

Abstract
Gram-negative bacteria use type IV secretion systems (T4SSs) for a variety of macromolecular transport processes that include the exchange of genetic material. The pKM101 plasmid encodes a T4SS similar to the well-studied model systems from Agrobacterium tumefaciens and Brucella suis. Here, we studied the structure and function of TraE, a homolog of VirB8 that is an essential component of all T4SSs. Analysis by X-ray crystallography revealed a structure that is similar to other VirB8 homologs but displayed an altered dimerization interface. The dimerization interface observed in the X-ray structure was corroborated using the bacterial two-hybrid assay, biochemical characterization of the purified protein, and in vivo complementation, demonstrating that there are different modes of dimerization among VirB8 homologs. Analysis of interactions using the bacterial two-hybrid and cross-linking assays showed that TraE and its homologs from Agrobacterium, Brucella, and Helicobacter pylori form heterodimers. They also interact with heterologous VirB10 proteins, indicating a significant degree of plasticity in the protein-protein interactions of VirB8-like proteins. To further assess common features of VirB8-like proteins, we tested a series of small molecules derived from inhibitors of Brucella VirB8 dimerization. These molecules bound to TraE in vitro, docking predicted that they bind to a structurally conserved surface groove of the protein, and some of them inhibited pKM101 plasmid transfer. VirB8-like proteins thus share functionally important sites, and these can be exploited for the design of specific inhibitors of T4SS function.
Keywords: bacterial conjugation, membrane protein, plasmid, protein secretion, protein-protein interaction, type IV secretion
Introduction
Plasmids of the IncN incompatibility group have a narrow host range mediating horizontal gene transfer among Enterobacteria. They code for replication, drug and metal resistance, and entry exclusion proteins. They also contribute to the transfer of clinically important antibiotic resistance genes (e.g. encoding carbapenem resistance in Klebsiella pneumoniae) (1). The clinical isolate R46 (2) as well as its deletion variant pKM101 are the most studied model systems (3). They encode genes sensitizing bacteria to radiation damage, and pKM101 gained notoriety because it was present in the Ames Salmonella strain used for the detection of mutagens (4). The transfer of antibiotic resistance genes by plasmid conjugation increasingly leads to problems in health care, and it is therefore important to better understand and to devise approaches to inhibit this process (5, 6).
Analysis of the sequence of the genes encoding the pKM101 conjugation machinery revealed striking similarities to type IV secretion systems (T4SSs)2 that transfer virulence factors from mammalian and plant pathogens to eukaryotic host cells (7, 8). The pKM101 conjugation machinery contains homologs to all of the 12 components of the most studied model, Agrobacterium tumefaciens; 11 of these proteins (TraL to TraG) are encoded in an operon similar to the VirB1–11 operon in Agrobacterium. Similar to other secretion systems of this class, pKM101 encodes homologs of putative ATPases (VirB4, VirB11, and VirD4), of core secretion system components (VirB1, VirB3, VirB6, VirB7, VirB8, VirB9, and VirB10), and of pilus components (VirB2 and VirB5). Whereas the biochemistry and the functions of individual T4SS components of the Agrobacterium and Brucella systems have been studied more thoroughly, structure biological approaches using co-expression of a subset of pKM101 core components (TraN, TraE, TraF, and TraO, homologs of VirB7 to VirB10) led to the first high resolution structure of the T4SS core complex using cryo-electron microscopy and X-ray crystallography (9–11). Surprisingly, co-expression of the VirB8 homolog TraE was not necessary for the formation of the TraN-TraF-TraO complex under these conditions, but VirB8 homologs are essential for the function of T4SSs, and they are thought to be assembly factors (12).
VirB8 homologs are small periplasmic proteins of about 25 kDa comprising a short N-terminal cytoplasmic region, one transmembrane helix, and a periplasmic region of 18 kDa. They are essential for all T4SSs in which they have been studied, and VirB8 was shown to be present in a helical arrangement around the cell in A. tumefaciens (13, 14). The results of extensive genetic and biochemical analyses suggest that VirB8-like proteins are assembly factors that undergo a series of mostly transient interactions with other T4SS components (15–21). The X-ray structures of the periplasmic domains of VirB8 homologs from A. tumefaciens (VirB8a) and from Brucella suis (VirB8b) were solved, and both proteins were predicted to form dimers of similar geometry via an α-helical region (22, 23). Interestingly, analysis of the TraM protein from the plasmid pIP501 conjugation system from Gram-positive Enterococci and of the TcpC protein from Clostridium perfringens showed that despite the absence of apparent sequence similarity, these proteins had a very similar fold (24, 25). However, these proteins form trimers, suggesting that VirB8-like proteins may be able to interact via different interfaces of their core structure. This notion is consistent with biochemical analysis suggesting that, in line with its predicted role as an assembly factor, VirB8 undergoes relatively weak protein-protein interactions with other T4SS components (19, 21, 26). Comparative analysis of different VirB8 homologs from Bartonella species showed homodimerization and a limited degree of heterodimer formation, suggesting a mechanistic solution preventing non-functional interactions of homologs that are simultaneously expressed in one organism (27). VirB8b was shown to interact with the close homolog TraJ from the pSB102 conjugation system, adding further evidence to the notion that VirB8 interactions are probably transient and may even be promiscuous (28, 29).
Here, we have extended the analysis of interactions between VirB8 homologs. We show that even distant homologs from different species interact and that this promiscuity extends to interactions with VirB10 homologs. Structural and biochemical analysis of TraE reveals a divergence as to the mode of dimerization compared with previously characterized homologs, underlining the cognate plasticity of this protein. Based on structural information and on previous work showing that VirB8b is a target for small molecule inhibitors (30, 31), we analyzed small molecules that bind to TraE and inhibit the conjugation of pKM101. We conclude that despite their divergent sequences and the transient nature of their interactions, VirB8-like proteins have common features that can be exploited for structure-based design of T4SS inhibitors.
Results
The Bacterial Two-hybrid Assay Shows Heterologous Interactions between Distantly Related VirB8 Homologs
To assess the capacity of VirB8 proteins to interact with homologs from other T4SSs, we expressed full-length VirB8a, VirB8b, TraE, and the VirB8 homolog CagV from Helicobacter pylori as fusions to the T25 and the T18 domains of Bordetella CyaA cytotoxin. The interactions were analyzed in the bacterial two-hybrid (BTH) assay using Escherichia coli cyaA deletion strain BTH101 and β-galactosidase activity as a readout fused to the cytoplasmic N terminus of the full-length proteins. The results show that all of these proteins homodimerize and that TraE also forms heterodimers with VirB8a, VirB8b, and CagV (Fig. 1A). Whereas the relative strength of the heterologous interactions was somewhat lower than that of TraE homodimerization, these results were nevertheless surprising because the sequence identities between the different homologs are low (14–35% as compared with TraE), the CagV protein from H. pylori being the most distantly related. In vitro cross-linking between the purified periplasmic domains of TraE, VirB8b, and CagV provided further evidence that these proteins have the capacity to form homomultimers as well as heteromultimers (Fig. 2). To test whether VirB8 proteins also interact in a heterologous fashion with other essential T4SS components from other organisms, we tested binding to VirB10 (19). Using this assay, TraE was shown to bind to VirB10 homologs from B. suis and from A. tumefaciens, further underlining its promiscuous binding capacity (Fig. 1B).
FIGURE 1.
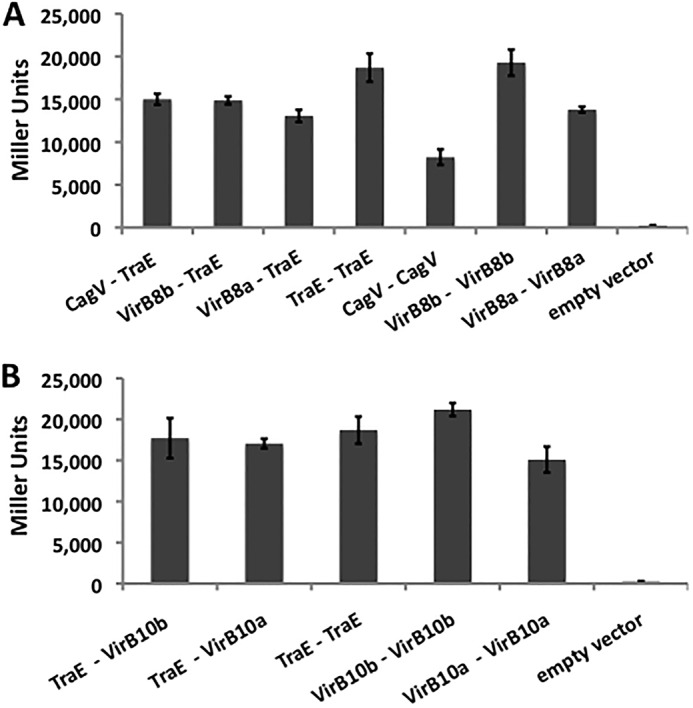
BTH analysis shows heterologous interactions between VirB8 homologs. The BTH assay was used to assess interactions between proteins fused to the adenylate cyclase domains, leading to production of β-galactosidase in E. coli reporter strains. A, interactions between VirB8 homologs from Agrobacterium (VirB8a), Brucella (VirB8b), Helicobacter (CagV), and pKM101 (TraE). B, interactions between VirB10 homologs from Agrobacterium (VirB10a), Brucella (VirB10b), and TraE. The homologous interactions between VirB8 proteins (A) and the homologous interactions between TraE and VirB10 proteins (B) were used as positive controls, whereas co-expression of the plasmid T25-TraE with an empty T18 fusion vector (A) or co-expression of the plasmid T25-VirB10b with an empty T18 fusion vector (B) was used as a negative control. Values and S.D. (error bars) were calculated from three independent experiments.
FIGURE 2.

TraE, VirB8b, and CagV have the capacity to form homomultimers and heteromultimers. A–C, SDS-PAGE analysis of the purified periplasmic domains of TraE (A), VirB8b (B), and CagV (C) in the absence (0 mm) and in the presence of the cross-linking agent DSS (0.4–2 mm). D, Western blot analysis with TraE-specific antiserum after SDS-PAGE of the purified periplasmic domains of TraE, CagV, and VirB8b following cross-linking with DSS (2 mm). Arrows, higher molecular weight complexes formed after cross-linking; TraE-CagV and TraE-VirB8b complexes in D migrate at higher molecular masses than TraE-TraE complexes due to the higher molecular masses of the heterologous interaction partners CagV and VirB8b.
Surface Plasmon Resonance Analysis of Interactions between CagV, TraE, and VirB8b
As an alternative approach to measure interactions between homologs CagV, TraE, and VirB8b, ligands were amine-coupled to individual flow cells of SPR chips, and then each was flowed overtop in solution (analyte). Single-cycle kinetic analyses (i.e. titration of low to high protein concentration without regeneration until the end of the series) showed that there was no nonspecific binding of bovine serum albumin (baseline control) to any of the reference or protein-immobilized surfaces (Fig. 3). When tested against CagV-coupled surfaces, TraE bound in a significant, dose-dependent manner with rapid association and dissociation kinetics (Fig. 3A). Similarly, the responses for CagV and TraE binding to TraE-coupled surfaces were significant (Fig. 3B); lower yet increasing signal responses with VirB8b were also detected. When tested in the reverse orientation, TraE bound to VirB8b-coupled surfaces in a dose-dependent manner, and VirB8b also interacted with itself (Fig. 3C). When analyzed according to a “steady-state affinity” model, the orthogonal SPR data provides direct evidence for CagV-TraE, TraE-TraE, and TraE-VirB8b interactions in the same low micromolar affinity range (Fig. 3D).
FIGURE 3.

A–C, representative single-cycle SPR analyses for tandem 1.56, 3.13, 6.25, 12.5, and 25 μm “analyte” injections (BSA (solid gray line), CagV (dashed black line), TraE (solid black line), and VirB8b (dashed gray line)) binding to amine-coupled “ligands” (CagV (A), TraE (B), and VirB8b (C); 300–900 RU each) at 25 μl/min. D, apparent equilibrium dissociation constants (KD) represent the average of at least three independent trials ± S.E.
Structural Analysis of TraE Reveals an Altered Mode of Dimerization
To gain high resolution insights into the molecular basis of the heterologous interactions, the X-ray structure of the periplasmic domain of TraE was determined by X-ray crystallography. Crystals of purified TraE diffracted up to 2.4 Å resolution (Table 1), and the structure was solved by molecular replacement using the homolog VirB8a that shares 25% amino acid identity as a search model. Structural comparison of TraE with its closest homologs VirB8a and VirB8b showed that the overall fold, comprising five α-helices on one side of the protein and four β-sheets on the opposite side, is similar. However, the orientations of the α-helices 1, 4, and 5 of TraE are somewhat different from VirB8a and VirB8b; the root mean square deviation is 0.66 Å compared with VirB8a and 1.67 Å compared with VirB8b (Fig. 4, A–C). The most interesting difference is that the dimer interface is clearly altered from that of VirB8a and VirB8b regarding the participating residues (Fig. 4D) and in its overall geometry (Fig. 5). The dimer interface buries a total of 844 Å2 for VirB8a and 925 Å2 for Virb8b, whereas the area of the TraE dimer interface is only 577 Å2. The second and third contact regions described in VirB8a and VirB8b structures are not present in the TraE dimer, and the two monomers are tilted in comparison with the more symmetrical arrangement of its homologs (Fig. 5). Despite these differences, in silico docking suggests that heterodimer formation of TraE with VirB8a and VirB8b is possible (Fig. 6), which is consistent with the results from the BTH assay (Fig. 1).
TABLE 1.
Data collection and refinement statistics for TraE
All values in parentheses are given for the highest resolution shell.
| Parameters | Values |
|---|---|
| Wavelength (Å) | 1.1 |
| Resolution range (Å) | 38.6–2.441 (2.529–2.441) |
| Space group | C 2 2 21 |
| Unit cell parameters: a (Å), b (Å), c (Å), α = β = γ = 90° | 112.049, 123.362, 109.8 |
| Total reflections | 184,794 |
| Unique reflections | 28,596 (2,826) |
| Multiplicity | 6.5 (6.0) |
| Completeness (%) | 99.98 (99.93) |
| Mean I/σ(I) | 32.7 (2.3) |
| Wilson B-factor | 46.46 |
| Rmerge | 0.121 |
| Rmeas | 0.132 |
| Reflections used for Rfree | 2,000 |
| Rworka | 0.2472 (0.3429) |
| Rfreeb | 0.2801 (0.3475) |
| No. of non-hydrogen atoms | 4,636 |
| Macromolecules | 4,569 |
| Water | 117 |
| Protein residues | 558 |
| Root mean square deviation | |
| Bonds (Å) | 0.004 |
| Angles (degrees) | 0.93 |
| Ramachandran favored (%)c | 99 |
| Ramachandran allowed (%)c | 1 |
| Ramachandran outliers (%)c | 0 |
| Clashscore | 4.21 |
| Average B-factor (Å2) | 38.20 |
| PDB number | 5I97 |
a Rwork = Σhkl‖Io(hkl)| − |Ic(hkl)‖/Σhkl|Io(hkl)|.
b Rfree = ΣhklΣT‖Io(hkl) − |Ic(hkl)‖/ΣhklΣT|Io(hkl)|, where T is a test data set randomly selected from the observed reflections before refinement. The test data set was not used throughout refinement and contained 7% of total unique reflections.
c Analyzed by Molprobity.
FIGURE 4.

Structural analysis of TraE dimerization. A, structure of the TraE dimer determined by X-ray crystallography (side view and top view). B, comparison of TraE and VirB8 from A. tumefaciens (PDB code 2CC3) monomer structures. C, comparison of TraE and VirB8 from B. suis (PDB code 4AKZ) monomer structures. D, four amino acids (Glu97, Gln105, Lys168, and Tyr214) implicated in the formation of interprotomer hydrogen bonds at the observed dimerization interface. RMSD, root mean square deviation.
FIGURE 5.

Comparison of the homodimer interface of the periplasmic forms of TraE, VirB8a, and VirB8b. A, sequence alignment of the periplasmic domains of TraE, VirB8a, and VirB8b with the amino acids at the homodimer interface (blue, residues present at the interface; yellow, residues making hydrogen bonds; green, residues making salt bridges). B, overall geometry of the VirB8a (PDB code 2CC3) homodimer structure as compared with VirB8b (PDB code 2BHM) and TraE.
FIGURE 6.

In silico docking to predict heterodimer formation between TraE, VirB8a, and VirB8b using the Zdock server (44). A, docking result showing potential TraE-VirB8a heterodimer. B, docking result showing potential TraE-VirB8b heterodimer.
The Predicted Dimerization Site Is Important for VirB8 Interactions in Vitro and for Its Functionality in Vivo
To assess whether the atypical dimerization site predicted in the X-ray structure is biologically significant, variants of residues Glu97, Gln105, Lys168, and Tyr214, which form hydrogen bonds across the dimer interface were constructed. Single amino acid changes, such as Y214A or K168A, had little or no effect on dimerization using the BTH assay. In contrast, dimerization of the variants E97A and Q105A and of those carrying double changes E97A/Q105A and K168A/Y214A was strongly reduced (Fig. 7A). Similarly, when variants of the periplasmic domain of TraE carrying single and double amino acid changes were analyzed by gel filtration, we found that the concentration-dependent dimerization observed in the wild type protein was significantly diminished if not abolished in both single and double variants (Fig. 7B). Finally, to assess functionality in the natural biological context, we tested the ability of TraE and its variants to complement conjugation of a non-polar pKM101::traE transposon insertion mutant. This analysis revealed that conjugation was markedly reduced or not detectable in the case of all single and double variants (Fig. 7C), despite the fact that the proteins were stably expressed. Taken together, these results support the biological relevance of the dimerization site predicted by X-ray crystallography. We next analyzed whether the structural information can be exploited for the design of inhibitors of TraE-dependent plasmid conjugation.
FIGURE 7.

Genetic and biochemical analysis shows the importance of TraE dimerization. A, the BTH assay was used to measure the interactions between fusion proteins of TraE variants and adenylate cyclase domains, leading to production of β-galactosidase in E. coli reporter strains. The dimerization of TraE wild type protein was used as a positive control, whereas co-expression of the plasmid T25-TraE with an empty T18 fusion vector was used as a negative control. Values and S.D. (error bars) were calculated from three independent experiments. B, gel filtration analysis at increasing concentrations of the periplasmic domain of TraE (diamonds) shows concentration-dependent dimer formation, whereas its variants E97A (squares), Y214A (triangles), and E97A/Q105A (crosses) show no evidence for dimer formation as a function of concentration. C, pKM101 conjugation assay to assess the capacity of TraE and of its variants at the dimerization site to complement a pKM101::traE transposon insertion plasmid (CFU, colony-forming units of recipient strain after plasmid transfer). The data represent averages and S.E. of three biological replicate cultures. Asterisks, statistically significant differences (p ≤ 0.0001, n = 3). Bottom, Western blot using anti-TraE antibody. Signals shown by an arrow correspond to the predicted molecular masse of TraE WT and variants.
Docking of VirB8b Inhibitors and of Novel Derivates to TraE
A common feature of VirB8 homologs is the presence of a surface groove located between the α-helical and the β-sheet faces of the protein, and it was hypothesized that this groove has a functional role in mediating protein interactions by VirB8 homologs (22, 26). We therefore explored whether putative binding by small molecules to this site in turn inhibits bacterial conjugation. Previous docking results showed that the most potent compounds that bound to this site in VirB8b were the inhibitors B8I-1, B8I-2, and B8I-3 (31). These and other small molecules also tested (31) were shown to bind the equivalent surface groove of TraE (Fig. 8). To further probe this binding site, we also synthesized seven novel analogues of compound B8I-2 (Fig. 9), which from our previous work inhibited Brucella virulence (31). Analogues of B8I-2 were designed to probe for enhanced interaction with the binding site, and in silico docking predicted that all of these compounds would bind to TraE (Fig. 8C). We therefore tested their impact on TraE interactions in vitro and in vivo.
FIGURE 8.

Docking of VirB8b inhibitors and of B8I-2 derivates to TraE. A, predicted binding of B8I-1 (orange), B8I-2 (blue), and B8I-3 (yellow) to TraE using AutoDock Vina. Amino acids Glu111, Lys140, and Thr157 form a hydrogen bond network in TraE. B, binding site of molecule BAR-072 (red) to TraE, as predicted by AutoDock Vina. In addition to the hydrogen bond network Glu111-Lys140-Thr157 contributing to ligand binding, Arg110 may form an additional hydrogen bond with BAR-072. C, schematic (green) and surface (gray) representations of the TraE monomer showing the common binding site for 17 of 18 tested small molecules, as predicted by AutoDock Vina. B8I-8 is the only compound predicted to bind in a surface groove in the back of the image. Atoms for each inhibitor were colored using a rainbow gradient based on the relative intermolecular binding energy with TraE (lowest energy shown in blue, highest energy shown in red).
FIGURE 9.

Chemical structures of small molecules tested by in silico docking, in vitro binding, and in vivo conjugation assays. Previously tested molecules (B8I-1 to B8I-16, UM-023 to UM-050) (31) and new B8I-2 analogues (BAR-068 to BAR-074) were used to probe the binding site.
Small Molecules Bind to TraE and Inhibit pKM101 Conjugation
To measure small molecule binding, we established an in vitro assay based on the quenching of tryptophan fluorescence following the addition of inhibitors to the purified periplasmic domain of TraE. Using this assay, we determined KD values from low to high micromolar range for the different molecules (Table 2). The lowest KD value of 2.7 μm was observed for the B8I-2 analogue BAR-072 (Fig. 10A). Using chemical cross-linking, we showed that the purified periplasmic domain of TraE forms dimers and higher molecular mass multimers, which is consistent with the results from the BTH assay (Fig. 10B). The addition of BAR-072 attenuated cross-linker-dependent formation of multimers, suggesting that binding by this analogue interferes with dimerization (Fig. 10, B and C). Finally, we assessed whether the TraE-binding small molecules have an effect on plasmid conjugation. Conjugation experiments were conducted between a pKM101-carrying donor (FM433) and a plasmid-free recipient (WL400) in the presence of the small molecules at 50 μm concentration. No effects on bacterial growth were observed at this concentration except for molecule B8I-3, which was not further pursued (data not shown). Analysis of plasmid transfer frequency showed that none of the original VirB8b inhibitors had a significant impact but that four of the tested molecules significantly reduced pKM101 transfer (B8I-16, BAR-072, BAR-073, and UM-024) (Fig. 11A). The strongest effect (10-fold reduction) was observed for molecule BAR-072, correlating with the lowest KD value observed by fluorescence quenching. None of the molecules that reduced pKM101 transfer impacted the conjugation of the unrelated control plasmid RP4, suggesting that their effects are specific for pKM101 and not due to a nonspecific effect on bacterial metabolism or viability (Fig. 11B).
TABLE 2.
Binding of small molecules to TraE measured using a fluorescence quenching assay
| Tested molecule | KDa |
|---|---|
| μm | |
| B8I-1 | 11.4 ± 0.6 |
| B8I-2 | 20.6 ± 1.4 |
| B8I-3 | 9.9 ± 0.5 |
| B8I-4 | 35.1 ± 3.1 |
| B8I-5 | 30.3 ± 2.8 |
| B8I-8 | 28.5 ± 2.5 |
| B8I-16 | 28.8 ± 2.2 |
| BAR-068 | 53.6 ± 3.9 |
| BAR-069 | 27.9 ± 2.4 |
| BAR-070 | 9.1 ± 0.3 |
| BAR-071 | 10.0 ± 0.4 |
| BAR-072 | 2.7 ± 0.1 |
| BAR-073 | 7.3 ± 0.6 |
| BAR-074 | 9.2 ± 0.4 |
| UM-0125823 | 9.6 ± 0.2 |
| UM-0125824 | 61.1 ± 5.5 |
| UM-0125848 | 14.4 ± 0.5 |
| UM-0125850 | 34.4 ± 1.6 |
a S.D. value derived from three replicas.
FIGURE 10.

Effect of BAR-072 on TraE. A, in vitro binding assay based on tryptophan quenching upon the addition of BAR-072 to the purified periplasmic domain of TraE. B, cross-linking of TraE with DSS (0.4 mm) in the presence of BAR-072 at given concentrations (0–1.6 mm, DMSO final concentration of 2.5%); arrows indicate higher molecular weight complexes formed after cross-linking of TraE. C, quantification of the formation of DSS-dependent cross-linking products of TraE (indicated by arrows) in the absence and in the presence of BAR-072 (0–1.6 mm). Formation of the two cross-linking products was normalized by comparison with the control without BAR-072 (100% is the control); the data represent averages and S.E. (error bars) of the mean of three replicates.
FIGURE 11.

Effect of TraE-binding small molecules on conjugation. A, conjugation assays between pKM101-carrying donor strain FM433 and plasmid-free recipient WL400 were conducted in the presence of 50 μm TraE-binding small molecules. B, RP4 conjugation assay in the presence of inhibitors of pKM101 transfer. Conjugation assays between plasmid RP4-carrying donor strain FM433 and plasmid-free recipient WL400 were conducted in the presence of 50 μm concentrations of the small molecules. The numbers of colony-forming units compared with a control experiment in the absence of the small molecules are shown; data represent averages and S.E. (error bars) of three biological replicate cultures. Asterisks, statistically significant differences (p ≤ 0.0001, n = 3).
Discussion
VirB8 homologs are essential components of all T4SSs in which they have been studied. They are believed to undergo a series of transient protein-protein interactions contributing to T4SS assembly (15–21). Although the sequence identities between VirB8 homologs are in the range of 20–40%, the overall protein fold is well conserved (22, 23, 27), and this was confirmed here by determination of the TraE structure. In some cases, due to high divergence of the primary sequence, VirB8 homologs were found only upon their structural analysis (24, 25, 32), suggesting strong evolutionary pressure to conserve protein overall fold, with functional specificity emerging at a later date. These data are consistent with the early hypothesis that VirB8 is an assembly factor that was based on its similarity to a nuclear transfer factor (NTF-2) (22). X-ray crystallographic approaches have shown that most studied VirB8 homologs form dimers. However, the more distantly related homologs from Gram-positive bacteria, such as Clostridium TcpC and Enterococcus TraM, crystallized as trimers, suggesting significant structural plasticity regarding VirB8-like protein multimerization. Whereas TraE crystallized as a dimer, the spatial arrangement of the two subunits was clearly distinct from that of its closest homologs, VirB8a and VirB8b. The biological significance of this dimerization site was verified by the analysis of variants at the interaction interface, suggesting that different spatial orientations are biologically significant even in the case of closely related VirB8 homologs (20, 26).
The fact that the VirB8 fold is so highly conserved underlines the importance of this protein as a T4SS assembly factor. Several studies have shown that VirB8 undergoes multiple interactions (15, 18, 19, 21, 26, 33), but until now only limited data were available on the molecular basis of these interactions. Considering that the periplasmic domain is relatively small (18 kDa), it is unlikely that VirB8 interactions are more than transient, which is consistent with the measured strength of interactions in the high nanomolar to micromolar range (19, 21). Nevertheless, specificity is important for ordered T4SS assembly, and a recent study on VirB8 homologue proteins from Bartonella and Rickettsia species suggests that structural barriers as well as differential expression prevent non-productive interactions between multiple VirB8 paralogs in these organisms (27). Whereas non-productive interactions are not pertinent in the case of VirB8 homologs from different bacteria, the high degree of interactions between VirB8 homologs from Agrobacterium, Brucella, pKM101, and H. pylori observed in the BTH assay as well as in vitro using cross-linking as well as SPR analysis was unexpected. In addition to homodimer formation, we observed heterologous binding to VirB10 homologs, indicating a high degree of plasticity of VirB8 interactions arising from the conserved protein fold. It would be of interest to test interactions with VirB8 homologs from Gram-positive bacteria in the future (TcpC, TraM) to determine whether the plasticity of interactions extends to trimer-forming homologs. Binding between VirB8 homologs in the BTH assay does not necessarily imply functional complementation in the natural biological context in which other additional protein-protein interactions are likely to be important. For example, despite the fact that TraJ from plasmid pSB201 shares 50% sequence identity and bound Brucella VirB8 in the BTH assay, complementation with the full-length protein was not possible (28, 29). Similarly, we found that Brucella VirB8 does not complement virB8 deletions in A. tumefaciens.3 The periplasmic domain from TraJ did complement to a limited extent when it was fused to the N-terminal domain of VirB8b, but expression of TraJ or of a TraJ-VirB8b fusion protein inhibited virulence. This is probably due to strong binding with VirB8b, suggesting that both the dimer and the free monomer forms are functionally important.
The biological role of the prominent hydrophobic surface groove between the β-sheet face of the protein and the α-helices present at the dimerization site has been the subject of debate, and it was hypothesized that it may be a protein-protein interaction site (22). Currently, there is no evidence for this notion, but we subsequently discovered that this surface groove is the binding site for small molecule inhibitors of VirB8 dimerization (30, 31). At first, this result was counterintuitive because the dimer interface is on the side opposite to the binding site. However, substituting amino acids at the inhibitor binding site reduced the potency of the inhibitors as well as dimerization, suggesting that conformational changes of this surface groove may be linked to dimerization. NMR analysis of VirB8b showed significant conformational changes between the monomeric and the dimeric state that are consistent with this notion.4 Here, we probed TraE with small molecules that had been shown to inhibit VirB8b dimerization (31) as well as derivatives that were synthesized to improve the interaction potency to binding site residues. Using a fluorescence quenching assay, we showed that the VirB8b inhibitors and newly synthesized compounds bound to TraE with affinities in the micromolar range despite apparent differences of the binding sites in the two proteins. Molecular docking suggested that almost all of the molecules bind to the surface groove between the β-sheet face and the α-helices of the protein that is equivalent to that in VirB8b. We determined a wide variety of binding affinities, and the molecule BAR-072 had the lowest KD value of 2.7 μm. Docking predicted that BAR-072 binds to four amino acids in the inhibitor-binding surface groove of TraE, whereas B8I-2 and other molecules only bind up to three amino acids, which could explain the difference in KD values (Fig. 8B). Interestingly, BAR-072 had a significant effect on T4SS function, leading to a 10-fold reduction of pKM101 transfer between bacteria. Further, molecule UM-024 that was weakly active against VirB8b also significantly inhibited pKM101 conjugation, whereas the most potent VirB8b inhibitor, B8I-2, had no effect on pKM101 conjugation, suggesting that differences in the inhibitor binding sites can be exploited for the design of specific molecules. The low degree of correlation between the KD values and the effects on pKM101 transfer (except for BAR-072) suggest the presence of an additional binding site or variations in the ability of the molecules to cross the cell envelope. Nevertheless, the observed attenuation of plasmid conjugation suggests for the first time that the inhibition of plasmid transfer is possible by targeting a specific component of a secretion/conjugation system. Our results also suggest that the surface groove targeted by VirB8b inhibitors may be suitable as a target for TraE inhibitors, pointing to a new paradigm enabling discovery of inhibitors targeting VirB8-like proteins in a wide variety of pathogens.
Experimental Procedures
Strains and Plasmids
The strains and plasmids used are listed in Table 3. E. coli strains XL-1 Blue or DH5α were used as hosts for cloning and mutagenesis. Strain BL21star (λDE3) was used for overexpression of TraE, VirB8a, VirB8b, and CagV; strain BTH101 was used for BTH assays.
TABLE 3.
Bacterial strains and plasmids
| Genotype/description | Source/reference | |
|---|---|---|
| Strains | ||
| DH5α | F− Φ80lacZΔM15 Δ (lacZYA-argF) U169 recA1 endA1 hsdR17 (rK−, mK+) phoA supE44 λ− thi-1 gyrA96 relA1 | Invitrogen |
| XL-1 Blue | recA1 endA1 gyrA96 thi-1 hsdR17 supE44 relA1 lac (F′ proAB lacIqZΔM15 Tn10 (Tetr)) | Agilent Technologies |
| BL21(DE3)star | F− ompT hsdSB (rB−, mB−) gal dcm rne131 (DE3) | Invitrogen |
| BTH101 | F- cya-99, araD139, galE15, galK16, rpsL1 (strr), hsdR2, mcrA1, mcrB1 | Ref. 40 |
| FM433 | SpcraraD139 Δ (argF-lac)U169 ptsF25 deoC1 relA1 flbB5301 rpsE13 Δ (srl-recA)306::Tn10, conjugation donor | Ref. 41 |
| WL400 | CmrStrraraD139 Δ (argF-lac)U169, ptsF25 deoC1 relA1 flbB5301 rpsL150 ΔselD204::cat, conjugation recipient | W. Leinfelder, unpublished data |
| Plasmids | ||
| pHT | kanr pET24d derivative T7 expression vector with N-terminal His6 tag and TEV protease cleavage site | Ref. 31 |
| pHTVirB8bp | kanr T7 promoter vector for the expression of His6-tagged periplasmic domain of B. suis VirB8 | Ref. 31 |
| pHTTraEp | kanr T7 promoter vector for the expression of His6-tagged periplasmic domain of pKM101 TraE | This work |
| pET21aCagVp | ampr T7 promoter vector for the expression of His6-tagged periplasmic domain of H. pylori CagV | This work |
| pHTTraEp E97A | pHTTraEp modified to encode TraE with amino acid change E97A | This work |
| pHTTraEp Y214A | pHTTraEp modified to encode TraE with amino acid change Y214A | This work |
| pHTTraEp E97A/Q105A | pHTTraEp modified to encode TraE with amino acid change E97A/Q105A | This work |
| pUT18 | pUC19 derivative including the T18 fragment (amino acids 225–399 of B. pertussis CyaA) N-terminal to the multiple cloning site | Ref. 40 |
| pKNT25 | pSU40 derivative including the T25 fragment (amino acids 1–224 of B. pertussis CyaA) N-terminal to the multiple cloning site | Ref. 40 |
| pUT18C | pUC19 derivative including the T18 fragment (amino acids 225–399 of B. pertussis CyaA) C-terminal to the multiple cloning site | Ref. 40 |
| pKT25 | pSU40 derivative including the T25 fragment (amino acids 1–224 of B. pertussis CyaA) C-terminal to the multiple cloning site | Ref. 40 |
| pUT18CVirB10b | ampr, pUT18C harboring 1,176-bp XbaI/KpnI virB10 fragment from B. suis (encoding full-length VirB10) | Ref. 30 |
| pKT25VirB10b | kanr, pKT25 harboring 1,176-bp XbaI/KpnI virB10 fragment from B. suis (encoding full-length VirB10) | Ref. 30 |
| pUT18CVirB10a | ampr, pUT18C harboring 1,134-bp XbaI/KpnI virB10 fragment from A. tumefaciens (encoding full-length VirB10) | This work |
| pKT25VirB10a | kanr, pKT25 harboring 1,134-bp XbaI/KpnI virB10 fragment from A. tumefaciens (encoding full-length VirB10) | This work |
| pUT18CVirB8b | ampr, pUT18C harboring 720-bp XbaI/KpnI virB8 fragment from B. suis (encoding full-length VirB8) | Ref. 19 |
| pKT25VirB8b | kanr, pKT25 harboring 720-bp XbaI/KpnI virB8 fragment from B. suis (encoding full-length VirB8) | Ref. 19 |
| pUT18CVirB8a | ampr, pUT18C harboring 711-bp XbaI/KpnI virB8 fragment from A. tumefaciens (encoding full-length VirB8) | This work |
| pKT25VirB8a | kanr, pKT25 harboring 711-bp XbaI/KpnI virB8 fragment from A. tumefaciens (encoding full-length VirB8) | This work |
| pUT18CCagV | ampr, pUT18C harboring 765-bp XbaI/KpnI cagV fragment from H. pylori (encoding full-length CagV) | This work |
| pKT25CagV | kanr, pKT25 harboring 765-bp XbaI/KpnI cagV fragment from H. pylori (encoding full-length CagV) | This work |
| pUT18CTraE | ampr, pUT18C harboring 699-bp XbaI/KpnI traE fragment from pKM101 (encoding full-length TraE) | This work |
| pKT25TraE | kanr, pKT25 harboring 699-bp XbaI/KpnI traE fragment from pKM101 (encoding full-length TraE) | This work |
| pUT18CTraE E97A | pUT18CB8 modified to encode TraE with amino acid change E97A | This work |
| pUT18CTraE Q105A | pUT18CB8 modified to encode TraE with amino acid change Q105A | This work |
| pUT18CTraE K168A | pUT18CB8 modified to encode TraE with amino acid change K168A | This work |
| pUT18CTraE Y214A | pUT18CB8 modified to encode TraE with amino acid change Y214A | This work |
| pUT18CTraE E97A/Q105A | pUT18CB8 modified to encode TraE with amino acid change E97A/Q105A | This work |
| pUT18CTraE K168A/Y214A | pUT18CB8 modified to encode TraE with amino acid change K168A/Y214A | This work |
| pKT25TraE E97A | pKT25B8 modified to encode TraE with amino acid change E97A | This work |
| pKT25TraE Q105A | pKT25B8 modified to encode TraE with amino acid change Q105A | This work |
| pKT25TraE K168A | pKT25B8 modified to encode TraE with amino acid change K168A | This work |
| pKT25TraE Y214A | pKT25B8 modified to encode TraE with amino acid change Y214A | This work |
| pKT25TraE E97A/Q105A | pKT25B8 modified to encode TraE with amino acid change E97A/Q105A | This work |
| pKT25TraE K168A/Y214A | pKT25B8 modified to encode TraE with amino acid change K168A/Y214A | This work |
| pTrc200 | Strr, Spcr, pVS1 derivative, LacIq, trc promoter expression vector | Ref. 42 |
| pTrc200TraE | pTrc200, traC PCR fragment cloned downstream of the trc promoter | This work |
| pTrc200TraE E97A | pTrc200TraE modified to encode TraE with amino acid change E97A | This work |
| pTrc200TraE Q105A | pTrc200TraE modified to encode TraE with amino acid change Q105A | This work |
| pTrc200TraE K168A | pTrc200TraE modified to encode TraE with amino acid change K168A | This work |
| pTrc200TraE Y214A | pTrc200TraE modified to encode TraE with amino acid change Y214A | This work |
| pTrc200TraE E97A/Q105A | pTrc200TraE modified to encode TraE with amino acid change E97A/Q105A | This work |
| pTrc200TraE K168A/Y214A | pTrc200TraE modified to encode TraE with amino acid change K168A/Y214A | This work |
| pKM101 | Ampr, mucA, mucB, TraI-TraIII region for DNA processing, DNA transfer, and entry exclusion | Ref. 43 |
| pKM101traE | Ampr, Kmr, pKM101traE1228::Tn5 | Ref. 3 |
Quantitation of Conjugative DNA Transfer
E. coli strains FM433 pKM101 (donor, ampicillin-resistant), FM433 pRP4 (donor, ampicillin-resistant), and WL400 (recipient, chloramphenicol-resistant) were grown in liquid LB medium at 37 °C (with 100 μg/ml for the pKM101- and RP4-carrying strains) to an A600 nm of 0.5–1, sedimented by centrifugation, and resuspended in an appropriate volume of LB medium without antibiotics. Equal amounts of donor and recipient strain (1 μl of each) were mixed on prewarmed LB agar and incubated for 2 h at 37 °C to enable conjugation, and the cells were then washed from the plate with 100 μl of liquid LB. To quantify conjugative transfer, dilutions of the conjugation mixture were plated on LB agar plates containing appropriate antibiotics for selection of pKM101-containing recipient strain WL400 (100 μg/ml ampicillin, 34 μg/ml chloramphenicol). To determine complementation with TraE variants, strain FM433 carrying a non-polar transposon insertion in the traE gene (pKM101traE1228::Tn5 (3)) was transformed with complementation vector pTrc200 (negative control), pTrc200TraE (positive control), or pTrc200 expressing TraE variants; complementation experiments were carried out as above. For analysis of the inhibition of conjugation with small molecules, the cells were cultivated on agar and in liquid media in the presence of a 50 μm concentration of the small molecules in the presence of 2.5% DMSO. The presence of DMSO and of the small molecules (with the exception of B8I-3) did not negatively impact bacterial growth.
DNA Isolation and Manipulation
Plasmid DNA was isolated using Qiagen Miniprep kits (Qiagen, Manchester, UK). Standard techniques were employed for the cloning, transformation, preparation, and restriction analysis of plasmid DNA from E. coli (34).
BTH Assay
Interactions between full-length TraE, VirB8a, VirB8b, CagV, VirB10a, VirB10b, and TraE variants were assessed in vivo using the BTH system as described (35). The genes encoding VirB8 homologs, TraE variants, and VirB10 homolog proteins were fused to the DNA sequences encoding the T18 and T25 fragments of the catalytic domain of Bordetella pertussis adenylate cyclase (AC), and they were co-expressed in BTH101 AC (cya)-deficient cells. The interactions were detected using functional complementation between the two catalytic AC fragments leading to cAMP and as a consequence to β-galactosidase production that was quantified using ortho-nitrophenyl-β-d-galactopyranoside as the substrate.
Protein Overexpression and Purification
For protein overproduction, E. coli strain BL21star (λDE3) carrying expression plasmids was grown under aerobic conditions at 37 °C in LB to exponential phase (A600 of 0.4–0.8), followed by the addition of 0.5 mm isopropyl-β-d-thiogalactopyranoside for TraE, TraE variants, and VirB8b and 1 mm isopropyl-β-d-thiogalactopyranoside for CagV to induce gene expression. Cultivation under aerobic conditions proceeded at 30 °C for 4 h after induction for TraE and its variants or at 25 °C for 16 h after induction for CagV and VirB8b. For purification, bacterial cells were harvested, resuspended in binding buffer (50 mm sodium phosphate, 300 mm NaCl, 40 mm imidazole, pH 7.4), and lysed using a One Shot cell disrupter (Constant Systems Inc.) at 27,000 p.s.i. Lysates were subsequently centrifuged twice at 15,000 rpm at 4 °C, filtered through a 0.45-μm membrane to remove cell debris, loaded onto a HisTrap nickel-chelate column (GE Healthcare), and eluted using a linear 50-ml gradient of 40–500 mm imidazole. The proteins were then desalted into TEV buffer (25 mm sodium phosphate, 125 mm NaCl, 5 mm DTT, pH 7.4) and subjected to cleavage of the N-terminal His6 tag using His6-tagged TEV protease in a ratio of 1:70 (TEV/protein) for 24 h at 20 °C. Following cleavage, the solution was diluted 10 times, centrifuged once at 13,000 rpm at 4 °C, and filtered through a 0.45-μm membrane to remove precipitate. Then it was passed over a HisTrap nickel-chelate column (GE Healthcare) to bind the tagged TEV protease, and the flow-through containing cleaved proteins was collected. The purified proteins were then desalted by PD-10 columns (GE Healthcare) into 20 mm HEPES, pH 7.4, 50 mm NaCl for TraE; 50 mm MES, pH 6.5, 100 mm NaCl for CagV; or 50 mm Tris, pH 7.4, 50 mm NaCl for VirB8b and stored at 4 °C. For crystallography, TraE was further purified by size exclusion chromatography using a Superdex 75 column (GE Healthcare). TraE protein concentrations were determined using the molar extinction coefficients at 280 nm of 27,390 m−1 cm−1 (with His6 tag) and of 25,900 m−1 cm−1 (without His6 tag). VirB8b protein concentrations were determined using the molar extinction coefficients at 280 nm of 34,380 m−1 cm−1 (with His6 tag) and of 32,890 m−1 cm−1 (without His6 tag). CagV protein concentrations were determined using the Bradford assay.
Crystallization of TraE and Data Collection
The TraE buffer was changed to 20 mm HEPES, 50 mm NaCl (pH 7.4), and the protein (15 mg/ml) was crystallized in 16% (w/v) PEG 10,000, 50 mm BisTris (pH 5.5), 100 mm ammonium acetate. The crystals were cryoprotected in 16% (w/v) PEG 10,000, 50 mm BisTris (pH 5.5), 100 mm ammonium acetate, and 20% ethylene glycol. The crystals were flash-frozen in liquid nitrogen, and the data were collected at beamline X25 of the National Synchrotron Light Source at Brookhaven National Laboratory. The intensity data were processed using the HKL2000 program.
Structure Determination of TraE
Structures were solved by molecular replacement using the structure of VirB8 from A. tumefaciens as a reference model (PDB code 2CC3) (23). Refinement was performed using Phenix software suite of program to the highest possible resolution (36) made possible by collecting diffraction data in 180° sweeps to take full advantage of the space group symmetry redundancy (C2221) to maximize signal/noise ratio. Electron density maps were calculated to the resolution indicated in Table 1 to ensure at least ∼90% completeness in the highest resolution shell with an I/σ(I) > 2. Final model statistics, calculated with Phenix, molprobity, and PROCHECK, are shown in Table 1. The atomic coordinates and structure factors for TraE have been deposited at the Protein Data Bank (PDB code 5I97). The model of the final structure has an Rcryst (Rfree) value of 0.237 (0.275). All figures were prepared using the program PyMOL.
Large Zone Gel Filtration Chromatography
The molecular masses of the purified periplasmic domain of TraE and of its variants were determined by gel filtration using large zone elution (37). To this effect, 200-μl aliquots with protein concentrations ranging from 12 to 240 μm were applied to a Superdex 75 column (GE Healthcare) in 20 mm HEPES, 50 mm NaCl, pH 7.4, at 4 °C. The void volume of the column was determined with blue dextran 2000, and the calibration curve was determined by using the Low Molecular Weight Gel Filtration Calibration Kit (GE Healthcare).
Analysis of Protein-Protein Interactions by Cross-linking
Chemical cross-linking with disuccinimidyl suberate (DSS; Pierce) was performed as described previously (18), and the formation of cross-linking products was quantified by SDS-PAGE, Western blotting, and using the ImageLab version 4.0 software (Bio-Rad).
Molecular Docking Analysis
In silico docking was performed using Autodock Vina (38) run through PyRx to manage the workflow and PyMOL to visualize the results. The chemical structures for each ligand were retrieved from the hit2lead.com website and converted to PDB format using openbabel software, followed by processing with Autodock Tools version 1.5.4 to assign Gasteiger charges, merging non-polar hydrogens, and to set torsional bonds. Docking runs were performed within a 47 × 52 × 33-Å rectangle search space surrounding the binding pocket, and output modes were ranked according to binding affinity. Autodock Vina identified molecular conformations with the best fit and strongest binding affinity (global minima) by a stochastic algorithm exploring surfaces/pockets of the rigid macromolecule, through an iterative series of local optimizations (changing shape, bond angles, and position of the ligand), evaluating both intermolecular (hydrophobic interactions, repulsions, hydrogen bonding, etc.) and intramolecular (torsion, rotational torque) factors.
Analysis of Small Molecule Binding by Fluorescence Spectroscopy
Changes in the intrinsic UV fluorescence emission of TraE upon binding of small molecules were recorded at 20 °C with a Cary Eclipse fluorometer (Varian) (λex, 295 nm; λem, 340 nm; 5-nm excitation and emission slit widths) in 20 mm HEPES, 50 mm NaCl, pH 7.4. The spectra were corrected for dilution effects, and the KD values were calculated from the ligand binding fluorescence data fitted to a single-site saturation curve with constant background using the Grafit version 6.0 software package.
Synthesis of Small Molecules
Small molecule derivates of B8I-2 were synthesized as described (31).
Surface Plasmon Resonance
Using label-free, real-time SPR, the interactions between CagV, TraE, and VirB8b were examined using a BIACORE 3000 system (GE Healthcare Bio-Sciences AB, Uppsala, Sweden; BIAcontrol version 4.1 operating software). For quality control, each batch of purified protein was pretested on an UltrafleXtreme MALDI-TOF/TOF system (Bruker Daltonics, Bremen, Germany) according to the manufacturer's recommendations (intact mass analysis in linear-positive mode as well as top-down sequencing (in-source decay) to verify the N-/C-terminal amino acid sequences of the intact proteins).
SPR experiments were performed on research-grade CM5 sensor chips (Biacore) at 25 °C using filtered (0.2 μm) and degassed HBS-EP buffer (10 mm Hepes, pH 7.4, 150 mm NaCl, 3 mm EDTA, 0.005% (v/v) Tween 20). Immobilized protein surfaces (10 μg/ml in 10 mm sodium acetate, pH 5.0 (CagV, TraE) or pH 4.0 (VirB8b)) were prepared using the Biacore Amine Coupling Kit as recommended by the manufacturer (final immobilization levels = 300–900 RU for each protein); corresponding reference surfaces were prepared in the absence of protein.
To assess binding specificity and affinity, single-cycle kinetic analyses were performed in which increasing concentrations of BSA (negative control), CagV, TraE, and VirB8b were titrated in tandem (0–25 μm; 2-fold dilution series) over reference and protein-immobilized surfaces at 25 μl/min in KINJECT mode (1-min association + 30-s dissociation). Between titration series, the surfaces were regenerated at 50 μl/min using two 30-s pulses of solution I (HBS-EP containing 1.0 m NaCl and 0.05% (v/v) Empigen) followed by the EXTRACLEAN and RINSE procedures. Due to the rapid steady-state binding kinetics observed for each interaction pair, the binding responses were independent of mass transport limitations, and all double-referenced data (39) presented are representative of duplicate injections acquired from at least three independent trials. To predict overall equilibrium dissociation constants (KD) for each “ligand,” steady-state binding responses (Req) were averaged near the end of each association phase, plotted as a function of “analyte” concentration (C), and then subjected to non-linear regression analysis (“Steady state affinity” model; BIAevaluation version 4.1 software). Complementary multicycle kinetic analyses (25 μl/min × 5-min association + 5-min dissociation) were performed in a similar manner and yielded consistent affinity constants.
Author Contributions
B. C. conducted and designed experiments and wrote the manuscript; J. S. designed experiments and revised the manuscript; M. A. H. conducted and designed experiments and revised the manuscript; M. S. conducted and designed experiments and revised the manuscript; J. S. designed experiments and revised the manuscript; C. B. designed experiments and wrote the manuscript.
Acknowledgments
We thank Dr. Pierre Lavallée and Benoit Jolicoeur (Université de Montréal, Department of Chemistry) for the synthesis of small molecules, Benoit Bessette for technical assistance, Dr. Durga Sivanesan for BTH constructs to measure for VirB8a and VirB10a BTH interactions, and Dr. Gerhard Multhaup (McGill University) for discussions on SPR experiments.
This work was supported by Canadian Institutes of Health Research (CIHR) Grant MOP-84239 and grants from the Natural Sciences and Engineering Research Council (NSERC), the NSERC-CREATE program on the Cellular Dynamics of Macromolecular Complexes (CDMC), the Bristol Myers Smith Research Chair in Molecular Biology at Université de Montréal, the Groupe d'études des proteines membranaires (GÉPROM), the Canada Foundation for Innovation (CFI), and the Fonds de recherche du Québec-Santé (FRQ-S) (to C. B.). The McGill SPR-MS facility receives infrastructure support from CFI. The authors declare that they have no conflicts of interest with the contents of this article.
The atomic coordinates and structure factors (code 5I97) have been deposited in the Protein Data Bank (http://wwpdb.org/).
B. Casu, J. Smart, M. A. Hancock, M. Smith, J. Sygusch, and C. Baron, unpublished results.
M. Sharifahmadian, J. Omichinski, and C. Baron, unpublished results.
- T4SS
- type IV secretion system
- AC
- adenylate cyclase
- BTH
- bacterial two-hybrid
- RU
- response units
- SPR
- surface plasmon resonance
- TEV
- tobacco etch virus
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- PDB
- Protein Data Bank
- DSS
- disuccinimidyl suberate.
References
- 1. Kayama S., Shigemoto N., Kuwahara R., Oshima K., Hirakawa H., Hisatsune J., Jové T., Nishio H., Yamasaki K., Wada Y., Ueshimo T., Miura T., Sueda T., Onodera M., Yokozaki M., et al. (2015) Complete nucleotide sequence of the IncN plasmid encoding IMP-6 and CTX-M-2 from emerging carbapenem-resistant Enterobacteriaceae in Japan. Antimicrob. Agents Chemother. 59, 1356–1359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mortelmans K. E., and Stocker B. A. (1979) Segregation of the mutator property of plasmid R46 from its ultraviolet-protecting property. Mol. Gen. Genet. 167, 317–327 [DOI] [PubMed] [Google Scholar]
- 3. Winans S. C., and Walker G. C. (1985) Conjugal transfer system of the IncN plasmid pKM101. J. Bacteriol. 161, 402–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. McCann J., Springarn N. E., Kobori J., and Ames B. N. (1975) Detection of carcinogens as mutagens: bacterial tester strains with R factor plasmids. Proc. Natl. Acad. Sci. U.S.A. 72, 979–983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Liu Y. Y., Wang Y., Walsh T. R., Yi L. X., Zhang R., Spencer J., Doi Y., Tian G., Dong B., Huang X., Yu L. F., Gu D., Ren H., Chen X., Lv L., et al. (2016) Emergence of plasmid-mediated colistin resistance mechanism MCR-1 in animals and human beings in China: a microbiological and molecular biological study. Lancet Infect. Dis. 16, 161–168 [DOI] [PubMed] [Google Scholar]
- 6. Kumarasamy K. K., Toleman M. A., Walsh T. R., Bagaria J., Butt F., Balakrishnan R., Chaudhary U., Doumith M., Giske C. G., Irfan S., Krishnan P., Kumar A. V., Maharjan S., Mushtaq S., Noorie T., et al. (2010) Emergence of a new antibiotic resistance mechanism in India, Pakistan, and the UK: a molecular, biological, and epidemiological study. Lancet Infect. Dis. 10, 597–602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pohlman R. F., Genetti H. D., and Winans S. C. (1994) Common ancestry between IncN conjugal transfer genes and macromolecular export systems of plant and animal pathogens. Mol. Microbiol. 14, 655–668 [DOI] [PubMed] [Google Scholar]
- 8. Winans S. C., Burns D. L., and Christie P. J. (1996) Adaptation of a conjugal transfer system for the export of pathogenic macromolecules. Trends Microbiol. 4, 64–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chandran V., Fronzes R., Duquerroy S., Cronin N., Navaza J., and Waksman G. (2009) Structure of the outer membrane complex of a type IV secretion system. Nature 462, 1011–1015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fronzes R., Schäfer E., Wang L., Saibil H. R., Orlova E. V., and Waksman G. (2009) Structure of a type IV secretion system core complex. Science 323, 266–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Trokter M., Felisberto-Rodrigues C., Christie P. J., and Waksman G. (2014) Recent advances in the structural and molecular biology of type IV secretion systems. Curr. Opin. Struct. Biol. 27, 16–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Baron C. (2006) VirB8: a conserved type IV secretion system assembly factor and drug target. Biochem. Cell Biol. 84, 890–899 [DOI] [PubMed] [Google Scholar]
- 13. Aguilar J., Zupan J., Cameron T. A., and Zambryski P. C. (2010) Agrobacterium type IV secretion system and its substrates form helical arrays around the circumference of virulence-induced cells. Proc. Natl. Acad. Sci. U.S.A. 107, 3758–3763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Aguilar J., Cameron T. A., Zupan J., and Zambryski P. (2011) Membrane and core periplasmic Agrobacterium tumefaciens virulence Type IV secretion system components localize to multiple sites around the bacterial perimeter during lateral attachment to plant cells. MBio 2, e00218–00211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Das A., Xie Y.-H. (2000) The Agrobacterium T-DNA transport pore proteins VirB8, VirB9, and VirB10 interact with one another. J. Bacteriol. 182, 758–763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kumar R. B., and Das A. (2001) Functional analysis of the Agrobacterium tumefaciens T-DNA transport pore protein VirB8. J. Bacteriol. 183, 3636–3641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Berger B. R., and Christie P. J. (1994) Genetic complementation analysis of the Agrobacterium tumefaciens virB operon: virB2 through virB11 are essential virulence genes. J. Bacteriol. 176, 3646–3660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yuan Q., Carle A., Gao C., Sivanesan D., Aly K. A., Höppner C., Krall L., Domke N., and Baron C. (2005) Identification of the VirB4-VirB8-VirB5-VirB2 pilus assembly sequence of type IV secretion systems. J. Biol. Chem. 280, 26349–26359 [DOI] [PubMed] [Google Scholar]
- 19. Sivanesan D., Hancock M. A., Villamil Giraldo A. M., and Baron C. (2010) Quantitative analysis of VirB8-VirB9-VirB10 interactions provides a dynamic model of type IV secretion system core complex assembly. Biochemistry 49, 4483–4493 [DOI] [PubMed] [Google Scholar]
- 20. Sivanesan D., and Baron C. (2011) The dimer interface of Agrobacterium tumefaciens VirB8 is important for type IV secretion system function, stability, and association of VirB2 with the core complex. J. Bacteriol. 193, 2097–2106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Villamil Giraldo A. M., Sivanesan D., Carle A., Paschos A., Smith M. A., Plesa M., Coulton J., and Baron C. (2012) Type IV secretion system core component VirB8 from Brucella binds to the globular domain of VirB5 and to a periplasmic domain of VirB6. Biochemistry 51, 3881–3890 [DOI] [PubMed] [Google Scholar]
- 22. Terradot L., Bayliss R., Oomen C., Leonard G. A., Baron C., and Waksman G. (2005) Structures of two core subunits of the bacterial type IV secretion system, VirB8 from Brucella suis and ComB10 from Helicobacter pylori. Proc. Natl. Acad. Sci. U.S.A. 102, 4596–4601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bailey S., Ward D., Middleton R., Grossmann J. G., and Zambryski P. (2006) Agrobacterium tumefaciens VirB8 structure reveals potential protein-protein interaction sites. Proc. Natl. Acad. Sci. U.S.A. 103, 2582–2587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Goessweiner-Mohr N., Grumet L., Arends K., Pavkov-Keller T., Gruber C. C., Gruber K., Birner-Gruenberger R., Kropec-Huebner A., Huebner J., Grohmann E., and Keller W. (2013) The 2.5 Å structure of the enterococcus conjugation protein TraM resembles VirB8 type IV secretion proteins. J. Biol. Chem. 288, 2018–2028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Porter C. J., Bantwal R., Bannam T. L., Rosado C. J., Pearce M. C., Adams V., Lyras D., Whisstock J. C., and Rood J. I. (2012) The conjugation protein TcpC from Clostridium perfringens is structurally related to the type IV secretion system protein VirB8 from Gram-negative bacteria. Mol. Microbiol. 83, 275–288 [DOI] [PubMed] [Google Scholar]
- 26. Paschos A., Patey G., Sivanesan D., Gao C., Bayliss R., Waksman G., O'callaghan D., and Baron C. (2006) Dimerization and interactions of Brucella suis VirB8 with VirB4 and VirB10 are required for its biological activity. Proc. Natl. Acad. Sci. U.S.A. 103, 7252–7257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gillespie J. J., Phan I. Q., Scheib H., Subramanian S., Edwards T. E., Lehman S. S., Piitulainen H., Rahman M. S., Rennoll-Bankert K. E., Staker B. L., Taira S., Stacy R., Myler P. J., Azad A. F., and Pulliainen A. T. (2015) Structural Insight into How Bacteria Prevent Interference between Multiple Divergent Type IV Secretion Systems. MBio. 6, e01867–e15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Patey G., Qi Z., Bourg G., Baron C., and O'Callaghan D. (2006) Swapping of periplasmic domains between Brucella suis VirB8 and a pSB102 VirB8 homologue allows heterologous complementation. Infect. Immun. 74, 4945–4949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bourg G., Sube R., O'Callaghan D., and Patey G. (2009) Interactions between Brucella suis VirB8 and its homolog TraJ from the plasmid pSB102 underline the dynamic nature of type IV secretion systems. J. Bacteriol. 191, 2985–2992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Paschos A., den Hartigh A., Smith M. A., Atluri V. L., Sivanesan D., Tsolis R. M., and Baron C. (2011) An in vivo high-throughput screening approach targeting the type IV secretion system component VirB8 identified inhibitors of Brucella abortus 2308 proliferation. Infect. Immun. 79, 1033–1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Smith M. A., Coincon M., Paschos A., Jolicoeur B., Lavallee P., Sygusch J., and Baron C. (2012) Identification of the binding site of Brucella VirB8 interaction inhibitors. Chem. Biol. 19, 1041–1048 [DOI] [PubMed] [Google Scholar]
- 32. Kuroda T., Kubori T., Thanh Bui X., Hyakutake A., Uchida Y., Imada K., and Nagai H. (2015) Molecular and structural analysis of Legionella DotI gives insights into an inner membrane complex essential for type IV secretion. Sci. Rep. 5, 10912–10926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kumar R. B., Xie Y.-H., and Das A. (2000) Subcellular localization of the Agrobacterium tumefaciens T-DNA transport pore proteins: VirB8 is essential for the assembly of the transport pore. Mol. Microbiol. 36, 608–617 [DOI] [PubMed] [Google Scholar]
- 34. Sambrook J., Fritsch E. F., and Maniatis T. (eds) (1989) Molecular Cloning: A Laboratory Manual, Cold Spring Harbor Laboratory, Cold Spring Harbor, NY [Google Scholar]
- 35. Villamil Giraldo A. M., Mary C., Sivanesan D., and Baron C. (2015) VirB6 and VirB10 from the Brucella type IV secretion system interact via the N-terminal periplasmic domain of VirB6. FEBS Lett. 589, 1883–1889 [DOI] [PubMed] [Google Scholar]
- 36. Adams P. D., Grosse-Kunstleve R. W., Hung L. W., Ioerger T. R., McCoy A. J., Moriarty N. W., Read R. J., Sacchettini J. C., Sauter N. K., and Terwilliger T. C. (2002) PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr. D Biol. Crystallogr. 58, 1948–1954 [DOI] [PubMed] [Google Scholar]
- 37. Valdes R. Jr., and Ackers G. K. (1979) Study of protein subunit association equilibria by elution gel chromatography. Methods Enzymol. 61, 125–142 [DOI] [PubMed] [Google Scholar]
- 38. Trott O., and Olson A. J. (2010) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 31, 455–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Myszka D. G. (1999) Improving biosensor analysis. J. Mol. Recognit. 12, 279–284 [DOI] [PubMed] [Google Scholar]
- 40. Karimova G., Dautin N., and Ladant D. (2005) Interaction network among Escherichia coli membrane proteins involved in cell division as revealed by bacterial two-hybrid analysis. J. Bacteriol. 187, 2233–2243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zinoni F., Heider J., and Böck A. (1990) Features of the formate dehydrogenase mRNA necessary for decoding of the UGA codon as selenocysteine. Proc. Natl. Acad. Sci. U.S.A. 87, 4660–4664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Schmidt-Eisenlohr H., Domke N., and Baron C. (1999) TraC of IncN plasmid pKM101 associates with membranes and extracellular high-molecular-weight structures in Escherichia coli. J. Bacteriol. 181, 5563–5571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Langer P. J., and Walker G. C. (1981) Restriction endonuclease cleavage map of pKM101: relationship to parental plasmid R46. Mol. Gen. Genet. 182, 268–272 [DOI] [PubMed] [Google Scholar]
- 44. Pierce B. G., Wiehe K., Hwang H., Kim B. H., Vreven T., and Weng Z. (2014) ZDOCK server: interactive docking prediction of protein-protein complexes and symmetric multimers. Bioinformatics 30, 1771–1773 [DOI] [PMC free article] [PubMed] [Google Scholar]