

Abstract
By inspection of the predicted proteome of the fungus Myceliophthora thermophila C1 for vanillyl-alcohol oxidase (VAO)-type flavoprotein oxidases, a putative oligosaccharide oxidase was identified. By homologous expression and subsequent purification, the respective protein could be obtained. The protein was found to contain a bicovalently bound FAD cofactor. By screening a large number of carbohydrates, several mono- and oligosaccharides could be identified as substrates. The enzyme exhibits a strong substrate preference toward xylooligosaccharides; hence it is named xylooligosaccharide oxidase (XylO). Chemical analyses of the product formed upon oxidation of xylobiose revealed that the oxidation occurs at C1, yielding xylobionate as product. By elucidation of several XylO crystal structures (in complex with a substrate mimic, xylose, and xylobiose), the residues that tune the unique substrate specificity and regioselectivity could be identified. The discovery of this novel oligosaccharide oxidase reveals that the VAO-type flavoprotein family harbors oxidases tuned for specific oligosaccharides. The unique substrate profile of XylO hints at a role in the degradation of xylan-derived oligosaccharides by the fungus M. thermophila C1.
Keywords: carbohydrate, crystal structure, flavin adenine dinucleotide (FAD), oligosaccharide, oxidase, CAZy, xylan, xylose, oxidase, carbohydrate processing
Introduction
Nature has developed a profusion of enzymes to produce, modify, and degrade carbohydrates. These enzymes usually outcompete chemical approaches for carbohydrate conversions as they are highly selective and effective. Enzymes involved in carbohydrate modifications are thoroughly studied as they can be handy biocatalytic tools in developing new biotechnological applications. Especially with the growing research efforts with the aim to develop efficient biorefinery processes, it is attractive to have a well equipped toolbox of enzymes active on carbohydrates (1).
The Carbohydrate Active enZymes (CAZy) database provides a very good overview on the classes and individual enzymes that are implicated in catalyzing carbohydrate chemistry. Although the database is very rich in non-redox enzymes (∼500,000 members), which include glycoside hydrolases and glycosyl transferases, relatively few redox enzymes are included (∼10,000 members). In fact, only in recent years has CAZy included the so-called auxiliary activity (AA) families, which represent the enzymes that assist in carbohydrate modifications through performing oxidations or reductions. One of these families is AA7, which contains flavoprotein oxidases acting on oligosaccharides (see “Auxiliary Activity Family 7” in the CAZy database). Currently, this family includes only a few characterized enzymes: glucooligosaccharide oxidase (GluO)2 from Acremonium strictum (2), chitooligosaccharide oxidase (ChitO) from Fusarium graminearum (3), and lactose oxidase (LaO) from Microdochium nivale (4). These fungal oxidases, all acting on different kinds of oligosaccharides, share a common structural fold as they belong to the vanillyl-alcohol oxidase (VAO) flavoprotein family (5). They also have in common that they selectively oxidize oligosaccharides at the C1 position (2, 3, 6). This results in the formation of the corresponding lactones, which typically undergo spontaneous hydrolysis. The electrons obtained through the oxidation of the carbohydrates are used by the enzyme to reduce molecular oxygen, thereby generating hydrogen peroxide. These redox reactions are facilitated by an FAD flavin cofactor that is covalently tethered to the enzymes via two covalent linkages.
Detailed studies on GluO and ChitO have revealed that these oxidases accept a wide range of mono- and oligosaccharides but are most efficient with oligosaccharides. Furthermore, they are able to discriminate between different types of oligosaccharides. For example, ChitO is very selective in oxidizing chitooligosaccharides, whereas it only poorly accepts a limited number of other oligosaccharides. Elucidation of the structures of GluO and LaO and model-inspired engineering of ChitO have revealed the molecular basis for the observed oligosaccharide selectivities of these oxidases. The structures display a rather open active site close to the surface, which allows binding of large oligosaccharides (7). As a result of the open character of the active site, the redox-active isoalloxazine moiety of the flavin cofactor is relatively solvent-exposed. This provides an explanation for why these flavoproteins have the flavin cofactor covalently anchored via two amino acids (8). Through the bicovalent linkage, the cofactor can still be positioned for catalyzing oxidations, whereas in other flavoproteins, the amino acids are used to bind and position the cofactor. The open active site architecture in oligosaccharide oxidases is different from carbohydrate oxidases that preferentially act on monosaccharides. For example, glucose oxidase and pyranose oxidase have very occluded active sites (9, 10). Still, the nature and positioning of a number of residues that form the carbohydrate binding groove dictate which oligosaccharides can bind in such a way that the flavin cofactor is able to oxidize the substrate. The role of these residues in substrate acceptance was clearly demonstrated by engineering ChitO: by replacing three residues, an efficient lactose oxidase could be generated from ChitO (11). This shows that the nature of the amino acids lining the carbohydrate binding groove in oligosaccharide oxidases determines the substrate specificity.
In this study, we report on the discovery, characterization, and crystal structures of a novel flavoprotein oxidase belonging to the VAO family, which is primarily active toward xylooligosaccharides (XOS): xylooligosaccharide oxidase (XylO) from the thermophilic fungus Myceliophthora thermophila C1. By determining the kinetic and catalytic properties and elucidating its crystal structure, our study provides detailed insight into the distinctive enzyme properties of XylO. The results also show that XylO can be used for generating XOS-based aldonic acids.
Results
Homology Analysis
We have recently identified a stretch of conserved residues that correlates with the presence of a bicovalently bound FAD in flavoprotein sequences that belong to the VAO flavoprotein family (12). As all currently known oligosaccharide oxidases contain FAD bound via a similar unique bicovalent attachment, we used this motif as a filter in a PHI-BLAST search. We decided to search specifically the predicted proteome of M. thermophila C1 using the ChitO sequence as query. The targeted fungus is a rich source of thermostable and biocatalytically interesting enzymes (13). Thirteen putative bicovalent flavoprotein sequences were found in the predicted proteome of this fungus, and through multiple sequence alignment, two of them were found to cluster with ChitO, GluO, and LaO. One of these two putative oxidases (GenBank accession number KX139007), which shared 41, 44, and 47% protein sequence identity with ChitO, GluO, and LaO, respectively, was selected for further studies. The corresponding gene encodes for a 497 amino acids protein whose first 16 amino acids encode for a signal sequence identifying the putative oxidase as an extracellular protein. The presence of the conserved FAD linking residues, His94 and Cys155 (Fig. 1), strongly suggests that the respective protein contains a bicovalently bound FAD.
FIGURE 1.

Partial sequence alignment of XylO, LaO, ChitO, and GluO showing the conservation around the two FAD-linking amino acids. The respective histidine and cysteine are in bold and in the consensus line.
Purification
From 50 ml of concentrated fermentation supernatant, ∼80 mg of yellow-colored XylO was purified after three steps of purification. Purified XylO runs as one single band on SDS-PAGE at ∼60 kDa. This does not agree with the predicted size of 53 kDa, which suggests that the protein has undergone post-translational modifications, potentially glycosylations. The protein displayed clear fluorescence when, upon SDS-PAGE, the polyacrylamide gel was illuminated by UV light. The fluorescence was more intense when incubating the gel for 5 min in 5% acetic acid, which confirmed the presence of covalently bound FAD. The purified enzyme displays a typical flavin spectrum, with absorbance maxima at 442 and 387 nm (Fig. 2). Upon the addition of 0.1% SDS, the flavin spectrum undergoes changes similar to the ones observed for unfolded berberine bridge enzyme (14), which also contains a bicovalently bound FAD cofactor.
FIGURE 2.

Spectrum of 52 μm native XylO (solid line) and unfolded XylO upon the addition of 0.1% SDS (dashed line), in 50 mm potassium phosphate buffer, pH 6.0.
Substrate Identification
To establish the substrate profile of XylO, 23 carbohydrates (glucose, galactose, fructose, mannose, xylose, l-arabinose, N-acetyl-d-glucosamine, sucrose, maltose, lactose, sorbitol, xylitol, raffinose, cellobiose, cellotetraose, α-cyclodextrin, β-cyclodextrin, arabitol, chitosan, chitin, starch, maltotetraose, and glycerol) were tested as substrates at 50 and 5 mm (for cyclodextrins, cellotetraose, and maltotetraose, the concentrations tested were 1 and 0.5 mm) using the HRP-based assay. After a 1-min incubation at 25 °C with 1 μm XylO, oxidase activity could be clearly observed for xylose, cellobiose, lactose, and arabinose through the formation of a clear pink color. A weak signal could also be detected for galactose, maltose, and cellotetraose. Interestingly, although l-arabinose did show some activity, 1,5-α-l-arabinobiose did not. Based on the observed substrate profile, the steady state kinetics parameters were determined for xylose, cellobiose, and lactose (Table 1). Intriguingly, although the kcat values are in the same order of magnitude when compared with other known oligosaccharide oxidases (3, 15–17), the Km values are several orders of magnitude higher. All oligosaccharide flavoprotein oxidases are known to perform better on oligosaccharides when compared with the monosaccharides. This is probably due to their more open active site on the surface and the presence of an oligosaccharide binding groove (3, 4, 7, 16). Because XylO displayed very poor activity on cellobiose and no detectable activity on maltotetraose, and because xylose had the lowest Km value among the tested carbohydrates (which did not include xylose-based oligosaccharides), as a next step, several XOS (xylobiose, xylotriose, and xylotetraose) were tested as substrates. This revealed that XylO is a highly efficient oxidase for XOS (Table 1). Although the kcat values were similar for all identified XylO substrates, the Km values dramatically decreased for all tested XOS. Although for xylose a Km of ∼400 mm was found, the Km values for xylobiose, xylotriose, and xylotetraose were ∼3 orders of magnitude lower (Table 1). With kcat values of 11.0–11.5 s−1 and Km values in the (sub)millimolar range, XylO displays similar kinetic properties as those reported for ChitO and GluO in combination with their best oligosaccharide substrates (3, 17). The kinetic data clearly show that XylO prefers pentose-based oligosaccharides over hexose-based oligosaccharides. This sets XylO apart from all other known oligosaccharide oxidases (16–20).
TABLE 1.
Steady state kinetic parameters of XylO determined for cellobiose, lactose, xylose, and xylooligosaccharides
Data are representative of three independent experiments.
| kcat | Km | kcat/Km | |
|---|---|---|---|
| s−1 | m | m−1 s−1 | |
| Cellobiose | 5.4 ± 0.3 | 0.342 ± 0.03 | 16 |
| Lactose | 4.8 ± 0.6 | 0.532 ± 0.071 | 9 |
| Xylose | 5.2 ± 0.4 | 0.359 ± 0.051 | 15 |
| Xylobiose | 11.5 ± 0.5 | 0.00115 ± 0.00008 | 10,000 |
| Xylotriose | 11.0 ± 0.3 | 0.00069 ± 0.00004 | 16,000 |
| Xylotetraose | 11.2 ± 0.3 | 0.00043 ± 0.00003 | 26,000 |
Although the HRP-based assay confirms that XylO is using dioxygen as electron acceptor, acting as an oxidase, we also determined the apparent affinity toward dioxygen. By analyzing oxygen depletion curves, a Km for oxygen of 0.13 mm could be determined. This shows that at standard atmospheric conditions (0.24 mm dioxygen in buffer solution), XylO is operating below the saturation of the electron acceptor.
General Enzyme Properties
Having identified efficient substrates for XylO, we determined several enzyme properties by using xylobiose as substrate. To determine the pH optimum for activity, oxidase activity over a pH range from 4 to 8 was measured (supplemental Fig. S3). This revealed that XylO is most active at pH 7, whereas it is still quite active at pH 6 (80%) and moderately active at pH 8 (33%).
M. thermophila strains are able to survive and replicate at temperatures higher than 45 °C (21–23). Therefore, many enzymes from this fungus have to tolerate relatively high temperatures. The thermostability of XylO was tested with the Thermofluor assay using SYPRO Orange as unfolding reporter dye (24). The apparent melting temperature was found to be 65 °C, which indicates that it is indeed a thermostable enzyme. The temperature optimum for activity was also determined between 20 and 60 °C using an excess of HRP to compensate for the loss of activity of this enzyme at higher temperatures (supplemental Fig. S4). The optimal temperature for enzyme activity is 30 °C, whereas it still displays 50% activity at 60 °C.
Product Identification
For determining the site of oxidation by XylO, the product formed upon conversion of xylobiose was determined. For product identification, NMR and GC methods were used. The NMR spectra revealed the presence of a disaccharide as product. The two monosaccharide units were labeled A and B according to the decreasing chemical shift values of their protons. The region (δ 4.6–3.0) in the 1D 1H NMR spectrum contained 11 well resolved signals (Fig. 3, Table 2). Based on observed 1H chemical shifts and 3J1,2 coupling constant values, residue A (3J1,2 8.5 Hz) was assigned to have the β-anomeric configuration. The 2D COSY spectrum also allowed the assignment of the sequential order of the chemical shifts belonging to residue A and residue B. All 13C chemical shifts could be assigned using a 2D 13C-1H HSQC spectrum in combination with 2D COSY (Fig. 3). The downfield shift of B-4 (δ 83.6) is indicative for 4-substitution of residue B. Further evidence for the interpretation of the HSQC spectrum and the determination of the glycosidic linkage is available in the HMBC spectrum, where intra-residual two- and three-bond 1H-13C couplings and inter-residual three-bond connectivities over the glycosidic linkages could be assigned (Fig. 3). The absence of anomeric signals for residue B fits with a xyluronic acid residue. The NMR data are fully consistent with the formation of xylobionic acid as oxidation product.
FIGURE 3.
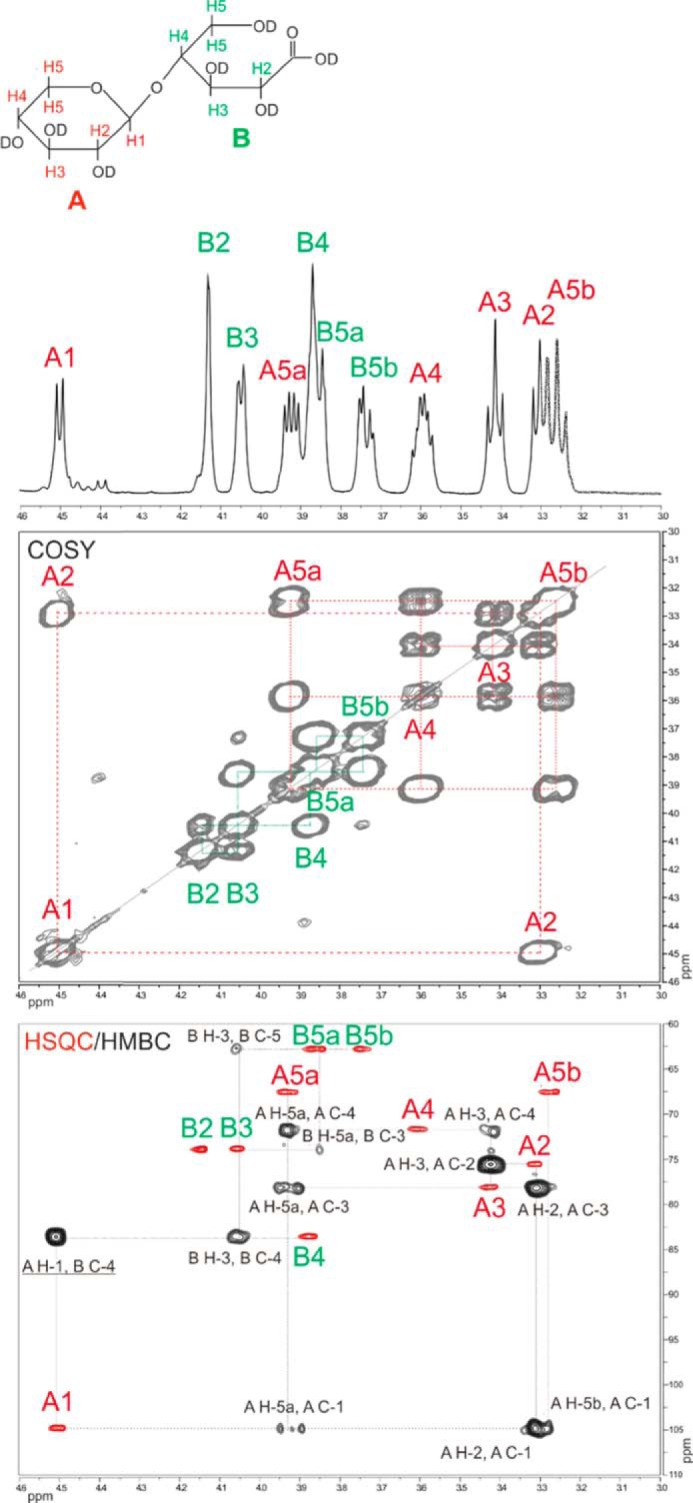
The 1D 1H NMR spectrum, the 2D 1H-1H COSY spectrum, and the 2D 13C-1H HSQC (red peaks)/HMBC (black) spectra of the product formed upon conversion of xylobiose by XylO, recorded in D2O at 25 °C. In the HMBC spectrum, the cross-peak confirming the glycosidic linkage is underlined. The structure of the xylobionate, including the atom annotation, is also presented.
TABLE 2.
Observed 1H and 13C chemical shifts (ppm) for the product formed upon conversion of xylobiose by XylO (D2O, 25 °C)
The ppm values are relative to the signal of internal acetone (δ 2.225 for 1H and δ 31.07 for 13C).
| Residue | H-1 | H-2 | H-3 | H-4 | H-5a | H-5b |
|---|---|---|---|---|---|---|
| C-1 | C-2 | C-3 | C-4 | C-5 | ||
| A (monosaccharide at non-reducing end) | 4.505 | 3.304 | 3.414 | 3.596 | 3.923 | 3.257 |
| 104.8 | 75.5 | 78.2 | 71.6 | 67.5 | 67.5 | |
| B (monosaccharide at reducing end) | 4.153 | 4.057 | 3.875 | 3.857 | 3.738 | |
| 73.9 | 73.8 | 83.6 | 62.8 | 62.8 |
The structure of the xylobionate was further confirmed by GC-MS analysis. The GC chromatogram of the intact xylobionate shows one major peak, and the respective MS spectrum showed a fragmentation pattern fitting xylobionate (supplemental Fig. S1). After methanolysis, the GC chromatogram of trimethylsilyl (TMS) derivatives showed four peaks, which were also identified by MS (supplemental Fig. S2). One minor peak corresponded with TMS derivatives of the methyl glycoside of α- and β-xylofuranosides, two peaks corresponded with the methyl glycosides of α- and β-xylopyranosides, and one peak corresponded with the methylated xyluronic acid. The GC data confirm that XylO oxidizes xylobiose into xylobionic acid by C1 oxidation of the reducing end.
Oxidation of Complex Substrates
In the literature, only a few studies report on using carbohydrate oxidases to oxidize complex polysaccharide substrates, i.e. LaO (4) and galactose oxidase (25). As XylO was found to act on XOS, we tested the oxidase with various complex carbohydrate-containing mixtures that vary in xylan content: beechwood xylan, wheat arabinoxylan, oat flour, rye whole grain flour, and barley flakes. The targeted mixtures (1% w/v) were first separated into soluble and insoluble fractions, and each of them was tested in duplicate. Using the HRP-based assay, specific activities toward the different potential substrate could be determined (Table 3). No activity was detected on insoluble substrates, suggesting the necessity for the reducing ends of the poly- or oligosaccharides to be in solution. Testing the soluble fractions of oat flour and barley flakes did not yield any oxidase activity, whereas the soluble fractions of the xylans and rye flour revealed significant oxidase activity. The observed preference is in line with the fact that, when compared with barley and oat, rye grains are relatively rich in arabinoxylan. In contrast, oat and barley are relatively rich in glucans (26, 27). Thus the observed activities are in agreement with the above reported substrate profile for XylO. To further probe the use of XylO for oxidizing xylan-derived XOS, we prepared hydrolyzed wheat arabinoxylan, which comprises a population of XOS of different lengths substituted with 2-,3- or 2,3-linked arabinose residues. The observation that hydrolysis of wheat arabinoxylan resulted in a 32-fold increase in XylO activity suggests that XylO is also able to accept arabinose-decorated XOS.
TABLE 3.
Oxidation activity of XylO on the soluble and insoluble fractions of complex natural substrates
The HRP-based assay was performed at 30 °C. ND, not detected. NT, not tested. Data are representative of three independent experiments.
| Specific activity (units/mg) |
||
|---|---|---|
| Soluble | Insoluble | |
| Beechwood xylan | 0.50 ± 0.06 | ND |
| Wheat arabinoxylan | 0.43 ± 0.08 | ND |
| Hydrolyzed wheat arabinoxylan | 13.8 ± 0.85 | NT |
| Oat flour | ND | ND |
| Rye whole grain flour | 1.03 ± 0.15 | ND |
| Barley flakes | ND | ND |
Overall Structure and Active Site of XylO
To understand the molecular basis by which XylO specifically accepts xylooligosaccharides, we set out to elucidate the crystal structure of XylO. Well diffracting crystals could be obtained using PEG6000 as precipitant, and the crystal structure of XylO was determined to a resolution of 1.93 Å using molecular replacement. The final model comprises amino acid residues 25–497, 1 FAD molecule, 7 GlcNAc molecules, 1 MES molecule, and 383 water molecules with R/Rfree 15.9/19.5% (Table 4). The XylO structure is composed of two distinct domains (Fig. 4A). The FAD binding domain (FAD domain) comprises residues 25–231 and 447–497, whereas the substrate binding domain (S domain) comprises residues 232–446. The FAD domain contains two subdomains, both of which have an α/β-fold. The first subdomain (residues 8–113) is composed of three parallel β strands (β1-β3), flanked on one side by two α-helices (α1-α2) and on the other side by one α-helix (α3). The second, larger subdomain (residues 114–231 and 447–497) contains five antiparallel β-strands (β4-β9), surrounded by six α-helices (α4-α7 and α13-α14). The FAD binding pocket is embedded between the two subdomains. The S domain contains a seven-stranded antiparallel β-sheet (β10-β16) flanked on the outside of the protein by four α-helices (α8-α9 and α11-α12) and on the inside by one α-helix (α10). N-Linked GlcNAc moieties could be modeled in electron density extending from Asn233, Asn245, and Asn289. Similarly, at Asn117 and Asn192, two GlcNAc residues were visible. Between the first and third α-helix in the first subdomain, a conserved disulfide bridge (Cys30-Cys79) is present, which probably stabilizes the N-terminal helix.
TABLE 4.
Data collection and refinement statistics
Values in parentheses are for the highest resolution shell.
| Diffraction data | Native | Xylose | Xylobiose |
|---|---|---|---|
| Wavelength (Å) | 1.54 | 1.54 | 1.54 |
| Resolution range (Å) | 53.6–1.93 | 53.5–1.79 | 53.4–1.80 |
| Space group | C2 | C2 | C2 |
| Cell dimensions (Å) a, b, c, β | 124.0, 59.4, 68.7, 90.6 | 123.3, 59.4, 68.5, 91.0 | 123.6, 59.2, 68.5, 90.8 |
| Unique reflections | 37,594 (2327) | 46,206 (2069) | 45,931 (2430) |
| Wilson B factor (Å2) | 7.1 | 12.8 | 11.3 |
| Completeness (%) | 99.4 (91.4) | 98.3 (78.6) | 95.6 (70.1) |
| Overall I/σ (I) | 11.8 (2.4) | 11.0 (2.6) | 11.4 (3.3) |
| Rmerge (%) | 16.3 (67.3) | 8.3 (40.7) | 7.8 (27.4) |
| Rpim (%) | 7.1 (32.9) | 4.9 (26.8) | 4.8 (20.8) |
| R/Rfree (%) | 15.9/19.5 | 17.3/20.7 | 15.2/18.4 |
| RMS deviations from ideal values | |||
| Bond lengths (Å) | 0.007 | 0.009 | 0.008 |
| Bond angles (°) | 1.30 | 1.39 | 1.45 |
| Protein residues | 473 | 473 | 473 |
| FAD molecules | 1 | 1 | 1 |
| Ligand molecules | 4 xylose | 4 xylobiose | |
| Water molecules | 383 | 405 | 400 |
| PDB accession ID | 5K8E | 5L6F | 5L6G |
FIGURE 4.

Crystal structure of XylO. A, overall structure with FAD domain in green and the substrate domain in red. The FAD flavin cofactor is in yellow sticks, while the MES molecule is in cyan sticks. The N-linked N-acetylglucosamine moieties are also highlighted in sticks (magenta). B, MES bound in the −2 substrate binding pocket. C, xylose bound in the −1 substrate binding pocket. D, xylobiose bound in the −1 and −2 substrate binding pockets. The calculated omit maps of the MES, xylose, and xylobiose molecules (gray mesh) are contoured at 3σ and were calculated with the ligands omitted using Phenix.polder.
The XylO structure is very similar to the structures of other structurally characterized VAO family members. The most similar structures are that of GluO from Acremonium strictum (28) (44% sequence identity, Protein Data Bank (PDB) code 2AXR) and LaO from M. nivale (48% sequence identity, PDB code 3RJ8). The RMSD of GluO with XylO is 1.1 Å on 445 Cα atoms, whereas the RMSD of LaO with XylO is 1.0 Å on 463 Cα atoms. Structural similarity to a lesser extent was observed with a glucose dehydrogenase from Phleum pratense (29) (PDB code 4PVE), the Bermuda grass isoallergen Cyn d 4 (PDB code 4DNS) (30), EncM from Streptomyces maritimus (31) (PDB code 3W8W), and a berberine bridge enzyme (ATBBE15) from Arabidopsis thaliana (32) (PDB code 4UD8). These proteins have 24–27% sequence identity when compared with XylO and superimpose with an RMSD of ∼1.9 Å. Among these structures, the FAD domains are very well conserved, and the S domains are less well conserved, reflecting different substrate specificities.
XylO contains a bicovalently linked FAD, 6-S-cysteinyl, 8α-N1-histidyl FAD. Such a covalently tethered FAD cofactor was first observed in GluO (7). The isoalloxazine ring of the cofactor is covalently linked with the C6 atom to the Sγ of Cys155 and with the 8α-methyl group to the Nδ1 of His94 (Fig. 4B). The access to the open active site is in a cleft in the S domain made up by the seven-stranded β-sheet and an α-helix loop structure (residues 315–341). XylO has a similar open carbohydrate binding groove as present in GluO and LaO, allowing mono-, di-, and oligosaccharides to bind with the non-reducing end of the sugar residue exposed to the solvent. Interestingly, at the entrance to the active site, the morpholino ring of a MES molecule is bound between Tyr325 and Tyr376, acting as a clamp and mimicking a monosaccharide binding at the −2 substrate site (Fig. 4B). Tyr325 and Trp376 stack on the morpholino ring with an interplanar distance of ∼4.0 Å. For obtaining a structure with a substrate bound, crystals were soaked with xylose and xylobiose. In the two substrate complex structures, xylose is bound in the −1 subsite, next to the flavin cofactor, whereas xylobiose occupies the −1 and −2 subsites (Fig. 4, C and D). The observation that substrate-soaked XylO crystals lost their yellow appearance suggests that the complexed structures reflect the complex of flavin-reduced XylO with bound xylose or xylobiose. The xylose C1 atom, the site of oxidation, is bound in front of the flavin N5 with a distance of 3.4 Å and an angle with the N5–N10 atoms of 108°. Such geometry of substrate binding is common among flavoprotein oxidases that catalyze alcohol oxidations by a hydride transfer from the substrate to the N5 of the flavin cofactor (33). The xylose O1 atom interacts with Tyr451-OH (2.5 Å), Glu412-OE1 (2.9 Å), and a water molecule. The endocyclic oxygen makes close contacts with the carboxylate group of Glu412 (2.3/3.1 Å). The equatorial OH2 group forms hydrogen bonds with Thr154-OG1 (2.7 Å), Arg272-NH1 (2.8 Å), the isoalloxazine O4 (3.4 Å), and a water molecule, and the equatorial OH3 and OH4 both form hydrogen bonds with two water molecules. The xylobiose C1 is positioned slightly closer to flavin N5 (at 3.2 Å) and has a slightly larger angle of 112° when compared with the xylose-bound structure. Interactions of OH1, O5, OH2, and OH3 with the protein are similar as in the xylose-bound structure. The xylose moiety in the −2 subsite has a π-π stacking interaction, similar to the morpholino ring of the MES molecule, and a hydrogen bond to one water molecule. The binding of xylobiose and MES induces no significant conformational change at the −2 subsite, except for the side chain of Tyr325. The phenyl ring rotates toward the bound sugar to optimize the stacking interactions. This rotation upon substrate binding is not observed in GluO and LaO, the latter having an Asn instead of Tyr325.
The active sites of XylO, GluO, and LaO are very similar. Tyr96, Thr154, Arg272, Tyr325 (Asn in LaO), and Tyr451 are identical among the three enzymes. Some other residues clearly differ among the three enzymes: Leu274 (Glu in GluO and Tyr in LaO), Tyr376 (Trp in GluO, Phe in LaO), Ile378 (Gln in GluO and LaO), and Glu412 (Gln in GluO and LaO). Tyr376-OH may prohibit binding of the C6-moiety of the aldohexoses in the active site as it is situated deeper in the −1 subsite when compared with the Trp and Phe residues in Glu and LaO, respectively. Furthermore, two hydrophobic residues, Leu274 and Ile378, reside in the −2 subsite and thereby contribute to the selectivity toward xylooligosaccharides. At the same positions in GluO and LaO, two polar residues, capable of accommodating the hydroxyl at the C6 position of the sugar moiety, are situated.
The substrate-soaked crystals also revealed auxiliary substrate binding sites at the XylO surface. Three additional binding sites for xylose are observed near Arg56-Phe426-Gly455, Trp182, and Asn369-Lys370. One additional binding site for xylobiose is detected near Trp259-Gly263 and the N terminus Asp28-Glu29-Arg41 from a symmetry-related XylO molecule. The second binding site is near Arg56-Phe426-Gly455. Interestingly the xylobiose in the third binding site near Glu104-Arg458-Glu484-Tyr487 has a stacking interaction with the xylobiose in the second binding site from a symmetry-related molecule.
Discussion
The XylO from M. thermophila C1 is the first carbohydrate oxidase discovered so far to be specific for xylooligosaccharides. The elucidation of several XylO crystal structures has provided insight into the molecular basis for its highly specific substrate preference. Although the overall structure and active site architecture of XylO resemble that of the so far characterized oligosaccharide oxidases, three residues in the substrate binding groove were found to be unique in XylO: Tyr376, Leu274, and Ile378. These residues seem to play a role in preventing aldohexoses to bind to XylO. Future mutagenesis studies on one of the known oligosaccharide oxidases might be able to prove our hypothesis.
Hemicellulose constitutes the second most abundant polysaccharide after cellulose in plant biomass. Among all hemicellulose components, xylan is the most abundant one. Xylan is an heteropolysaccharide with a β-(1,4)-linked xylose-based backbone, often carrying other carbohydrate or aromatic moieties such as l-arabinose, d-galactose, feruloyl, and glucuronic acid residues or acetyl moieties. By treating the xylan portion of hemicellulose chemically or enzymatically or by a combination of the two, XOS can be generated. Depending on the source of the xylan, the generated XOS can vary in degree of polymerization and substitutions (34). XOS are currently used as valuable dietary fibers and prebiotics (35). Because of the configuration of their glycosidic bonds, XOS resist hydrolysis by salivary and intestinal digestive enzymes and arrive unmodified in the colon, where they are fermented by anaerobic bacteria. XOS are also known to act as xylanase and cellulase inhibitors (36–38). For efficient degradation of plant biomass, efficient degradation or modification of XOS is desirable. As M. thermophila C1 is known to be equipped with an enzyme arsenal to degrade plant biomass, XylO may be part of a strategy to take out the inhibitory effects of XOS. The highly specific substrate acceptance profile of XylO toward XOS would provide the fungus a good biocatalytic tool to alleviate cellulase inhibition during biomass conversion. Except for a role in degrading or modifying hydrolyzed xylan, the oxidase may also serve as a hydrogen peroxide-producing biocatalyst. Many fungi, including M. thermophila strains, secrete peroxidases, which fully depend on the in situ production of hydrogen peroxide for their activity. Therefore, being a secreted oxidase, XylO may utilize XOS to serve extracellular peroxidases. It is interesting to note that the role of XylO does not seem to be generic among fungi as only very few close homologs can be found in the genome sequence database. Clearly, future studies are needed to elucidate the exact role of XylO.
Through its rather narrow substrate range, XylO may develop as a valuable biocatalyst. Given its exquisite specificity for XOS and very poor activity with cellobiose, XylO might be used in xylanase activity assays similar to the cellulose and chitinase assays that we previously described (39). By the combination of ChitO and HRP, we developed a fast and sensitive assay to determine the activity of chitinases and cellulases, exploiting the specificity of ChitO for the products of hydrolysis of chitin and cellulose (39). Due to the low Km values of XylO for XOS substrates, a sensitive xylanase assay involving XylO should be feasible. Furthermore, XylO could be used to derivatize XOS. XylO introduces a carboxylic moiety to any xylooligosaccharide, resulting in the production of aldonic acids, which may represent valuable compounds (40). Future studies will reveal the true potential of XylO as biocatalyst.
Experimental Procedures
Bioinformatic Analysis
The protein sequence of ChitO (accession number: XP_391174.1) was used to perform PHI-BLAST with the pattern GXCX6GX4GG on the genome of M. thermophila C1 using the non-redundant genome database of NCBI. The used sequence pattern will limit the hits for protein sequences predicted to contain a bicovalent flavin cofactor (12). Signal sequence identification was performed through the SignalP server (41).
Cloning and Expression
The putative oligosaccharide oxidase encoding gene (GenBankTM accession number: KX139007) was amplified from genomic M. thermophila C1 DNA and cloned into a C1 expression vector. The expression cassette, consisting of the gene of interest under the control of the Pcbh1 promoter and the cbh terminator, was obtained from the vector and used to transform a low protease/(hemi-)cellulase-free M. thermophila C1 expression host in a co-transformation with the auxotrophic marker pyr5 (42). Sixty randomly integrated transformants were grown in microtiter plates (43) and screened for XylO production levels in the culture broth by analyzing the flavin fluorescence upon SDS-PAGE (44). The transformant showing the highest levels of fluorescent polypeptide was grown in a fed-batch fermentation to produce XylO on a larger scale. The strain was grown aerobically in 2-liter fermenters in mineral medium, containing glucose as carbon source, ammonium sulfate as nitrogen source, and trace elements for the essential salts. After biomass formation, the enzyme was produced under glucose-limiting conditions at pH 6.0 and 32 °C (45). The supernatant containing the enzyme was centrifuged and filtrated to remove cell biomass, concentrated 4-fold, and dialyzed against 20 mm acetate, pH 5.0.
Purification
The concentrated culture supernatant was exchanged with 20 mm Tris/HCl, pH 7.6, and then loaded on a HiTrap Q-Sepharose column (5 ml) pre-equilibrated with the same buffer. The purification was performed on an ÄKTA system while monitoring absorbance values at 280 and 445 nm to detect flavoproteins. After applying the protein sample, the column was washed with buffer until the A280 reached the baseline. Subsequent elution was performed by using a gradient (0–2 m NaCl in 20 mm Tris/HCl, pH 7.6) in 60 min at 3 ml/min. Fractions containing XylO were pooled and buffer-exchanged to 50 mm acetate buffer, pH 5.6. The sample was diluted 1:2 with a solution of 50 mm acetate buffer, pH 5.6, containing 3.0 m ammonium sulfate and incubated at 4 °C while stirring for 40 min. Centrifugation at 4000 × g for 30 min allowed the removal of precipitated proteins. After filtration of the supernatant through a 0.45-μm filter, it was applied to a HiTrap Phenyl-Sepharose column (5 ml) pre-equilibrated with 50 mm acetate buffer, pH 5.6, containing 1.5 m ammonium sulfate. The column was washed with the equilibration buffer until the absorbance at 280 nm reached the baseline. Elution was performed by using a gradient (1.5–0 m ammonium sulfate in 50 mm acetate buffer, pH 5.6) in 60 min at 3 ml/min. Fractions that displayed absorbance at 445 nm (eluting at about 1.4 m ammonium sulfate) were pooled, concentrated, and loaded on a Superdex 200 column. For pre-equilibration and elution of the Superdex column, a 50 mm acetate buffer, pH 5.6, containing 100 mm NaCl was used. The fractions containing XylO activity were pooled and concentrated.
Crystallization, Data Collection, Structure Determination, and Refinement
Initial vapor diffusion crystallization experiments were performed using a Mosquito crystallization robot (TTP Labtech). In a typical experiment, 0.1 μl of screening solution was added to 0.1 μl of protein solution (9.6 mg/ml) on a 96-well MRC2 plate (Molecular Dimensions); reservoir wells contained 50 μl of screening solution. The screening solutions used for the experiments were PACT and JCSG+ (Molecular Dimensions). XylO crystals appeared after 3–4 days of incubation at 294 K in solutions containing PEG with pH 4.5–6.0. Crystallization conditions were optimized using hanging-drop set-ups with 20% PEG6000, 0.2 m ammonium chloride, and 0.1 m MES, pH 6.0, as precipitant, and drops containing 1 μl of protein solution and 1 μl of reservoir solution. Crystals grown from sodium acetate buffer at pH 5.5 showed better morphology but diffracted worse.
Before data collection, crystals were briefly soaked in a cryoprotectant solution, consisting of 20% glycerol, 20% PEG6000, 0.2 m ammonium chloride, and 0.1 m MES, pH 6.0. Ligand complexes were obtained by soaking in 2 m xylose or xylobiose, replacing glycerol as cryoprotectant, for 1 min. X-ray diffraction data were collected on an in-house MarDTB Goniostat System using Cu-Kα radiation from a Bruker Microstar H rotating-anode generator equipped with Helios MX mirrors. Intensity data were processed using XDS (46). XylO crystals belong to space group C2 with one monomer of 54 kDa in the asymmetric unit. The Vm is 2.3 Å3/Da (47) with a solvent content of 46%. Data collection statistics are listed in Table 4. The structure of the XylO was determined by the molecular replacement method using Phaser (48) with mixed model coordinates of GluO (7) (PDB code 1ZR6) and LaO (PDB code 3RJ8) as the search model.
ARP/wARP (49) was used for automatic building and the model was refined with REFMAC5 (50). Coot (51) was used for manual rebuilding and map inspection. In 2mFo − DFc and mFo − DFc maps, electron density was present for N-glycosylation and the ligands xylose and xylobiose. The quality of the models was analyzed with MolProbity (52). Five translation/libration/screw groups were used in the last rounds of refinement. Atomic coordinates and experimental structure factor amplitudes have been deposited in the PDB (entries 5K8E for the holoprotein structure, 5L6F for the xylose-bound structure, and 5L6G for the xylobiose-bound structure). To exclude the bulk solvent around the omitted regions, omit maps were calculated using the Polder option from the Phenix software suite.
Analysis of Kinetic and Stability Properties
All experiments were performed in 50 mm potassium phosphate buffer, pH 6.0. The absorbance spectrum of XylO was recorded before and after the addition of 0.1% (w/v) SDS. The extinction coefficient of XylO was determined using the extinction coefficient of 6-S-cysteinyl-bound FMN (ϵ445 = 11.6 mm−1 cm−1): 11.5 mm−1 cm−1 (53). The oxidase activity of XylO was detected by coupling H2O2 production to the oxidation of 4-aminoantipyrine and 3,5-dichloro-2-hydroxybenzenesulfonic acid by HRP. The formation of the resulting pink/purple-colored product can be followed at 515 nm (ϵ515 = 28 mm−1 cm−1). The reaction mixture contained 50 mm potassium-phosphate buffer, pH 6.0, 0.1 mm 4-aminoantipyrine, 1.0 mm 3,5-dichloro-2-hydroxybenzenesulfonic acid, 4.0 units of HRP, and 30 nm XylO. Substrate screening was performed using 23 different carbohydrates comprising mono-, di-, tetra-, poly-, and cyclooligosaccharides. Potential substrates were tested in duplicate using concentrations of 50 and 5.0 mm for mono- and disaccharides, and 1.0 and 0.5 mm for tetra- and cyclooligosaccharides.
Enzyme kinetic parameters were determined at 25 °C for several carbohydrates using different ranges of substrate concentrations: glucose (30 mm–1.5 m), cellobiose (7.0–262 mm), lactose (14–525 mm), xylose (10–560 mm), xylobiose (0.05–1.0 mm), xylotriose (0.03–2.0 mm), and xylotetraose (0.025–1.0 mm). For each substrate, 12 different substrate concentrations were measured at least in triplicate. Initial rates were obtained by measuring product formation for 1 min every 2 s. The temperature optimum for activity was measured in duplicate in a temperature range from 20 to 60 °C with 25 nm purified XylO and 5.0 mm xylobiose. Reactions were performed as described above in duplicate with the only difference that 127 units/ml of HRP were used so that HRP would not be the limiting factor in the assay at high temperatures. Relative activities to the highest kobs were plotted against temperature.
The pH optimum was determined using the universal buffer solution designed by Britton and Robinson (54) from pH 4.1 to 8.3. kobs values were measured by monitoring the initial dioxygen consumption rates with 400 nm purified XylO and 5.0 mm xylobiose in duplicate for each pH value. Dioxygen concentrations were monitored with REDFLASH sensor spots and a FireSting O2 detector and light source (PyroScience, Aachen, Germany). For measuring dioxygen concentration-dependent kinetic data, the depletion of molecular oxygen was monitored at pH 7.0 in Britton and Robinson buffer using 400 nm purified XylO, 5.0 mm xylobiose, and 0.24 mm dioxygen as starting concentrations. The dioxygen depletion kinetic data were analyzed using the Michaelis-Menten formula (55).
The thermostability of XylO was assessed with the Thermofluor assay using SYPRO Orange as dye (24). The method is based on the fluorescence increase of the dye SYPRO Orange upon binding to the hydrophobic interior of proteins, which becomes exposed as the protein unfolds. The fluorescence increase was monitored by recording the emission at 575 nm while exciting at 490 nm and increasing the temperature by 1 °C/min. 20 μl of purified XylO (1 mg/ml) were tested in triplicate in 50 mm acetate buffer, pH 5.6. The apparent melting temperature was determined by plotting the first derivative of the observed fluorescence versus temperature (56).
Oxidation of Complex Substrates
Wheat flour, rye whole grain flour, oat flour, and barley flakes were purchased at the local supermarket. Xylan from beechwood was obtained from TCI Chemicals, and wheat arabinoxylan was from MEGAZYMES. Hydrolyzed wheat arabinoxylan was prepared by incubating 50 mg/ml wheat arabinoxylan in 50 mm MES, pH 5.6, with 8.5 units of β-xylanase M1 from Trichoderma viride (MEGAZYMES) at 50 °C upon overnight shaking at 700 rpm. Subsequently, the samples were spun down, and 20 μl of the supernatant were used for the HRP-based assay.
Solutions of 4% (w/v) in 50 mm acetate buffer, pH 5.6, of the above mentioned flours, xylan, and wheat arabinoxylan were incubated at room temperature while shaking for 48 h. Samples were centrifuged for 45 min at 3000 × g, and the insoluble part was washed three times in acetate buffer. Subsequently, the sample was resuspended in a volume to achieve 20% (w/v). Samples at 0.1 and 1% of both the soluble and insoluble fractions were tested using the above described HRP assay using 30 nm XylO. Also, 1% of insoluble substrates were incubated overnight at 37 °C while shaking at 2000 rpm with 1.0 μm XylO. The samples were then spun down, and the supernatant was tested with the HRP assay to detect the formation of hydrogen peroxide.
Oxygen Measurements
The affinity for oxygen was determined using 400 nm XylO and 5 mm xylobiose in an air-tight cuvette. The oxygen concentration was measured during catalysis with REDFLASH sensor spots and a FireSting O2 detector and light source (PyroScience). The slope of the oxygen decrease in time was plotted against the concentration of oxygen. The curve was fitted using a hyperbolic function: v = kcat × [O2]/Kox + [O2].
NMR and GC-MS Analysis
Conversion of 10 mm xylobiose was performed for 210 min at 50 °C in 50 mm potassium phosphate buffer, pH 6.0, using 1.0 μm XylO. NMR samples were exchanged twice with D2O (99.9 atom %, Cambridge Isotope Laboratories, Inc., Andover, MA) with intermediate lyophilization and finally dissolved in 0.6 ml of D2O. 1D/2D 500-MHz 1H NMR spectra and 125-MHz 13C NMR spectra were recorded in D2O, containing acetone as internal standard (δ1H 2.225, δ13C 31.07). 1D and 2D NMR spectra were recorded on a Varian Inova 500 spectrometer (NMR Department, University of Groningen, The Netherlands) at a probe temperature of 25 °C. Suppression of the HOD signal was achieved by applying a Wet1D pulse sequence. 2D COSY data were collected in 200 increments of 4000 complex data points. Natural abundance 2D 13C-1H HSQC and HMBC experiments were recorded without decoupling during acquisition of the 1H free induction decay. For GC-MS analysis, the product was analyzed intact, as well as after methanolysis (1.0 m methanolic HCl, 24 h, 85 °C) as a TMS ethers derivative (1:1:5 hexamethyldisilazane, trimethylchlorosilane, pyridine; 30 min, room temperature) by GC-EI-MS (where EI is electron ionization) on a QP2010 Plus instrument (Shimadzu, 's-Hertogenbosch, The Netherlands), using a ZB-1HT column (Phenomenex B.V., Utrecht, The Netherlands). Separation was achieved using a temperature gradient: 140–240 °C.
Author Contributions
A. R. F. performed the bioinformatics analysis that led to the identification of XylO. A. R. F. performed and designed the experimental work with help in the interpretation of the data by M. W. F. A. R. F. wrote the first draft of the manuscript. M. W. F. wrote the final version. H. J. R. crystallized and resolved the 3D structure of XylO and wrote the corresponding part in this manuscript. J. M. D. performed the experimental part to identify the product of XylO. J. M. D. and S. S. v. L. analyzed the NMR and GC/MS results of the conversion and wrote the corresponding part in the manuscript. A. S. C. V. performed the experimental work concerning the cloning and expression of XylO. M. J. K. wrote the cloning and expression section in this manuscript. M. J. K., J. V., and M. W. F. contributed with concept and design of the studies and with interpretation of the data.
Supplementary Material
This work was supported by the Netherlands Organisation for Scientific Research (NWO) in the framework of the Technology Areas for Sustainable Chemistry (TASC) Technology Area Biomass. The authors declare that they have no conflicts of interest with the contents of this article.

This article contains supplemental Figs. S1–S4.
The nucleotide sequence(s) reported in this paper has been submitted to the GenBankTM/EBI Data Bank with accession number(s) KX139007.
The atomic coordinates and structure factors (codes 5K8E, 5L6F, and 5L6G) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- GluO
- glucooligosaccharide oxidase
- XylO
- xylooligosaccharide oxidase
- VAO
- vanillyl alcohol oxidase
- LaO
- lactose oxidase
- ChitO
- chitooligosaccharide oxidase
- XOS
- xylooligosaccharide(s)
- TMS
- trimethylsilyl
- HMBC
- heteronuclear multiple bond correlation
- HSQC
- heteronuclear single quantum coherence
- RMSD
- root mean square deviation
- FAD domain
- FAD binding domain
- S domain
- substrate binding domain.
References
- 1. van Hellemond E. W., Leferink N. G. H., Heuts D. P. H. M., Fraaije M. W., and van Berkel W. J. H. (2006) Occurrence and biocatalytic potential of carbohydrate oxidases. Adv. Appl. Microbiol. 60, 17–54 [DOI] [PubMed] [Google Scholar]
- 2. Lin S. F., Yang T. Y., Inukai T., Yamasaki M., and Tsai Y. C. (1991) Purification and characterization of a novel glucooligosaccharide oxidase from Acremonium strictum T1. Biochim. Biophys. Acta 1118, 41–47 [DOI] [PubMed] [Google Scholar]
- 3. Heuts D. P. H. M., Janssen D. B., and Fraaije M. W. (2007) Changing the substrate specificity of a chitooligosaccharide oxidase from Fusarium graminearum by model-inspired site-directed mutagenesis. FEBS Lett. 581, 4905–4909 [DOI] [PubMed] [Google Scholar]
- 4. Xu F., Golightly E. J., Fuglsang C. C., Schneider P., Duke K. R., Lam L., Christensen S., Brown K. M., Jørgensen C. T., and Brown S. H. (2001) A novel carbohydrate:acceptor oxidoreductase from Microdochium nivale. Eur. J. Biochem. 268, 1136–1142 [DOI] [PubMed] [Google Scholar]
- 5. Leferink N. G. H., Heuts D. P. H. M., Fraaije M. W., and van Berkel W. J. H. (2008) The growing VAO flavoprotein family. Arch. Biochem. Biophys. 474, 292–301 [DOI] [PubMed] [Google Scholar]
- 6. Nordkvist M., Nielsen P. M., and Villadsen J. (2007) Oxidation of lactose to lactobionic acid by a Microdochium nivale carbohydrate oxidase: kinetics and operational stability. Biotechnol. Bioeng. 97, 694–707 [DOI] [PubMed] [Google Scholar]
- 7. Huang C.-H., Lai W.-L., Lee M.-H., Chen C.-J., Vasella A., Tsai Y.-C., and Liaw S.-H. (2005) Crystal structure of glucooligosaccharide oxidase from Acremonium strictum: a novel flavinylation of 6-S-cysteinyl, 8α-N1-histidyl FAD. J. Biol. Chem. 280, 38831–38838 [DOI] [PubMed] [Google Scholar]
- 8. Heuts D. P. H. M., Scrutton N. S., McIntire W. S., and Fraaije M. W. (2009) What's in a covalent bond? On the role and formation of covalently bound flavin cofactors. FEBS J. 276, 3405–3427 [DOI] [PubMed] [Google Scholar]
- 9. Kujawa M., Ebner H., Leitner C., Hallberg B. M., Prongjit M., Sucharitakul J., Ludwig R., Rudsander U., Peterbauer C., Chaiyen P., Haltrich D., and Divne C. (2006) Structural basis for substrate binding and regioselective oxidation of monosaccharides at C3 by pyranose 2-oxidase. J. Biol. Chem. 281, 35104–35115 [DOI] [PubMed] [Google Scholar]
- 10. Hecht H. J., Kalisz H. M., Hendle J., Schmid R. D., and Schomburg D. (1993) Crystal structure of glucose oxidase from Aspergillus niger refined at 2.3 Å resolution. J. Mol. Biol. 229, 153–172 [DOI] [PubMed] [Google Scholar]
- 11. Ferrari A. R., Lee M., and Fraaije M. W. (2015) Expanding the substrate scope of chitooligosaccharide oxidase from Fusarium graminearum by structure-inspired mutagenesis. Biotechnol. Bioeng. 112, 1074–1080 [DOI] [PubMed] [Google Scholar]
- 12. Kopacz M. M., and Fraaije M. W. (2014) Turning a monocovalent flavoprotein into a bicovalent flavoprotein by structure-inspired mutagenesis. Bioorg. Med. Chem. 22, 5621–5627 [DOI] [PubMed] [Google Scholar]
- 13. Kolbusz M. A., Di Falco M., Ishmael N., Marqueteau S., Moisan M.-C., Baptista C. da S., Powlowski J., and Tsang A. (2014) Transcriptome and exoproteome analysis of utilization of plant-derived biomass by Myceliophthora thermophila. Fungal Genet. Biol. 72, 10–20 [DOI] [PubMed] [Google Scholar]
- 14. Winkler A., Hartner F., Kutchan T. M., Glieder A., and Macheroux P. (2006) Biochemical evidence that berberine bridge enzyme belongs to a novel family of flavoproteins containing a bi-covalently attached FAD cofactor. J. Biol. Chem. 281, 21276–21285 [DOI] [PubMed] [Google Scholar]
- 15. Lee M.-H., Lai W.-L., Lin S.-F., Hsu C.-S., Liaw S.-H., and Tsai Y.-C. (2005) Structural characterization of glucooligosaccharide oxidase from Acremonium strictum. Appl. Environ. Microbiol. 71, 8881–8887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kiryu T., Nakano H., Kiso T., and Murakami H. (2008) Purification and characterization of a carbohydrate:acceptor oxidoreductase from Paraconiothyrium sp. that produces lactobionic acid efficiently. Biosci. Biotechnol. Biochem. 72, 833–841 [DOI] [PubMed] [Google Scholar]
- 17. Vuong T. V., Vesterinen A.-H., Foumani M., Juvonen M., Seppälä J., Tenkanen M., and Master E. R. (2013) Xylo- and cello-oligosaccharide oxidation by gluco-oligosaccharide oxidase from Sarocladium strictum and variants with reduced substrate inhibition. Biotechnol. Biofuels. 6, 148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Baminger U., Subramaniam S. S., Renganathan V., and Haltrich D. (2001) Purification and characterization of cellobiose dehydrogenase from the plant pathogen Sclerotium (Athelia) rolfsii. Appl. Environ. Microbiol. 67, 1766–1774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ludwig R., Salamon A., Varga J., Zámocky M., Peterbauer C. K., Kulbe K. D., and Haltrich D. (2004) Characterisation of cellobiose dehydrogenases from the white-rot fungi Trametes pubescens and Trametes villosa. Appl. Microbiol. Biotechnol. 64, 213–222 [DOI] [PubMed] [Google Scholar]
- 20. Schou C., Christensen M. H., and Schülein M. (1998) Characterization of a cellobiose dehydrogenase from Humicola insolens. Biochem. J. 330, 565–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Karnaouri A., Topakas E., Paschos T., Taouki I., and Christakopoulos P. (2013) Cloning, expression and characterization of an ethanol tolerant GH3 β-glucosidase from Myceliophthora thermophila. PeerJ. 1, e46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Topakas E., Moukouli M., Dimarogona M., and Christakopoulos P. (2012) Expression, characterization and structural modelling of a feruloyl esterase from the thermophilic fungus Myceliophthora thermophila. Appl. Microbiol. Biotechnol. 94, 399–411 [DOI] [PubMed] [Google Scholar]
- 23. van Gool M. P., van Muiswinkel G. C. J., Hinz S. W. A., Schols H. A., Sinitsyn A. P., and Gruppen H. (2012) Two GH10 endo-xylanases from Myceliophthora thermophila C1 with and without cellulose binding module act differently towards soluble and insoluble xylans. Bioresour. Technol. 119, 123–132 [DOI] [PubMed] [Google Scholar]
- 24. Ericsson U. B., Hallberg B. M., Detitta G. T., Dekker N., and Nordlund P. (2006) Thermofluor-based high-throughput stability optimization of proteins for structural studies. Anal. Biochem. 357, 289–298 [DOI] [PubMed] [Google Scholar]
- 25. Parikka K., Leppänen A.-S., Pitkänen L., Reunanen M., Willför S., and Tenkanen M. (2010) Oxidation of polysaccharides by galactose oxidase. J. Agric. Food Chem. 58, 262–271 [DOI] [PubMed] [Google Scholar]
- 26. Theander O., Westerlund E., Åman P., and Graham H. (1989) Plant cell walls and monogastric diets. Anim. Feed Sci. Technol. 23, 205–225 [Google Scholar]
- 27. Knudsen K. E. B. (1997) Carbohydrate and lignin contents of plant materials used in animal feeding. Anim. Feed Sci. Technol. 67, 319–338 [Google Scholar]
- 28. Huang C.-H., Winkler A., Chen C.-L., Lai W.-L., Tsai Y.-C., Macheroux P., and Liaw S.-H. (2008) Functional roles of the 6-S-cysteinyl, 8α-N1-histidyl FAD in glucooligosaccharide oxidase from Acremonium strictum. J. Biol. Chem. 283, 30990–30996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zafred D., Steiner B., Teufelberger A. R., Hromic A., Karplus P. A., Schofield C. J., Wallner S., and Macheroux P. (2015) Rationally engineered flavin-dependent oxidase reveals steric control of dioxygen reduction. FEBS J. 282, 3060–3074 [DOI] [PubMed] [Google Scholar]
- 30. Huang T.-H., Peng H.-J., Su S.-N., and Liaw S.-H. (2012) Various cross-reactivity of the grass pollen group 4 allergens: crystallographic study of the Bermuda grass isoallergen Cyn d 4. Acta Crystallogr. D Biol. Crystallogr. 68, 1303–1310 [DOI] [PubMed] [Google Scholar]
- 31. Teufel R., Miyanaga A., Michaudel Q., Stull F., Louie G., Noel J. P., Baran P. S., Palfey B., and Moore B. S. (2013) Flavin-mediated dual oxidation controls an enzymatic Favorskii-type rearrangement. Nature. 503, 552–556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Daniel B., Pavkov-Keller T., Steiner B., Dordic A., Gutmann A., Nidetzky B., Sensen C. W., van der Graaff E., Wallner S., Gruber K., and Macheroux P. (2015) Oxidation of monolignols by members of the Berberine Bridge Enzyme family suggests a role in plant cell wall metabolism. J. Biol. Chem. 290, 18770–18781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fraaije M. W., and Mattevi A. (2000) Flavoenzymes: diverse catalysts with recurrent features. Trends Biochem. Sci. 25, 126–132 [DOI] [PubMed] [Google Scholar]
- 34. Jain I., Kumar V., and Satyanarayana T. (2015) Xylooligosaccharides: an economical prebiotic from agroresidues and their health benefits. Indian J. Exp. Biol. 53, 131–142 [PubMed] [Google Scholar]
- 35. Samanta A. K., Jayapal N., Jayaram C., Roy S., Kolte A. P., Senani S., and Sridhar M. (2015) Xylooligosaccharides as prebiotics from agricultural by-products: production and applications. Bioact. Carbohydrates Diet. Fibre 5, 62–71 [Google Scholar]
- 36. Royer J. C., and Nakas J. P. (1991) Purification and characterization of two xylanases from Trichoderma longibrachiatum. Eur. J. Biochem. 202, 521–529 [DOI] [PubMed] [Google Scholar]
- 37. Baumann M. J., Borch K., and Westh P. (2011) Xylan oligosaccharides and cellobiohydrolase I (TrCel7A) interaction and effect on activity. Biotechnol. Biofuels. 4, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Qing Q., and Wyman C. E. (2011) Supplementation with xylanase and β-xylosidase to reduce xylo-oligomer and xylan inhibition of enzymatic hydrolysis of cellulose and pretreated corn stover. Biotechnol Biofuels. 4, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ferrari A. R., Gaber Y., and Fraaije M. W. (2014) A fast, sensitive and easy colorimetric assay for chitinase and cellulase activity detection. Biotechnol. Biofuels. 7, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Foumani M., Vuong T. V., MacCormick B., and Master E. R. (2015) Enhanced polysaccharide binding and activity on linear β-glucans through addition of carbohydrate-binding modules to either terminus of a glucooligosaccharide oxidase. PLoS ONE 10, e0125398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Petersen T. N., Brunak S., von Heijne G., and Nielsen H. (2011) SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat. Methods. 8, 785–786 [DOI] [PubMed] [Google Scholar]
- 42. Visser H., Joosten V., Punt P. J., Gusakov A. V., Olson P. T., Joosten R., Bartels J., Visser J., Sinitsyn A. P., Emalfarb M. A., Verdoes J. C., and Wery J. (2011) Development of a mature fungal technology and production platform for industrial enzymes based on a Myceliophthora thermophila isolate, previously known as Chrysosporium lucknowense C1. Ind. Biotechnol. 7, 214–223 [Google Scholar]
- 43. Verdoes J. C., Punt P. J., Burlingame R., Bartels J., Dijk R. van, Slump E., Meens M., Joosten R., and Emalfarb M. (2007) A dedicated vector for efficient library construction and high throughput screening in the hyphal fungus Chrysosporium lucknowense. Ind. Biotechnol. 3, 48–57 [Google Scholar]
- 44. Nishikimi M., Kiuchi K., and Yagi K. (1977) Detection of l-gulono-γ-lactone oxidase on SDS-polyacrylamide gels by the fluorescence of its covalently bound flavin. FEBS Lett. 81, 323–325 [DOI] [PubMed] [Google Scholar]
- 45. Punt, Peter J., Burlingame, Richard P., Pynnonen, Christine M., Olson, Phillip T., Wery J., Visser, Johannes H., Emalfarb, Mark A., Visser J., and Verdoes, Jan C. (September 23, 2010) Chrysosporium lucknowense protein production system. World Intellectual Property Organization patent 2,010,107,303 [Google Scholar]
- 46. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Matthews B. W. (1968) Solvent content of protein crystals. J. Mol. Biol. 33, 491–497 [DOI] [PubMed] [Google Scholar]
- 48. McCoy A. J. (2007) Solving structures of protein complexes by molecular replacement with Phaser. Acta Crystallogr. D Biol. Crystallogr. 63, 32–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Langer G., Cohen S. X., Lamzin V. S., and Perrakis A. (2008) Automated macromolecular model building for X-ray crystallography using ARP/wARP version 7. Nat. Protoc. 3, 1171–1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Murshudov G. N., Skubák P., Lebedev A. A., Pannu N. S., Steiner R. A., Nicholls R. A., Winn M. D., Long F., and Vagin A. A. (2011) REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 67, 355–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Chen V. B., Arendall W. B. 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., and Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kasprzak A. A., Papas E. J., and Steenkamp D. J. (1983) Identity of the subunits and the stoichiometry of prosthetic groups in trimethylamine dehydrogenase and dimethylamine dehydrogenase. Biochem. J. 211, 535–541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Britton H. T. S., and Robinson R. A. (1931) Universal buffer solutions and the dissociation constant of veronal. J. Chem. Soc. 10.1039/jr9310001456 [DOI] [Google Scholar]
- 55. Dijkman W. P., and Fraaije M. W. (2014) Discovery and characterization of a 5-hydroxymethylfurfural oxidase from Methylovorus sp. strain MP688. Appl. Environ. Microbiol. 80, 1082–1090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Schallmey M., Floor R. J., Hauer B., Breuer M., Jekel P. A., Wijma H. J., Dijkstra B. W., and Janssen D. B. (2013) Biocatalytic and structural properties of a highly engineered halohydrin dehalogenase. Chembiochem 14, 870–881 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.