

Abstract
Agonist-induced activation of Rho GTPase signaling leads to endothelial cell (EC) permeability and may culminate in pulmonary edema, a devastating complication of acute lung injury. Cingulin is an adaptor protein first discovered in epithelium and is involved in the organization of the tight junctions. This study investigated the role of cingulin in control of agonist-induced lung EC permeability via interaction with RhoA-specific activator GEF-H1. The siRNA-induced cingulin knockdown augmented thrombin-induced EC permeability monitored by measurements of transendothelial electrical resistance and endothelial cell permeability for macromolecules. Increased thrombin-induced permeability in ECs with depleted cingulin was associated with increased activation of GEF-H1 and RhoA detected in pulldown activation assays. Increased GEF-H1 association with cingulin was essential for down-regulation of thrombin-induced RhoA barrier disruptive signaling. Using cingulin-truncated mutants, we determined that GEF-H1 interaction with the rod + tail domain of cingulin was required for inactivation of GEF-H1 and endothelial cell barrier preservation. The results demonstrate the role for association of GEF-H1 with cingulin as the mechanism of RhoA pathway inactivation and rescue of EC barrier after agonist challenge.
Keywords: endothelium, lung injury, permeability, Rho (Rho GTPase), signal transduction, vascular biology
Introduction
Endothelial cell (EC)2 lining forms a semi-permeable, dynamically regulated barrier to fluids, macromolecules, and cell elements present in the circulation. Increased vascular EC permeability caused by vasoactive agonists (1, 2), excessive mechanical forces such as high magnitude cyclic stretch related to lung mechanical ventilation (3), or increased vascular stiffness (4, 5) is a cardinal feature of many pathological conditions, including brain and lung edema, atherosclerosis, pulmonary hypertension, and other syndromes (6–9).
The serine protease thrombin triggers blood coagulation, but it also directly increases pulmonary EC permeability and plays a major role in the pathogenesis of brain and pulmonary edema (10, 11) by activating the RhoA GTPase signaling cascade (12, 13). Activated RhoA, via its effector, Rho-associated kinase (Rho kinase), induces assembly of stress fibers and focal adhesions (14–16). Rho kinase may directly catalyze myosin light chain (MLC) phosphorylation or act indirectly via inactivation of myosin light chain phosphatase (MYPT1) by phosphorylation at Thr695, Ser894, and Thr850 (13, 17). Together, these mechanisms cause actomyosin stress fiber formation, MLC phosphorylation, and cell contraction, leading to increased EC permeability. Agonist-induced vascular EC permeability is a reversible process. However, mechanisms of local regulation of barrier-disruptive RhoA signaling and barrier dysfunction remain incompletely understood. Guanine nucleotide exchange factor H1 (GEF-H1) exhibits Rho-specific activity (18) and may localize on microtubules (18) or at cell-cell junctions (19). In a microtubule-bound state, the guanine-exchange activity of GEF-H1 is suppressed. GEF-H1 release from microtubules stimulates Rho-specific GEF activity (20). In contrast, GEF-H1 targeting to the tight junctions (TJ) in polarized kidney epithelial cells was associated with down-regulation of RhoA signaling (19). Tight junctions consist of transmembrane proteins occludin and claudin, which interact with the intracellular proteins zonula occludens (ZO), afadin, cingulin, and others (21, 22).
Cingulin is a protein localized in the cytoplasmic compartment of the TJ in epithelial and endothelial cells and is involved in TJ reassembly (23). The cingulin forms a parallel homodimer of two 140-kDa subunits, each comprising a globular Head domain, a coiled-coil “rod,” and a small globular tail (24). The cingulin Head domain interacts with TJ structural proteins ZO-1, ZO-2, ZO-3, myosin, and afadin, the protein interacting with both tight junctions and adherens junctions (24–26). Although early studies suggested a role of cingulin in epithelial tight junction formation due to its role in sequestration of GEF-H1 and inhibition of RhoA signaling (27), later studies suggested that cingulin is not directly required for the formation of tight junctions and epithelial polarity of the Madin-Darby canine kidney epithelial cell line (28, 29). Cingulin involvement in the regulation of the Rho pathway of agonist-induced vascular EC permeability remains unexplored. This study tested the hypothesis that increased cingulin interaction with GEF-H1 after stimulation with barrier-disruptive agonists is required for down-regulation of Rho signaling and preservation of the EC barrier.
Results
Down-regulation of Cingulin Augments Thrombin-induced Endothelial Barrier Disruption
Roles of cingulin in the maintenance of basal barrier properties of the pulmonary EC monolayer and in permeability changes in response to barrier-disruptive agonist stimulation were evaluated in experiments with siRNA-induced cingulin knockdown. Studies in canine kidney epithelial cells did not show significant effects of cingulin overexpression on TJ formation and epithelial polarity under basal conditions (28). Previous studies on Madin-Darby canine kidney cell monolayers looked at the effect of cingulin KO/depletion on transepithelial electrical resistance (27, 30). In human pulmonary ECs, introduction of cingulin-specific siRNA did not affect basal levels of transendothelial electrical resistance (TER) in comparison with cells transfected with nonspecific RNA. Basal TER levels were 981 ± 62 ohm for HPAEC transfected with non-specific-RNA and 964 ± 91 ohm for ECs transfected with cingulin-specific siRNA. Measurements of TER changes over 60 h after transfection did not reveal significant difference between control and cingulin-depleted ECs (Fig. 1A). Knockdown efficiency was monitored by Western blotting analysis of cingulin protein content in cell lysates (Fig. 1A).
FIGURE 1.

Cingulin knockdown exacerbates thrombin-induced EC hyperpermeability. Human lung ECs were transfected with a 200 nm pool of four cingulin-specific siRNAs (si-CGN), two single cingulin-specific siRNAs (si-CGN1 and si-CGN2), or nonspecific (ns) RNA. A, measurements of TER were performed in unstimulated ECs over time. siRNA-induced cingulin protein depletion was confirmed by Western blotting. B, at the time point indicated by the arrow, control or cingulin-depleted ECs were stimulated by thrombin (0.1 unit/ml), and TER measurements were performed over time. Results are representative of four independent experiments. C and D, cells grown on glass coverslips (C) or 96-well plates (D) with immobilized biotinylated gelatin (0.25 mg/ml) were stimulated with vehicle (Veh) or thrombin (Thr) for 10 min followed by addition of FITC-avidin (25 μg/ml, 3 min). Unbound FITC-avidin was removed, and FITC fluorescence signal was visualized by fluorescence microscopy; bar, 5 μm (C). Alternatively, fluorimetric analysis of the FITC fluorescence signal in control and agonist-stimulated ECs plated in 96-well microplates was performed using the Victor X5 multilabel plate reader (see “Experimental Procedures”). Normalized data are expressed as means ± S.D.; n = 5, *, p < 0.05 (D). AU, arbitrary units.
In turn, EC stimulation with sub-maximal concentration of edemagenic agonist thrombin caused rapid decline in TER reflecting EC barrier compromise. This effect was exacerbated in EC monolayers with depleted cingulin. Cingulin knockdown in these experiments was achieved by EC transfection with a pool of four cingulin-specific siRNAs (Fig. 1B, left panel) or two additional single cingulin-specific siRNAs (Fig. 1B, right panel), as described under “Experimental Procedures.” Basal TER values in nonspecific RNA controls and cingulin-depleted ECs were 1371 ± 156 and 1297 ± 183 ohm, respectively. Cingulin knockdown compromised recovery of EC barrier after thrombin challenge, as compared with thrombin-stimulated ECs without cingulin knockdown.
Visualization of EC monolayer permeability for macromolecules using FITC-labeled avidin as tracer (see “Experimental Procedures”) confirmed the results of TER measurements and demonstrated augmented permeability response to thrombin in ECs with cingulin knockdown (Fig. 1C). The quantitative analysis of thrombin-induced EC permeability for FITC-labeled avidin was performed by fluorimetric measurements of control and thrombin-stimulated ECs grown in 96-well plates after addition of FITC-labeled avidin (Fig. 1D). The results show augmented permeability response to thrombin in ECs with depleted cingulin.
Analysis of thrombin-induced remodeling of actin cytoskeleton and cell junctions, developed after 5 min of thrombin treatment (acute phase), showed increased stress fiber formation and paracellular gap formation in ECs with cingulin knockdown (Fig. 2A). Quantitative analysis of paracellular gaps in non-stimulated and thrombin-stimulated ECs (Fig. 2B) showed that cingulin knockdown did not affect the overall number of gaps observed in basal conditions, but it markedly increased the total area of paracellular gaps formed after thrombin challenge. These effects were accompanied by disruption of VE-cadherin-positive adherens junctions in ECs with cingulin knockdown, as compared with cells treated with nonspecific RNA (Fig. 2C).
FIGURE 2.
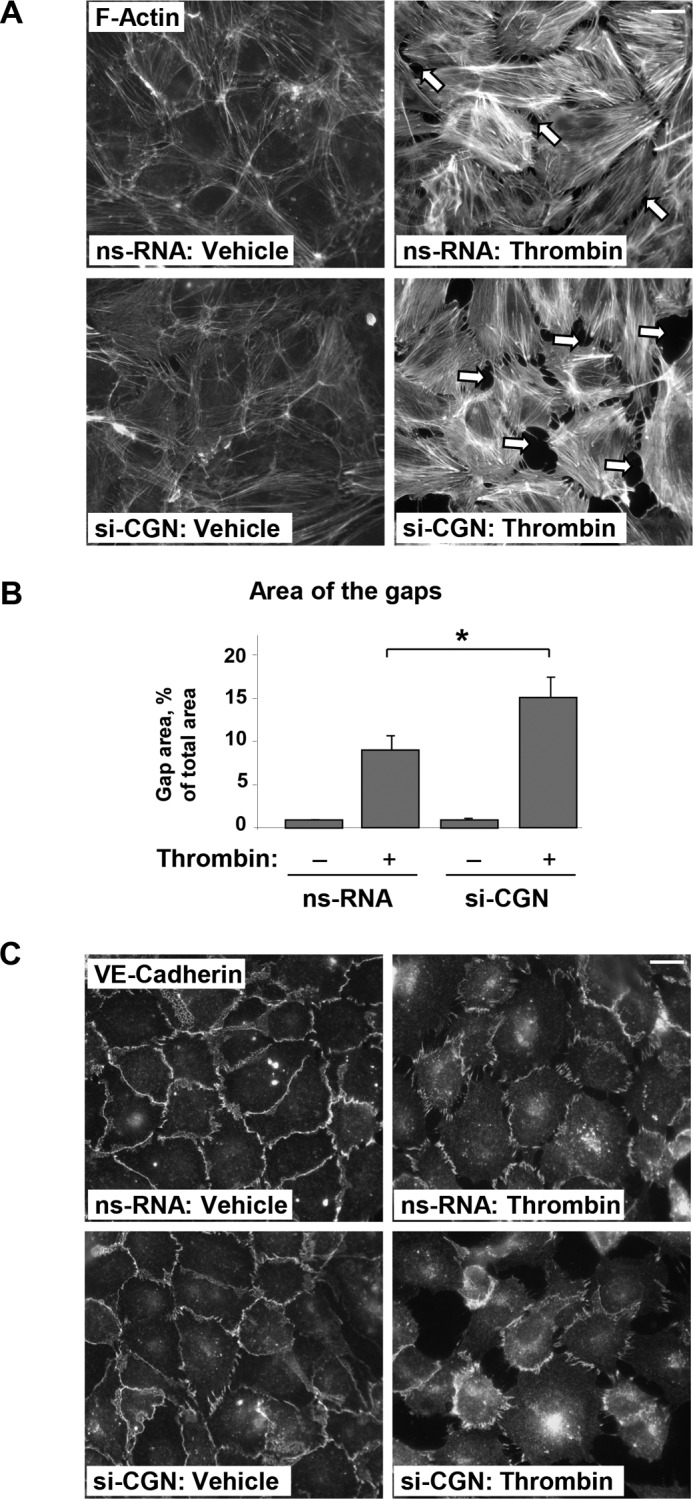
Effects of cingulin knockdown on thrombin-induced EC monolayer disruption. HPAECs transfected with cingulin-specific siRNA or nonspecific RNA (nsRNA) were stimulated with thrombin for 5 min. A and B, effects of cingulin knockdown on thrombin-induced cytoskeletal remodeling. F-actin was visualized by immunofluorescence staining with Texas Red phalloidin; bar, 10 μm. Paracellular gaps are marked by arrows (A). Bar graphs represent quantitative analysis of gap formation in control and treated HPAECs. Data are expressed as means ± S.D.; n = 3; *, p < 0.05 (B). C, cell junction remodeling was monitored by immunofluorescence staining with VE-cadherin antibody. Shown are representative results of three independent experiments; bar, 10 μm.
Dynamic actin cytoskeleton remodeling is critical to formation of cell protrusions, re-establishment of cell-cell contacts, and EC monolayer integrity. LifeAct, a 17-amino acid peptide, interacts with F-actin structures in eukaryotic cells, does not interfere with actin dynamics, and has been widely used to track down actin cytoskeletal remodeling in live cells (31). We used ECs expressing GFP-tagged LifeAct to visualize the effect of cingulin knockdown on the cytoskeletal dynamics during EC barrier recovery after thrombin challenge. Monitoring of neighbor cells expressing GFP-LifeAct using time-lapse live imaging microscopy showed that thrombin induced rapid disruption of cell-cell contacts and formation of intercellular gaps. It was seen as early as 1 min after thrombin challenge followed by re-establishment of cell junctions and resealing of intercellular gaps by 8–10 min after thrombin stimulation (Fig. 3, upper panels). In contrast, disruption of cell-cell contacts caused by thrombin was not recovered in ECs with cingulin knockdown (Fig. 3, lower panels).
FIGURE 3.
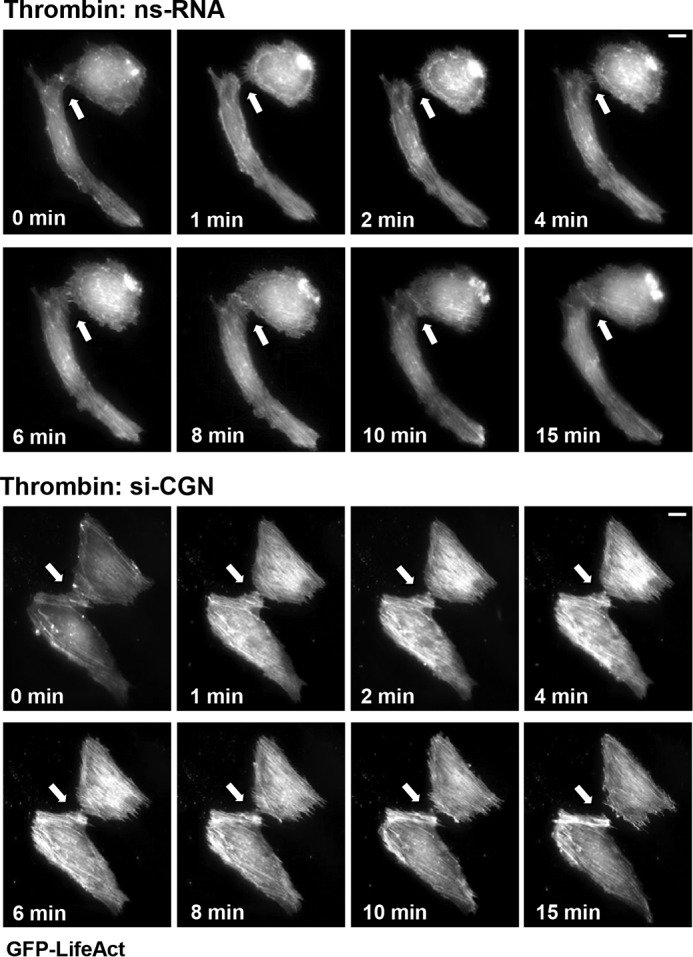
Effects of cingulin knockdown on thrombin-induced cytoskeletal remodeling. HPAECs transfected with cingulin-specific siRNA or nonspecific RNA (ns-RNA) were stimulated with thrombin. Live cell imaging of the cells expressing GFP-LifeAct is shown. Snapshots depict thrombin-induced actin rearrangement and cell contraction in control and cingulin-depleted cells; bar, 5 μm. Arrows indicate cell-cell interface areas. Shown are representative results of three independent experiments.
Cingulin Modulates Thrombin-induced GEF-H1 and Rho Activation and Associates with GEF-H1
Previous reports described RhoA-specific guanine nucleotide exchange factor GEF-H1 as a cingulin-binding partner in epithelial cells (32, 33). Activation of the RhoA pathway is a major mechanism of thrombin-induced pulmonary EC permeability (34). The following experiments tested the involvement of cingulin in thrombin-induced activation of the GEF-H1-RhoA pathway. We used three distinct cingulin-specific siRNA sets as follows: a pool of four cingulin-specific siRNAs and two additional cingulin-specific single RNAs to achieve cingulin knockdown.
Cingulin knockdown increased GEF-H1 activation by thrombin, which was measured in pulldown assays with mutated RhoA interacting with activated GEF (Fig. 4A). Additional GEF-H1 activation in ECs with cingulin knockdown led to increased activation of RhoA (Fig. 4B) and phosphorylation of the Rho kinase downstream target, myosin light chains (Fig. 4C).
FIGURE 4.

Effects of cingulin knockdown on thrombin-induced Rho pathway activation. Pulmonary ECs were transfected with a pool of four cingulin-specific siRNA, two additional single cingulin-specific siRNAs, or nonspecific RNA (nsRNA) followed by thrombin stimulation for 5 min. A, GEF-H1 activation was evaluated by GEF pulldown assay with immobilized RhoG17A and evaluated by increased GEF-H1 association with activated Rho. Western blotting detection of GEF-H1 in corresponding total lysates was used as a normalization control. Cingulin knockdown was confirmed by Western blotting of total lysates with cingulin antibody. Bar graphs depict quantitative densitometry analysis of Western blotting data; data are expressed as means ± S.D.; n = 4, *, p < 0.05. B, Rho activation was determined by Rho-GTP pulldown assay. Content of activated Rho was normalized to the total Rho content in EC lysates. Cingulin knockdown was confirmed by Western blotting of total lysates with cingulin antibody. Results of densitometry are shown as means ± S.D.; n = 3, *, p < 0.05. C, phosphorylation of MLC in ECs with cingulin knockdown achieved using three distinct cingulin-specific siRNA sets was detected by Western blotting with phospho-specific antibody. Equal protein loading was confirmed by re-probing of membranes with MLC antibody. Cingulin knockdown was confirmed by Western blotting of total lysates with cingulin antibody. Results of densitometry are shown as means ± S.D.; n = 4, *, p < 0.05. D, HPAECs stimulated with thrombin for the indicated periods of time were used for reciprocal co-immunoprecipitation (IP) assays with GEF-H1 (left panel) and cingulin (right panel) antibodies followed by Western blotting detection of cingulin and GEF-H1. Results are representative of four independent experiments. IP, immunoprecipitation; RDU, relative density units.
Experiments with reciprocal co-immunoprecipitation of endogenous GEF-H1 and cingulin from non-transfected pulmonary ECs showed that thrombin stimulation induced time-dependent and reversible association of endogenous cingulin and GEF-H1, which reached a peak at 15 min and returned to basal levels by 60 min post-thrombin (Fig. 4D), the time corresponding to the point of EC barrier restoration (Fig. 1B).
Analysis of Cingulin Domains Interacting with GEF-H1
Cingulin has a domain structure represented by three major domains as follows: a globular 49-kDa N-terminal Head region (amino acids 1–346), a central 105-kDa coiled-coil Rod region (amino acids 347–1155), and a C-terminal 4-kDa Tail (amino acids 1156–1203) (35). Characterization of cingulin domains involved in cingulin/GEF-H1 interaction and their role in agonist-induced EC permeability was performed in the next experiments. Interaction of endogenous GEF-H1 with recombinant c-Myc-tagged full-length cingulin or its domains ectopically expressed in pulmonary ECs was further evaluated in immunoprecipitation studies and pulldown assays from human pulmonary ECs with ectopic expression of recombinant cingulin variants or GEF-H1.
We expressed in pulmonary ECs the c-Myc-tagged full-length cingulin, the cingulin rod + tail (R + T) or cingulin Head domains, and we examined their association with endogenous GEF-H1 under non-stimulated conditions. Co-immunoprecipitation assays showed increased association of endogenous GEF-H1 with cingulin full-length and R + T domains (Fig. 5A). Reciprocal co-immunoprecipitation assays with GEF-H1 antibodies confirmed GEF-H1 association with full-length and R + T cingulin domain, but not with the Head domain (Fig. 5B). Ectopic expression of recombinant cingulin variants in human pulmonary ECs was verified by Western blotting analysis of total cell lysates with c-Myc antibody (Fig. 5B, right panel). The direct nature of cingulin/GEF-H1 interaction was confirmed in the study by Aijaz et al. (32), which showed a direct binding of recombinant His6-pleckstrin homology domain of GEF-H1 to a GST fusion with cingulin residues 782–1025, within the coiled-coil domain. The next experiments investigated the functional role of cingulin/GEF-H1 interactions and their physiological regulation.
FIGURE 5.

Effects of cingulin knockdown on thrombin-induced Rho pathway activation. Pulmonary ECs were transfected with cingulin full-length (FL), Head (H), or rod + tail (R + T) cingulin domains. Control transfections were performed with empty vector (Em. Vec.). A, cingulin association with endogenous GEF-H1 was examined by immunoprecipitation (IP) assays with c-Myc tag antibody. GEF-H1 content in immunoprecipitates was determined by probing with corresponding antibody. Probing for c-Myc was used as a normalization control. B, immunoprecipitation assays were performed using GEF-H1 antibody. Presence of overexpressed cingulin in immunoprecipitates was determined by probing with c-Myc antibody. Probing for endogenous GEF-H1 was used as a normalization control (left panels). Ectopic expression of cingulin domains in pulmonary ECs was verified by immunoblotting using c-Myc antibody (right panels). Results are representative of four independent experiments.
GEF-H1 under basal conditions associates with microtubules, whereas microtubule disassembly causes GEF-H1 release to the cytoplasm and its activation (20). We used this GEF-H1 property to test whether free GEF-H1 released to the cytosol interacts with bacterially expressed purified cingulin. We expressed full-length GST-tagged cingulin in the bacterial system and conjugated it with agarose beads. Analysis of the cingulin/GEF-H1 interaction was performed by pulldown of endogenous GEF-H1 from lysates of pulmonary ECs stimulated with thrombin known to cause partial microtubule disassembly and from cells stimulated with nocodazole causing total microtubule disassembly (36). The results show increased immobilization of GEF-H1 on cingulin-conjugated beads from thrombin-stimulated EC lysates and more robust immobilization of GEF-H1 from the cell lysates of nocodazole-treated cells (Fig. 6A). These results strongly suggest that the interaction of recombinant bacterially expressed cingulin with GEF-H1 correlates with the amount of free cytosolic GEF-H1.
FIGURE 6.
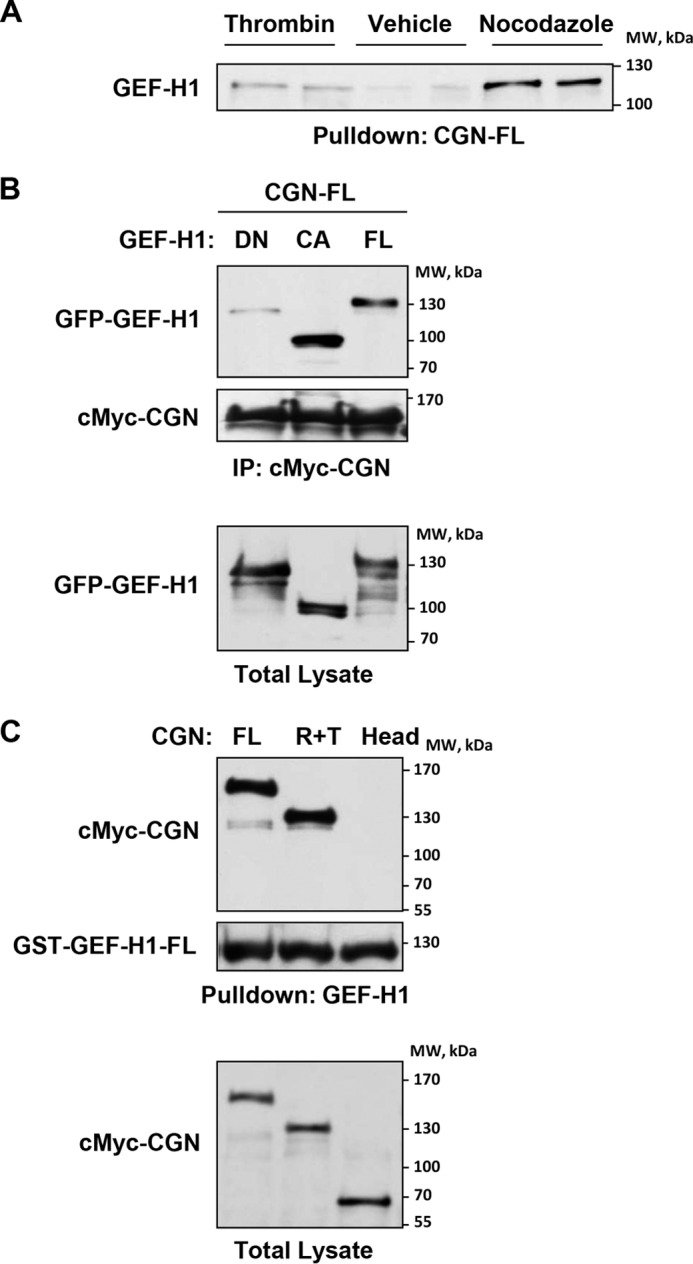
Effects of cingulin knockdown on thrombin-induced Rho pathway activation. A, EC lysates stimulated with vehicle, thrombin (0.1 units/ml), or nocodazole (0.5 μm) were incubated with agarose beads conjugated with bacterially expressed full-length cingulin. After washing, immobilized endogenous GEF-H1 was detected by immunoblotting. B, ECs were co-transfected with full-length (FL), constitutively active (CA), or dominant-negative (DN) GEF-H1 mutants and full-length cingulin. Cingulin/GEF-H1 association was examined by immunoprecipitation (IP) assays with c-Myc antibody. GEF-H1 content in immunoprecipitates was monitored by probing with GFP antibody. Probing with c-Myc antibody was used as normalization control (upper panels). Ectopic expression of GEF-H1 mutants in pulmonary ECs was verified by immunoblotting using GFP antibody (lower panel). Results are representative of three independent experiments. C, cingulin full-length (FL), Head (H), or rod + tail (R + T) domains were ectopically expressed in lung ECs. Cell lysates were incubated with agarose beads conjugated with recombinant bacterially expressed full-length GEF-H1. After washing steps, immobilized cingulin was detected by immunoblotting with c-Myc antibody. Probing membranes with GST antibody was used as a normalization control (upper panels). Ectopic expression of cingulin domains in pulmonary ECs was verified by immunoblotting using c-Myc antibody (lower panel). Results are representative of three independent experiments.
To investigate whether the GEF-H1 activated state is important for GEF-H1/cingulin interaction, we co-expressed cingulin with GEF-H1 wild type, dominant-negative, or constitutively activated mutants in pulmonary ECs. Co-immunoprecipitation assays using c-Myc antibody showed strong association of c-Myc-tagged cingulin with wild type and constitutively activated GEF-H1, but not with dominant-negative GEF-H1 mutant (Fig. 6B). Of note, we observed more robust cingulin interaction with constitutively activated GEF-H1 with deleted microtubule-binding domain, as compared with full-length GEF-H1. Control immunoblotting analysis of the levels of recombinant GEF-H1 variants in total cell lysates showed comparable levels of protein expression (Fig. 6B, bottom panel).
Finally, pulldown assays using immobilized bacterially expressed full-length GEF-H1 and lysates from pulmonary ECs expressing cingulin mutants showed GEF-H1 interactions with cingulin full-length and R + T variants but not with the Head domain (Fig. 6C). Taken together, these results demonstrate that cingulin interacts with GEF-H1 via its R + T domain, and this interaction increases under thrombin-stimulated conditions. This interaction is further enhanced by the GEF-H1-activated state.
Cingulin R + T Domain Attenuates Thrombin-induced Activation of GEF-H1-RhoA Signaling and EC Permeability
Functional role of cingulin/GEF-H1 interactions in modulation of Rho signaling and EC responses to vasoactive agonists was further evaluated in experiments with ectopic expression of cingulin-truncated mutants in pulmonary ECs.
Analysis of GEF-H1 activation in ECs expressing cingulin Head domain showed time-dependent increase in GEF-H1 activity caused by thrombin stimulation. In contrast, expression of cingulin R + T domain inhibited thrombin-induced GEF-H1 activation (Fig. 7A). In agreement with the effects on thrombin-induced GEF-H1 activation, expression of cingulin R + T domain suppressed thrombin-induced activation of RhoA (Fig. 7B) and phosphorylation of Rho kinase effectors, myosin light chain phosphatase (MYPT1) and MLC (Fig. 7C). These inhibitory effects on thrombin-induced Rho signaling were not observed in ECs with ectopic expression of cingulin Head domain.
FIGURE 7.

Effect of overexpressed cingulin domains on thrombin-induced Rho pathway activation. Pulmonary ECs were transfected with c-Myc-tagged Head (H) or rod + tail (R + T) cingulin followed by thrombin stimulation for 5 min. A, GEF-H1 activation was evaluated by GEF pulldown assay. Western blotting detection of GEF-H1 in corresponding total lysates was used as normalization control. Bar graphs depict quantitative densitometry analysis of Western blotting data; data are expressed as means ± S.D.; n = 3, *, p < 0.05. B, Rho activation was evaluated using Rho-GTP pulldown assay and normalized to the total Rho content in cell lysates. Results of densitometry shown as means ± S.D.; n = 3, *, p < 0.05. C, Western blotting analysis of thrombin-induced MYPT1 and MLC phosphorylation. Results of densitometry shown as means ± S.D.; n = 4, *, p < 0.05. Ectopic expression of c-Myc-tagged Head and rod + tail cingulin in pulmonary ECs was verified by immunoblotting using anti-c-Myc antibody. Probing for β-actin was used as a normalization control. Results are representative of thee to five independent experiments. nsRNA, nonspecific RNA. RDU, relative density units.
Effects of expression of cingulin R + T and Head domains on thrombin-induced permeability and cytoskeletal remodeling were further examined by measurements of transendothelial electrical resistance and immunofluorescence studies. Basal TER values in non-transfected controls, cells expressing head and R + T domains were 1056 ± 78, 1103 ± 154, and 1131 ± 177 ohm, respectively. In contrast to non-transfected ECs and cells expressing the cingulin Head domain, expression of cingulin R + T mutant abolished EC permeability response to thrombin (Fig. 8A). Consistent with the observed attenuation of thrombin-induced EC barrier dysfunction, ectopic expression of the cingulin R + T domain attenuated thrombin-induced F-actin stress fiber formation. In contrast, expression of cingulin Head domain did not affect F-actin arrangement in the ECs stimulated with thrombin (Fig. 8B).
FIGURE 8.

Effect of overexpressed cingulin domains on thrombin-induced EC barrier function. HPAECs were transfected with c-Myc-tagged Head (H), rod + tail (R + T) cingulin, or were left non-transfected (non-TF), followed by stimulation with thrombin. A, at the time point indicated by the arrow, cells were stimulated with thrombin, and TER measurements were performed over time. Bar graph depicts EC permeability changes at the time point corresponding to the maximal thrombin response. Results are presented as means ± S.D.; *, p < 0.05; n = 4. B, cytoskeletal remodeling in response to thrombin was analyzed in transfected ECs by immunofluorescence staining for F-actin. Co-staining with c-Myc antibody was performed to detect cingulin-expressing cells. Results are representative of thee independent experiments; bar, 5 μm.
Discussion
This study provides the first evidence of a direct role for cingulin in dynamic regulation of agonist-induced pulmonary EC permeability and describes the mechanism of EC barrier regulation by cingulin. Cingulin was first discovered as a protein localized at the cytoplasmic surface of tight junctions in intestinal epithelial cells (37) and later identified in vascular endothelial cells (38). Studies in kidney epithelial cells showed that cingulin gene disruption did not affect TJ formation, epithelial polarity, or maintenance of TJ organization (30). However, cingulin was shown to interact with GEF-H1 and reduce GEF-H1 basal activity in vitro (32, 33). Despite the exciting findings that cingulin may be involved in controlling Rho activity, a functional role of cingulin in the regulation of EC permeability by vasoactive agonists has not yet been identified.
A recent study (39) has described cingulin expression in human lung, brain, and skin endothelial cells. Cingulin overexpression in cultured ECs induced TJ formation and strengthened the endothelial barrier, although cingulin knock-out only minimally increased basal EC permeability to low molecular weight tracers, without affecting basal permeability to high molecular weight molecules (39). These results are consistent with our data showing no significant differences in basal macromolecular permeability between control and cingulin knockdown ECs. This study further demonstrates that cingulin knockdown increased thrombin-induced disruption of cell junctions and permeability response in pulmonary ECs, which was monitored by measurements of transendothelial electrical resistance and direct analysis of EC monolayer permeability for FITC-avidin. In live imaging experiments, cingulin knockdown exacerbated thrombin-induced actin stress fiber formation and disruption of adherens junctions. Resealing of junctions (Fig. 3) in those experiments occurred by 8–10 min after thrombin addition in control but not si-CGN cells. The TER measurements in Fig. 1B show a recovery of the TER after 30 min. These differences in timing may be explained by the fact that primary EC cultures, in contrast to immortalized cell lines, exhibit natural variability of cell responses, including recovery times. Therefore, the most useful comparisons are those made within the same experiment between control and stimulated conditions. Also, the timing of cytoskeletal remodeling observed in single cells may not exactly reflect the timing of general permeability response by the cell monolayer. For this reason, the timing of morphological experiments and experiments with TER measurements may be not an exact match. The effects of cingulin depletion on cell response to thrombin were linked to augmented GEF-H1 and RhoA activation in thrombin-activated pulmonary ECs with cingulin knockdown, suggesting a predominant signaling role, rather than a structural role for cingulin in the agonist-induced control of EC barrier.
Pulldown studies using affinity-purified cingulin domains expressed in the bacterial system or cingulin domains ectopically expressed in the primary human lung endothelial cells revealed cingulin R + T domain as a GEF-H1-binding domain involved in negative regulation of GEF-H1 activity. Expression of R + T domain attenuated thrombin-induced activation of GEF-H1 and Rho and alleviated thrombin-induced EC monolayer disruption and hyper-permeability. These results complemented experiments with cingulin knockdown and further supported the role of cingulin as a molecular brake of agonist-induced RhoA activation. Overexpression of R + T cingulin mutant did not significantly affect basal RhoA activity levels (Fig. 7) but suppressed thrombin-induced RhoA activation. As a negative regulator of RhoA signaling, cingulin acts by suppressing the activity of GEF-H1, which is one of several GEFs regulating RhoA (40). Because GEF-H1 plays a major role in thrombin-induced Rho activation, its inhibition by the cingulin mutant has a dramatic effect on thrombin-induced permeability. However, other instances of Rho activation may engage other GEFs, and the role of cingulin in those conditions may not be as dramatic as in thrombin-stimulated conditions.
We also cannot exclude other functions of cingulin in vascular ECs, such as a structural role in regulation of tight junction integrity. We did not observe significant effects of cingulin depletion on cell morphology or basal permeability in ECs isolated from pulmonary large vessels. However, the structural role of tight junctions in permeability control becomes increasingly important in microvascular compartments, specifically in brain capillary beds, where tight junctions are believed to play a major role in mass transport control (9).
The direct analysis of GEF-H1 activation in this study unequivocally showed GEF-H1-specific mechanism of Rho regulation by cingulin in thrombin-stimulated endothelial cells. In addition to GEF-H1, cingulin has been reported to interact with another Rho-specific GEF, p114RhoGEF, in certain types of epithelial cells (41). Whether cingulin harbors more than one Rho-specific GEF in vascular ECs and whether this interaction has functional implications remain to be investigated.
It is possible that after EC stimulation with agonists, such as thrombin, causing microtubule destabilization, GEF-H1 is released from microtubules and triggers a barrier-disruptive Rho pathway (42). At this point, released GEF-H1 becomes accessible to cingulin, and this interaction leads to inhibition of GEF-H1 nucleotide exchange activity. These events lead to Rho inactivation and recovery of the endothelial barrier. The transient nature of agonist-induced cingulin-GEF-H1 association strongly suggests additional regulatory mechanisms of cingulin/GEF-H1 interactions, possibly via post-translational modification of cingulin, GEF-H1, or both proteins. These remaining questions warrant further investigation.
Restoration of integrity of endothelial and epithelial monolayers is also regulated by other mechanisms. For example, thrombin stimulation of vascular ECs causes barrier disruption, but it simultaneously triggers auto-recovery mechanisms by activating Rap1-Rac1-dependent down-regulation of Rho signaling, stimulation of peripheral cytoskeletal dynamics, and resealing of intercellular gaps (43). Other mechanisms involve activation of the negative regulator of RhoA, p190RhoGAP, at cell junctions and focal adhesions (44, 45), or cell junction-restricted activation of Rac1-specific GEF Trio in epithelial monolayers (23). The abundance of negative feedback of Rho regulation mechanisms in vascular ECs, including the GEF-H1-cingulin axis, emphasizes a critical role for small GTPase activity switch as a part of biphasic vascular endothelial permeability response to a variety of vasoactive stimuli, which ensures dynamic regulation of EC barrier in vivo.
In conclusion, the results of this study demonstrate for the first time a functional role of cingulin in the regulation of agonist-induced vascular endothelial permeability by the mechanism, which involves interaction of the cingulin R + T domain with RhoA activator GEF-H1 leading to down-regulation of the GEF-H1/RhoA signaling pathway and initiation of EC barrier restoration. Augmentation of thrombin-induced Rho activation and EC permeability by molecular inhibition of cingulin suggests its role as an important modulator of Rho signaling in vascular ECs, which may be essential for fine-tuning of permeability responses to various bioactive molecules.
Experimental Procedures
Cell Culture and Reagents
HPAECs were obtained from Lonza (East Rutherford, NJ) and used for experiments at passages 5–7. Texas Red-conjugated phalloidin (catalog no. T7471, lot no. 983923) was from Invitrogen; Alexa Fluor 488 anti-mouse (A11029, lot 1550911) and anti-rabbit (A11034, lot 1531670) were obtained from Molecular Probes (Eugene, OR). Antibodies to cingulin (sc-365264, lot 63014), GST (sc-138, lot I1009), RhoA (sc-179, lot J0113), VE-cadherin (sc-9989, lot B2310), and c-Myc tag (sc-40, lot B1115) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Antibodies to phospho-MYPT1 (36-003, lot 2474979) were from Millipore (Billerica, MA); antibodies to phospho-MLC (3674, lot 5) were obtained from Cell Signaling (Beverly, MA). Unless otherwise specified, all biochemical reagents, including β-actin antibody (A5441, lot 064M4789V), were obtained from Sigma.
siRNA and DNA Transfections
For pre-designed standard purity cingulin-specific siRNAs, a set of four (E-014025-00, Homo sapiens; sequences: A-014025-13, GCAAUAAGCUGAUAGAUGG; A-014025-14, CCUCUAGCACAAAAUAUGA; A-014025-15, CUCUCAGGUCAAGGGAUUU; and A-014025-16, GGACCAGGGUGAAGAUUUA) and two additional siRNAs, cingulin1, J-014025-11 (sequence: AGGGGAAUGUUAUGGGUAA) and cingulin2, J-014025-10 (sequence: GCAGAGAACAAGAAGCGUU), were ordered from Dharmacon (Lafayette, CO). Transfection of ECs with siRNA was performed as described previously (46). Typically, the efficiency of siRNA transfection exceeds 80%, as tested in our previous reports (47, 48). The pooled data from Western blotting analyses of cingulin knockdown efficiency in different experiments did not reveal significant differences between all tested siRNAs and resulted in 83.3 ± 11.0, 84.7 ± 10.1, and 86.1 ± 7.6% decrease in protein expression for cingulin1, cingulin2, and CGN pool, respectively. Nonspecific and non-targeting RNA was used as a control treatment (D-001210-01, sequence: UAGCGACUAAACACAUCAA). After 72 h of transfection, cells were used for experiments or harvested for Western blot verification of specific protein depletion. Plasmids encoding human enhanced GFP-tagged GEF-H1 full-length (1–894), constitutively active (1–572), and dominant-negative (DH mutant) mutants have been described previously (20). Plasmid encoding GFP-tagged LifeAct was provided by L. Phillipson (University of Chicago, IL). Plasmids encoding Myc-tagged full-length cingulin and deletion mutants were provided by S. Citi (University of Geneva, Switzerland) and used for transient transfections according to the protocols described previously (42, 46). After 24 h of transfection, cells were treated with the agonist of interest and used for experiments.
Measurement of Endothelial Permeability
The cellular barrier properties were analyzed by measurements of TER across confluent human pulmonary artery endothelial monolayers using an electrical cell-substrate impedance sensing system (Applied Biophysics, Troy, NY) as described previously (36, 42). Variations in absolute TER basal resistance are typical for this type of experiment and depends on the EC growth rate and time in cell culture. Importantly, comparisons between experimental groups discussed in this work were always done using the same cell batch. Of note, basal TER values in experiments with siRNA and DNA transfections are different, because they depend on time after cell plating on electrodes (e.g. 72 or 24 h post-transfection time). Express macromolecule permeability testing assay (XPerT) was recently developed in our group (49) is and currently available from Millipore (Vascular Permeability Imaging Assay, catalog no. 17-10398). This assay is based on high affinity binding of avidin-conjugated FITC-labeled tracer to the biotinylated extracellular matrix proteins immobilized on the bottom of culture dishes after the EC barrier is compromised by treatment with a barrier-disruptive agonist. XPerT permeability assays were performed in 96-well plates, and fluorimetric analysis of the FITC-labeled tracer reflecting EC monolayer permeability for macromolecules was performed using the Victor X5 multilabel plate reader (PerkinElmer Life Sciences). Alternatively, visualization of EC monolayer permeability was performed in HPAECs plated on glass coverslips coated with biotinylated gelatin followed by agonist stimulation, incubation with FITC-avidin tracer, fluorescence microscopy, and imaging analysis as described previously (49, 50).
Imaging Studies
Endothelial monolayers plated on glass coverslips were subjected to immunofluorescence staining as described previously (51). Texas Red phalloidin was used to visualize F-actin. VE-cadherin antibody was used to visualize adherens junctions. Slides were analyzed using a Nikon video imaging system (Nikon, Tokyo, Japan). Images were processed with ImageJ software (National Institutes of Health, Bethesda, MD) and Adobe Photoshop 7.0 (Adobe Systems, San Jose, CA) software. Quantitative analysis of paracellular gap formation was performed as described previously (42, 52). For live cell imaging and time-lapse tracking of actin dynamics, cells were plated on MatTek dishes (MatTek, Ashland, MA) and transfected with GFP-LifeAct. Images were acquired with ×100 NA 1.45 oil objective in a 3I Marianas Yokogawa-type spinning disk confocal system equipped with a CO2 chamber and a heated stage. Time-lapse images were taken with 15-s intervals for 15 min.
Pulldown Assays
GST-tagged cingulin or GEF-H1 in pGEX vector was used for bacterial expression in BL21-AI Escherichia coli strain. GST fusion proteins were isolated (53) using glutathione resin (Clontech) and stored as a 50% glycerol slurry. After stimulation with agonist, endothelial monolayers were washed with PBS and incubated on ice for 15 min with lysis buffer (50 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1.5 mm MgCl2, 1 mm EDTA, 1% Triton X-100, and 10% glycerol) supplemented with protease (catalog no. 04693159001) and phosphatase (catalog no. 04906837001) inhibitor mixtures (Roche Applied Science). Lysate was clarified by centrifugation and incubated with glutathione resin loaded with GST-tagged proteins (2 h, 4 °C). Then, the resin was collected by centrifugation and washed three times with lysis buffer, and an amount of the protein of interest bound to the beads was evaluated by Western blotting analysis. Activation of RhoA GTPase in pulmonary endothelial cell culture was analyzed using GTPase in vitro pulldown assay kit available from Millipore (Billerica, MA). Active GEF-H1 was affinity precipitated from cell lysates using the RhoA(G17A) mutant, which cannot bind nucleotide and therefore has high affinity for activated GEFs (54).
Immunoprecipitation and Western Blotting
After agonist stimulation, cells were washed in cold PBS and lysed on ice with cold TBS/Nonidet P-40 lysis buffer (20 mm Tris, pH 7.4, 150 mm NaCl, 1% Nonidet P-40) supplemented with protease and phosphatase inhibitor mixtures (Roche Applied Science). Clarified lysates were then incubated with corresponding antibodies overnight at 4 °C, washed 3–4 times with TBS/Nonidet P-40 lysis buffer, and the complexes were analyzed by Western blotting using appropriate antibodies. Protein extracts were separated by SDS-PAGE, transferred to polyvinylidene fluoride (PVDF) membrane, and probed with specific antibodies. Equal protein loading was verified by re-probing membranes with antibody to β-actin or specific protein of interest. Equal protein loading of samples was confirmed by measurements of protein concentration in cell lysates before SDS-PAGE and Western blotting analysis.
Statistical Analysis
Results are expressed as means ± S.D. of three to five independent experiments. Stimulated samples were compared with controls by unpaired Student's t tests. For multiple-group comparisons, a one-way variance analysis followed by the post hoc Fisher's test were used. p < 0.05 was considered statistically significant.
Author Contributions
A. A. B. designed the study and wrote the paper. Y. T. performed pulldown and immunoprecipitation studies and Western blotting analysis of phosphorylated proteins. G. G. performed pulldown and immunoprecipitation studies. X. T. performed imaging studies. A. S. S. performed imaging studies. N. S. performed TER measurements. S. C. was involved in discussion of the results. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
We thank K. Szaszi (St. Michael's Hospital, Toronto, Canada) for sharing RhoG17A construct. We thank Sophia Son for superb laboratory assistance.
This work was supported by National Institutes of Health Grants HL107920 and HL130431 from NHLBI and National Institutes of Health Grant GM114171 from NIGMS. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- EC
- endothelial cell
- MLC
- myosin light chain
- HPAEC
- human pulmonary artery endothelial cell
- ZO
- zonula occludens
- TER
- transendothelial electrical resistance
- R + T
- rod + tail
- TJ
- tight junction.
References
- 1. Lo S. K., Perlman M. B., Niehaus G. D., and Malik A. B. (1985) Thrombin-induced alterations in lung fluid balance in awake sheep. J. Appl. Physiol. 58, 1421–1427 [DOI] [PubMed] [Google Scholar]
- 2. Garcia J. G., Verin A. D., and Schaphorst K. L. (1996) Regulation of thrombin-mediated endothelial cell contraction and permeability. Semin. Thromb. Hemost. 22, 309–315 [DOI] [PubMed] [Google Scholar]
- 3. Gawlak G., Tian Y., O'Donnell J. J. 3rd, Tian X., Birukova A. A., and Birukov K. G. (2014) Paxillin mediates stretch-induced Rho signaling and endothelial permeability via assembly of paxillin-p42/44MAPK-GEF-H1 complex. FASEB J. 28, 3249–3260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Birukova A. A., Tian X., Cokic I., Beckham Y., Gardel M. L., and Birukov K. G. (2013) Endothelial barrier disruption and recovery is controlled by substrate stiffness. Microvasc. Res. 87, 50–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mammoto A., Mammoto T., Kanapathipillai M., Wing Yung C., Jiang E., Jiang A., Lofgren K., Gee E. P., and Ingber D. E. (2013) Control of lung vascular permeability and endotoxin-induced pulmonary oedema by changes in extracellular matrix mechanics. Nat. Commun. 4, 1759. [DOI] [PubMed] [Google Scholar]
- 6. Dejana E., Orsenigo F., Molendini C., Baluk P., and McDonald D. M. (2009) Organization and signaling of endothelial cell-to-cell junctions in various regions of the blood and lymphatic vascular trees. Cell Tissue Res. 335, 17–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Le Guelte A., Dwyer J., and Gavard J. (2011) Jumping the barrier: VE-cadherin, VEGF and other angiogenic modifiers in cancer. Biol. Cell 103, 593–605 [DOI] [PubMed] [Google Scholar]
- 8. Birukov K. G., Zebda N., and Birukova A. A. (2013) Barrier enhancing signals in pulmonary edema. Compr. Physiol. 3, 429–484 [DOI] [PubMed] [Google Scholar]
- 9. Tietz S., and Engelhardt B. (2015) Brain barriers: Crosstalk between complex tight junctions and adherens junctions. J. Cell Biol. 209, 493–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bogatcheva N. V., Garcia J. G., and Verin A. D. (2002) Molecular mechanisms of thrombin-induced endothelial cell permeability. Biochemistry 67, 75–84 [DOI] [PubMed] [Google Scholar]
- 11. Mehta D., and Malik A. B. (2006) Signaling mechanisms regulating endothelial permeability. Physiol. Rev. 86, 279–367 [DOI] [PubMed] [Google Scholar]
- 12. Burridge K., and Wennerberg K. (2004) Rho and Rac take center stage. Cell 116, 167–179 [DOI] [PubMed] [Google Scholar]
- 13. van Nieuw Amerongen G. P., van Delft S., Vermeer M. A., Collard J. G., and van Hinsbergh V. W. (2000) Activation of RhoA by thrombin in endothelial hyperpermeability: role of Rho kinase and protein tyrosine kinases. Circ. Res. 87, 335–340 [DOI] [PubMed] [Google Scholar]
- 14. Kaibuchi K., Kuroda S., and Amano M. (1999) Regulation of the cytoskeleton and cell adhesion by the Rho family GTPases in mammalian cells. Annu. Rev. Biochem. 68, 459–486 [DOI] [PubMed] [Google Scholar]
- 15. Geiger B., and Bershadsky A. (2001) Assembly and mechanosensory function of focal contacts. Curr. Opin. Cell Biol. 13, 584–592 [DOI] [PubMed] [Google Scholar]
- 16. Katsumi A., Orr A. W., Tzima E., and Schwartz M. A. (2004) Integrins in mechanotransduction. J. Biol. Chem. 279, 12001–12004 [DOI] [PubMed] [Google Scholar]
- 17. Fukata Y., Amano M., and Kaibuchi K. (2001) Rho-Rho kinase pathway in smooth muscle contraction and cytoskeletal reorganization of non-muscle cells. Trends Pharmacol. Sci. 22, 32–39 [DOI] [PubMed] [Google Scholar]
- 18. Ren Y., Li R., Zheng Y., and Busch H. (1998) Cloning and characterization of GEF-H1, a microtubule-associated guanine nucleotide exchange factor for Rac and Rho GTPases. J. Biol. Chem. 273, 34954–34960 [DOI] [PubMed] [Google Scholar]
- 19. Benais-Pont G., Punn A., Flores-Maldonado C., Eckert J., Raposo G., Fleming T. P., Cereijido M., Balda M. S., and Matter K. (2003) Identification of a tight junction-associated guanine nucleotide exchange factor that activates Rho and regulates paracellular permeability. J. Cell Biol. 160, 729–740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Krendel M., Zenke F. T., and Bokoch G. M. (2002) Nucleotide exchange factor GEF-H1 mediates cross-talk between microtubules and the actin cytoskeleton. Nat. Cell Biol. 4, 294–301 [DOI] [PubMed] [Google Scholar]
- 21. Miyoshi J., and Takai Y. (2005) Molecular perspective on tight-junction assembly and epithelial polarity. Adv. Drug. Deliv. Rev. 57, 815–855 [DOI] [PubMed] [Google Scholar]
- 22. Bazzoni G. (2006) Endothelial tight junctions: permeable barriers of the vessel wall. Thromb. Haemost. 95, 36–42 [PubMed] [Google Scholar]
- 23. Citi S., Guerrera D., Spadaro D., and Shah J. (2014) Epithelial junctions and Rho family GTPases: the zonular signalosome. Small GTPases 5, 1–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cordenonsi M., D'Atri F., Hammar E., Parry D. A., Kendrick-Jones J., Shore D., and Citi S. (1999) Cingulin contains globular and coiled-coil domains and interacts with ZO-1, ZO-2, ZO-3, and myosin. J. Cell Biol. 147, 1569–1582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tsukita S., Katsuno T., Yamazaki Y., Umeda K., Tamura A., and Tsukita S. (2009) Roles of ZO-1 and ZO-2 in establishment of the belt-like adherens and tight junctions with paracellular permselective barrier function. Ann. N.Y. Acad. Sci. 1165, 44–52 [DOI] [PubMed] [Google Scholar]
- 26. Ogita H., Rikitake Y., Miyoshi J., and Takai Y. (2010) Cell adhesion molecules nectins and associating proteins: Implications for physiology and pathology. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 86, 621–629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Guillemot L., and Citi S. (2006) Cingulin regulates claudin-2 expression and cell proliferation through the small GTPase RhoA. Mol. Biol. Cell 17, 3569–3577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Paschoud S., and Citi S. (2008) Inducible overexpression of cingulin in stably transfected MDCK cells does not affect tight junction organization and gene expression. Mol. Membr. Biol. 25, 1–13 [DOI] [PubMed] [Google Scholar]
- 29. Guillemot L., Schneider Y., Brun P., Castagliuolo I., Pizzuti D., Martines D., Jond L., Bongiovanni M., and Citi S. (2012) Cingulin is dispensable for epithelial barrier function and tight junction structure, and plays a role in the control of claudin-2 expression and response to duodenal mucosa injury. J. Cell Sci. 125, 5005–5014 [DOI] [PubMed] [Google Scholar]
- 30. Guillemot L., Hammar E., Kaister C., Ritz J., Caille D., Jond L., Bauer C., Meda P., and Citi S. (2004) Disruption of the cingulin gene does not prevent tight junction formation but alters gene expression. J. Cell Sci. 117, 5245–5256 [DOI] [PubMed] [Google Scholar]
- 31. Riedl J., Crevenna A. H., Kessenbrock K., Yu J. H., Neukirchen D., Bista M., Bradke F., Jenne D., Holak T. A., Werb Z., Sixt M., and Wedlich-Soldner R. (2008) Lifeact: a versatile marker to visualize F-actin. Nat. Methods 5, 605–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Aijaz S., D'Atri F., Citi S., Balda M. S., and Matter K. (2005) Binding of GEF-H1 to the tight junction-associated adaptor cingulin results in inhibition of Rho signaling and G1/S phase transition. Dev. Cell 8, 777–786 [DOI] [PubMed] [Google Scholar]
- 33. Citi S., Paschoud S., Pulimeno P., Timolati F., De Robertis F., Jond L., and Guillemot L. (2009) The tight junction protein cingulin regulates gene expression and RhoA signaling. Ann. N.Y. Acad. Sci. 1165, 88–98 [DOI] [PubMed] [Google Scholar]
- 34. Beckers C. M., van Hinsbergh V. W., and van Nieuw Amerongen G. P. (2010) Driving Rho GTPase activity in endothelial cells regulates barrier integrity. Thromb. Haemost. 103, 40–55 [DOI] [PubMed] [Google Scholar]
- 35. Citi S., D'Atri F., and Parry D. A. (2000) Human and Xenopus cingulin share a modular organization of the coiled-coil rod domain: predictions for intra- and intermolecular assembly. J. Struct. Biol. 131, 135–145 [DOI] [PubMed] [Google Scholar]
- 36. Birukova A. A., Adyshev D., Gorshkov B., Bokoch G. M., Birukov K. G., and Verin A. D. (2006) GEF-H1 is involved in agonist-induced human pulmonary endothelial barrier dysfunction. Am. J. Physiol. Lung Cell Mol. Physiol. 290, L540–L548 [DOI] [PubMed] [Google Scholar]
- 37. Citi S., Sabanay H., Jakes R., Geiger B., and Kendrick-Jones J. (1988) Cingulin, a new peripheral component of tight junctions. Nature 333, 272–276 [DOI] [PubMed] [Google Scholar]
- 38. Corada M., Mariotti M., Thurston G., Smith K., Kunkel R., Brockhaus M., Lampugnani M. G., Martin-Padura I., Stoppacciaro A., Ruco L., McDonald D. M., Ward P. A., and Dejana E. (1999) Vascular endothelial-cadherin is an important determinant of microvascular integrity in vivo. Proc. Natl. Acad. Sci. U.S.A. 96, 9815–9820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Schossleitner K., Rauscher S., Gröger M., Friedl H. P., Finsterwalder R., Habertheuer A., Sibilia M., Brostjan C., Födinger D., Citi S., and Petzelbauer P. (2016) Evidence that cingulin regulates endothelial barrier function in vitro and in vivo. Arterioscler. Thromb. Vasc. Biol. 36, 647–654 [DOI] [PubMed] [Google Scholar]
- 40. Cherfils J., and Zeghouf M. (2013) Regulation of small GTPases by GEFs, GAPs, and GDIs. Physiol. Rev. 93, 269–309 [DOI] [PubMed] [Google Scholar]
- 41. Terry S. J., Zihni C., Elbediwy A., Vitiello E., Leefa Chong San I. V., Balda M. S., and Matter K. (2011) Spatially restricted activation of RhoA signalling at epithelial junctions by p114RhoGEF drives junction formation and morphogenesis. Nat. Cell Biol. 13, 159–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Birukova A. A., Birukov K. G., Smurova K., Adyshev D., Kaibuchi K., Alieva I., Garcia J. G., and Verin A. D. (2004) Novel role of microtubules in thrombin-induced endothelial barrier dysfunction. FASEB J. 18, 1879–1890 [DOI] [PubMed] [Google Scholar]
- 43. Birukova A. A., Tian X., Tian Y., Higginbotham K., and Birukov K. G. (2013) Rap-afadin axis in control of Rho signaling and endothelial barrier recovery. Mol. Biol. Cell 24, 2678–2688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Holinstat M., Knezevic N., Broman M., Samarel A. M., Malik A. B., and Mehta D. (2006) Suppression of RhoA activity by focal adhesion kinase-induced activation of p190RhoGAP: role in regulation of endothelial permeability. J. Biol. Chem. 281, 2296–2305 [DOI] [PubMed] [Google Scholar]
- 45. Zebda N., Tian Y., Tian X., Gawlak G., Higginbotham K., Reynolds A. B., Birukova A. A., and Birukov K. G. (2013) Interaction of p190RhoGAP with C-terminal domain of p120-catenin modulates endothelial cytoskeleton and permeability. J. Biol. Chem. 288, 18290–18299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Birukova A. A., Fu P., Xing J., Yakubov B., Cokic I., and Birukov K. G. (2010) Mechanotransduction by GEF-H1 as a novel mechanism of ventilator-induced vascular endothelial permeability. Am. J. Physiol. Lung Cell Mol. Physiol. 298, L837–L848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Birukova A. A., Alekseeva E., Mikaelyan A., and Birukov K. G. (2007) HGF attenuates thrombin-induced permeability in the human pulmonary endothelial cells by Tiam1-mediated activation of the Rac pathway and by Tiam1/Rac-dependent inhibition of the Rho pathway. FASEB J. 21, 2776–2786 [DOI] [PubMed] [Google Scholar]
- 48. Birukova A. A., Shah A. S., Tian Y., Moldobaeva N., and Birukov K. G. (2016) Dual role of vinculin in barrier-disruptive and barrier-enhancing endothelial cell responses. Cell. Signal. 28, 541–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Dubrovskyi O., Birukova A. A., and Birukov K. G. (2013) Measurement of local permeability at subcellular level in cell models of agonist- and ventilator-induced lung injury. Lab. Invest. 93, 254–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tian X., Tian Y., Gawlak G., Sarich N., Wu T., and Birukova A. A. (2014) Control of vascular permeability by atrial natriuretic peptide via GEF-H1-dependent mechanism. J. Biol. Chem. 289, 5168–5183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Birukova A. A., Cokic I., Moldobaeva N., and Birukov K. G. (2009) Paxillin is involved in the differential regulation of endothelial barrier by HGF and VEGF. Am. J. Respir. Cell Mol. Biol. 40, 99–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Birukova A. A., Smurova K., Birukov K. G., Usatyuk P., Liu F., Kaibuchi K., Ricks-Cord A., Natarajan V., Alieva I., Garcia J. G., and Verin A. D. (2004) Microtubule disassembly induces cytoskeletal remodeling and lung vascular barrier dysfunction: role of Rho-dependent mechanisms. J. Cell. Physiol. 201, 55–70 [DOI] [PubMed] [Google Scholar]
- 53. Vikis H. G., and Guan K. L. (2004) Glutathione S-transferase-fusion based assays for studying protein-protein interactions. Methods Mol. Biol. 261, 175–186 [DOI] [PubMed] [Google Scholar]
- 54. García-Mata R., Wennerberg K., Arthur W. T., Noren N. K., Ellerbroek S. M., and Burridge K. (2006) Analysis of activated GAPs and GEFs in cell lysates. Methods Enzymol. 406, 425–437 [DOI] [PubMed] [Google Scholar]