

Abstract
Microglia are the immune effector cells that are activated in response to pathological changes in the central nervous system. Microglial activation is accompanied by the alteration of integrin expression on the microglia surface. However, changes of integrin expression upon chemoattractant (ADP) stimulation still remain unknown. In this study, we investigated whether ADP induces the alteration of integrin species on the cell surface, leading to changes in chemotactic ability on different extracellular matrix proteins. Flow cytometry scans and on-cell Western assays showed that ADP stimulation induced a significant increase of α6 integrin-GFP, but not α5, on the surface of microglia cells. Microglia also showed a greater motility increase on laminin than fibronectin after ADP stimulation. Time lapse microscopy and integrin endocytosis assay revealed the essential role of calcium-independent phospholipase A2 activity for the recycling of α6 integrin-GFP from the endosomal recycling complex to the plasma membrane. Lack of calcium-independent phospholipase A2 activity caused a reduced rate of focal adhesion formation on laminin at the leading edge. Our results suggest that the alteration of integrin-mediated adhesion may regulate the extent of microglial infiltration into the site of damage by controlling their chemotactic ability.
Keywords: chemotaxis, focal adhesion, integrin, microglia, phospholipase A
Introduction
Microglia are the immune effector cells in the central nervous system (CNS). Under normal conditions, microglia exist as nonmigratory ramified cells. The ramified morphology of resting microglia is rapidly transformed into a motile ameboid form after pathological stimuli, driving the migration of microglia cells toward lesion sites (1–3). There is a growing body of evidence that the extracellular matrix (ECM)2 and integrins are important for modulating microglial behavior. ECM influences microglial behavior as fibronectin (FN) and laminin (LN) often show opposite effects on microglial morphology, adhesion, and activation. FN promotes transformation of amoeboid microglia into ramified microglia, whereas LN causes the reverse transformation (4). FN also promotes increased secretion of amyloid precursor protein by microglial cells, whereas LN and collagen have the opposite effect (5). It has been demonstrated that microglia attach strongly to FN and vitronectin but only weakly to LN and astrocyte ECM and that LN exerts a dominant antiadhesive effect on microglial adhesion (6).
Integrins are transmembrane receptors that mediate cell adhesion to the ECM, a fundamental cellular process that regulates cell growth, differentiation, and motility (7). All integrins are αβ heterodimers. The α subunits vary in size between 120 and 180 kDa and are each noncovalently associated with a β subunit (90–110 kDa) (8). Ligand binding to integrin triggers integrin clustering as well as the formation, disassembly, and reorganization of stress fibers and focal adhesion (FA) complexes. These structural changes are involved in the regulation of cell adhesion, cell migration, and cell division (8). Engagement of integrin with ligands also elicits the activation of a number of signaling pathways, regulating cytoskeletal organization, cell migration, differentiation, and death (9).
Microglial activation is accompanied by the alteration of integrin expression. Inflammatory cytokines cause increased expression of α4β1, α5β1, and Mac-1 integrins (10). The intensity of expression of Mac-1, LFA-1, and β2 was enhanced on reactive microglia in Alzheimer's disease tissue (11). An increase of LFA-1 expression was also detected on activated microglia located close to the edge of demyelinating lesions of multiple sclerosis (12). Microglial adhesion to LN and astrocyte ECM is increased by proinflammatory cytokines such as tumor necrosis factor (TNF) and interferon-γ (IFN-γ) (13). Because reduction of LFA-1 integrin expression markedly attenuates microglial migration and activation during neuroinflammation (14), the alteration of integrin-mediated adhesion may regulate the extent of microglial infiltration into the site of damage. Extracellular ATP or ADP released from damaged cells and surrounding astrocytes could induce microglia chemotaxis and membrane ruffling through Gi/o-coupled P2Y12 receptor in microglia (15). Inflammatory cytokines increased expression of α4β1, α5β1, and Mac-1 integrins on microglia (10), but changes of integrin expression upon P2Y12 receptor activation were not well understood. How ADP exerts such a major influence on microglial integrin expression is an open question. In this study, we investigated whether ADP can induce an alteration of integrin species on the cell surface that leads to changes in chemotactic ability on different ECM substrates. We also examined the role of iPLA2 in the regulation of α6 integrin recycling upon ADP stimulation.
Results
Integrin α6 Expression on Microglia Surface and Motility on LN Is Up-regulated upon ADP Stimulation
To determine whether the expression level of α5 or α6 integrin changes upon ADP stimulation, we used FACS analysis to compare cell surface levels of integrins in BV2 cells at 15 min after ADP stimulation. As shown in Fig. 1A, the α5 integrin expression level on cells stimulated with ADP was comparable with the level of control cells not treated with ADP. In contrast, the α6 integrin expression level was significantly higher than the level of control cells. The increase of α6 integrin expression upon ADP stimulation was confirmed by on-cell Western assay (Fig. 1B) using BV2 cells that were stimulated with ADP and fixed with formaldehyde at different time points. Non-permeabilized BV2 cells were stained with α5 or α6 antibodies, so only cell surface antigens are detected. Consistent with the FACS data, the expression of α6, but not α5, was significantly increased in 2 and 5 min. The increase of α6 expression was quite variable at 10 min as indicated by no statistical significance. P2Y12 receptor activation by ADP results in an increase of cell velocity and net distance of movement, both on FN and LN (Fig. 1C). The increase of motility on LN, however, is significantly greater than on FN, presumably due to the increase of α6 integrin expression on the microglia surface. Thus, the increase of specific integrins on the surface might have different impacts on microglia adhesion to different ECM proteins. Adhesion strength and migration of cells are dependent upon the number and size of FAs (17). Examination of FAs by paxillin immunostaining revealed a striking difference in cells adhered on FN or LN (Fig. 1D). In control cells, cells on FN show many FAs, both around the periphery of cells and inside the cell where stress fibers are anchored. Upon ADP stimulation of cells adhered on FN, the number of FAs was significantly increased. Their average size also increased although mainly in the cell interior. For cells on LN, however, the increase in number of FAs was exclusively in the periphery, suggesting that LN might positively affect effective cell displacement and migration potential compared with FN substrate that might induce loss of cell polarity and increased adhesion. To further investigate the role of P2Y12 receptor activation in the regulation of FA attachments, we examined changes of cell attachment/adhesion by an electric cell-substrate impedance sensing (ECIS) instrument that measures adhesion strength by sensing the change of impedance of a small electrode to alternating current flow (18). Cell impedance increased drastically when ADP was injected into the ECIS chambers of control BV2 microglia cells on FN, indicating increased adhesion strength that resulted from the increase of FA number and size (Fig. 1E). The increase of impedance on LN was slower and weaker than on FN, consistent with limited increase of FA around the periphery of cells.
FIGURE 1.

A, change of integrin expression on BV2 microglia upon ADP stimulation. Integrin α subunit expression level on the microglia surface was examined with flow cytometry. Flow cytometry profiles showing microglial α5 or α6 integrin expression in the absence or presence of ADP for 20 min are shown. Note that ADP increases microglial α6 expression but not α5 integrin. B, changes of integrin expression on the cell surface after ADP stimulation for 20 min were quantified by an on-cell Western assay with near-IR dyes in non-permeabilized cells using antibodies against α5 and α6 and the Odyssey IR imaging system. Quantification from four independent triplicated experiments is shown in the graphs. Error bars represent S.E. Change of α-tubulin is shown as a housekeeping protein control. C, the increase of BV2 microglia motility, represented as mean velocity and net distance, upon ADP stimulation was significantly greater on LN than on FN. Mean velocity and net distance were measured by tracking cell migration from movies taken by live cell imaging of 10 cells from three different culture dishes. **, p < 0.01 versus non-ADP by t test. The α6 and α5 data shown in C and Fig. 3C are the same. D, changes of the size and number of FAs upon ADP stimulation in cells plated on FN or LN (10 μg). Cells on FN have more and bigger FAs, whereas cells on LN only show the increase of FA around the periphery of the cell. Scale bar, 10 μm. E, increase of adhesion strength of microglia cells plated on FN or LN upon ADP stimulation was measured by continuous ECIS readings of impedance. ECIS readings of BV2 cells on FN and LN are the same in Fig. 7. Cont, control; Tub, tubulin.
iPLA2 Activity Is Required for the Trafficking and Localization of α6 Integrin-Green Fluorescent Protein (GFP)
To investigate the mechanism by which α6 integrin expression on microglia is regulated, we expressed α6 integrin fused to GFP (α6-GFP) in microglia. To keep the expression of α6-GFP at moderate levels, we first knocked down endogenous mouse α6 integrin using short hairpin RNA (shRNA) (α6-KD cells; Fig. 2A) and then expressed human α6 integrin fused to GFP. α6-KD cells clearly exhibited defects in adhering and spreading on LN, and the expression of α6-GFP rescued the adhesive defects of α6-KD cells to the level of control BV2 cells (Fig. 2B). The α6-GFP also localized with their endogenous counterparts to focal adhesions in cells plated on LN but not on FN (Fig. 2C). We also observed large and small α6-GFP vesicles in protrusions on the leading edge that were more prominent in cells on FN.
FIGURE 2.

A, knockdown of endogenous (mouse) α6 integrin with shRNA. The Western blot of α6 integrin in α6-KD cells shows a significant reduction of α6 integrin. B, cell adhesion/spreading assay on LN to determine whether human α6-GFP expressed in α6-KD cells is functional. A 96-well plate was coated with 3 μg/ml LN at 4 °C overnight. 1 × 104 cells were added to each well and incubated for a specific time period. After washing off non-attached cells, four non-overlapping images from each well were taken with a 10× objective, and the percentage of cells (% cells adhered) that show partially or fully spread morphologies among total cells attached on the substrate after certain times of incubation was determined. 12 images in three different wells from the one culture are shown. Expression of human α6-GFP can rescue the adhesion/spreading defects of α6-KD cells. A specific iPLA2 inhibitor, BEL, also inhibits cell adhesion/spreading on LN. Error bars represent S.E. C, localization of α6 integrin-GFP was examined with live cell imaging in cells plated on different matrix proteins. Localization of α6 integrin-GFP was observed on vesicles and membrane protrusions (arrow) in cells plated on FN. α6 integrin-GFP localizes to focal contacts and adhesions (arrow) in cells plated on LN.
Given the relatively rapid increase of α6 integrin after ADP stimulation, the increase might result more from active recycling than from active translation. In our previous study, we demonstrated that iPLA2 activity increases up to 3 times upon ADP stimulation and that inhibition of iPLA2 by 4-bromoenol lactone (BEL), a very potent and specific suicidal inhibitor for iPLA2, or iPLA2 knockdown suppressed endosomal recycling (19). We hypothesized that iPLA2 activity might also be required for the increase of α6 integrin expression on the microglia surface upon ADP stimulation. Consistent with this hypothesis, the increase of α6 integrin expression observed by FACS can be blocked by BEL (Fig. 3A), suggesting that increase of α6 integrin expression requires iPLA2 activity. Consistent with this, the increase of α6 integrin on the cell surface was significantly lower in cells in which iPLA2 was knocked down (iPLA2-KD cells). On-cell Western assays also showed that iPLA2-KD cells had substantially decreased levels of α6 integrin surface expression but exhibited only moderate inhibition of α5 integrin expression on the cell surface (Fig. 3C). Immunofluorescence staining of paxillin showed a significant reduction of FA number in iPLA2-KD cells, presumably due to the decreased levels of α6 integrin surface expression (Fig. 3D). We examined the subcellular localization of α6 integrin-GFP in cells treated with BEL or in iPLA2-KD cells. The α6 integrin-GFP is predominantly associated with vesicular structures in the perinuclear area, and α6 integrin-GFP vesicles moved from a perinuclear region to the base of the lamellipodia (Fig. 3E). Upon ADP stimulation, a large fraction of α6 integrin-GFP vesicles was translocated and resided near the cortical membrane. In contrast, localization of α6 integrin-GFP vesicles is tightly confined in the perinuclear region in iPLA2-KD cells. Even after ADP stimulation, most of the α6 integrin-GFP vesicles remained in the perinuclear area. Taken together, these observations suggest the presence of a defect in α6 integrin trafficking to the plasma membrane in iPLA2-KD cells. We examined the defect of α6 integrin recycling in more detail by performing an integrin endocytosis assay on iPLA2-KD cells. Cell surface α6 integrin was labeled with anti-α6 integrin antibody for 45 min at 4 °C, a temperature that halts endocytosis. Cells were then switched to 37 °C to resume endocytosis and incubated for 30 min, allowing the internalization of antibody bound to cell surface integrin. Staining of internalized α6 integrin with secondary antibody revealed that most of the internalized α6 was localized in the perinuclear area, showing a colocalization with Rab11, a marker for the endosomal recycling compartment (ERC) in BV2 cells (Fig. 4, A and B). Upon ADP stimulation for 10 min, internalized α6 integrin was associated with vesicles near the plasma membrane, presumably en route to recycling back to the plasma membrane. However, this recycling of α6 integrin is essentially absent in iPLA2-KD cells and BV2 cells treated with BEL as most internalized α6 integrin was still retained in the ERC after ADP stimulation, suggesting that iPLA2 activity is required for the recycling of α6 integrin-GFP.
FIGURE 3.

A, flow cytometry analysis of α6 integrin expression on the cell surface. Inhibition of iPLA2 with 5 μm BEL abolishes the increase of α6 integrin expression on the cell surface upon ADP stimulation. The graph shows average of three independent flow analyses. Error bars represent S.E. **, p < 0.01 versus control by ANOVA. B, flow cytometry analysis of α6 integrin expression on the iPLA2-KD cell surface. C, quantification of α6 integrin expression on the cell surface in iPLA2-KD cells by an on-cell Western assay. The increase of surface expression of α6 integrin, but not α5 integrin, upon ADP stimulation was significantly reduced in iPLA2-KD cells. Averages of three to four independent experiments that were triplicated are shown. **, p < 0.01 versus iPLA2-KD by ANOVA. D, knockdown of iPLA2 causes a significant inhibition of FA formation on LN. BV2 and iPLA2-KD cells were plated on coverslips coated with 10 μg/ml LN. Immunostaining was performed on fixed cells with anti-paxillin antibody. The number of FAs was measured from the images. **, p < 0.01 versus BV2 by t test. E, ADP stimulation leads to the localization of α6 integrin-GFP vesicles to membrane protrusions or the leading edge (arrow) in BV2 cells. In contrast, α6-GFP-containing vesicles remain in a perinuclear region even after ADP stimulation in iPLA2-KD cells. The graph shows quantification of fluorescence intensity from 10 individual cells in three culture dishes before ADP stimulation. The ratio of GFP intensity in the peripheral area to perinuclear area is shown. The perinuclear area was defined as a concentric circle around the nucleus with a diameter twice as big. 10 individual cells in three culture dishes were examined. *, p < 0.05 versus BV2 by t test; scale bar indicates 10 μm. Cont, control.
FIGURE 4.

A, integrin endocytosis assay to examine the recycling α6 integrin. BV2 and iPLA-KD cells were plated on LN-coated coverslips, and an integrin endocytosis assay was performed as described under “Experimental Procedures.” In untreated BV2 and iPLA-KD cells, endocytosed α6 integrin was found predominantly in a perinuclear vesicular structure. Following ADP treatment, α6 integrin localization changed to plasma membrane or vesicles in the periphery of BV2 cells, but this change is absent in iPLA-KD cells. Scale bar, 10 μm. The ratios of fluorescence intensities in the perinuclear area and cellular periphery from 10 cells were determined and are shown in the graph. Error bars represent S.E. **, p < 0.01 versus BV2 by ANOVA. B, endocytosed α6 integrin is colocalized with mCherry-Rab11 in the perinuclear region, but the localization of α6 integrin changes rapidly to the periphery of the cell upon ADP stimulation. However, in iPLA2-KD or BEL-treated BV2 cells, α6 integrin remains colocalized with Rab11 in the perinuclear region even after ADP stimulation.
iPLA2 Activity Is Required for FA Formation at the Leading Edge
To examine how the defect of α6 integrin recycling affects dynamic changes of FA during cell migration, we used time lapse video microscopy to examine FA on the leading edge of microglia cells expressing α6 integrin-GFP (Fig. 5A). After ADP stimulation, we were able to observe incorporation of α6 integrin-GFP into newly forming adhesions in BV2 cells on LN. In both BEL-treated BV2 cells and iPLA2-KD cells, newly organizing α6-containg adhesions were essentially absent, indicating that iPLA2 activity is required for the formation of new adhesions, presumably via the regulation of trafficking of α6 integrin-GFP to FAs upon ADP stimulation. This result was strengthened by the measurement of FA assembly rate using paxillin-GFP in live cells. We analyzed FA dynamics by examining the increase of paxillin-GFP in FAs in cells on FN or LN using time lapse video microscopy for 20 min after ADP stimulation (Fig. 5B). The formation of paxillin-GFP-containing FAs in the newly protruding regions or the cell edge of the cells transfected with paxillin-GFP was examined. The rate constants for formation and disassembly were determined from the slope of graphs of paxillin-GFP intensities over the time. The rate of paxillin-GFP assembly on FN was not significantly different from the rate on LN in BV2 cells. In contrast, paxillin-GFP assembly on LN in iPLA2-KD cells was almost abolished, whereas assembly on FN was moderately affected, consistent with the role of iPLA2 in α6 integrin expression and trafficking.
FIGURE 5.

Reduced rate of the formation and growth of focal adhesions on LN in cells treated with BEL or iPLA2-KD cells. A, α6-KD cells were transfected with α6 integrin-GFP and plated on 10 μg/ml LN. Cells were stimulated with ADP, and FA formation in cells was imaged for 20 min by capturing images every 6 s. In control BV2 cells, α6 integrin-GFP localizes in small foci at the edge of the lamellipodium (indicated with arrows), and their sizes grow over time with the leading edge progression. However, cells treated with 5 μm BEL lack the formation of these foci. Scale bar, 5 μm. B, the growth of FA was examined by paxillin-GFP imaging. Arrows indicate representative adhesion assembly, and arrows with dashed line indicate representative adhesion disassembly in a control cell plated on LN. C, the rate of FA growth was assessed by imaging GFP-paxillin in cells plated on FN or LN. The size of FA measured with GFP-paxillin was plotted against time, and the rate constants for FA formation were determined from the slope of these graphs. Measurements of slopes were obtained for 10–15 individual adhesions from four to five cells and are shown in the graph. Error bars represent S.E. **, p < 0.01 versus BV2 on LN by ANOVA.
Reduced Motility of α6 Integrin-GFP Vesicles in iPLA2-KD Cells
Individual α6-GFP vesicles were then traced to determine the path, distance traveled, and velocity of vesicles. Movements of vesicles in control or iPLA2-KD cells were captured at 6-s intervals for 15 min and traced manually. Fig. 6B shows the representative paths of α6 integrin-GFP vesicle movements and velocity profiles of individual vesicles in different cell types. As shown in Fig. 6, B and C, most α6 integrin-GFP vesicles showed a diffuse-and-go movement in which diffusional movements are interrupted by directional movements over 0.5 μm. The α6-GFP vesicles traveled with an average velocity of 8.06 μm/min in BV2 cells. In contrast, α6-GFP vesicles in iPLA2-KD cells maintained slow and diffusional movement without any direction, and their average velocity was 3.03 μm/min. Vesicles in BEL-treated BV2 cells showed similar velocity. Vesicles in iPLA2-KD cells appear to show a largely random pattern of motion with rare and transient association with microtubules, indicated by the absence of movements with high velocity.
FIGURE 6.

Tracking of α6-GFP vesicle movements in control or iPLA2-KD cells by live cell imaging. A, images of cells expressing α6 integrin-GFP were acquired at 6-s intervals for 15 min, and tracks of vesicles were traced. B, six representative paths taken by vesicles carrying α6 integrin-GFP are shown. C, histograms of the velocity of α6 integrin-GFP vesicles. Trajectories for 15–20 α6-GFP vesicles from four independent movies were traced. The speed was calculated from the distance between two successive positions, and speeds of vesicles were sorted into bins by tallying how many speeds there are in each bin range. The frequency of high speeds is clearly lower in iPLA2-KD or BEL-treated BV2 cells.
Microglia Pretreated with ADP Exhibit Higher Motility and Chemotaxis on LN
To examine whether the increase of α6 integrin on the microglia surface has an impact on the adhesion and motility of microglia, we first monitored changes of adhesion strength upon ADP stimulation using an ECIS system (Fig. 7A). When ADP was injected into the ECIS chambers of BV2 microglia cells on both FN and LN, cell impedance increased drastically and remained elevated for 2 h. The increase of impedance of iPLA2-KD cells on FN was slower but reached a level comparable with control cells. However, impedance of iPLA2-KD cells on LN did not increase at all, suggesting that ADP might have greater impact on α6 integrin expression on the cell surface. Interestingly, impedance of iPLA2-KD cells on LN decreased below the level before ADP stimulation, presumably due to the endocytosis of surface α6 integrin. Examination of random and chemotactic motility of cells treated with BEL also revealed that iPLA2 activity has greater impact on the increase of motility on LN than on FN. Upon ADP stimulation, random motility of microglia increased significantly more on LN than on FN as both mean velocity and net distance of movement were increased about 200% on LN compared with a 90% increase on FN (Fig. 7B). This result is consistent with the increase of FAs in the cell periphery. This increase can be blocked by BEL inhibition of iPLA2. Chemotactic motility was measured using Transwell chamber membranes coated with LN. Cells challenged with ADP for 1 h before they were plated on the Transwell chamber membranes exhibited very high migration to the bottom well even without a chemoattractant cue, presumably due to high basal motility. Chemotaxis of microglia challenged with ADP was greatly improved but was significantly reduced in iPLA2-KD cells (Fig. 7C). Potential off-target effects of the shRNA were ruled out by rescue of the chemotaxis defect of iPLA2-KD cells by expression of human iPLA2-GFP (data not shown). This result suggests that the increase of α6 integrin on the microglia surface upon ADP stimulation would have a positive impact on the chemotactic ability of microglia.
FIGURE 7.
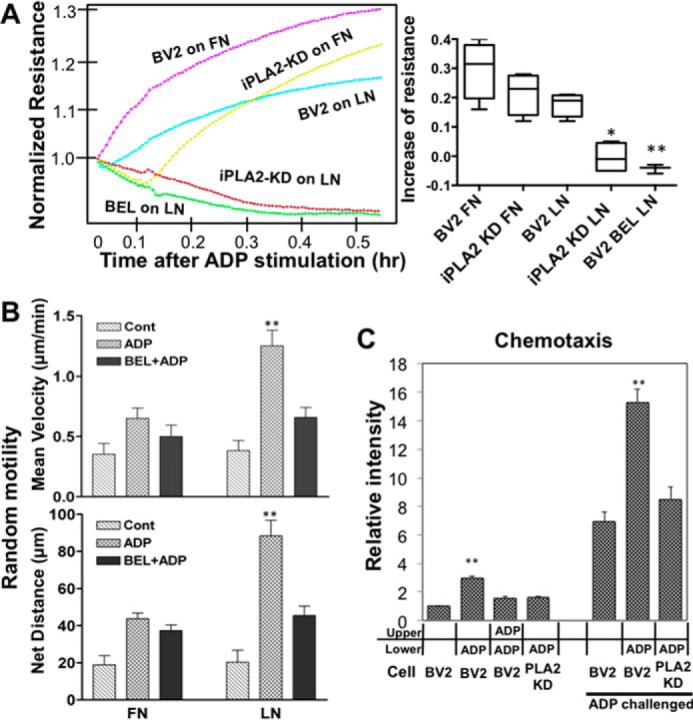
A, continuous ECIS readings of iPLA2-KD cells plated on LN upon ADP stimulation shows no increase in adhesion strength. Quantification of maximum increase of resistance from four independent ECIS readings is shown in the graph. Error bars represent S.E. *, p < 0.05; **, p < 0.01 versus BV2 on LN by ANOVA. B, increase of microglia motility upon ADP stimulation on FN or LN. Random motility on individual ECM substrates was examined. Microglia exhibited higher motility on LN upon ADP stimulation. **, p < 0.01 versus control and BEL + ADP by t test. C, Transwell chemotaxis assay. Transwell chamber membranes were coated with LN, and cells were plated. Cells were then assayed for migration toward 100 μm ADP. Cells pretreated with ADP for 30 min exhibited significantly greater chemotactic ability compared with non-treated cells. Knockdown of iPLA2 abolished this increase of chemotaxis. Four duplicated independent assays were quantified. **, p < 0.01 versus BV2 and KD + ADP. Cont, control.
Discussion
Alteration of integrin-mediated adhesion may regulate the extent of microglial infiltration into the site of neuronal injury by controlling their motility. In this study, we demonstrated that ADP stimulation induced a significant increase in the expression of α6 integrin, but not α5, on the surface of microglia cells. An integrin endocytosis assay also revealed that iPLA2 activity is required for the recycling of internalized α6 integrin back to the plasma membrane. Time lapse microscopy also clearly revealed the essential role of iPLA2 activity for the recycling of α6 integrin from the ERC to the plasma membrane via recycling endosomes.
We demonstrated in this study that microglia pretreated with ADP exhibited higher chemotactic motility, which can be abolished by the inhibition of iPLA2 activity. There is a growing body of evidence that iPLA2 plays a key role in the regulation of chemotaxis. Strong chemotaxis defects are observed only when both the PI3K and PLA2 pathways are disrupted in Dictyostelium (20). Recent studies also showed that monocyte chemotaxis toward monocyte chemoattractant protein-1 requires iPLA2 activity (21, 22). Our previous study also demonstrated that iPLA2 activity plays an important role in the regulation of microglia chemotaxis (19).
Internalized integrins recycle back to the cell surface along two different routes. Integrins, such as α5β1 and αLβ2, are known to enter the ERC before being recycled to the plasma membrane (long loop), which is regulated by Rab11a. Rab25 (or Rab11c), a member of the Rab11 family, has been reported to be physically associated with α5β1 integrin, modulating recycling of α5β1 and invasive migration of ovarian tumor cells (23). Rab4 has been shown to regulate recycling of integrin αvβ3 from early endosomes in a “short loop” pathway (23). The arrest of α6 integrin recycling at the ERC suggests that α6 integrin is recycled via a long loop. Our observation that α6 integrin remains at the ERC in iPLA2-KD cells suggests the possibility that the formation of recycling vesicles from the ERC might be defective in these cells. Recent studies shed light on the role of PLA2 in the regulation of the formation of membrane tubules and membrane fusion events in the secretory and endocytic pathways (24). iPLA2 activity has been suggested to be associated with the formation of Golgi membrane tubules in response to brefeldin A, and these tubules have been suggested to function in various trafficking pathways (25). Based on these observations, it would be reasonable to conclude that iPLA2 activity might be required for the formation of α6 integrin-GFP vesicles from the ERC. However, the number of α6-GFP vesicles in iPLA2-KD cells is comparable with that in control cells, suggesting that the recycling defect of α6 integrin-GFP vesicles is not simply due to the lack of vesicle formation. iPLA2 has been shown previously to play a role in the regulation of casein-containing secretory vesicles. In that study, treatment of cells with BEL caused casein to accumulate in the perinuclear area of the cell, suggesting that the transport of milk proteins to the apical side of the cell was partly hindered (26). Various PLA2 antagonists caused a block in the endocytic recycling pathway of transferrin or transferrin receptor (27). These reports are consistent with our results and suggest that iPLA2 activity might be required for the trafficking of recycling vesicles to the plasma membrane. Our time lapse video microscopy revealed a reduction in motility of α6 integrin-GFP vesicles in iPLA2-KD cells. Fatty acids released by iPLA2 activity might be required for the interaction between a motor protein and cargo membrane. Kinesin motor proteins facilitate vesicle formation/trafficking by pulling membrane tubules or vesicles along on microtubules. The tail domains of some kinesins can interact with phospholipids. Unc104/KIF1A has a pleckstrin homology (PH) domain located at the C-terminal tail that directly interacts with PI 4,5-bisphosphate and transports PI 4,5-bisphosphate-containing vesicles (28, 29). The PX motif of KIF16B binds PI 3-phosphate and localizes to membranes bearing this lipid in vivo (30). Tracking of α6-GFP vesicles in iPLA2β-KD cells clearly revealed a lack of directionality and slow speed of vesicle movements. Kinectin accumulation at sites of clustered integrins has been reported (31), and iPLA2 activity might be required for the recycling of integrin vesicles by controlling the interaction between kinectin and integrin.
Cell migration is a highly coordinated and integrated process, including numerous factors leading to migration. Microglia on an LN substrate adopt a less activated, poorly adhesive phenotype and show reduced levels of expression of the activation markers (32). It has also been shown that microglial adhesion to LN is mediated entirely by the α6β1 integrin, and microglial adhesion to LN was increased significantly by the proinflammatory cytokines (6, 33). However, cytokines did not change α6β1 expression levels but altered the activation state of α6β1, mediated by a PKC-dependent mechanism. Our study revealed that activation of P2Y12 receptor with ADP also results in an increase of α6 integrin on the microglia surface and a concomitant increase of adhesion and motility on LN. Enhanced microglial adhesion to LN might serve to promote microglial adhesion and entry into areas of CNS injury. LN is transiently expressed by neurons inside the ischemic core 24 h after a stroke has occurred as an acute reaction of the brain to ischemia (34). Interestingly, peptides derived from the α chain of LN caused a significant reduction of leukocyte accumulation and infarct size in rats subjected to 1 h of cerebral ischemia (35), suggesting that increased adhesion/motility on LN is prerequisite for leukocyte migration. Our results described in this study also suggest that microglial adhesion and migration into the LN-rich injury site would be promoted by the increase of α6β1 integrin expression that is a consequence of ADP stimulation.
Experimental Procedures
Cell Culture and Transfection
BV2 microglia cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% FBS and penicillin-streptomycin (Gibco). Lentivirus-mediated shRNAs were used for gene knockdown. MISSION pLKO.1 shRNA clones were from Sigma and contained hairpin sequences that were used for iPLA2 knockdown (NM_016915.2-2305s1c1) and for α6 integrin knockdown (NM_008397.2-679s1c1). Lentivirus-infected cells were selected for stable cell lines in the presence of puromycin. α6 integrin-GFP construct was made by amplifying human α6 integrin (transcript variant 1) cDNA with primers (forward, GTTTCTCGAGAAAATGGCCGCCGCCGGGCAGCTGTG; reverse, GTTTGGATCCCGTGAGTAGCTTTCATTTTCGTTCCACTTTG) and cloned into pEGFP-N1 at the XhoI and BamHI sites. Cells were transfected with mCherry-Rab11 and α6 integrin-GFP using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions and cultured for 12 h. All experiments were performed 32 h after transfection. Potential off-target effects of the iPLA2 shRNA were ruled out by showing rescue of the chemotaxis defect of iPLA2-KD cells by expressing human iPLA2-GFP, which is immune to RNAi.
Migration Assay
Mean velocity and net distance were measured by tracking cell migration from movies taken by live cell imaging. Chemotaxis assays were performed as described previously (16). Briefly, Transwell chamber membranes (6.5-mm diameter, 8-μm pore size; Corning, Corning, NY) were coated with FN (3 μg/ml) for 8 h. For the chemotaxis assay, either serum-free DMEM or DMEM containing 100 μm ADP was added to the lower chamber. Cells starved for 4 h and suspended in serum-free DMEM were added to the upper chamber. After further incubation for 6 h, non-migrating cells were removed from the upper chamber with a cotton swab, and cells that had migrated to the lower surface of the membrane were fixed with 3.7% formaldehyde for 10 min and stained with 0.2% crystal violet. Cells were imaged, and intensity of staining was measured using ImageQuant software.
On-cell Western Assays
BV2 microglia cells were plated on 24-well tissue culture plates and cultured to 80% confluence. After starvation for 4 h and ADP stimulation for 30 min, cells were then fixed with 3.7% formaldehyde and incubated with PBS containing 5% BSA for blocking nonspecific binding sites. For labeling, cells were incubated for 1 h at 4 °C with a 1:200 dilution of primary antibody against α5 or α6 integrin (BD Biosciences). Samples were washed for 10 min each with PBS and then incubated for 1 h at 4 °C in a 1:10,000 dilution of the secondary antibody, IRDye 680 goat anti-mouse (LI-COR Biosciences, Lincoln, NE). Cells were then washed three times in PBS for 10 min each and imaged using the LI-COR Odyssey infrared imaging system (LI-COR Biosciences). The intensity of the 700-nm infrared signal for each well was quantified using the LI-COR Odyssey infrared imaging system software. The mean intensity of cells incubated only with the secondary antibody was subtracted from the intensity of cells to correct for any background signal not related to integrin staining.
Flow Cytometry
Microglial cells were starved for 4 h and stimulated with ADP for 30 min. Cells were then detached with cell suspension buffer (Gibco) and pelleted. Cells were suspended at 106 cells/ml in FACS staining buffer (PBS containing 2% FBS and 0.2% sodium azide) and stained with FITC-labeled anti-integrin antibodies for 30 min in the dark at 4 °C. Suitable isotype controls were used for calibration. Cells were washed and resuspended in staining buffer and processed through a FACScan flow cytometer before analysis.
ECIS
An ECIS impedance instrument and consumable 8-well arrays were obtained from Applied Biophysics, Inc. (Troy, NY). The base of the device has an array of gold film electrodes that connect the ECIS electronics to each of the 8 wells. For each assay, BV2 control or iPLA2-KD cells (1 × 105 cells) were plated onto a well coated with FN or LN. The 250-μm-diameter gold electrode in each well measured the impedance through alternating current flow over a period of several hours. Measurements were made at 4000 Hz. For ADP stimulation, 100 μl of 100 μm ADP was injected into a well.
Integrin Endocytosis Assay
Cells (with or without transfection of mCherry-Rab11) were grown overnight on coverslips at 60–70% confluence in DMEM containing 10% FBS and antibiotics (penicillin and streptomycin) and then starved for 4 h in serum-free medium. Cell surface α6 integrin was labeled by incubating cells with anti-α6 integrin antibody for 45 min at 4 °C to inhibit endocytosis. Cells were then incubated at 37 °C for 30 min to resume endocytosis and allow the internalization of antibody bound to cell surface integrin. Cells were then stimulated for 15 min with 100 μm ADP, washed with PBS, fixed with 3.7% formaldehyde at 37 °C, permeabilized with 0.2% Triton X-100, labeled with Alexa Fluor 488-conjugated secondary antibody to stain endocytosed α6 integrin, mounted, and examined with a spinning disc confocal fluorescence microscope.
Author Contributions
S.-H. L., N. S., and N. L. performed experiments. S. S. analyzed the data. S.-H. L. and C. Y. C. conceived the experiments, analyzed the data, and wrote the paper.
Acknowledgments
We thank members of the Chung laboratory, Margaret Means, and Dr. Kenneth Woycechowsky for useful discussions and critical reading of the manuscript.
This work was supported by National Institutes of Health Grant GM68097 (to C. Y. C.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- ECM
- extracellular matrix
- FN
- fibronectin
- LN
- laminin
- iPLA2
- calcium-independent phospholipase A2 (group VIA)
- FA
- focal adhesion
- ECIS
- electric cell-substrate impedance sensing
- KD
- knockdown
- BEL
- 4-bromoenol lactone
- ERC
- endosomal recycling compartment
- PI
- phosphatidylinositol
- ANOVA
- analysis of variance.
References
- 1. Hanisch U. K. (2002) Microglia as a source and target of cytokines. Glia 40, 140–155 [DOI] [PubMed] [Google Scholar]
- 2. Kreutzberg G. W. (1996) Microglia: a sensor for pathological events in the CNS. Trends Neurosci. 19, 312–318 [DOI] [PubMed] [Google Scholar]
- 3. Stence N., Waite M., and Dailey M. E. (2001) Dynamics of microglial activation: a confocal time-lapse analysis in hippocampal slices. Glia 33, 256–266 [PubMed] [Google Scholar]
- 4. Chamak B., and Mallat M. (1991) Fibronectin and laminin regulate the in vitro differentiation of microglial cells. Neuroscience 45, 513–527 [DOI] [PubMed] [Google Scholar]
- 5. Mönning U., Sandbrink R., Weidemann A., Banati R. B., Masters C. L., and Beyreuther K. (1995) Extracellular matrix influences the biogenesis of amyloid precursor protein in microglial cells. J. Biol. Chem. 270, 7104–7110 [DOI] [PubMed] [Google Scholar]
- 6. Milner R., and Campbell I. L. (2002) Cytokines regulate microglial adhesion to laminin and astrocyte extracellular matrix via protein kinase C-dependent activation of the α6β1 integrin. J. Neurosci. 22, 1562–1572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Howe A., Aplin A. E., Alahari S. K., and Juliano R. L. (1998) Integrin signaling and cell growth control. Curr. Opin. Cell Biol. 10, 220–231 [DOI] [PubMed] [Google Scholar]
- 8. Hynes R. O. (1992) Integrins: versatility, modulation, and signaling in cell adhesion. Cell 69, 11–25 [DOI] [PubMed] [Google Scholar]
- 9. Mitra S. K., Hanson D. A., and Schlaepfer D. D. (2005) Focal adhesion kinase: in command and control of cell motility. Nat. Rev. Mol. Cell Biol. 6, 56–68 [DOI] [PubMed] [Google Scholar]
- 10. Yu N., Zhang X., Magistretti P. J., and Bloom F. E. (1998) IL-1-α and TNF-α differentially regulate CD4 and Mac-1 expression in mouse microglia. Neuroimmunomodulation 5, 42–52 [DOI] [PubMed] [Google Scholar]
- 11. Akiyama H., and McGeer P. L. (1990) Brain microglia constitutively express β-2 integrins. J. Neuroimmunol. 30, 81–93 [DOI] [PubMed] [Google Scholar]
- 12. Boddeke E. W., Meigel I., Frentzel S., Biber K., Renn L. Q., and Gebicke-Härter P. (1999) Functional expression of the fractalkine (CX3C) receptor and its regulation by lipopolysaccharide in rat microglia. Eur. J. Pharmacol. 374, 309–313 [DOI] [PubMed] [Google Scholar]
- 13. Liesi P., Kaakkola S., Dahl D., and Vaheri A. (1984) Laminin is induced in astrocytes of adult brain by injury. EMBO J. 3, 683–686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ullrich O., Diestel A., Eyüpoglu I. Y., and Nitsch R. (2001) Regulation of microglial expression of integrins by poly(ADP-ribose) polymerase-1. Nat. Cell Biol. 3, 1035–1042 [DOI] [PubMed] [Google Scholar]
- 15. Davalos D., Grutzendler J., Yang G., Kim J. V., Zuo Y., Jung S., Littman D. R., Dustin M. L., and Gan W. B. (2005) ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 8, 752–758 [DOI] [PubMed] [Google Scholar]
- 16. Lee S., and Chung C. Y. (2009) Role of VASP phosphorylation for the regulation of microglia chemotaxis via the regulation of focal adhesion formation/maturation. Mol. Cell. Neurosci. 42, 382–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Critchley D. R. (2000) Focal adhesions—the cytoskeletal connection. Curr. Opin. Cell Biol. 12, 133–139 [DOI] [PubMed] [Google Scholar]
- 18. Schmidt D., Trübenbach J., König C. W., Brieger J., Duda S., Claussen C. D., and Pereira P. L. (2003) Radiofrequency ablation ex vivo: comparison of the efficacy of impedance control mode versus manual control mode by using an internally cooled clustered electrode. Rofo 175, 967–972 [DOI] [PubMed] [Google Scholar]
- 19. Lee S. H., Schneider C., Higdon A. N., Darley-Usmar V. M., and Chung C. Y. (2011) Role of iPLA2 in the regulation of Src trafficking and microglia chemotaxis. Traffic 12, 878–889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chen L., Iijima M., Tang M., Landree M. A., Huang Y. E., Xiong Y., Iglesias P. A., and Devreotes P. N. (2007) PLA2 and PI3K/PTEN pathways act in parallel to mediate chemotaxis. Dev. Cell 12, 603–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Carnevale K. A., and Cathcart M. K. (2001) Calcium-independent phospholipase A2 is required for human monocyte chemotaxis to monocyte chemoattractant protein 1. J. Immunol. 167, 3414–3421 [DOI] [PubMed] [Google Scholar]
- 22. Mishra R. S., Carnevale K. A., and Cathcart M. K. (2008) iPLA2β: front and center in human monocyte chemotaxis to MCP-1. J. Exp. Med. 205, 347–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Caswell P. T., and Norman J. C. (2006) Integrin trafficking and the control of cell migration. Traffic 7, 14–21 [DOI] [PubMed] [Google Scholar]
- 24. Brown W. J., Chambers K., and Doody A. (2003) Phospholipase A2 (PLA2) enzymes in membrane trafficking: mediators of membrane shape and function. Traffic 4, 214–221 [DOI] [PubMed] [Google Scholar]
- 25. de Figueiredo P., Drecktrah D., Katzenellenbogen J. A., Strang M., and Brown W. J. (1998) Evidence that phospholipase A2 activity is required for Golgi complex and trans Golgi network membrane tubulation. Proc. Natl. Acad. Sci. U.S.A. 95, 8642–8647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Péchoux C., Boisgard R., Chanat E., and Lavialle F. (2005) Ca2+-independent phospholipase A2 participates in the vesicular transport of milk proteins. Biochim. Biophys. Acta 1743, 317–329 [DOI] [PubMed] [Google Scholar]
- 27. de Figueiredo P., Doody A., Polizotto R. S., Drecktrah D., Wood S., Banta M., Strang M. S., and Brown W. J. (2001) Inhibition of transferrin recycling and endosome tubulation by phospholipase A2 antagonists. J. Biol. Chem. 276, 47361–47370 [DOI] [PubMed] [Google Scholar]
- 28. Klopfenstein D. R., Tomishige M., Stuurman N., and Vale R. D. (2002) Role of phosphatidylinositol(4,5)bisphosphate organization in membrane transport by the Unc104 kinesin motor. Cell 109, 347–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Klopfenstein D. R., and Vale R. D. (2004) The lipid binding pleckstrin homology domain in UNC-104 kinesin is necessary for synaptic vesicle transport in Caenorhabditis elegans. Mol. Biol. Cell 15, 3729–3739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hoepfner S., Severin F., Cabezas A., Habermann B., Runge A., Gillooly D., Stenmark H., and Zerial M. (2005) Modulation of receptor recycling and degradation by the endosomal kinesin KIF16B. Cell 121, 437–450 [DOI] [PubMed] [Google Scholar]
- 31. Tran H., Pankov R., Tran S. D., Hampton B., Burgess W. H., and Yamada K. M. (2002) Integrin clustering induces kinectin accumulation. J. Cell Sci. 115, 2031–2040 [DOI] [PubMed] [Google Scholar]
- 32. Milner R., and Campbell I. L. (2002) The integrin family of cell adhesion molecules has multiple functions within the CNS. J. Neurosci. Res. 69, 286–291 [DOI] [PubMed] [Google Scholar]
- 33. Milner R., and Campbell I. L. (2006) Increased expression of the β4 and α5 integrin subunits in cerebral blood vessels of transgenic mice chronically producing the pro-inflammatory cytokines IL-6 or IFN-α in the central nervous system. Mol. Cell. Neurosci. 33, 429–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jucker M., Tian M., and Ingram D. K. (1996) Laminins in the adult and aged brain. Mol. Chem. Neuropathol. 28, 209–218 [DOI] [PubMed] [Google Scholar]
- 35. Yanaka K., Camarata P. J., Spellman S. R., Skubitz A. P., Furcht L. T., and Low W. C. (1997) Laminin peptide ameliorates brain injury by inhibiting leukocyte accumulation in a rat model of transient focal cerebral ischemia. J. Cereb. Blood Flow Metab. 17, 605–611 [DOI] [PubMed] [Google Scholar]