- atomic force microscopy
- MTT
- 3,(4,5-dimethylthiazol-2-yl)2,5-diphenyltetrazolium bromide
- STD
- saturation transfer difference
- TEM
- transmission electron microscopy
- trNOESY
- transferred nuclear Overhauser effect spectroscopy
- ThT
- thioflavin T
- CSP
- chemical shift perturbation
- TOCSY
- total correlation spectroscopy
- SA
- simulated annealing
- r.m.s.d.
- roof mean square deviation.
References
- 1. Benilova I., Karran E., and De Strooper B. (2012) The toxic Aβ oligomer and Alzheimer's disease: an emperor in need of clothes. Nat. Neurosci. 15, 349–357 [DOI] [PubMed] [Google Scholar]
- 2. Dobson C. M. (2001) The structural basis of protein folding and its links with human disease. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 356, 133–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Eisenberg D., and Jucker M. (2012) The amyloid state of proteins in human diseases. Cell 148, 1188–1203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kodali R., and Wetzel R. (2007) Polymorphism in the intermediates and products of amyloid assembly. Curr. Opin. Struct. Biol. 17, 48–57 [DOI] [PubMed] [Google Scholar]
- 5. Woods R. J., Alarcón J., McVey E., and Pettis R. J. (2012) Intrinsic fibrillation of fast-acting insulin analogs. J. Diabetes Sci. Technol. 6, 265–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Guilhem I., Leguerrier A. M., Lecordier F., Poirier J. Y., and Maugendre D. (2006) Technical risks with subcutaneous insulin infusion. Diabetes Metab. 32, 279–284 [DOI] [PubMed] [Google Scholar]
- 7. Gupta Y., Singla G., and Singla R. (2015) Insulin-derived amyloidosis. Indian J. Endocrinol. Metab. 19, 174–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nagase T., Iwaya K., Iwaki Y., Kotake F., Uchida R., Oh-i T., Sekine H., Miwa K., Murakami S., Odaka T., Kure M., Nemoto Y., Noritake M., and Katsura Y. (2014) Insulin-derived amyloidosis and poor glycemic control: A case series. Am. J. Med. 127, 450–454 [DOI] [PubMed] [Google Scholar]
- 9. Rahuel-Clermont S., French C. A., Kaarsholm N. C., Dunn M. F., and Chou C. I. (1997) Mechanisms of stabilization of the insulin hexamer through allosteric ligand interactions. Biochemistry 36, 5837–5845 [DOI] [PubMed] [Google Scholar]
- 10. Whittingham J. L., Edwards D. J., Antson A. A., Clarkson J. M., and Dodson G. G. (1998) Interactions of phenol and m-cresol in the insulin hexamer, and their effect on the association properties of B28 Pro-Asp insulin analogues. Biochemistry 37, 11516–11523 [DOI] [PubMed] [Google Scholar]
- 11. Derewenda U., Derewenda Z., Dodson E. J., Dodson G. G., Reynolds C. D., Smith G. D., Sparks C., and Swenson D. (1989) Phenol stabilizes more helix in a new symmetrical zinc insulin hexamer. Nature 338, 594–596 [DOI] [PubMed] [Google Scholar]
- 12. Ahmad A., Millett I. S., Doniach S., Uversky V. N., and Fink A. L. (2003) Partially folded intermediates in insulin fibrillation. Biochemistry 42, 11404–11416 [DOI] [PubMed] [Google Scholar]
- 13. Ahmad A., Millett I. S., Doniach S., Uversky V. N., and Fink A. L. (2004) Stimulation of insulin fibrillation by urea-induced intermediates. J. Biol. Chem. 279, 14999–15013 [DOI] [PubMed] [Google Scholar]
- 14. Hua Q. X., and Weiss M. A. (2004) Mechanism of insulin fibrillation: the structure of insulin under amyloidogenic conditions resembles a protein-folding intermediate. J. Biol. Chem. 279, 21449–21460 [DOI] [PubMed] [Google Scholar]
- 15. Ciszak E., Beals J. M., Frank B. H., Baker J. C., Carter N. D., and Smith G. D. (1995) Role of C-terminal B-chain residues in insulin assembly: the structure of hexameric LysB28ProB29-human insulin. Structure 3, 615–622 [DOI] [PubMed] [Google Scholar]
- 16. Olsen H. B., Ludvigsen S., and Kaarsholm N. C. (1998) The relationship between insulin bioactivity and structure in the NH2-terminal A-chain helix. J. Mol. Biol. 284, 477–488 [DOI] [PubMed] [Google Scholar]
- 17. Groenning M., Frokjaer S., and Vestergaard B. (2009) Formation mechanism of insulin fibrils and structural aspects of the insulin fibrillation process. Curr. Protein Pept. Sci. 10, 509–528 [DOI] [PubMed] [Google Scholar]
- 18. Dodson G., and Steiner D. (1998) The role of assembly in insulin's biosynthesis. Curr. Opin. Struct. Biol. 8, 189–194 [DOI] [PubMed] [Google Scholar]
- 19. Gibson T. J., and Murphy R. M. (2006) Inhibition of insulin fibrillogenesis with targeted peptides. Protein Sci. 15, 1133–1141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Murray B., Rosenthal J., Zheng Z., Isaacson D., Zhu Y., and Belfort G. (2015) Cosolute effects on amyloid aggregation in a nondiffusion limited regime: intrinsic osmolyte properties and the volume exclusion principle. Langmuir 31, 4246–4254 [DOI] [PubMed] [Google Scholar]
- 21. Selivanova O. M., Suvorina M. Y., Dovidchenko N. V., Eliseeva I. A., Surin A. K., Finkelstein A. V., Schmatchenko V. V., and Galzitskaya O. V. (2014) How to determine the size of folding nuclei of protofibrils from the concentration dependence of the rate and lag-time of Aggregation. II. Experimental application for insulin and LysPro insulin: aggregation morphology, kinetics, and sizes of nuclei. J. Phys. Chem. B 118, 1198–1206 [DOI] [PubMed] [Google Scholar]
- 22. Choudhary S., Kishore N., and Hosur R. V. (2015) Inhibition of insulin fibrillation by osmolytes: mechanistic insights. Sci. Rep. 5, 17599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Valipour M., Maghami P., Habibi-Rezaei M., Sadeghpour M., Khademian M. A., Mosavi K., Sheibani N., and Moosavi-Movahedi A. A. (2015) Interaction of insulin with methyl tert-butyl ether promotes molten globule-like state and production of reactive oxygen species. Int. J. Biol. Macromol. 80, 610–614 [DOI] [PubMed] [Google Scholar]
- 24. Martí-Aluja I., and Larrechi M. S. (2013) MCR-ALS analysis of insulin aggregation/association processes: influence of biochemical variables. Chemom. Intell. Lab. Syst. 127, 49–54 [Google Scholar]
- 25. Crespo R., Villar-Alvarez E., Taboada P., Rocha F. A., Damas A. M., and Martins P. M. (2016) What can the kinetics of amyloid fibril formation tell about off-pathway aggregation? J. Biol. Chem. 291, 2018–2032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Erlkamp M., Grobelny S., Faraone A., Czeslik C., and Winter R. (2014) Solvent effects on the dynamics of amyloidogenic insulin revealed by neutron spin echo spectroscopy. J. Phys. Chem. B 118, 3310–3316 [DOI] [PubMed] [Google Scholar]
- 27. Smirnovas V., and Winter R. (2008) Revealing different aggregation pathways of amyloidogenic proteins by ultrasound velocimetry. Biophys. J. 94, 3241–3246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Javid N., Vogtt K., Krywka C., Tolan M., and Winter R. (2007) Capturing the interaction potential of amyloidogenic proteins. Phys. Rev. Lett. 99, 028101 [DOI] [PubMed] [Google Scholar]
- 29. Smirnovas V., Winter R., Funck T., and Dzwolak W. (2006) Protein amyloidogenesis in the context of volume fluctuations: a case study on insulin. Chemphyschem 7, 1046–1049 [DOI] [PubMed] [Google Scholar]
- 30. Wang S-H., Dong X-Y., and Sun Y. (2012) Effect of (−)-epigallocatechin-3-gallate on human insulin fibrillation/aggregation kinetics. Biochem. Eng. J. 63, 38–49 [Google Scholar]
- 31. Wang J. B., Wang Y. M., and Zeng C. M. (2011) Quercetin inhibits amyloid fibrillation of bovine insulin and destabilizes preformed fibrils. Biochem. Biophys. Res. Commun. 415, 675–679 [DOI] [PubMed] [Google Scholar]
- 32. Gong H., He Z., Peng A., Zhang X., Cheng B., Sun Y., Zheng L., and Huang K. (2014) Effects of several quinones on insulin aggregation. Sci. Rep. 4, 5648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Banerjee V., Kar R. K., Datta A., Parthasarathi K., Chatterjee S., Das K. P., and Bhunia A. (2013) Use of a small peptide fragment as an inhibitor of insulin fibrillation process: a study by high and low resolution spectroscopy. PLoS ONE 8, e72318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mishra N. K., Krishna Deepak R. N., Sankararamakrishnan R., and Verma S. (2015) Controlling in vitro insulin amyloidosis with stable peptide conjugates: a combined experimental and computational study. J. Phys. Chem. B 119, 15395–15406 [DOI] [PubMed] [Google Scholar]
- 35. Viet M. H., Siposova K., Bednarikova Z., Antosova A., Nguyen T. T., Gazova Z., and Li M. S. (2015) In silico and in vitro study of binding affinity of tripeptides to amyloid β fibrils: implications for Alzheimer's disease. J. Phys. Chem. B 119, 5145–5155 [DOI] [PubMed] [Google Scholar]
- 36. Gasteiger E., Hoogland C., Gattiker A., Duvaud S., Wilkins M. R., Appel R. D., and Bairoch A. (2005) Protein Identification and Analysis Tools on the ExPASy Server in Proteomics Protocols Handb. (Walker J. M., Ed.) pp. 571–607, Humana Press, Totowa, N. J. [Google Scholar]
- 37. Biancalana M., and Koide S. (2010) Molecular mechanism of Thioflavin-T binding to amyloid fibrils. Biochim. Biophys. Acta 1804, 1405–1412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lee C.-C., Nayak A., Sethuraman A., Belfort G., and McRae G. J. (2007) A three-stage kinetic model of amyloid fibrillation. Biophys. J. 92, 3448–3458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tu L.-H., Young L. M., Wong A. G., Ashcroft A. E., Radford S. E., and Raleigh D. P. (2015) Mutational analysis of the ability of resveratrol to inhibit amyloid formation by islet amyloid polypeptide: critical evaluation of the importance of aromatic-inhibitor and histidine-inhibitor interactions. Biochemistry 54, 666–676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Patel H. R., Pithadia A. S., Brender J. R., Fierke C. A., and Ramamoorthy A. (2014) In search of aggregation pathways of IAPP and other amyloidogenic proteins: finding answers through NMR spectroscopy. J. Phys. Chem. Lett. 5, 1864–1870 [DOI] [PubMed] [Google Scholar]
- 41. Brender J. R., Krishnamoorthy J., Messina G. M. L., Deb A., Vivekanandan S., La Rosa C., Penner-Hahn J. E., and Ramamoorthy A. (2013) Zinc stabilization of prefibrillar oligomers of human islet amyloid polypeptide. Chem. Commun. (Camb.) 49, 3339–3341 [DOI] [PubMed] [Google Scholar]
- 42. Yesuvadian R., Krishnamoorthy J., Ramamoorthy A., and Bhunia A. (2014) Potent c-secretase inhibitors / modulators interact with amyloid-β fibrils but do not inhibit fibrillation: a high-resolution NMR study. Biochem. Biophys. Res. Commun. 447, 590–595 [DOI] [PubMed] [Google Scholar]
- 43. Milojevic J., Raditsis A., and Melacini G. (2009) Human serum albumin inhibits Aβ fibrillization through a “monomer-competitor” mechanism. Biophys. J. 97, 2585–2594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Smith R. A., Nabok A., Blakeman B. J., Xue W. F., Abell B., and Smith D. P. (2015) Analysis of toxic amyloid fibril interactions at natively derived membranes by ellipsometry. PLoS ONE 10, e0132309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jakhria T., Hellewell A. L., Porter M. Y., Jackson M. P., Tipping K. W., Xue W. F., Radford S. E., and Hewitt E. W. (2014) β2-Microglobulin amyloid fibrils are nanoparticles that disrupt lysosomal membrane protein trafficking and inhibit protein degradation by lysosomes. J. Biol. Chem. 289, 35781–35794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lee Y. J., Savtchenko R., Ostapchenko V. G., Makarava N., and Baskakov I. V. (2011) Molecular structure of amyloid fibrils controls the relationship between fibrillar size and toxicity. PLoS ONE 6, e20244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Xue W.-F., Hellewell A. L., Hewitt E. W., and Radford S. E. (2010) Fibril fragmentation in amyloid assembly and cytotoxicity: when size matters. Prion 4, 20–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Jiménez J. L., Nettleton E. J., Bouchard M., Robinson C. V., Dobson C. M., and Saibil H. R. (2002) The protofilament structure of insulin amyloid fibrils. Proc. Natl. Acad. Sci. U.S.A. 99, 9196–9201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hua Q. X., and Weiss M. A. (1990) Toward the solution structure of human insulin: sequential 2D 1H NMR assignment of a des-pentapeptide analogue and comparison with crystal structure. Biochemistry 29, 10545–10555 [DOI] [PubMed] [Google Scholar]
- 50. Hua Q. X., and Weiss M. A. (1991) Comparative 2D NMR studies of human insulin and des-pentapeptide insulin: sequential resonance assignment and implications for protein dynamics and receptor recognition. Biochemistry 30, 5505–5515 [DOI] [PubMed] [Google Scholar]
- 51. Ivanova M. I., Sievers S. A., Sawaya M. R., Wall J. S., and Eisenberg D. (2009) Molecular basis for insulin fibril assembly. Proc. Natl. Acad. Sci. U.S.A. 106, 18990–18995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Matthes D., Daebel V., Meyenberg K., Riedel D., Heim G., Diederichsen U., Lange A., and de Groot B. L. (2014) Spontaneous aggregation of the insulin-derived steric zipper peptide VEALYL results in different aggregation forms with common features. J. Mol. Biol. 426, 362–376 [DOI] [PubMed] [Google Scholar]
- 53. Bhunia A., Bhattacharjya S., and Chatterjee S. (2012) Applications of saturation transfer difference NMR in biological systems. Drug Discov. Today 17, 505–513 [DOI] [PubMed] [Google Scholar]
- 54. Banerjee V., and Das K. P. (2012) Modulation of pathway of insulin fibrillation by a small molecule helix inducer 2,2,2-trifluoroethanol. Colloids Surf. B Biointerfaces 92, 142–150 [DOI] [PubMed] [Google Scholar]
- 55. Hackl E. V., Darkwah J., Smith G., and Ermolina I. (2015) Effect of acidic and basic pH on Thioflavin T absorbance and fluorescence. Eur. Biophys. J. 44, 249–261 [DOI] [PubMed] [Google Scholar]
- 56. Mayer M., and Meyer B. (1999) Characterization of ligand binding by saturation transfer difference NMR spectroscopy. Angew. Chem. Int. Ed. 38, 1784–1788 [DOI] [PubMed] [Google Scholar]
- 57. Mayer M., and Meyer B. (2001) Group epitope mapping by saturation transfer difference NMR to identify segments of a ligand in direct contact with a protein receptor. J. Am. Chem. Soc. 123, 6108–6117 [DOI] [PubMed] [Google Scholar]
- 58. Ghosh A., Ratha B. N., Gayen N., Mroue K. H., Kar R. K., Mandal A. K., and Bhunia A. (2015) Biophysical characterization of essential phosphorylation at the flexible C-terminal region of C-Raf with 14-3-3ζ protein. PLoS ONE 10, e0135976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kar R. K., Gazova Z., Bednarikova Z., Mroue K. H., Ghosh A., Zhang R., Ulicna K., Siebert H.-C., Nifantiev N. E., and Bhunia A. (2016) Evidence for inhibition of lysozyme amyloid fibrillization by peptide fragments from human lysozyme: a combined spectroscopy, microscopy, and docking study. Biomacromolecules 17, 1998–2009 [DOI] [PubMed] [Google Scholar]
- 60. Feig M., Karanicolas J., and Brooks C. L. 3rd (2004) MMTSB tool set: enhanced sampling and multiscale modeling methods for applications in structural biology. J. Mol. Graph. Model. 22, 377–395 [DOI] [PubMed] [Google Scholar]
- 61. Lorenzen K., Schwörer M., Tröster P., Mates S., and Tavan P. (2012) Optimizing the accuracy and efficiency of fast hierarchical multipole expansions for MD simulations. J. Chem. Theory Comput. 8, 3628–3636 [DOI] [PubMed] [Google Scholar]
- 62. Kurouski D., Washington J., Ozbil M., Prabhakar R., Shekhtman A., and Lednev I. K. (2012) Disulfide bridges remain intact while native insulin converts into amyloid fibrils. PLoS ONE 7, e36989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Dominguez C., Boelens R., and Bonvin A. M. (2003) HADDOCK: a protein-protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc. 125, 1731–1737 [DOI] [PubMed] [Google Scholar]
- 64. van Zundert G. C., Rodrigues J. P., Trellet M., Schmitz C., Kastritis P. L., Karaca E., Melquiond A. S., van Dijk M., de Vries S. J., and Bonvin A. M. (2016) The HADDOCK2.2 web server: user-friendly integrative modeling of biomolecular complexes. J. Mol. Biol. 428, 720–725 [DOI] [PubMed] [Google Scholar]
- 65. Jenssen H., and Aspmo S. I. (2008) Serum stability of peptides in Peptide-Based Drug Design, Methods in Molecular Biology (Otvos L., Ed.) Vol. 494, pp. 171–186, Humana Press, New York, N. Y. [DOI] [PubMed] [Google Scholar]
- 66. Goddard T. D., and Kneller D. G. (2014) SPARKY (Linux 2.6.18) 3.115, University of California, San Francisco [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.