

Abstract
The programed death-1 (PD-1)–programed death ligand-1 (PD-L1) and PD-L2 co-inhibitory pathway has been implicated in the evasion strategies of Mycobacterium tuberculosis. Specifically, M. tuberculosis-induced PD-L1 orchestrates expansion of regulatory T cells and suppression of Th1 response. However, the role of PD pathway in regulating Th17 response to M. tuberculosis has not been investigated. In the present report, we demonstrate that M. tuberculosis and M. tuberculosis-derived antigen fractions have differential abilities to mediate human monocyte- and dendritic cell (DC)-mediated Th17 response and were independent of expression of PD-L1 or PD-L2 on aforementioned antigen-presenting cells. Importantly, we observed that blockade of PD-L1 or PD-1 did not significantly modify either the frequencies of Th17 cells or the production of IL-17 from CD4+ T cells though IFN-γ response was significantly enhanced. On the contrary, IL-1β from monocytes and DCs were critical for the Th17 response to M. tuberculosis. Together, our results indicate that IL-1β, but not members of the programed death pathway, is critical for human Th17 response to M. tuberculosis.
Keywords: PD-L1, PD-1, Mycobacterium tuberculosis, dendritic cells, monocytes, Th17, IL-23, IL-1β
Introduction
Tuberculosis caused by Mycobacterium tuberculosis still remains a global threat with an estimated 1.5 million deaths annually. Cellular immunity plays a critical role in mediating the protection against tuberculosis. Indeed, IFN-γ-producing CD4+ T helper type 1 cells are critical for the control of M. tuberculosis in humans and murine models (1–4). Thus, general paradigm for tuberculosis vaccines has largely focused on enhancing the Th1/IFN-γ response (5, 6). However, despite enhancing IFN-γ response, the recombinant MVA85A vaccine failed to protect infants from tuberculosis (7). Therefore, it is pertinent to decipher the role played by other CD4+ T cell subsets and their cytokines in mediating immunity against M. tuberculosis.
Th17 cells that express transcription factor RORC and secrete archetype cytokine IL-17A represent a distinct subset of CD4+ T cells. Th17 cells also produce IL-17F, IL-21, IL-22, GM-CSF, and IL-26 and mediate pro-inflammatory responses (8–11). Th17 cells are associated with the pathogenesis of several autoimmune and inflammatory diseases (12–14). Evolutionarily, Th17 response is conserved to mediate protection at mucosal surfaces and against extracellular pathogens (10, 15). Recent reports have also indicated that Th17 response may play a crucial role in mediating protection against intracellular pathogens, such as Francisella tularensis and Chlamydia muridarum (16–18). These data thus indicate the diverse role of Th17 cells in various physiopathologies.
Mycobacterium tuberculosis employs a plethora of mechanisms to suppress both innate and adaptive immune responses. The role of Th17 response to M. tuberculosis is largely pursued in mice, and it remains highly controversial (19–25). Recent reports in tuberculosis patients indicate that active disease and its severity are associated with low Th17 response (26, 27). Of note, anti-tuberculosis therapy is associated with enhanced Th17 response, suggesting that M. tuberculosis suppresses Th17 response as one of the immune evasion mechanisms (28).
Programed death-1 (PD-1)–programed death ligand-1 (PD-L1)/PD-L2 pathway occupies a unique place in the immune evasion strategies employed by M. tuberculosis. Recent data highlight the role of PD-1–PD-L1/PD-L2 axis in modulating regulatory T cell (Treg) and Th1 response to M. tuberculosis (29–33). Whether this pathway also regulates Th17 response to M. tuberculosis is not known. Therefore, in the present study, we have evaluated the role of PD pathway members (PD-L1, PD-L2, and PD-1) in mediating human monocyte- and dendritic cell (DC)-mediated Th17 response to M. tuberculosis.
Several reports have shown that DCs promote Th17 responses to either M. tuberculosis or its antigens (34–37). We found that monocytes and DCs have differential capacity to promote Th17 response to M. tuberculosis and M. tuberculosis-derived antigens. Notably, a prominent IL-17 response was mediated by DCs in comparison to monocytes. Although both monocytes and DCs did not express PD-L2, PD-L1 was significantly enhanced upon stimulation with M. tuberculosis. Similarly, M. tuberculosis stimulation of monocyte/DC–CD4+ cocultures also lead to significant increase in the frequency of PD-1+CD4+ T cells. Importantly, blocking PD-L1 or PD-1 neither significantly altered the frequencies of Th17 cells nor augmented IL-17 secretion from CD4+ T cells. Analysis of key Th17-polarizing cytokines indicated that the production of IL-1β was crucial in the establishment of Th17 response to M. tuberculosis. These results thus reveal that the outcome of Th17 response to M. tuberculosis is dictated by the capacity of human innate cells to secrete key Th17-polarizing cytokine (IL-1β) and not expression of members of the PD pathway.
Materials and Methods
Antibodies
FITC-conjugated mAbs to CD86 [clone 2331 (FUN-1)], CD274 (clone MIH1), PE-conjugated mAbs to pSTAT3 (clone 4/P-STAT3), CD80 (clone L307.4), PD-L2 (clone 2D3/B7-H2), antigen-presenting cell (APC)-conjugated mAbs to HLA-DR (clone G46-6), PD-1 (clone MIH4), Alexa 700-conjugated mAb to CD4 (clone RPA-T4), and BV421-conjugated mAb to CD4 were from BD Biosciences (Le Pont de Claix, France). PE-conjugated mAbs to IL-17A (clone eBio64CAP17), human–mouse RORγt (AFKJS-9), APC-conjugated mAb to FoxP3 (clone 236A/E7), and Fixable Vibility Dye eFluor® 506 were from eBioscience (Paris, France). PE-conjugated mAb to CD40 (clone MAB89) was from Beckman Coulter (Villepinte, France). Blocking mAb to human PD-L1 (clone MIH1) and isotype control mAb were from eBioscience. Alexa-488 conjugated mAb to IL-10 (clone JES59D7) and blocking mAb to PD-1 (clone EH12.2H7) were from Biolegend (London, UK).
M. tuberculosis Antigens
γ-irradiated M. tuberculosis (strain H37Rv) and M. tuberculosis cell wall, cell membrane cytoplasmic fractions were obtained from BEI resources NIAID, NIH.
Purification of Immune Cells
Peripheral blood mononuclear cells (PBMCs) were obtained from buffy bags of healthy donors by Ficoll density gradient centrifugation. Buffy bags of the healthy blood donors were purchased from Centre Necker-Cabanel, Etablissement Français du Sang, Paris, France. Ethical committee permission was obtained for the use of buffy bags of healthy donors (Institut National de la Santé et de la Recherche-EFS ethical committee convention 15/EFS/012). Monocytes and autologous CD4+ T cells were isolated from PBMCs by positive selection using the human CD14 and the CD4 MicroBeads (Miltenyi Biotec, Paris, France), respectively. The cell purity was more than 97%.
Generation of DCs
Monocytes (0.5 × 106 cells/ml) were cultured in the presence of granulocyte-macrophage colony-stimulating factor (GM-CSF; 1,000 IU/106 cells) and IL-4 (500 IU/106 cells) (both cytokines from Miltenyi Biotec) for 5 days to obtain immature monocyte-derived DCs (38). The differentiation of DCs was confirmed by flow cytometry.
Stimulation of Monocytes and DCs with M. tuberculosis and Their Fractions
Monocytes or DCs (0.5 × 106/ml) were cultured with (20 μg/ml) γ-irradiated M. tuberculosis or M. tuberculosis-derived cell wall, cell membrane, or cytoplasmic fractions (10 μg/ml) for 24 h. Activation of DCs and monocytes was assessed based on the expression of HLA-DR, CD40, CD80, and CD86 by using fluorescence-conjugated mAbs. In addition, induction of PD-L1 and PD-L2 on these cells was also analyzed.
Monocyte–CD4+ T Cell and DC–CD4+ T Cell Cocultures
Monocytes or DCs (10,000 cells/200 μl/well) were cocultured with autologous CD4+ T cells at a ratio of 1:10 in U-bottom 96-well plates and stimulated with (20 μg/ml) γ-irradiated M. tuberculosis or M. tuberculosis-derived cell wall, cell membrane, or cytoplasmic fractions (10 μg/ml) for 5 days. After 5 days, cell-free supernatants were collected, and T cells were activated with phorbol myristate acetate (50 ng/ml) and ionomycin (500 ng/ml, Sigma-Aldrich, France), along with GolgiStop (BD Biosciences), for 4 h. For the analysis of IL-17+CD4+ T cells and IL-10+CD4+ T cells in the cocultures, surface staining was performed with fluorescence-conjugated mAbs to CD4. Then, cells were fixed, permeabilized using intracellular staining kit (eBioscience), and incubated at 4°C with fluorescence-conjugated mAbs to IL-17A, IL-10, pSTAT3, and RORC. Samples were acquired by using LSR II (BD Biosciences) flow cytometry, and data were analyzed by BD FACS DIVA software (BD Biosciences).
For the analysis of FoxP3+CD4+ T in the cocultures, surface staining was performed with fluorescence-conjugated mAb to CD4. Then, cells were fixed, permeabilized using intracellular staining kit (eBioscience), and incubated at 4°C with fluorescence-conjugated mAb to FoxP3.
For the analysis of PD-1 on CD4+ T cells, surface staining was performed with fluorescence-conjugated mAbs to CD4 and PD-1.
PD-L1 and PD-1 Blocking Experiment
Autologous monocyte–CD4+ T cell and DC–CD4+ T cell cocultures were stimulated with M. tuberculosis for 18 h. Anti-PD-L1 (10 μg/ml), anti-PD-1 (10 μg/ml), or isotype control mAbs were then added to the coculture. After 5 days, frequency of IL-17A+CD4+ T cells and IL-17 secretion were analyzed.
Validation of Role for Innate Cytokines in M. tuberculosis-Mediated Th17 Responses
Dendritic cells (10,000 cells/200 μl/well) were cocultured with autologous CD4+ T cells at a ratio of 1:10 in U-bottom 96-well plates and stimulated with (20 μg/ml) γ-irradiated M. tuberculosis or M. tuberculosis-derived cytoplasmic fractions (10 μg/ml) either alone or in the presence of (10 ng/ml) rhIL-1β (R&D systems, Lille, France) or rhIL-23 (PeproTech, Neuilly-Sur-Seine, France) for 5 days. After 5 days, cell-free supernatants were collected, and T cells were analyzed for Th17 responses by intracellular staining as described earlier.
Quantification of Cytokines
IL-17A, IFN-γ, IL-6, IL-1β, and IL-23 in the cell-free supernatants of monocyte–CD4+ T cell and DC–CD4+ T cell cocultures were quantified by ELISA (Ready-SET-Go, eBioscience).
Statistical Analysis
Statistical analyses were performed by two-way non-parametric Mann–Whitney test or one-way ANOVA (Kruskal–Wallis test or Holm–Sidak’s multiple comparisons test) as indicated using Prism 6 software. P < 0.05 was considered significant.
Results
Human Monocytes Promote Th17 Response to M. tuberculosis
Tuberculosis is associated with an expansion of immunomodulatory CD16+ monocyte population (39). Monocytes have been implicated in the establishment of Th17 response in autoimmune diseases (40, 41) and infection (42). We first investigated the ability of human monocytes to promote Th17 response to M. tuberculosis. We found that M. tuberculosis-stimulated monocytes significantly enhanced both frequency of IL-17A+CD4+ T cells (Figures 1A,B) and the amount of secretion of IL-17A (Figure 1C). On further examination of downstream Th17 signaling events in CD4+ T cells, we found that M. tuberculosis-stimulated monocytes significantly enhanced the frequency of pSTAT3 (Figure 1D) and RORC (Figure 1E). These results thus suggest that human monocytes have the ability to promote Th17 response to M. tuberculosis.
Figure 1.

Human monocytes stimulated with M. tuberculosis enhance Th17 response. Human peripheral blood monocytes were cocultured with autologous CD4+ T cells at a ratio of 1:10 in X-vivo medium for 5 days with or without γ-irradiated M. tuberculosis (Mtb). Th17 cells were analyzed by flow cytometry by combination of surface staining for CD4 and intracellular staining for IL-17A, pSTAT3, and ROR-γt. IL-17A in the cell-free supernatants was quantified by ELISA. (A,B) Representative dot plots showing the frequencies of CD4+IL-17A+ T cells and (B) median ± SEM data from eight donors. (C) The amount of secretion of IL-17A (median ± SEM, n = 8). (D) Percentage of CD4+ T cells positive for pSTAT3 (median ± SEM, n = 4). (E) Percentage of CD4+ T cells positive for ROR-γt (median ± SEM, n = 4). *P < 0.05; ***P < 0.001; as determined by Mann–Whitney test.
Different Antigen Fractions of M. tuberculosis Have Similar Capacity to Induce Monocyte-Mediated Th17 Response
Mycobacterium tuberculosis possesses a plethora of antigens to modulate immune response. Most of these antigens are either located in the cell wall, cell membrane, or cytosol. Hence, we investigated whether these different antigens of M. tuberculosis have similar or distinct ability to mount monocyte-mediated Th17 response. All these antigenic fractions induced similar level of activation of monocytes as shown by the significantly augmented expressions of CD80, CD86, and CD40 (Figure S1 in Supplementary Material). Consistent with the activation status of monocytes, all the antigen fractions, i.e., cell wall, cell membrane, and cytoplasmic fractions significantly enhanced the frequency of IL-17A+CD4+ T cells (Figures 2A,B) and the secretion of IL-17A (Figure 2C). However, we observed no significant differences in the extent of Th17 response mediated by different fractions of M. tuberculosis. Our data thus indicate that all the antigen fractions of M. tuberculosis have similar ability to promote monocyte-mediated Th17 responses.
Figure 2.

Different antigen fractions of M. tuberculosis have similar capacity to amplify human monocyte-mediated Th17 response. Monocytes were cocultured with autologous CD4+ T cells either alone or were stimulated with M. tuberculosis-derived cell wall (CW), cell membrane (CM), or cytoplasmic (Cyt) fractions for 5 days. Th17 cells were analyzed by flow cytometry by combination of surface staining for CD4 and intracellular staining for IL-17A. IL-17A in the cell-free supernatants was quantified by ELISA. (A,B) Representative dot plots showing the frequencies of CD4+IL-17A+ T cells and (B) mean ± SEM data from six independent donors. (C) The amount of secretion of IL-17A (mean ± SEM, n = 6). *P < 0.05; **P < 0.01; as determined by one-way ANOVA.
Dendritic Cells Differentially Promote Th17 Response to M. tuberculosis and Its Antigen Fractions
Dendritic cells play a critical role in mediating protection to M. tuberculosis by priming T cell response. Hence, we investigated the capacity of DCs to promote Th17 response to M. tuberculosis and its antigen fractions. Our results indicate that DCs have the capacity to enhance Th17 response to M. tuberculosis (Figures 3A–C). However, in contrast to monocytes, human DCs displayed differential ability in stimulating Th17 response to different antigen fractions of M. tuberculosis. Thus, cell wall antigen fraction substantially enhanced Th17 response and was comparable to that induced by M. tuberculosis bacteria (Figures 3A–C). Although cell membrane fraction also enhanced Th17 response, it was lower than that observed with M. tuberculosis bacteria and cell wall fraction. Surprisingly, cytoplasmic fraction did not significantly enhance either frequencies of IL-17A+CD4+ T cells or the production of IL-17A (Figures 3A–C).
Figure 3.

Human dendritic cells differentially promote Th17 response to M. tuberculosis and its antigen fractions. Human monocyte-derived DCs were cocultured with autologous CD4+ T cells at a ratio of 1:10 in X-vivo medium alone or with γ-irradiated M. tuberculosis (Mtb) or M. tuberculosis-derived cell wall (CW), cell membrane (CM), or cytoplasmic (Cyt) fractions for 5 days. Th17 cells were analyzed by flow cytometry by combination of surface staining for CD4 and intracellular staining for IL-17A. IL-17A in the cell-free supernatants was quantified by ELISA. (A,B) Representative dot plots showing the frequencies of CD4+IL-17A+ T cells and (B) mean ± SEM data from seven independent donors. (C) The amount of secretion of IL-17A (mean ± SEM, n = 7). (D,E) Frequencies of IL-10+CD4+ T cells or FoxP3+CD4+ Treg cells in DC–T cell cocultures stimulated with various antigens of M. tuberculosis (mean ± SEM, n = 5). *P < 0.05; **P < 0.01; ***P < 0.001; ns, not significant; as determined by one-way ANOVA.
We confirm that low-level induction of Th17 response by cytoplasmic fraction was not because of lack of induction of DC maturation. In fact, DCs stimulated with cytoplasmic fraction significantly enhanced DC maturation markers HLA-DR, CD80, CD86, and CD40 and was similar to those observed with M. tuberculosis bacteria, cell wall, and cell membrane fractions (Figure S2 in Supplementary Material).
Further, the inability of cytoplasmic fraction to promote DC-mediated Th17 response was not due to increased frequency of suppressor T cells, such as IL-10+CD4+ T cells or FoxP3+CD4+ Treg cells. In fact, the frequency of IL-10+CD4+ T cells remain unaltered upon stimulation with M. tuberculosis or its antigen fractions (Figure 3D). Consistent with the previous findings, M. tuberculosis and its antigen fractions including cytoplasmic fraction significantly enhanced the frequency of FoxP3+CD4+ Tregs (Figure 3E). However, the extent of stimulation of Treg response was similar among all three antigen fractions of M. tuberculosis.
Differential Expression of PD-L1 on Monocytes and DCs upon Stimulation with M. tuberculosis and Its Antigen Fractions
M. tuberculosis employs several strategies to modulate effector CD4 T cell response (43). Several recent reports have indicated that PD-1–PD-L1/PD-L2 axis has a pivotal role in the regulation of T cell response to M. tuberculosis (29, 31, 33). Therefore, to decipher the role of PD-L1/PD-L2 in regulating Th17 response to M. tuberculosis, we first investigated the ability of M. tuberculosis and its different antigen fractions in modulating PD-L1 and PD-L2 expression on monocytes and DCs. Our results reveal that M. tuberculosis bacteria, cell wall fraction, and cell membrane fractions significantly induce PD-L1 expression on both monocytes and DCs (Figures 4A,B). However, significant induction of PD-L1 by cytoplasmic fraction was observed only in monocytes but not in DCs (Figures 4A,B). Unstimulated monocytes and DCs did not express PD-L2 and was not altered upon stimulation with either M. tuberculosis or its different antigen fractions (Figures 4C,D).
Figure 4.

Differential expression of PD-L1 and PD-L2 on monocytes and dendritic cells upon stimulation with M. tuberculosis and its antigen fractions. Monocytes or monocyte-derived DCs were either cultured alone or stimulated with γ-irradiated M. tuberculosis or M. tuberculosis-derived cell wall (CW), cell membrane (CM), or cytoplasmic (Cyt) fractions for 24 h. Surface expressions of PD-L1 (A,B) and PD-L2 (C,D) were analyzed by flow cytometry. (A,C) Percentage of monocytes expressing PD-L1 and PD-L2 (mean ± SEM, n = 5). (B,D) Percentage of DCs expressing PD-L1 and PD-L2 (mean ± SEM, n = 5). **P < 0.01; ***P < 0.001; ns, not significant; as determined by one-way ANOVA.
PD-L1 on Human Innate Cells Regulates Th1, But Not Th17, Response to M. tuberculosis
PD-1–PD-L1 interaction has been reported to inhibit Th1 response to M. tuberculosis. To explore if PD-L1 also checks Th17 response to M. tuberculosis, we employed blocking antibodies to PD-L1. We found that blocking PD-L1 either in the monocyte–CD4+ T cell coculture or in the DC–CD4+ T cell coculture did not significantly modify either the frequencies of IL-17A+CD4+ T cells or the secretion of IL-17A (Figures 5A–F). These data are in line with the fact that lack of induction of PD-L1 on DCs by cytoplasmic fraction of M. tuberculosis was not associated with enhanced Th17 responses (Figures 3 and 4B). Whereas, same cytoplasmic fraction promoted Th17 response despite it induced significant expression of PD-L1 on monocytes (Figures 2 and 4A). Taken together, these results imply that PD-L1 does not function as negative regulator of Th17 response to M. tuberculosis.
Figure 5.

PD-L1 on human innate cells is dispensable for regulating Th17 response to M. tuberculosis. Monocytes or DCs were cocultured with autologous CD4+ T cells either alone or with γ-irradiated M. tuberculosis. After 18 h, PD-L1 blocking mAb or isotype control mAb were added, and cultures were maintained for 5 days. Th17 cells were analyzed by flow cytometry by combination of surface staining for CD4 and intracellular staining for IL-17A. IL-17A in the cell-free supernatants was quantified by ELISA. (A–C) Representative dot plots showing the frequencies of CD4+IL-17A+ T cells (A), frequencies of CD4+IL-17A+ T cells (mean ± SEM, n = 5) (B), and amounts of IL-17A production (mean ± SEM, n = 6) (C) in monocyte–CD4+ T cell cocultures. (D–F) Representative dot plots showing the frequencies of CD4+IL-17A+ T cells (D), frequencies of CD4+IL-17A+ T cells (mean ± SEM, n = 6) (E), and amounts of IL-17A production (mean ± SEM, n = 6) (F) in DC–CD4+ T cell cocultures. *P < 0.05; **P < 0.01; ***P < 0.001; ns, not significant; as determined by one-way ANOVA.
It was important to demonstrate that observed phenomenon is not due to inefficient PD-L1 blocking. Previous results have demonstrated that PD-L1 negatively regulates IFN-γ responses. Therefore, to demonstrate the efficiency of PD-L1 blocking, we have assessed the secretion of IFN-γ in the monocyte–CD4+ T cell and DC–CD4+ T cell cocultures. Consistent with the previous data, blockade of PD-L1 significantly augmented the production of IFN-γ from CD4+ T cells (Figures 6A,B).
Figure 6.
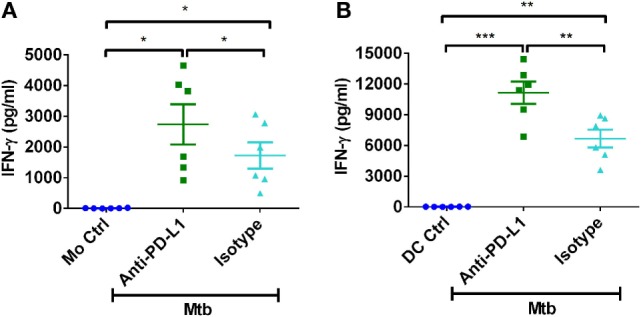
PD-L1 on human innate cells regulates Th1 response to M. tuberculosis. Monocytes or DCs were cocultured with autologous CD4+ T cells either alone or with γ-irradiated M. tuberculosis. After 18 h, PD-L1 blocking mAb or isotype control mAb were added, and cultures were maintained for 5 days. Amounts of secretion of IFN-γ in monocyte–CD4+ T cell cocultures (A) and in DC–CD4+ T cell cocultures (mean ± SEM, n = 6) (B). *P < 0.05; **P < 0.01; ***P < 0.001; ns, not significant; as determined by one-way ANOVA.
PD-1 Is Dispensable for the Regulation of Th17 Response to M. tuberculosis
Programed death ligand-1 signals via PD-1 on CD4+ T cells, and recently it was reported that Mycobacterium-induced PD-1 coordinates suppression of Th17 response in tuberculosis patients (28). We confirm that M. tuberculosis significantly enhances the frequency of PD-1+ T cells in the DC/monocyte–CD4+ T cell cocultures (Figures 7A,D). However, blockade of PD-1 did not significantly alter either the frequencies of IL-17A+CD4+ T cells or the secretion of IL-17A (Figures 7B,C,E,F). Similar to PD-L1 experiments, blockade of PD-1 also lead to increased IFN-γ responses. Together, these data indicate that human Th17 response to M. tuberculosis is not under the control of PD pathway.
Figure 7.

PD-1 is dispensable for the regulation of Th17 response to M. tuberculosis. Monocytes or DCs were cocultured with autologous CD4+ T cells either alone or with γ-irradiated M. tuberculosis. After 18 h, PD-1 blocking mAb or isotype control mAb were added, and cultures were maintained for 5 days. (A–C) Frequency of PD-1+CD4+ T cells (mean ± SEM, n = 4) (A), frequency of CD4+IL-17A+ T cells (mean ± SEM, n = 6) (B), and amounts of IL-17A production (mean ± SEM, n = 6) (C) in the monocyte–CD4+ T cell cocultures. (D–F) Frequency of PD-1+CD4+ T cells (mean ± SEM, n = 4) (D), frequency of CD4+IL-17A+ T cells (mean ± SEM, n = 6) (E), and amounts of IL-17A production (mean ± SEM, n = 6) (F) in the DC–CD4+ T cell cocultures. *P < 0.05; **P < 0.01; ns, not significant; as determined by one-way ANOVA.
IL-1β Is Critical for Mediating Th17 Response to M. tuberculosis
Upon encountering with pathogens, APCs secrete various cytokines that dictate the outcome of T cell response. Particularly, IL-6, IL-1β, and IL-23 derived from innate immune cells play a crucial role in establishing and sustaining Th17 response. In addition, IL-21 and TGF-β also have important role in Th17 priming (8, 14). Thus, to decipher the mechanism of differential Th17 response to M. tuberculosis and its fractions by monocytes and DCs, we analyzed the Th17-polarizing cytokines secreted by these innate cells. IL-21 was not produced by monocytes and DCs upon stimulation with M. tuberculosis and its antigen fractions. Similarly, basal level of TGF-β was not significantly altered by M. tuberculosis. However, upon stimulation with M. tuberculosis and its antigen fractions, both innate cells secreted large quantities of IL-6 (Figures 8A,D) and moderate amounts of IL-1β (Figures 8B,E). Of note, monocytes produced significantly lower amounts (~10-fold) of IL-23 as compared to DCs (Figures 8C,F). Interestingly, consistent with the lack of role in the modulation of Th17 response, none of these innate cytokines were significantly altered upon blockade of PD-L1 (Figure S3 in Supplementary Material).
Figure 8.

Production of Th17 priming cytokines by human innate cells upon stimulation with M. tuberculosis or its antigen fractions. Monocytes or DCs were cocultured with autologous CD4+ T cells either alone or stimulated with γ-irradiated M. tuberculosis or M. tuberculosis-derived cell wall (CW), cell membrane (CM), or cytoplasmic (Cyt) fractions for 5 days. Cell-free supernatants were analyzed for the secretion of innate cytokines by ELISA. (A–C) IL-6, IL-1β, and IL-23 production by monocytes (mean ± SEM, n = 5) and (D–F) IL-6, IL-1β, and IL-23 production by DCs (mean ± SEM, n = 6). *P < 0.05; **P < 0.01; ***P < 0.001; ns, not significant; as determined by one-way ANOVA.
Despite induction of large quantities of IL-6 in DCs similar to those induced by M. tuberculosis or its other antigen fractions, lack of Th17 response by cytoplasmic fraction provide a pointer toward dispensability of this cytokine in M. tuberculosis-mediated Th17 response. The similar amount of IL-1β production by stimulated monocytes explains elicitation of Th17 response by all the antigen fractions of M. tuberculosis. On the contrary, cytoplasmic fraction was inferior to cell wall and cell membrane antigen fractions in promoting IL-1β and IL-23 by DCs, and this could explain the lack of Th17 responses observed in DCs upon stimulation with cytoplasmic fraction. Therefore, to establish a role for IL-1β and IL-23 in M. tuberculosis-mediated Th17 responses, we supplemented IL-1β or IL-23 to the DC–CD4+ T cell cocultures that are stimulated with cytoplasmic fraction of M. tuberculosis. We found that supplementation of IL-1β enabled cytoplasmic fraction-stimulated DCs to mount strong Th17 response (Figures 9A,B), while IL-23 had no effect (Figures 9C,D). Our results thus reveal that innate Th17-polarizing cytokine IL-1β but not members of the PD pathway. dictates the outcome of human Th17 response to M. tuberculosis and its antigen fractions.
Figure 9.
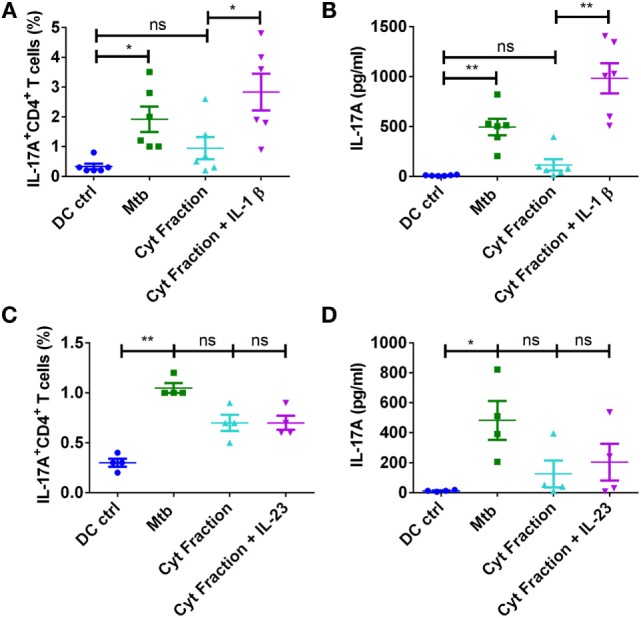
IL-1β is critical for mediating Th17 response to M. tuberculosis. DCs were cocultured with autologous CD4+ T cells either alone or stimulated with γ-irradiated M. tuberculosis, M. tuberculosis-derived cytoplasmic (Cyt) fraction, or Cyt fraction in combination with exogenous IL-1β (A,B) or IL-23 (C,D) for 5 days. Frequencies of CD4+IL-17A+ T cells (mean ± SEM, n = 4–6) (A,C) and amounts of IL-17A production (mean ± SEM, n = 4–6) (B,D) in DC–CD4+ T cell cocultures. *P < 0.05; **P < 0.01; ns, not significant; as determined by one-way ANOVA.
Discussion
M. tuberculosis-loaded aerosols that enter the lungs interact with resident phagocytes, which include alveolar macrophages and DCs. Different subpopulations of DCs are known to coexist in the human lungs (44). Circulating monocytes form a major reservoir for tissue macrophages and different subsets of DCs (45–47). Moreover, monocytes are known to give rise to DCs in vivo at mucosal surfaces, such as skin (48) and lungs (49, 50), thus suggesting an in vivo relevance of monocyte-derived DCs in mediating immune response to mucosal pathogens, such as M. tuberculosis. In comparison to other infections, there is a delay in the onset of adaptive immune response upon M. tuberculosis infection allowing the bacteria to form a niche in the lungs (51). While it is clear that Th1/IFN-γ response is indispensable for the protection against tuberculosis, the role of Th17/IL-17 in mediating protection is unclear.
Initial studies in mice suggested that the IL-23/Th17 axis is dispensable for the overall protection against M. tuberculosis challenge but is required for enhancing the formation of granuloma in the absence of an active IL-12/Th1 axis (52). Surprisingly, Th17 response mediated protection to a highly virulent M. tuberculosis strain HN878 by activating macrophages and curtailing bacterial burden in the lungs. However, it was dispensable to the less virulent strains of M. tuberculosis thus ascribing a protective role for Th17 to emerging virulent strains (53). Earlier reports in tuberculosis patients indicated that virulent strains, such as multi-drug resistant M. tuberculosis, strongly induce Th17 response. However, enhanced IL-17 was correlated to the severity of the disease and high bacterial burden in the lungs, suggesting a detrimental role for Th17 response in humans (54). More recent reports have demonstrated that tuberculosis patients display lower antigen-specific Th17 response. Incidentally, anti-tuberculosis therapy not only enhanced Th1 response but also augmented Th17 response (28). Furthermore, individuals with biallelic RORC mutations with a defective Th17 response are associated with a compromised Th1 response and are susceptible to fungal and mycobacterial infections (55). Thus, it appears that the protective roles of Th17 response may vary depending on the stage of infection. Th17 response contributes to vaccine-mediated protection and during the earlier stages of infection by recruiting lymphocytes and promoting Th1 response. However, chronic exposure or during the later stages of infection it may be detrimental due to neutrophil recruitment that can mediate tissue damage leading to immunopathology.
Previous studies have shown that DCs can mediate Th17 response to M. tuberculosis by signaling through TLR-2, dectin-1, DC-SIGN, and mannose receptors (36, 53, 56). In the present study, we show that monocytes and DCs have differential capacity to modulate Th17 responses to M. tuberculosis and M. tuberculosis-derived antigen fractions. Notably, DCs evoked a much stronger IL-17A production from CD4+ T cells than monocytes. This might be due to the capacity of DCs to secrete large quantities of IL-23 than monocytes. IL-23 plays an important role in stabilizing and sustaining the ensuing Th17 response. Interestingly, unlike monocytes that promoted Th17 response to cell wall, cell membrane, and cytoplasmic fractions of M. tuberculosis, DCs displayed differential response to these antigen fractions. Thus, cell wall fraction triggered strong DC-mediated IL-17A production from CD4+ T cells; cell membrane fraction promoted intermediate IL-17A response, and the cytoplasmic fraction did not significantly modulate Th17 response. As immuno suppressor T cells have the ability to inhibit effector T cell responses, we surmised whether increased frequency of IL-10+CD4+ T cells or FoxP3+CD4+ Treg cells was responsible for low-level induction of DC-mediated Th17 responses by cytoplasmic fraction of M. tuberculosis (57–60). However, IL-10+CD4+ T cells were not induced under any stimulatory conditions. Although cytoplasmic fraction significantly enhanced the frequency of FoxP3+CD4+ Treg cells, this increase was similar to that observed with M. tuberculosis and other antigen fractions.
Suppression of cellular immunity reckons a major evasion strategy employed by M. tuberculosis. Of the numerous evasion strategies employed by M. tuberculosis, the exploitation of PD-L1/PD-L2–PD-1 axis occupies a central role due to its implication in the expansion of Treg response and suppression of effector Th1 response (61, 62). Considering that induced Tregs and Th17 responses are reciprocally regulated, the possible role of PD-L1/PD-L2 in regulating Th17 response has not been questioned. It is well established that M. tuberculosis infection induces PD-L1 on APCs and PD-1 on T cells. Interaction of PD-L1 with PD-1 promotes Treg differentiation and expansion by activating SHP1/2. Induced SHP1/2 suppresses STAT1 functions, abrogates IFN-γ production, and abolishes its inhibitory effect on FOXP3 leading to Treg expansion (63). Since PD-1 axis plays a critical role in mediating immune tolerance and loss of which can predispose to inflammatory conditions and autoimmune diseases (64, 65), it is deleterious to completely abrogate PD-L1–PD-1 signaling during M. tuberculosis infection. This is evidenced by the fact that PD-L1-deficient and PD-1-deficient mice are susceptible to M. tuberculosis infection and display exacerbated inflammation (66, 67).
In the present study, we found that monocytes stimulated with M. tuberculosis and its antigen fractions significantly induce PD-L1 expression on human monocytes. On the contrary, only cell wall and cell membrane fractions induced PD-L1 expression on DCs, whereas the cytoplasmic fraction failed to enhance PD-L1 on DCs. PD-L2 was not expressed on both monocytes and DCs under any stimulatory conditions. The inability of cytoplasmic fraction to induce PD-L1 on DCs was not due to its lack of stimulatory capacity as cytoplasmic fraction inducted DC maturation similar to that of cell wall and cell membrane fractions.
To decipher the role PD-L1 in regulating Th17 response to M. tuberculosis, we employed blocking antibodies to PD-L1. Our results demonstrate that PD-L1 blockade did not significantly alter the frequencies of Th17 cells. On the other hand, IFN-γ production was significantly enhanced when PD-L1 was blocked, thus confirming the previous data on role of PD axis in regulating Th1 responses (31, 32). However, we did observe a marginal (~50 pg/ml) but insignificant increase in IL-17 production upon PD-L1 blockade. As Th17 cells and Tregs are reciprocally regulated and moreover Tregs are known to suppress Th17 response (66, 68, 69), it is likely that blockade of PD-L1 can indirectly favor Th17 response to a certain extent. This could explain minimal augmentation of IL-17A production that we observed upon PD-L1 blockade.
In addition to PD-L1, M. tuberculosis significantly enhanced PD-1 on CD4+ T cells. However, PD-1 blockade had no repercussion on either monocyte- or DC-mediated Th17 response. Our data contradict a recent report that indicated that Mycobacterium-induced PD-1 orchestrates suppression of Th17 response in tuberculosis patients (28). This discrepancy might be due to the fact that previous report focused on active disease where prolonged infection can enhance additional co-stimulatory molecules that might not be present during the earlier phases of stimulation. For example, APCs from active disease express both the co-stimulatory molecules PD-L1 and PD-L2 (31), whereas our current data and previous reports show that DCs from healthy individuals stimulated with M. tuberculosis both at the transcript level as well as the protein level induce only PD-L1 and not PD-L2 (30, 33). It is essential to note that we have used γ-irradiated M. tuberculosis or M. tuberculosis-derived cell wall, cell membrane, or cytoplasmic fractions in our experiments, and these conditions may not completely mimic in vivo situation of infection with live bacteria. Therefore, further work is necessary to decipher the role of PD-1–PD-L1/PD-L2 pathway in tuberculosis. Nevertheless, our report indicates that PD pathway does not contribute to negative regulation of human Th17 response to M. tuberculosis. Also, cytoplasmic fraction of M. tuberculosis failed to induce Th17 response despite lack of induction of PD-L1 on DCs.
To decipher the possible mechanism that explains inability of cytoplasmic fraction to promote DC-mediated Th17 response, we analyzed key Th17-polarizing cytokines secreted by monocytes and DCs. Our results indicate that cytoplasmic fraction failed to induce IL-1β, the key cytokine that promotes Th17 response. The amounts of secretion of IL-1β and Th17 responses were significantly correlated. Importantly, exogenous supplementation of IL-1β was sufficient to significantly augment Th17 response by the cytoplasmic fraction. These results indicate that inability of cytoplasmic fraction to induce the production of IL-1β from DCs resulted in its failure to prime a robust Th17 response. Further work on mechanisms underlying the differential production of IL-1β from monocytes and DCs upon stimulation with cytoplasmic fraction is warranted (70). Distinct expression of pattern-recognition receptors that sense cytoplasmic fraction on monocytes and DCs might have resulted in the differential IL-1β secretion upon stimulation with the cytoplasmic fraction. Therefore, antigens that come in direct contact with innate immune cells, such as the cell wall antigens, are attractive candidates for future tuberculosis vaccines. It is important to note that IL-1β is a critical mediator of immunity to M. tuberculosis (71, 72). Our results suggest that this function of IL-1β is in part via enhancement of Th17 responses. As tuberculosis patients were reported to exhibit lower antigen-specific Th17 response (28), supplementing IL-1β might enhance Th17 response.
Taken together, our report demonstrates that DCs have differential capacity to mediate Th17 response to various antigen fractions of M. tuberculosis. Therefore, cell wall antigens of M. tuberculosis constitute potential subunit vaccine candidates. Importantly, we demonstrate that PD pathway is critical for regulating Th1, but not Th17, response to M. tuberculosis. IL-1β however is necessary for promoting a prominent Th17 response to M. tuberculosis.
Author Contributions
ES-V and JB designed the research. ES-V, VS, MD, AK, CS, ML, and CG performed the research. ES-V, VS, MD, AK, CS, ML, CG, SK, and JB contributed to data analyses and data interpretation. ES-V and JB wrote the manuscript. ES-V, VS, MD, AK, CS, ML, CG, SK, and JB revised the manuscript critically for important intellectual content and approved the final version.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank NIH Biodefense and Emerging Infections Research Resources Repository, NIAID, NIH, for kindly supplying the cell wall, cell membrane, cytoplasmic fractions, and gamma-irradiated whole cells of Mycobacterium tuberculosis.
Funding
Supported by Institut National de la Santé et de la Recherche Médicale (INSERM), Université Pierre et Marie Curie, and Université Paris Descartes; grant from the Indo-French Center for Promotion of Advanced Research (Reference No: 4803-1); and fellowships from Indo-French Center for Promotion of Advanced Research (ES-V and AK).
Supplementary Material
The Supplementary Material for this article can be found online at http://journal.frontiersin.org/article/10.3389/fimmu.2016.00465.
M. tuberculosis and its antigen fractions enhance costimulatory molecules on monocytes. Monocytes (0.5 × 106/ml) were either cultured alone or stimulated with γ-irradiated M. tuberculosis (Mtb) or M. tuberculosis-derived cell wall (CW), cell membrane (CM), or cytoplasmic (Cyt) fractions for 24 h. (A–D) Surface expression of CD80 [mean fluorescence intensity (MFI), CD86 (% positive cells), CD40 (MFI), and HLA-DR (MFI)] (mean ± SEM, n = 5) was analyzed by flow cytometry. **P < 0.01; ns, not significant; as determined by one-way ANOVA.
M. tuberculosis-derived cytoplasmic fraction induce maturation of DCs. DCs (0.5 × 106/ml) were either cultured alone or stimulated with γ-irradiated M. tuberculosis (Mtb) or M. tuberculosis-derived cell wall (CW), cell membrane (CM), or cytoplasmic (Cyt) fractions for 24 h. (A–D) Surface expression of CD80 [mean fluorescence intensity (MFI), CD86 (% positive cells), CD40 (MFI), and HLA-DR (MFI)] (mean ± SEM, n = 4) was analyzed by flow cytometry. *P < 0.05; **P < 0.01; ***P < 0.001; as determined by one-way ANOVA.
Production of Th17-polarizing cytokines by human innate cells upon PD-L1 blockade. Monocytes or DCs were cocultured with autologous CD4+ T cells either alone or stimulated with γ-irradiated M. tuberculosis (Mtb). After 18 h, PD-L1 blocking mAb or isotype control mAb were added, and cultures were maintained for 5 days. Cell-free supernatants were analyzed for the secretion of innate cytokines by ELISA. (A–C) IL-6, IL-1β, and IL-23 production by monocytes and (D–F) IL-6, IL-1β, and IL-23 production by DCs (mean ± SEM, n = 7). *P < 0.05; **P < 0.01; ***P < 0.001; ns, not significant; as determined by one-way ANOVA.
References
- 1.Flynn JL, Chan J. Immunology of tuberculosis. Annu Rev Immunol (2001) 19:93–129. 10.1146/annurev.immunol.19.1.93 [DOI] [PubMed] [Google Scholar]
- 2.Cooper AM. Cell-mediated immune responses in tuberculosis. Annu Rev Immunol (2009) 27:393–422. 10.1146/annurev.immunol.021908.132703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bustamante J, Boisson-Dupuis S, Abel L, Casanova JL. Mendelian susceptibility to mycobacterial disease: genetic, immunological, and clinical features of inborn errors of IFN-gamma immunity. Semin Immunol (2014) 26:454–70. 10.1016/j.smim.2014.09.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Prezzemolo T, Guggino G, La Manna MP, Di Liberto D, Dieli F, Caccamo N. Functional signatures of human CD4 and CD8 T cell responses to Mycobacterium tuberculosis. Front Immunol (2014) 5:180. 10.3389/fimmu.2014.00180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Skeiky YA, Sadoff JC. Advances in tuberculosis vaccine strategies. Nat Rev Microbiol (2006) 4:469–76. 10.1038/nrmicro1419 [DOI] [PubMed] [Google Scholar]
- 6.Ottenhoff TH, Kaufmann SH. Vaccines against tuberculosis: where are we and where do we need to go? PLoS Pathog (2012) 8:e1002607. 10.1371/journal.ppat.1002607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tameris MD, Hatherill M, Landry BS, Scriba TJ, Snowden MA, Lockhart S, et al. Safety and efficacy of MVA85A, a new tuberculosis vaccine, in infants previously vaccinated with BCG: a randomised, placebo-controlled phase 2b trial. Lancet (2013) 381:1021–8. 10.1016/S0140-6736(13)60177-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 cells. Annu Rev Immunol (2009) 27:485–517. 10.1146/annurev.immunol.021908.132710 [DOI] [PubMed] [Google Scholar]
- 9.Annunziato F, Cosmi L, Liotta F, Maggi E, Romagnani S. Defining the human T helper 17 cell phenotype. Trends Immunol (2012) 33:505–12. 10.1016/j.it.2012.05.004 [DOI] [PubMed] [Google Scholar]
- 10.Meller S, Di Domizio J, Voo KS, Friedrich HC, Chamilos G, Ganguly D, et al. T(H)17 cells promote microbial killing and innate immune sensing of DNA via interleukin 26. Nat Immunol (2015) 16:970–9. 10.1038/ni.3211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stephen-Victor E, Fickenscher H, Bayry J. IL-26: an emerging proinflammatory member of the IL-10 cytokine family with multifaceted actions in antiviral, antimicrobial, and autoimmune Responses. PLoS Pathog (2016) 12:e1005624. 10.1371/journal.ppat.1005624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kotake S, Udagawa N, Takahashi N, Matsuzaki K, Itoh K, Ishiyama S, et al. IL-17 in synovial fluids from patients with rheumatoid arthritis is a potent stimulator of osteoclastogenesis. J Clin Invest (1999) 103:1345–52. 10.1172/JCI5703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fujimoto M, Serada S, Mihara M, Uchiyama Y, Yoshida H, Koike N, et al. Interleukin-6 blockade suppresses autoimmune arthritis in mice by the inhibition of inflammatory Th17 responses. Arthritis Rheum (2008) 58:3710–9. 10.1002/art.24126 [DOI] [PubMed] [Google Scholar]
- 14.Maddur MS, Miossec P, Kaveri SV, Bayry J. Th17 cells: biology, pathogenesis of autoimmune and inflammatory diseases, and therapeutic strategies. Am J Pathol (2012) 181:8–18. 10.1016/j.ajpath.2012.03.044 [DOI] [PubMed] [Google Scholar]
- 15.Khader SA, Gaffen SL, Kolls JK. Th17 cells at the crossroads of innate and adaptive immunity against infectious diseases at the mucosa. Mucosal Immunol (2009) 2:403–11. 10.1038/mi.2009.100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bai H, Cheng J, Gao X, Joyee AG, Fan Y, Wang S, et al. IL-17/Th17 promotes type 1 T cell immunity against pulmonary intracellular bacterial infection through modulating dendritic cell function. J Immunol (2009) 183:5886–95. 10.4049/jimmunol.0901584 [DOI] [PubMed] [Google Scholar]
- 17.Lin Y, Ritchea S, Logar A, Slight S, Messmer M, Rangel-Moreno J, et al. Interleukin-17 is required for T helper 1 cell immunity and host resistance to the intracellular pathogen Francisella tularensis. Immunity (2009) 31:799–810. 10.1016/j.immuni.2009.08.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Khader SA, Gopal R. IL-17 in protective immunity to intracellular pathogens. Virulence (2010) 1:423–7. 10.4161/viru.1.5.12862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Khader SA, Bell GK, Pearl JE, Fountain JJ, Rangel-Moreno J, Cilley GE, et al. IL-23 and IL-17 in the establishment of protective pulmonary CD4+ T cell responses after vaccination and during Mycobacterium tuberculosis challenge. Nat Immunol (2007) 8:369–77. 10.1038/ni1449 [DOI] [PubMed] [Google Scholar]
- 20.Khader SA, Cooper AM. IL-23 and IL-17 in tuberculosis. Cytokine (2008) 41:79–83. 10.1016/j.cyto.2007.11.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Scriba TJ, Kalsdorf B, Abrahams DA, Isaacs F, Hofmeister J, Black G, et al. Distinct, specific IL-17- and IL-22-producing CD4+ T cell subsets contribute to the human anti-mycobacterial immune response. J Immunol (2008) 180:1962–70. 10.4049/jimmunol.180.3.1962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cooper AM. Editorial: be careful what you ask for: is the presence of IL-17 indicative of immunity? J Leukoc Biol (2010) 88:221–3. 10.1189/jlb.0310146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cruz A, Fraga AG, Fountain JJ, Rangel-Moreno J, Torrado E, Saraiva M, et al. Pathological role of interleukin 17 in mice subjected to repeated BCG vaccination after infection with Mycobacterium tuberculosis. J Exp Med (2010) 207:1609–16. 10.1084/jem.20100265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gallegos AM, van Heijst JW, Samstein M, Su X, Pamer EG, Glickman MS. A gamma interferon independent mechanism of CD4 T cell mediated control of M. tuberculosis infection in vivo. PLoS Pathog (2011) 7:e1002052. 10.1371/journal.ppat.1002052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Satchidanandam V, Kumar N, Jumani RS, Challu V, Elangovan S, Khan NA. The glycosylated Rv1860 protein of Mycobacterium tuberculosis inhibits dendritic cell mediated TH1 and TH17 polarization of T cells and abrogates protective immunity conferred by BCG. PLoS Pathog (2014) 10:e1004176. 10.1371/journal.ppat.1004176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen X, Zhang M, Liao M, Graner MW, Wu C, Yang Q, et al. Reduced Th17 response in patients with tuberculosis correlates with IL-6R expression on CD4+ T cells. Am J Respir Crit Care Med (2010) 181:734–42. 10.1164/rccm.200909-1463OC [DOI] [PubMed] [Google Scholar]
- 27.Perreau M, Rozot V, Welles HC, Belluti-Enders F, Vigano S, Maillard M, et al. Lack of Mycobacterium tuberculosis-specific interleukin-17A-producing CD4+ T cells in active disease. Eur J Immunol (2013) 43:939–48. 10.1002/eji.201243090 [DOI] [PubMed] [Google Scholar]
- 28.Bandaru A, Devalraju KP, Paidipally P, Dhiman R, Venkatasubramanian S, Barnes PF, et al. Phosphorylated STAT3 and PD-1 regulate IL-17 production and IL-23 receptor expression in Mycobacterium tuberculosis infection. Eur J Immunol (2014) 44:2013–24. 10.1002/eji.201343680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Periasamy S, Dhiman R, Barnes PF, Paidipally P, Tvinnereim A, Bandaru A, et al. Programmed death 1 and cytokine inducible SH2-containing protein dependent expansion of regulatory T cells upon stimulation with Mycobacterium tuberculosis. J Infect Dis (2011) 203:1256–63. 10.1093/infdis/jir011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Trinath J, Maddur MS, Kaveri SV, Balaji KN, Bayry J. Mycobacterium tuberculosis promotes regulatory T-cell expansion via induction of programmed death-1 ligand 1 (PD-L1, CD274) on dendritic cells. J Infect Dis (2012) 205:694–6. 10.1093/infdis/jir820 [DOI] [PubMed] [Google Scholar]
- 31.Singh A, Mohan A, Dey AB, Mitra DK. Inhibiting the programmed death 1 pathway rescues Mycobacterium tuberculosis-specific interferon gamma-producing T cells from apoptosis in patients with pulmonary tuberculosis. J Infect Dis (2013) 208:603–15. 10.1093/infdis/jit206 [DOI] [PubMed] [Google Scholar]
- 32.Stephen-Victor E, Saha C, Sharma M, Holla S, Balaji KN, Kaveri SV, et al. Inhibition of programmed death 1 ligand 1 on dendritic cells enhances Mycobacterium-mediated interferon gamma (IFN-gamma) production without modulating the frequencies of IFN-gamma-producing CD4+ T cells. J Infect Dis (2015) 211:1027–9. 10.1093/infdis/jiu532 [DOI] [PubMed] [Google Scholar]
- 33.Holla S, Stephen-Victor E, Prakhar P, Sharma M, Saha C, Udupa V, et al. Mycobacteria-responsive sonic hedgehog signaling mediates programmed death-ligand 1- and prostaglandin E2-induced regulatory T cell expansion. Sci Rep (2016) 6:24193. 10.1038/srep24193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Balboa L, Kviatcovsky D, Schierloh P, Garcia M, de la Barrera S, Sasiain MD. Monocyte-derived dendritic cells early exposed to Mycobacterium tuberculosis induce an enhanced T helper 17 response and transfer mycobacterial antigens. Int J Med Microbiol (2016). 10.1016/j.ijmm.2016.06.004 [DOI] [PubMed] [Google Scholar]
- 35.Choi HG, Kim WS, Back YW, Kim H, Kwon KW, Kim JS, et al. Mycobacterium tuberculosis RpfE promotes simultaneous Th1- and Th17-type T-cell immunity via TLR4-dependent maturation of dendritic cells. Eur J Immunol (2015) 45:1957–71. 10.1002/eji.201445329 [DOI] [PubMed] [Google Scholar]
- 36.Sondergaard JN, Laursen JM, Rosholm LB, Brix S. Mycobacterium tuberculosis promotes Th17 expansion via regulation of human dendritic cells toward a high CD14 and low IL-12p70 phenotype that reprograms upon exogenous IFN-gamma. Int Immunol (2014) 26:705–16. 10.1093/intimm/dxu085 [DOI] [PubMed] [Google Scholar]
- 37.Chatterjee S, Dwivedi VP, Singh Y, Siddiqui I, Sharma P, Van Kaer L, et al. Early secreted antigen ESAT-6 of Mycobacterium tuberculosis promotes protective T helper 17 cell responses in a toll-like receptor-2-dependent manner. PLoS Pathog (2011) 7:e1002378. 10.1371/journal.ppat.1002378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maddur MS, Sharma M, Hegde P, Stephen-Victor E, Pulendran B, Kaveri SV, et al. Human B cells induce dendritic cell maturation and favour Th2 polarization by inducing OX-40 ligand. Nat Commun (2014) 5:4092. 10.1038/ncomms5092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lastrucci C, Benard A, Balboa L, Pingris K, Souriant S, Poincloux R, et al. Expansion of a motile, permissive and immunomodulatory CD16+ monocyte population via the IL-10/STAT3 axis. Cell Res (2015) 25:1333–51. 10.1038/cr.2015.123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stamp LK, Easson A, Pettersson L, Highton J, Hessian PA. Monocyte derived interleukin (IL)-23 is an important determinant of synovial IL-17A expression in rheumatoid arthritis. J Rheumatol (2009) 36:2403–8. 10.3899/jrheum.081304 [DOI] [PubMed] [Google Scholar]
- 41.Corvaisier M, Delneste Y, Jeanvoine H, Preisser L, Blanchard S, Garo E, et al. IL-26 is overexpressed in rheumatoid arthritis and induces proinflammatory cytokine production and Th17 cell generation. PLoS Biol (2012) 10:e1001395. 10.1371/journal.pbio.1001395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Olliver M, Hiew J, Mellroth P, Henriques-Normark B, Bergman P. Human monocytes promote Th1 and Th17 responses to Streptococcus pneumoniae. Infect Immun (2011) 79:4210–7. 10.1128/IAI.05286-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Goldberg MF, Saini NK, Porcelli SA. Evasion of innate and adaptive immunity by Mycobacterium tuberculosis. Microbiol Spectr (2014) 2:MGM2-0005-2013. 10.1128/microbiolspec.MGM2-0005-2013 [DOI] [PubMed] [Google Scholar]
- 44.van Haarst JM, de Wit HJ, Drexhage HA, Hoogsteden HC. Distribution and immunophenotype of mononuclear phagocytes and dendritic cells in the human lung. Am J Respir Cell Mol Biol (1994) 10:487–92. 10.1165/ajrcmb.10.5.8179911 [DOI] [PubMed] [Google Scholar]
- 45.Leon B, Lopez-Bravo M, Ardavin C. Monocyte-derived dendritic cells. Semin Immunol (2005) 17:313–8. 10.1016/j.smim.2005.05.013 [DOI] [PubMed] [Google Scholar]
- 46.Serbina NV, Jia T, Hohl TM, Pamer EG. Monocyte-mediated defense against microbial pathogens. Annu Rev Immunol (2008) 26:421–52. 10.1146/annurev.immunol.26.021607.090326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cheong C, Matos I, Choi JH, Dandamudi DB, Shrestha E, Longhi MP, et al. Microbial stimulation fully differentiates monocytes to DC-SIGN/CD209(+) dendritic cells for immune T cell areas. Cell (2010) 143:416–29. 10.1016/j.cell.2010.09.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ginhoux F, Tacke F, Angeli V, Bogunovic M, Loubeau M, Dai XM, et al. Langerhans cells arise from monocytes in vivo. Nat Immunol (2006) 7:265–73. 10.1038/ni1307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jakubzick C, Tacke F, Ginhoux F, Wagers AJ, van Rooijen N, Mack M, et al. Blood monocyte subsets differentially give rise to CD103+ and CD103− pulmonary dendritic cell populations. J Immunol (2008) 180:3019–27. 10.4049/jimmunol.180.5.3019 [DOI] [PubMed] [Google Scholar]
- 50.Osterholzer JJ, Chen GH, Olszewski MA, Curtis JL, Huffnagle GB, Toews GB. Accumulation of CD11b+ lung dendritic cells in response to fungal infection results from the CCR2-mediated recruitment and differentiation of Ly-6C high monocytes. J Immunol (2009) 183:8044–53. 10.4049/jimmunol.0902823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Urdahl KB, Shafiani S, Ernst JD. Initiation and regulation of T-cell responses in tuberculosis. Mucosal Immunol (2011) 4:288–93. 10.1038/mi.2011.10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Khader SA, Pearl JE, Sakamoto K, Gilmartin L, Bell GK, Jelley-Gibbs DM, et al. IL-23 compensates for the absence of IL-12p70 and is essential for the IL-17 response during tuberculosis but is dispensable for protection and antigen-specific IFN-gamma responses if IL-12p70 is available. J Immunol (2005) 175:788–95. 10.4049/jimmunol.175.2.788 [DOI] [PubMed] [Google Scholar]
- 53.Gopal R, Monin L, Slight S, Uche U, Blanchard E, Fallert Junecko BA, et al. Unexpected role for IL-17 in protective immunity against hypervirulent Mycobacterium tuberculosis HN878 infection. PLoS Pathog (2014) 10:e1004099. 10.1371/journal.ppat.1004099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Basile JI, Geffner LJ, Romero MM, Balboa L, Sabio YGC, Ritacco V, et al. Outbreaks of Mycobacterium tuberculosis MDR strains induce high IL-17 T-cell response in patients with MDR tuberculosis that is closely associated with high antigen load. J Infect Dis (2011) 204:1054–64. 10.1093/infdis/jir460 [DOI] [PubMed] [Google Scholar]
- 55.Okada S, Markle JG, Deenick EK, Mele F, Averbuch D, Lagos M, et al. IMMUNODEFICIENCIES. Impairment of immunity to Candida and Mycobacterium in humans with bi-allelic RORC mutations. Science (2015) 349:606–13. 10.1126/science.aaa4282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zenaro E, Donini M, Dusi S. Induction of Th1/Th17 immune response by Mycobacterium tuberculosis: role of dectin-1, Mannose Receptor, and DC-SIGN. J Leukoc Biol (2009) 86:1393–401. 10.1189/jlb.0409242 [DOI] [PubMed] [Google Scholar]
- 57.Gerosa F, Nisii C, Righetti S, Micciolo R, Marchesini M, Cazzadori A, et al. CD4(+) T cell clones producing both interferon-gamma and interleukin-10 predominate in bronchoalveolar lavages of active pulmonary tuberculosis patients. Clin Immunol (1999) 92:224–34. 10.1006/clim.1999.4752 [DOI] [PubMed] [Google Scholar]
- 58.Boer MC, Joosten SA, Ottenhoff TH. Regulatory T-cells at the interface between human host and pathogens in infectious diseases and vaccination. Front Immunol (2015) 6:217. 10.3389/fimmu.2015.00217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shafiani S, Tucker-Heard G, Kariyone A, Takatsu K, Urdahl KB. Pathogen-specific regulatory T cells delay the arrival of effector T cells in the lung during early tuberculosis. J Exp Med (2010) 207:1409–20. 10.1084/jem.20091885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Babu S, Bhat SQ, Kumar NP, Kumaraswami V, Nutman TB. Regulatory T cells modulate Th17 responses in patients with positive tuberculin skin test results. J Infect Dis (2010) 201(1):20–31. 10.1086/648735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Latchman Y, Wood CR, Chernova T, Chaudhary D, Borde M, Chernova I, et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat Immunol (2001) 2:261–8. 10.1038/85330 [DOI] [PubMed] [Google Scholar]
- 62.Sharpe AH, Wherry EJ, Ahmed R, Freeman GJ. The function of programmed cell death 1 and its ligands in regulating autoimmunity and infection. Nat Immunol (2007) 8:239–45. 10.1038/ni1443 [DOI] [PubMed] [Google Scholar]
- 63.Amarnath S, Mangus CW, Wang JC, Wei F, He A, Kapoor V, et al. The PDL1-PD1 axis converts human TH1 cells into regulatory T cells. Sci Transl Med (2011) 3:111ra120. 10.1126/scitranslmed.3003130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nishimura H, Nose M, Hiai H, Minato N, Honjo T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity (1999) 11:141–51. 10.1016/S1074-7613(00)80089-8 [DOI] [PubMed] [Google Scholar]
- 65.Wang J, Yoshida T, Nakaki F, Hiai H, Okazaki T, Honjo T. Establishment of NOD- Pdcd1-/- mice as an efficient animal model of type I diabetes. Proc Natl Acad Sci U S A (2005) 102:11823–8. 10.1073/pnas.0505497102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ogino H, Nakamura K, Ihara E, Akiho H, Takayanagi R. CD4+CD25+ regulatory T cells suppress Th17-responses in an experimental colitis model. Dig Dis Sci (2011) 56:376–86. 10.1007/s10620-010-1286-2 [DOI] [PubMed] [Google Scholar]
- 67.Lazar-Molnar E, Chen B, Sweeney KA, Wang EJ, Liu W, Lin J, et al. Programmed death-1 (PD-1)-deficient mice are extraordinarily sensitive to tuberculosis. Proc Natl Acad Sci U S A (2010) 107:13402–7. 10.1073/pnas.1007394107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.McGovern JL, Nguyen DX, Notley CA, Mauri C, Isenberg DA, Ehrenstein MR. Th17 cells are restrained by Treg cells via the inhibition of interleukin-6 in patients with rheumatoid arthritis responding to anti-tumor necrosis factor antibody therapy. Arthritis Rheum (2012) 64:3129–38. 10.1002/art.34565 [DOI] [PubMed] [Google Scholar]
- 69.Omenetti S, Pizarro TT. The Treg/Th17 Axis: a dynamic balance regulated by the gut microbiome. Front Immunol (2015) 6:639. 10.3389/fimmu.2015.00639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Krishnan N, Robertson BD, Thwaites G. Pathways of IL-1beta secretion by macrophages infected with clinical Mycobacterium tuberculosis strains. Tuberculosis (Edinb) (2013) 93:538–47. 10.1016/j.tube.2013.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Novikov A, Cardone M, Thompson R, Shenderov K, Kirschman KD, Mayer-Barber KD, et al. Mycobacterium tuberculosis triggers host type I IFN signaling to regulate IL-1beta production in human macrophages. J Immunol (2011) 187:2540–7. 10.4049/jimmunol.1100926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bourigault ML, Segueni N, Rose S, Court N, Vacher R, Vasseur V, et al. Relative contribution of IL-1alpha, IL-1beta and TNF to the host response to Mycobacterium tuberculosis and attenuated M. bovis BCG. Immun Inflamm Dis (2013) 1:47–62. 10.1002/iid3.9 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
M. tuberculosis and its antigen fractions enhance costimulatory molecules on monocytes. Monocytes (0.5 × 106/ml) were either cultured alone or stimulated with γ-irradiated M. tuberculosis (Mtb) or M. tuberculosis-derived cell wall (CW), cell membrane (CM), or cytoplasmic (Cyt) fractions for 24 h. (A–D) Surface expression of CD80 [mean fluorescence intensity (MFI), CD86 (% positive cells), CD40 (MFI), and HLA-DR (MFI)] (mean ± SEM, n = 5) was analyzed by flow cytometry. **P < 0.01; ns, not significant; as determined by one-way ANOVA.
M. tuberculosis-derived cytoplasmic fraction induce maturation of DCs. DCs (0.5 × 106/ml) were either cultured alone or stimulated with γ-irradiated M. tuberculosis (Mtb) or M. tuberculosis-derived cell wall (CW), cell membrane (CM), or cytoplasmic (Cyt) fractions for 24 h. (A–D) Surface expression of CD80 [mean fluorescence intensity (MFI), CD86 (% positive cells), CD40 (MFI), and HLA-DR (MFI)] (mean ± SEM, n = 4) was analyzed by flow cytometry. *P < 0.05; **P < 0.01; ***P < 0.001; as determined by one-way ANOVA.
Production of Th17-polarizing cytokines by human innate cells upon PD-L1 blockade. Monocytes or DCs were cocultured with autologous CD4+ T cells either alone or stimulated with γ-irradiated M. tuberculosis (Mtb). After 18 h, PD-L1 blocking mAb or isotype control mAb were added, and cultures were maintained for 5 days. Cell-free supernatants were analyzed for the secretion of innate cytokines by ELISA. (A–C) IL-6, IL-1β, and IL-23 production by monocytes and (D–F) IL-6, IL-1β, and IL-23 production by DCs (mean ± SEM, n = 7). *P < 0.05; **P < 0.01; ***P < 0.001; ns, not significant; as determined by one-way ANOVA.