

Summary
Periodontitis is a chronic inflammatory condition characterized by destruction of non‐mineralized and mineralized connective tissues. It is initiated and maintained by a dysbiosis of the bacterial biofilm adjacent to teeth with increased prevalence of Gram‐negative microorganisms. Nucleotide‐binding oligomerization domain containing 1 (NOD1) is a member of the Nod‐like receptors (NLRs) family of proteins that participate in the activation of the innate immune system, in response to invading bacteria or to bacterial antigens present in the cytoplasm. The specific activating ligand for NOD1 is a bacterial peptidoglycan derived primarily from Gram‐negative bacteria. This study assessed the role of NOD1 in inflammation‐mediated tissue destruction in the context of host–microbe interactions. We used mice with whole‐genome deletion of the NOD1 gene in a microbe‐induced periodontitis model using direct injections of heat‐killed Gram‐negative or Gram‐negative/Gram‐positive bacteria on the gingival tissues. In vitro experiments using primary bone‐marrow‐derived macrophages from wild‐type and NOD1 knockout mice provide insight into the role of NOD1 on the macrophage response to Gram‐negative and Gram‐negative/Gram‐positive bacteria. Microcomputed tomography analysis indicated that deletion of NOD1 significantly aggravated bone resorption induced by Gram‐negative bacteria, accompanied by an increase in the numbers of osteoclasts. This effect was significantly attenuated by the association with Gram‐positive bacteria. In vitro, quantitative PCR arrays indicated that stimulation of macrophages with heat‐killed Gram‐negative bacteria induced the same biological processes in wild‐type and NOD1‐deficient cells; however, expression of pro‐inflammatory mediators was increased in NOD1‐deficient cells. These results suggest a bone‐sparing role for NOD1 in this model.
Keywords: bone resorption, host–microbe interactions, inflammation, innate immunity, NOD1
Abbreviations
- BMDM
bone‐marrow‐derived macrophages
- BV
bone (mineralized tissue) volume
- CARD
caspase‐activating and recruitment domain
- DAMPs
damage‐associated molecular patterns
- KO
knockout (gene deletion)
- LPS
lipopolysaccharide
- MAMPs
microbial‐derived molecular patterns
- NLR
nucleotide‐binding domain and Leucine‐rich repeat‐containing Receptor
- NOD1
nucleotide‐binding oligomerization domain containing 1
- NOD2
nucleotide‐binding oligomerization domain containing 2
- PRRs
pathogen‐recognition receptors
- qPCR
quantitative (real‐time) polymerase chain reaction
- ROI
region of interest
- TLR
Toll‐like receptor
- WT
wild‐type
Introduction
Periodontitis is a chronic inflammatory condition initiated and maintained by a highly complex microbiota organized in a biofilm present adjacent to the teeth that represents the most prevalent lytic disease of bone in humans.1 Besides the obvious relevance for oral health as a major cause of tooth loss, accumulating evidence indicates that this persistent chronic inflammation caused by sustained bacterial challenge in the surroundings of teeth may influence other systemic conditions of great morbidity, including diabetes, cardiovascular diseases/atherosclerosis and rheumatoid arthritis.2 Moreover, we think that, similarly to the study of host–microbe interactions in intestinal disease models, knowledge derived from experimental periodontitis models may provide important insights into the regulation of the immune response to microorganisms.
The identification and study of pathogen‐recognition receptors (PRRs) in the past 15 years has led to a great advance in the understanding of the mechanisms by which the host response recognizes microbially derived molecular patterns (MAMPs), including antigens and virulence factors. Importantly, this knowledge has been continuously increased with the identification of mechanisms and novel functions for the PRRs, including the sensing of host‐derived damage‐associated molecular patterns (DAMPs). NOD1 (CARD4) and NOD2 (CARD15) were the first described and remain the most studied PRRs from the nucleotide‐binding domain and leucine‐rich repeat‐containing receptor (NLR) family that includes 22 genes in humans and over 30 genes in mice.3, 4 NOD1 belongs to the NLRC family of NLRs, characterized by a caspase‐activating and recruitment domain (CARD) at the N‐terminal. It was initially described as a cytosolic sensor of innate immune cells that recognizes peptidoglycan, more specifically d‐glutamyl‐meso‐diaminopimelic acid present in most Gram‐negative bacteria and a few Gram‐positive bacteria including Bacillus and Listeria;5 however, recent evidence suggests that NOD1 activation may be triggered by other non‐bacterial and host‐derived ligands and affect autophagy and adaptive immune responses;6, 7 as well as possibly playing a role in conditions such as asthma and inflammatory bowel diseases.8
Iinformation on the role of NOD1 in modulating host–microbe interactions in periodontitis is scarce, but activation of NOD1 and NOD2 by direct injections of Eschierchia coli‐ or Staphylococcus aureus‐derived peptidoglycan synergistically enhanced lipopolysaccharide (LPS) ‐induced alveolar bone loss in mice in vivo and also RANKL‐induced osteoclastogenesis in vitro.9 Recently, NOD1 KO (knockout) mice were shown to be less susceptible to ligature‐induced periodontitis.10 In this study, we assessed the role of NOD1 in a microbe induced experimental periodontitis model, with inflammatory bone resorption and connective tissue inflammation as main outcomes. We further explore the role of NOD1 in the response of macrophages to Gram‐negative (pathogenic, periodontitis‐associated) and Gram‐positive (commensal, associated with periodontal health) microorganisms.
Materials and methods
Animal use
We used wild‐type (WT) C57BL/6 mice and NOD1 knockout mice backcrossed to C57BL/6 background for eight generations.11, 12 All animals used in these studies were between 8 and 10 weeks of age. Killing was always performed by cervical dislocation and the Institutional Committee on the Use of Experimental Animals approved the study protocol. Primary bone marrow‐derived macrophages (BMDM) were obtained from bone marrow flushed from tibias and femurs, differentiated and expanded in the presence of macrophage colony stimulating factor, as previously described.13 Host–microbe interactions in vivo were studied using a microbe ‐induced model of experimental periodontitis. In this model, 3 μl of a suspension of PBS containing 109 colony‐forming units/ml of heat‐killed Gram‐negative Aggregatibacter actinomycetemcomitans (Aa, JP2 clone, serotype b – obtained from the private collection of Dr Mario J. Avila Campos‐ICB/USP, Brazil) associated with periodontitis combined or not with 109 colony‐forming units/ml of Gram‐positive Lactobacillus fermentum (Lf, serotype 36/ATCC 9338 – obtained from the private collection of Dr Mario J. Avila Campos‐ICB/USP, Brazil) was directly injected into the palatal gingival tissues surrounding the upper molar teeth of the mice, three times/week for 4 weeks. These in vivo studies used 14 WT and 14 NOD1 KO mice. Killing was performed 2 days after the last injection. Six mice of each genotype were vehicle controls and received bilateral injections of sterile PBS in the palatal aspect of upper first molars; whereas eight mice received injections with heat‐killed bacteria.
Assessment of alveolar bone loss and inflammation
Immediately after death, tissue blocks including the upper molars and surrounding tissues were carefully dissected from the animals, rinsed in PBS and fixed in 4% paraformaldehyde for 18 hr at 4°. These samples were then rinsed in distilled water, transferred to 70% ethanol and maintained at 4°. Microcomputed tomography scanning of these samples was done on a Skyscan (Skyscan, Aartselaar, Belgium) at a resolution of 18 μm and three‐dimensional images were reconstructed, spacially re‐oriented in a standardized orientation and analysed using the equipment's software (nrecon/dataviewer/ctan/ctvol, Skyscan, Aartselaar, Belgium). A standardized region of interest (ROI) of 2·5 mm3 was positioned on the three‐dimensional images using anatomical landmarks as reference points, and the fraction of the volume of the ROI occupied by mineralized tissue (Bone/mineralized tissue volume, BV%) was be determined using a standard threshold for detection of mineralized tissues (which include tooth roots in the ROI). Considering that the variation in the volume of similar tooth roots in different animals is negligible, a decrease in the BV% in the ROI indicates bone loss.
After scanning, the same tissue blocks used on the microcomputed tomography analysis were decalcified in 0·5 m EDTA (pH 8·0) and submitted to routine processing for paraffin embedding. Five‐micrometer thick, semi‐serial sections were obtained on the mesio‐distal plane and stained with haematoxylin & eosin for descriptive assessment of inflammation by an experienced examiner blinded to the experimental groups according to a severity score system (0, no significant inflammation; 1, mild inflammation; 2, moderate inflammation; 3, severe inflammation). A minimum of six equally‐spaced semi‐serial sections spanning 500 μm of the bucco‐lingual diameter of the specimens was used. Sections from three or four different animals of each genotype were assessed. Scorings were performed three times with a minimum interval of 2 weeks between the assessments and the most prevalent score was used.
Immunohistochemical detection of TRAP (Tartrate‐resistant acid phosphatase) was performed using a goat polyclonal antibody (sc‐30833; Santa Cruz Biotechnology, Dallas, TX) and a biotin–streptavidin–DAB visualization system (LSAB2+, Dako, Carpinteria, CA, USA). A minimum of six equally spaced semi‐serial sections of each experimental condition (PBS, Aa or Aa+Lf injections) from three different animals of each genotype were stained. TRAP+ cells containing two or more nuclei present in the vicinity of the bone surface were considered osteoclasts. The number of osteoclasts in a linear extension of 400 μm from the palatal aspect of the first molar was counted by a trained examiner blind to the experimental groups.
In vitro studies
Bone marrow‐derived macrophages (1 × 106 cells/well in 12‐well plates) were grown overnight in RPMI‐1640 supplemented with penicillin/streptomycin and 10% heat‐inactivated fetal bovine serum. After a 6‐hr de‐induction in medium containing 0·2% heat‐inactivated fetal bovine serum these cells were stimulated with 1 × 106 colony‐forming units/ml of heat‐killed Aa (1 : 1 ratio bacteria : cells) for 6 hr to assess the early macrophage response. Negative controls were treated with the same volume of PBS vehicle used to resuspend the bacteria. A total of six samples (unstimulated, Aa and Aa+Lf‐stimulated for both WT and NOD1 −/− macrophages) were obtained from each experiment. Three independent experiments were performed, each experiment using cells obtained from two to four different mice of each genotype.
RT‐quantitative PCR and quantitative PCR arrays
Total RNA from BMDM derived from WT and NOD1 KO mice was harvested 6 hr after stimulation with the bacteria or vehicle using an affinity column system (RNeasy micro; Qiagen, Hilden, Germany), From three independent experiments using cells from different animals, we prepared three pools of RNA from macrophages derived from WT mice and three pools of RNA derived from NOD1 KO mice by combining 300 ng of total RNA isolated from samples of each one of the three independent experiments (each pooled sample had a total concentration of 900 ng of total RNA). These six pools (three from BMDM derived from WT mice and three from BMDM derived from NOD1 KO mice) represented the following experimental conditions: non‐stimulated; Aa‐stimulation; and Aa+Lf‐stimulation. The cDNA was synthesized from each of these six 900 ng pools of total RNA using the reagents and procedure indicated by the supplier of the PCR‐based arrays (RT2 First Strand cDNA kit,; SABiosciences/Qiagen, Frederick, MD, USA). Expression of 84 genes related with innate immunity in each sample was investigated using quantitative PCR (qPCR) ‐based arrays (RT2 Profiler™ PCR Array Mouse Toll‐Like Receptor Signaling Pathway; SABiosciences/Qiagen, Frederick, MD, USA) performed according to the instructions of the supplier on a StepOne Plus qPCR thermocycler (Applied Biosystems, Foster City, CA) using the indicated cycling conditions (10 min at 95°, followed by 40 cycles of 15 seconds at 95° and 60 seconds at 60°). Analysis of the qPCR arrays was performed as indicated by the supplier (SA Biosciences, Frederick, MD, USA). Briefly, the cycle threshold (Ct) values obtained from the qPCR thermocycler software were exported into the online analysis tool provided by the supplier of the array, which included the selection of the proper normalizing/house‐keeping gene (from a panel of six constitutively expressed genes included in the arrays) and yielded the results of gene regulation as a fold change relative to the indicated control sample (in this case, non‐stimulated WT macrophages). The purpose of the analysis was to assess the relative regulation of the 84 target genes using the profile of gene expression observed in non‐stimulated macrophages from WT mice.
Array data were verified by RT‐qPCR analysis of two candidate genes that showed regulation in the array analyses. Verification was performed using cDNA prepared using 500 ng of RNA isolated from each sample (non‐stimulated, Aa‐stimulated; Aa+Lf‐stimulated) of all three independent experiments (i.e., three sets of independent data/experimental condition/genotype) using random hexamers as primers (High‐capacity cDNA synthesis kit; Applied Biosystems). The qPCR was performed on the same thermocycler (StepOne Plus; Applied Biosystems) using pre‐designed and optimized sets of primers and probe using TaqMan chemistry (TaqMan gene expression assays, TaqMan Fast qPCR master Mix; Applied Biosystems). Data were normalized to the expression of β‐actin and analysed by the ΔΔCt method.
Data analysis
The statistical analysis aimed at comparing the results obtained in each experimental condition (control/non‐stimulated, Aa‐stimulated and Aa+Lf‐stimulated) between different genotypes (WT and NOD1 KO) in instances where at least three data points from independent experiments were available. These comparisons were performed using non‐paired t‐tests with Welch's correction for unequal variances, assuming complete independence between the results of the two genotypes. For these analyses, statistical significance was set at 95% (*P < 0·05). The qPCR focused array data obtained with pooled samples was analysed using an online bioinformatics tool, DAVID (Database for Annotation, Visualization and Integrated Discovery – http://david.abcc.ncifcrf.gov),14, 15 in an exploratory manner consistent with a hypothesis‐generating study.16 The purpose of these analyses was to assess how the gene functional clusters that were markedly regulated in macrophages stimulated with heat‐killed bacteria were affected by the deficiency of NOD1 gene.
Results
Inflammation and bone resorption increased upon stimulation with Gram‐negative bacteria in NOD1 KO mice. Concomitant stimulation with Gram‐positive bacteria attenuated inflammation and bone resorption
In the Gram‐negative bacteria‐induced experimental periodontitis model, deletion of NOD1 significantly accentuated alveolar bone resorption; which is supported by a significant increase of osteoclast numbers (Fig. 1) and an aggravation of inflammation observed histologically (Fig. 2). However, the simultaneous stimulation with Gram‐positive bacteria in NOD1 KO mice significantly attenuated bone resorption, inflammation and also reduced osteoclast numbers. Considering that NOD2 is present in NOD1 KO mice, and also that its ligand, muramyl‐dipeptide, is present in most bacteria, particularly Gram‐positive, this attenuation effect may be the result of increased NOD2 activation.
Figure 1.
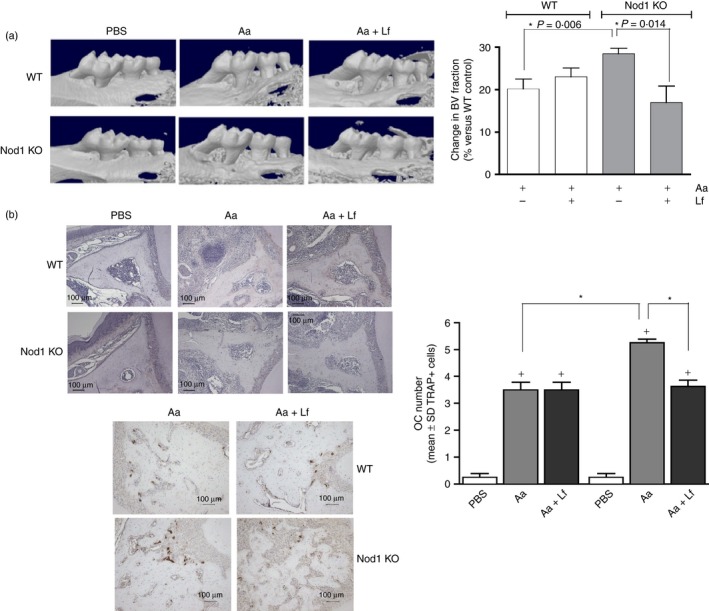
Role of nucleotide‐binding oligomerization domain containing 1 (NOD1) in inflammatory bone resorption and osteoclastogenesis in vivo. Microcomputed tomography analysis of the hemimaxillae of wild‐type (WT) and NOD1 knockout (KO) mice subjected to injections of PBS vehicle, heat‐killed Aggregatibacter actinomycetemcomitans (Aa) or heat‐killed Aa associated with heat‐killed Lactobacillus fermentum (Aa+Lf) reveals that inflammatory bone resorption induced by Aa injections alone is significantly increased in NOD1 KO mice. Concomitant injection of Gram‐positive Lf in the gingival tissues markedly attenuates bone resorption. (a) Depicts representative three‐dimensional images reconstructed from microcomputed tomography scans of the hemi‐maxillae. The graph represents the average and standard deviation of the relative reduction in mineralized tissue content [the ‘bone volume’ (BV) fraction] in the standardized region of interest (ROI) assessed in comparison to the BV fraction of the ROI in PBS‐injected WT control samples (set to 100%). Asterisk (*) indicates significant difference between the indicated pairs of bars by unpaired Student's t‐tests with Welch's correction for unequal variances. Samples from at least six different animals were analysed for each group. (b) Left, top: representaive sections (40× magnification) of hemi‐maxillae stained with eosin to provide an overview of the ROI. Osteoclasts were counted on TRAP‐stained images from the apex of the palatal root until the area subjacent to the major palatine artery and nerve. Left, bottom: representative images (100× magnification) of immunohistochemical staining for TRAP, identifying osteoclasts in tissue sections from WT and NOD1 KO mice subjected to Aa or Aa+Lf injections. The graph depicts the results of osteoclast counting according to the genetic background and presence or absence of disease induction. Plus sign (+) indicates a significant difference (unpaired Student's t‐test with Welch's correction for unequal variances) in comparison with the osteoclast number in sections from control (PBS‐injected) WT mice. Asterisk (*) indicates significant difference between bracketed pairs of bars (unpaired Student's t‐test with Welch's correction for unequal variances).
Figure 2.
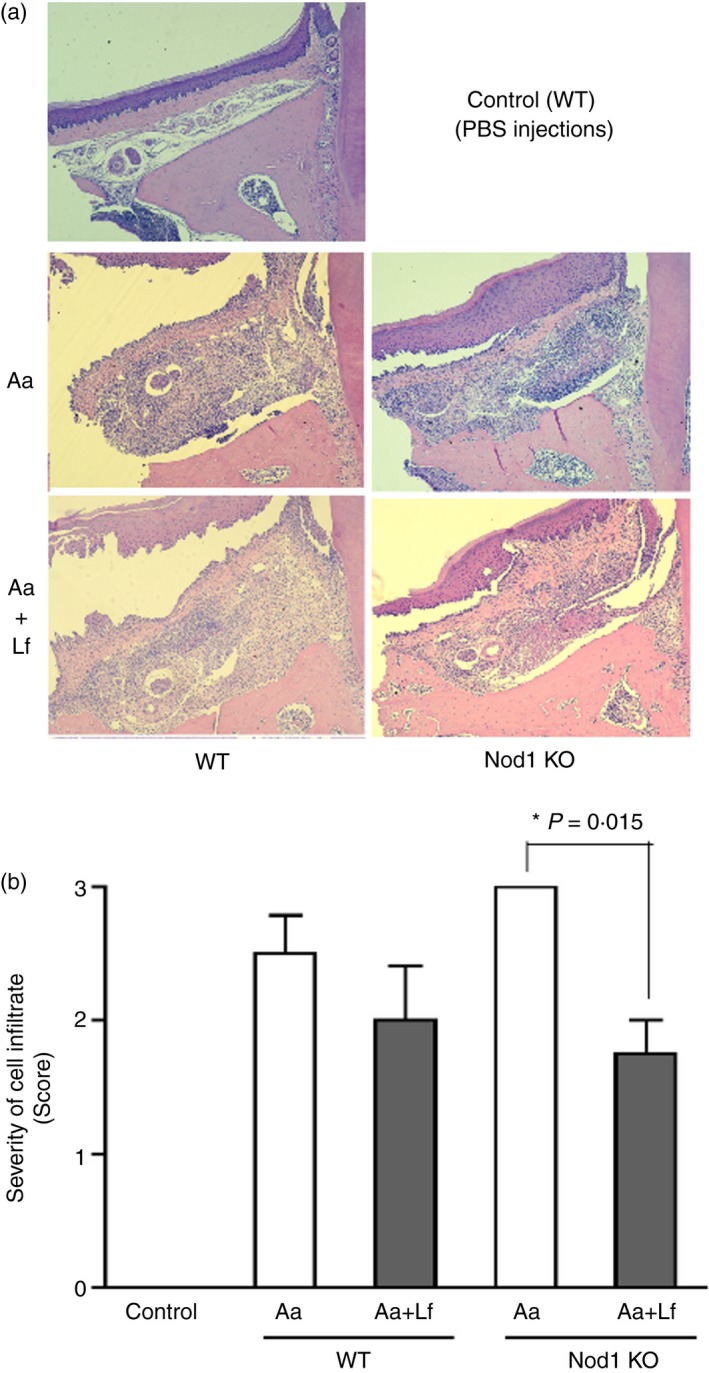
Representative images of 5 μm haematoxylin & eosin‐stained sections in the bucco‐lingual (frontal) plane depicting the palatal aspect of upper first molars, according to the genotype of the animal and experimental condition [PBS, heat‐killed Aggregatibacter actinomycetemcomitans (Aa) or heat‐killed Aa associated with heat‐killed Lactobacillus fermentum (Aa+Lf) injections]. (a) Bar graph presents the assessment of inflammation (cellular infiltrate) severity by the scoring system according to the genotype and experimental condition. Asterisk (*) indicates significant difference between the indicated pair of bars by unpaired Student's t‐test with Welch's correction for unequal variances (b). Samples from at least six different animals were analysed for each group and these representative images were obtained at 100 × magnification.
NOD1 deficiency in macrophages increased mRNA expression of pro‐inflammatory genes induced by bacterial stimulation. Concomitant stimulation with Gram‐positive bacteria increased expression of interleukin‐10 mRNA and attenuated the expression of macrophage activation‐related genes
To further investigate the overall effect of NOD1 deletion on macrophage response to periodontopathogenic and non‐disease‐associated Gram‐positive bacteria presenting both NOD1 and NOD2 ligands, we used a focused qPCR array, investigating the relative modulation of the expression of 84 genes associated with host–microbe interactions. Interestingly, in the absence of stimulation, NOD1 −/− cells showed higher expression of Toll‐like receptors (TLR) 4, 5, 7, 8 and 9 (but not of TLR1, 2, 3 and 6) in comparison with non‐stimulated WT macrophages, which may suggest a compensatory mechanism for the diminished microbial‐sensing capability of NOD1 −/− cells. In support of this possibility, besides the expression of PRRs, expression of Ly86 and Ly96, which are important for activation of macrophages by TLR4/LPS, were also up‐regulated in non‐stimulated NOD1 −/− macrophages (Fig. 3).
Figure 3.
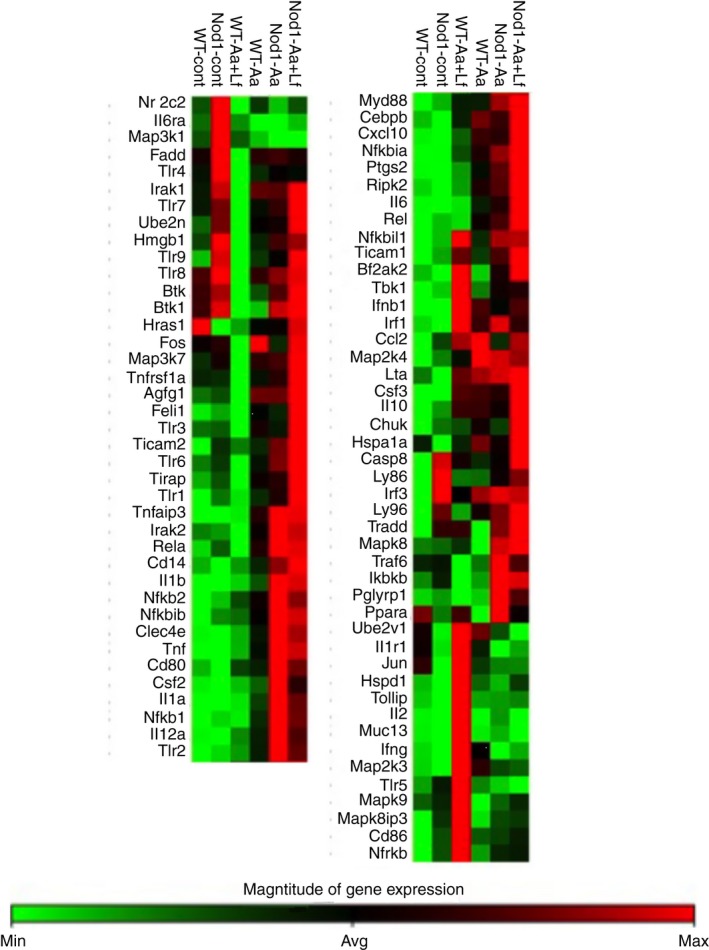
Heat map of the expression of 84 genes associated with innate immunity in bone‐marrow‐derived macrophages from wild‐type (WT) and NOD1 knockout (KO) mice stimulated with heat‐killed Aggregatibacter actinomycetemcomitans (Aa) alone or associated with Gram‐positive Lactobacillus fermentum (Aa+Lf) for 6 hr. Total RNA was harvested and the cDNA was used in quantitative PCR‐based arrays. These data are based on the analysis of cDNA prepared from pooled RNA samples harvested from three independent experiments (cells obtained from a minimum of three animals in each independent experiment).
Upon stimulation with Gram‐negative Aa, NOD1 −/− macrophages showed a clear increase in the expression of several pro‐inflammatory genes in comparison with WT macrophages, including macrophage activation‐related markers (CD80, CD14, Csf2) nuclear factor‐κB signalling‐related intermediates (Nfkb1, Nfkb2, Nfkbia, Nfkbib, Rel) and inflammatory cytokines (Il1b, Tnf, Il12a) (Fig. 3). Interestingly, concomitant stimulation of NOD1 −/− macrophages with Gram‐positive and Gram‐negative bacteria attenuated the expression of some pro‐inflammatory cytokines (Il12a, Il1a), whereas the expression of other inflammatory cytokines remained unchanged and up‐regulated (IL1b, Tnf) or was even further increased (Ptgs2, Il6) in comparison with Gram‐negative stimulation alone. Notably, stimulation of NOD1 −/− macrophages with Gram‐negative and Gram‐positive bacteria concomitantly did increase the expression of anti‐inflammatory cytokine interleukin‐10 (Il10) and attenuated the expression of some macrophage activation‐related markers (CD80, Csf2) in comparison with NOD1 −/− macrophages stimulated with heat‐killed Aa only (Fig. 3, Table 2); but the expression of nuclear factor‐κB signalling‐related intermediates was not modulated in comparison with Gram‐negative stimulation alone. These results were validated by RT‐qPCR for Il6 and tumour necrosis factor‐α (Tnf) expression, which was performed using cDNA prepared from the different RNA samples isolated from the three independent experiments that were pooled to be used on the arrays (Fig. 4). In this confirmatory experiment, Tnf and Il6 expression were significantly increased after Aa and Aa+Lf stimulation; however, in spite of a noticeable increase of TNF expression in NOD1‐deficient macrophages in comparison to WT cells, this change was not statistically significant.
Table 2.
List of genes presenting at least fourfold up‐regulation in their expression in macrophages from wild‐type (WT) and NOD1 knockout (NOD1 –/–) mice after a 6‐hr stimulation with heat‐killed Aggregatibacter actinomycetemcomitans (Aa) associated or not with heat‐killed Lactobacillus fermentum (Lf) in comparison to WT untreated control
| Gene name | Aa simulation | Aa+Lf stimulation | ||
|---|---|---|---|---|
| WT | NOD1−/− | WT | NOD1−/− | |
| Ccl2 | 4·40 | 4·00 | 4·03 | |
| Cd14 | 4·45 | |||
| Cd86 | 5·01 | |||
| Clec4e | 5·08 | 10·68 | 9·52 | |
| Csf2 | 6·71 | 26·89 | 17·08 | |
| Csf3 | 16·08 | 14·54 | 16·86 | 22·76 |
| Cxcl10 | 15·60 | 14·62 | 5·92 | 22·85 |
| Ifnb1 | 12·92 | 9·22 | 17·67 | 12·96 |
| Ifng | 6·18 | 11·92 | ||
| Il10 | 13·36 | 11·04 | 13·42 | 19·36 |
| Il12a | 15·28 | 37·58 | 6·67 | 27·24 |
| Il1a | 90·94 | 281·95 | 12·79 | 217·05 |
| Il1b | 45·80 | 166·39 | 12·34 | 159·89 |
| Il2 | 19·11 | |||
| Il6 | 16·56 | 24·77 | 40·05 | |
| Irf1 | 5·29 | 5·45 | ||
| Lta | 4·29 | 4·52 | ||
| Muc13 | 11·19 | |||
| Nfkb1 | 6·59 | 5·40 | ||
| Nfkb2 | 5·39 | 9·16 | 7·79 | |
| Nfkbia | 7·90 | 10·83 | 4·31 | 12·57 |
| Nfkbib | 4·27 | 7·45 | 7·12 | |
| Ptgs2 | 19·23 | 30·43 | 6·28 | 40·60 |
| Rel | 4·83 | 6·44 | ||
| Ripk2 | 4·97 | |||
| Tlr1 | 4·28 | 6·45 | ||
| Tlr2 | 8·12 | 18·21 | 14·10 | |
| Tlr9 | 4·36 | |||
| Tnf | 29·23 | 61·02 | 7·55 | 57·45 |
| Tnfaip3 | 8·32 | 12·89 | 12·67 | |
Data obtained from the analysis of quantitative PCR arrays performed using cDNA prepared from pools of RNA isolated from bone‐marrow‐derived macrophages from six different mice in each experimental condition.
Figure 4.
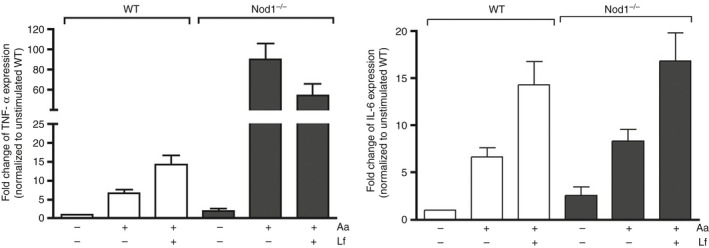
Validation of quantitative PCR array data by RT‐qPCR array using cDNA prepared from RNA obtained from bone‐marrow‐derived macrophages from wild‐type (WT) and NOD1 knockout (KO) mice in three independent experiments (four to six mice of each genotype were used). These were the same RNA samples combined to prepare the pool of RNA used in the array analysis. Target gene expression was normalized to β‐actin expression and fold regulation was calculated by comparison with the normalized gene expression in WT unstimulated control macrophages.
Functional clusters of genes modulated by heat‐killed bacteria in macrophages are similar, but in NOD1‐deficient macrophages the expression of Th1‐associated cytokines and TLR receptor genes are more markedly attenuated than in Receptor‐Interacting Serine‐Threonine Kinase 2 (RIP2)‐deficient macrophages
To obtain a better understanding of the functional role of NOD1 in the macrophage response to Gram‐negative bacteria alone and associated with Gram‐positive bacteria, we initially assessed the overall effect of NOD1 deficiency by extracting terms defining biological function of the list of genes up‐regulated in stimulated macrophages using an online bioinformatics application (DAVID). To be considered ‘up‐regulated’, we used a minimum of fourfold increase over the expression level in unstimulated controls (or twofold increase on a log2 scale), which is more stringent than the guidelines for studies using treated primary cells in a microarray approach for hypothesis‐generating studies.16 This stringent criterion was used to reduce Type I error due to the use of pooled samples in the qPCR array analysis, at the cost of accepting some false‐negatives. In defining the functional gene clusters, DAVID parameters were also set to the highest stringency setting to generate fewer functional clusters including more tightly associated genes in each cluster. The ‘score’ represents the overall enrichment score for the gene cluster, with scores > 2 considered of biological significance.14, 15 To reduce the possibility of spurious associations, we have considered only the gene clusters associated with an enrichment score > 3. Table 1 indicates that overall the biological functions defined by clusters formed from the list of genes up‐regulated in macrophages after stimulation with heat‐killed Aa is very similar in WT and NOD1 −/− macrophages, with a greater enrichment score in the first cluster of genes functionally defined as inflammation‐associated genes.
Table 1.
Comparison of the top five functional effects caused by Aggregatibacter actinomycetemcomitans (Aa) and Aggregatibacter actinomycetemcomitans plus Lactobacillus fermentum (Aa+Lf) stimulation of bone marrow‐derived macrophages from wild‐type (WT) and NOD1 knockout (NOD1 –/–) mice
| Gene cluster | Aa stim/WT | Aa stim/NOD1−/− | ||
|---|---|---|---|---|
| Functional annotation | Score | Functional annotation | Score | |
| 1 | Defence response, inflammatory response, positive regulation of multicellular organism process, response to wounding | 10·78 | Immune response, defence response, inflammatory response, response to wounding | 17·54 |
| 2 | Immune response, cytokine, cytokine activity, macrophage, lymphokine | 8·52 | Cytokine, cytokine activity, macrophage, lymphokine, JAK‐STAT signalling pathway | 8·53 |
| 3 | Haematopoietic cell lineage, JAK‐STAT signalling activity, growth factor activity | 6·40 | Response to bacterium, regulation of interleukin‐6 production, response to molecule of bacterial origin, positive regulation of NF‐κB transcription factor activity | 6·16 |
| 4 | Response to bacterium, regulation of interleukin‐6 production, response to molecule of bacterial origin, positive regulation of cytokine production | 5·92 | Positive regulation of protein kinase cascade, positive regulation of cytokine production, positive regulation of signal transduction | 5·98 |
| 5 | Immunoregulation, acute‐phase response, acute inflammatory response, homeostatic process | 5·23 | Macrophage, immunoregulation, negative regulation of transport, acute inflammatory response | 5·18 |
| Gene cluster | Aa+Lf stim/WT | Aa+Lf stim/NOD1−/− | ||
|---|---|---|---|---|
| Functional annotation (top 5) | Score | Functional annotation (top 5) | Score | |
| 1 | Immune response, cytokine, cytokine activity, JAK‐STAT signalling pathway, cytokine network | 11·66 | Regulation of IκB/NF‐κB cascade, response to bacterium, regulation of interleukin‐6 production, regulation of cellular response to stress | 8·97 |
| 2 | Defence response, inflammatory response, response to wounding | 10·09 | Regulation of transcription factor activity, innate immune response‐activating signal transduction, defence response to Gram‐positive bacterium | 6·93 |
| 3 | Macrophage, immune system development, leucocyte differentiation | 4·57 | Regulation of tumour necrosis factor production, response to lipoteichoic acid | 6·34 |
| 4 | Lymphokine, regulation of cytokine production, response to bacterium, immunoregulation | 4·49 | Cytokine, cytokine activity, macrophage, lymphokine, interleukin‐10 anti‐inflammatory signalling pathway | 6·32 |
| 5 | Signal, disulphide bond, glycoprotein | 4·24 | Immune system development, leucocyte differentiation | 5·47 |
The functional‐describing terms and enrichment scores (Fisher Exact Statistics in DAVID system, referring to one‐tail Fisher Exact Probability Value used for gene‐enrichment analysis) were obtained from the lists of up‐regulated genes expressed by Aa‐ and Aa+Lf‐stimulated macrophages using the Database for Annotation, Visualization and Integrated Discovery (DAVID) algorithm.
On the other hand, the comparison of functional clusters of genes up‐regulated by stimulation with the association of heat‐killed Aa and Lf in macrophages from WT and NOD1 KO mice (Table 1) shows some differences in the functional defining terms of the gene clusters, particularly the inclusion genes related with nuclear factor‐κB cascade in NOD1 −/− macrophages [versus Janus kinase/signal transducer and activator of transcription (JAK‐STAT) in WT macrophages], and the presence of gene clusters functionally defined by Il10 anti‐inflammatory signalling, as well as by TNF production and response to Gram‐positive bacterium/lipoteichoic acid in the NOD1 −/− macrophages only. Collectively, this information may be interpreted as an indication that upon stimulation with both Gram‐positive and Gram‐negative bacteria, deficiency of NOD1 shifts the signalling from the JAK‐STAT to the nuclear factor‐κB pathway and also results in the the activation of the IL‐10 anti‐inflammatory pathway.
Assessment of the pathways affected using the Kyoto Encyclopaedia of Genes and Genomes (KEGG) also indicated a great similarity in both the percentage of genes and the log10 of the P values, indicating the statistical significance for the association in the top 20 KEGG terms defining the functions and utilities of genes that were up‐regulated in macrophages by heat‐killed Aa in macrophages obtained from WT and NOD1 KO mice (Fig. 5).
Figure 5.
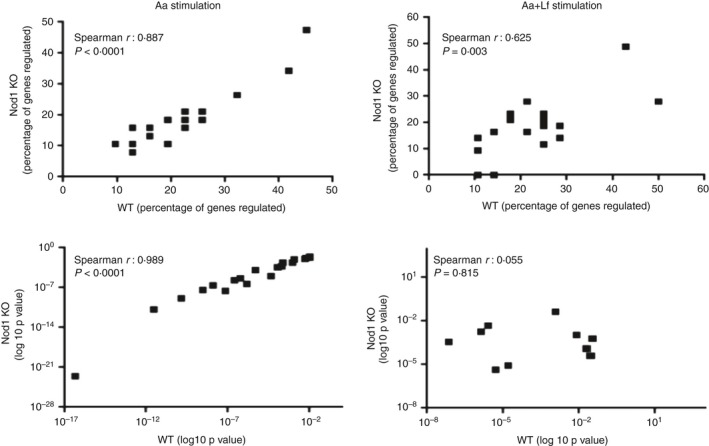
Correlations of percentage of genes included in each functional annotation term and of the enrichment statistical values (log10 P value) obtained from DAVID Chart Reports. The correlation plots measure the annotation agreement between genes up‐regulated by heat‐killed Aggregatibacter actinomycetemcomitans (Aa) alone or associated with Gram‐positive Lactobacillus fermentum (Aa+Lf) stimulation in WT and NOD1‐deficient macrophages. The gene hit percentages and enrichment P values of top enriched terms between the lists of up‐regulated genes show very strong correlation upon Aa stimulation. The comparison of WT and NOD1‐deficient macrophages upon Aa+Lf stimulation shows a weak correlation between the percentage of genes up‐regulated and no correlation between the P values, indicating a qualitative difference in the response of NOD1‐deficient macrophages upon Gram‐negative and Gram‐positive stimulation.
Table 2 presents the data on the fold change up‐regulation of genes according to the genotype of the mice and the type of bacterial stimulation to provide further insight into the role of NOD1 in regulating the macrophage response to Gram‐negative and Gram‐negative/Gram‐positive bacteria from both an intensity/quantitative and qualitative standpoint.
NOD1 deficiency markedly increased the expression of some pro‐inflammatory cytokines upon heat‐killed Aa stimulation in comparison with WT macrophages, notably Il1α and Il1β, Il12, Tnf and Ptgs2 (murine analogue of Cox‐2). On the other hand, there was a marked inhibition of Ppara expression (−8·81‐fold) in Aa‐stimulated WT macrophages that was not observed in the comparison between WT control and Aa‐stimulated NOD1 −/− macrophages, suggesting that this inhibition of Ppara gene expression requires functional NOD1. Moreover, when both Aa and Lf were associated, expression of Elk1, Fadd, Irak2, Tlr7 and Tlr8 were all inhibited more than twofold in WT macrophages in comparison with control (non‐stimulated) WT macrophages; whereas none of the 84 genes assessed in the qPCR array were markedly inhibited in NOD1 −/− macrophages stimulated with Aa+Lf, stressing the role of NOD1 in the down‐regulation of these genes. Interestingly, stimulation with the association Aa+Lf reduced the expression of Il1α and Il1β, Il6, Tnf and PTGS2 in WT macrophages in comparison with stimulation with Aa alone in WT macrophages, which supports the hypothesis of functional NOD1 being required for the down‐regulation of these genes. In fact, NOD1‐deficient macrophages did show a discrete attenuation of Il1α and Il1β gene expression with the association of Aa and Lf; however, expression of Il6, Il10, Tnf and Ptgs2 were in fact further increased in comparison to NOD1‐deficient macrophages stimulated with Aa only.
Discussion
We showed that genomic deletion of NOD1 in vivo accentuates inflammation‐driven bone resorption in an experimental periodontitis model, representative of host–microbe interactions. This effect was supported by significantly increased numbers of osteoclasts in NOD1 KO mice, indicating an influence of NOD1 on osteoclastogenesis. Interestingly, the association of a Gram‐positive, NOD2‐activating microorganism (Lf) in the model attenuated the bone resorption and reduced both the cellular infiltrate and the number of osteoclasts in the diseased microenvironment. This influence may be either directly affecting the osteoclast precursor cells (mostly macrophage/monocytes) or indirectly modulating the external signals regulating osteoclast differentiation. A possible indication of the latter possibility is the up‐regulation of Il10 gene expression in NOD1 −/− macrophages stimulated with Aa+Lf in comparison with NOD1 −/− cells stimulated with Aa only, which was not observed in WT cells (Table 2). Gram‐positive bacteria of the Lactobacillus genus were shown to induce Il10 when used as vaccine adjuvants in a murine asthma model.17 Similarly, there is in vivo and in vitro evidence that administration of probiotic Gram‐positive bacteria (including Lactobacillus species) attenuate colitis in murine experimental models by decreasing pro‐inflammatory cytokine expression.18, 19 However, this induction of Il10 by Gram‐positive bacteria is dependent on the bacterial species and strain, as Lactobacillus crispatus (strain M206119) aggravates experimental dextran‐induced colitis in mice by increasing expression of pro‐inflammatory genes and decreasing IL10.20
Interestingly, our in vitro data using BMDM shows a marked increase on the expression of Csf2/Gcsf in NOD1‐deficient cells upon microbial stimulation, and there is evidence for increased osteoclast differentiation in mice upon in vivo administration of granulocyte colony‐stimulating factor alone.21 Moreover, muramyl‐dipeptide (which can activate both NOD1 and NOD2) has been shown to enhance osteoclastogenesis induced by LPS and inflammatory cytokines in co‐cultures of osteoblasts and haematopoietic cells by modulating NOD2‐mediated RANKL mRNA expression in osteoblasts.22 It is important to bear in mind that NOD1 KO animals and the macrophages derived from these animals present functional NOD2.
In fact, NOD1 KO mice still present all other PRRs, including TLRs and NLRs, and so the effect on inflammatory bone resorption may have shifted the MAMP and DAMP signalling to other pathways, modulating the cytokine network and hence the phenotypical characteristics of immune cells and the inflammatory microenvironment. Alternatively, NOD1 may have a direct inhibitory role in osteoclastogenesis and its deletion allowed other signalling pathways to exert more influence on this process. In fact, simultaneous stimulation of osteoclast precursor cells with RANKL and NOD1 agonist inhibited osteoclast differentiation in vitro.9
We used heat‐killed microorganisms to avoid colonization issues and also bias introduced by bacterially secreted products; moreover, there is evidence indicating that osteoclast differentiation induced in mice by injecting live or heat‐killed bacteria in mice is similar23 and also that both live and heat‐killed bacteria can activate NOD signalling.24 NOD1 was originally considered a cytosolic PRR, therefore especially suited for the detection of invading microorganisms; however, it has been shown that viability or even the presence of bacteria is not necessary for the stimulation of NOD125 and even with live bacteria the intracellular localization of bacteria is also not a requisite for the activation of NOD1.26 These findings support the functional relevance of NOD1 in non‐phagocytosing cells; whereas it is also possible that in our model the specific NOD1 and NOD2 ligands gained access to the cytoplasm of macrophages and neutrophils by phagocytosis or pinocytosis.27
The microbially induced periodontitis model we used is characterized by sustained stimulation over a period of 4 weeks, so there are multiple cell types involved in the microenvironment, including stromal cells (osteoblasts, fibroblasts, epithelial cells) and neutrophils, macrophages, dendritic cells, T and B cells. The effect observed in vivo is the net outcome of NOD1 genomic deletion affecting all these cell types, which also present other TLRs, particularly TLR2 and TLR4 that are activated by major MAMPs of the microorganisms used; respectively, lipoteichoic acid (from Gram‐positive Lf) and LPS (from Gram‐negative Aa). Even in the specific cell type assessed in vitro, gene expression of various TLRs, particularly of TLR4 and its co‐activators Ly86 and Ly96, was increased in unstimulated macrophages. Moreover, over the course of the 4‐week experimental period, host‐derived ligands from damaged tissue and cells may also activate PRRs as danger signals and add another layer of complexity to the interpretation of the observations. Specifically, TLR ligands, interferon‐γ and TNF that accumulate over time in a chronic infectious/inflammatory setting may also activate NOD signalling.5, 28 Even though these facts preclude a direct mechanistic interpretation of the results, it provides an in vivo perspective of the pathological relevance of NOD1 in inflammation‐driven bone resorption, involving a sustained exposure to Gram‐positive and Gram‐negative bacteria. The current study must be considered an initial study that will be further expanded to assess the relative relevance of NOD1 gene in stromal/resident cells and leucocytes and bone‐marrow‐derived cells for the host response in this host–microbe interaction model.
Interestingly, a recent publication presents completely opposite results using another model of experimental periodontitis. Using the ligature‐induced model in mice, Jiao et al.10 showed reduced alveolar bone loss in NOD1 KO mice. The authors report a decrease in the expression of inflammatory cytokine mRNA and neutrophil infiltration, associating these effects with the increased prevalence of an endogenous Gram‐negative bacterium (NI1060) with great genetic similarity to A. actinomycetemcomitans. The obvious differences in the experimental models (ligature for 10 days, versus sustained stimulation with heat‐killed bacteria over 4 weeks) may account for the diametrically opposite findings. Moreover, we have not assessed the potential role of the endogenous microbiota, but we assume that since in our model we injected the microorganisms directly into the tissues, the possible role of endogenous microbiota in our results, if any, was minimal. A possible drawback of the current model is the periodontal bone breakdown caused by the trauma associated with the injections, although we have controlled for this possible bias by injecting the same volume of PBS (used to resuspend the bacteria) in the same administration regimen.
To obtain insight into the role of NOD1 in the modulation of the cell response to Gram‐negative and Gram‐positive bacteria, we chose to use BMDM, as these are professional antigen‐processing and presenting innate immune cells, represent the prototypical NOD‐expressing cells and are also osteoclast precursors. The comprehensive assessment of the regulation of gene expression by RT‐qPCR focused arrays indicated that microbial stimulation induced a marked increase on the gene expression of some inflammatory cytokines in NOD1‐deficient macrophages in comparison with WT macrophages, most notably IL1α, IL1β, TNF and IL12a, whereas interferon‐γ was up‐regulated only in WT macrophages. This supports the in vivo finding of an increased inflammatory infiltrate and osteoclast numbers/bone resorption in NOD1 KO mice.
Recent evidence indicates that NOD1 may bind/interact with different intracellular molecules, and the effects associated with NOD1 deficiency may be derived from the interference with the positive or negative regulatory function of these interacting partners, such as BID (BH3 Interacting Domain Death Agonist)29 and CENTβ1 (Centaurin beta 1).30 Future studies will assess the role of other interacting partners of NOD1, besides RIP2, that may play a role in mediating its functions.
In summary, we report on the aggravation of inflammation‐driven bone resorption in NOD1 KO mice subjected to a Gram‐negative microorganism‐induced model of experimental periodontitis, suggesting that NOD1 may have a role in modulating osteoclast differentiation in vivo, either directly or indirectly. We also show that association of a Gram‐positive microorganism significantly attenuated bone resorption induced by Gram‐negative bacteria in NOD1 KO mice. Lack of NOD1 renders macrophages even more responsive to Gram‐negative microbial stimulation and concomitant stimulation with Gram‐positive bacteria attenuates this enhanced response. We conclude that NOD1 plays an important attenuating role in inflammation and bone resorption associated with host–microbe interactions in experimental periodontitis in vivo.
Author contributions
JACS performed the experiments, analysed data and collaborated in the writing of the manuscript; SCTF and FAC collaborated in the performance of experiments; MJAC provided the microorganisms and collaborated in data interpretation and writing of the manuscript; LCS collaborated in data analysis and in the writing of the manuscript; DSZ provided the experimental animals, collaborated in data interpretation and in the writing of the manuscript; DTG collaborated in data interpretation and in the writing of the manuscript; CRJ designed the study, collaborated in the performance of experiments, data analysis and writing of the manuscript.
Disclosures
All authors of this paper state that they have no financial or commercial conflicts of interest.
Acknowledgements
The authors wish to thank research technician Leandro Alves dos Santos (Department of Diagnosis and Surgery, School of Dentistry at Araraquara‐Univ Estadual Paulista/UNESP) for his technical assistance in the histological processing and obtaining the sections. Funding support to CRJ was received from the Sao Paulo State Research Foundation (FAPESP) grants # 2010/05783‐5 and # 2010/05632‐7 and support from NIDCR grants R01DE017732 and R01DE021921 to DTG.
References
- 1. Pacios S, Kang J, Galicia J, Gluck K, Patel H, Ovaydi‐Mandel A et al Diabetes aggravates periodontitis by limiting repair through enhanced inflammation. FASEB J 2012; 26:1423–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hajishengallis G. Periodontitis: from microbial immune subversion to systemic inflammation. Nat Rev Immunol 2015; 15:30–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ting JP, Willingham SB, Bergstralh DT. NLRs at the intersection of cell death and immunity. Nat Rev Immunol 2008; 8:372–9. [DOI] [PubMed] [Google Scholar]
- 4. Schroder K, Tschopp J. The inflammasomes. Cell 2010; 140:821–32. [DOI] [PubMed] [Google Scholar]
- 5. Moreira LO, Zamboni DS. NOD1 and NOD2 signaling in infection and inflammation. Front Immunol 2012; 3:328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Travassos LH, Carneiro LA, Ramjeet M, Hussey S, Kim YG, Magalhaes JG et al Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat Immunol 2010; 11:55–62. [DOI] [PubMed] [Google Scholar]
- 7. Mercier A, Clement R, Harnois T, Bourmeyster N, Faivre JF, Findlay I et al The β1‐subunit of Na(v)1.5 cardiac sodium channel is required for a dominant negative effect through α‐α interaction. PLoS ONE 2012; 7:e48690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kufer TA, Sansonetti PJ. NLR functions beyond pathogen recognition. Nat Immunol 2011; 12:121–8. [DOI] [PubMed] [Google Scholar]
- 9. Kishimoto T, Kaneko T, Ukai T, Yokoyama M, Ayon Haro R, Yoshinaga Y et al Peptidoglycan and lipopolysaccharide synergistically enhance bone resorption and osteoclastogenesis. J Periodontal Res 2012; 47:446–54. [DOI] [PubMed] [Google Scholar]
- 10. Jiao Y, Darzi Y, Tawaratsumida K, Marchesan JT, Hasegawa M, Moon H et al Induction of bone loss by pathobiont‐mediated Nod1 signaling in the oral cavity. Cell Host Microbe 2013; 13:595–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kobayashi KS, Chamaillard M, Ogura Y, Henegariu O, Inohara N, Nunez G et al Nod2‐dependent regulation of innate and adaptive immunity in the intestinal tract. Science 2005; 307:731–4. [DOI] [PubMed] [Google Scholar]
- 12. Kobayashi K, Inohara N, Hernandez LD, Galan JE, Nunez G, Janeway CA et al RICK/Rip2/CARDIAK mediates signalling for receptors of the innate and adaptive immune systems. Nature 2002; 416:194–9. [DOI] [PubMed] [Google Scholar]
- 13. Marim FM, Silveira TN, Lima DS Jr, Zamboni DS. A method for generation of bone marrow‐derived macrophages from cryopreserved mouse bone marrow cells. PLoS ONE 2010; 5:e15263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. da Huang W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 2009; 37:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. da Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 2009; 4:44–57. [DOI] [PubMed] [Google Scholar]
- 16. Ley Md. Cytokine Protocols. 2nd edn Totowa, NJ; London: Humana; Springer [distributor], 2012, p.1 v. [Google Scholar]
- 17. Van Overtvelt L, Lombardi V, Razafindratsita A, Saint‐Lu N, Horiot S, Moussu H et al IL‐10‐inducing adjuvants enhance sublingual immunotherapy efficacy in a murine asthma model. Int Arch Allergy Immunol 2008; 145:152–62. [DOI] [PubMed] [Google Scholar]
- 18. Saba K, Denda‐Nagai K, Irimura T. A C‐type lectin MGL1/CD301a plays an anti‐inflammatory role in murine experimental colitis. Am J Pathol 2009; 174:144–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Roselli M, Finamore A, Nuccitelli S, Carnevali P, Brigidi P, Vitali B et al Prevention of TNBS‐induced colitis by different Lactobacillus and Bifidobacterium strains is associated with an expansion of γδT and regulatory T cells of intestinal intraepithelial lymphocytes. Inflamm Bowel Dis 2009; 15:1526–36. [DOI] [PubMed] [Google Scholar]
- 20. Zhou FX, Chen L, Liu XW, Ouyang CH, Wu XP, Wang XH et al Lactobacillus crispatus M206119 exacerbates murine DSS‐colitis by interfering with inflammatory responses. World J Gastroenterol 2012; 18:2344–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hirbe AC, Uluckan O, Morgan EA, Eagleton MC, Prior JL, Piwnica‐Worms D et al Granulocyte colony‐stimulating factor enhances bone tumor growth in mice in an osteoclast‐dependent manner. Blood 2007; 109:3424–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yang S, Takahashi N, Yamashita T, Sato N, Takahashi M, Mogi M et al Muramyl dipeptide enhances osteoclast formation induced by lipopolysaccharide, IL‐1α, and TNF‐α through nucleotide‐binding oligomerization domain 2‐mediated signaling in osteoblasts. J Immunol 2005; 175:1956–64. [DOI] [PubMed] [Google Scholar]
- 23. Zubery Y, Dunstan CR, Story BM, Kesavalu L, Ebersole JL, Holt SC et al Bone resorption caused by three periodontal pathogens in vivo in mice is mediated in part by prostaglandin. Infect Immun 1998; 66:4158–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pudla M, Kananurak A, Limposuwan K, Sirisinha S, Utaisincharoen P. Nucleotide‐binding oligomerization domain‐containing protein 2 regulates suppressor of cytokine signaling 3 expression in Burkholderia pseudomallei‐infected mouse macrophage cell line RAW 264·7. Innate Immun 2011; 17:532–40. [DOI] [PubMed] [Google Scholar]
- 25. Chen G, Shaw MH, Kim YG, Nunez G. NOD‐like receptors: role in innate immunity and inflammatory disease. Ann Rev Pathol 2009; 4:365–98. [DOI] [PubMed] [Google Scholar]
- 26. Hasegawa M, Yang K, Hashimoto M, Park JH, Kim YG, Fujimoto Y et al Differential release and distribution of Nod1 and Nod2 immunostimulatory molecules among bacterial species and environments. J Biol Chem 2006; 281:29054–63. [DOI] [PubMed] [Google Scholar]
- 27. Kapetanovic R, Nahori MA, Balloy V, Fitting C, Philpott DJ, Cavaillon JM et al Contribution of phagocytosis and intracellular sensing for cytokine production by Staphylococcus aureus‐activated macrophages. Infect Immun 2007; 75:830–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Takahashi Y, Isuzugawa K, Murase Y, Imai M, Yamamoto S, Iizuka M et al Up‐regulation of NOD1 and NOD2 through TLR4 and TNF‐α in LPS‐treated murine macrophages. J Vet Med Sci 2006; 68:471–8. [DOI] [PubMed] [Google Scholar]
- 29. Yeretssian G, Correa RG, Doiron K, Fitzgerald P, Dillon CP, Green DR et al Non‐apoptotic role of BID in inflammation and innate immunity. Nature 2011; 474:96–9. [DOI] [PubMed] [Google Scholar]
- 30. Yamamoto‐Furusho JK, Barnich N, Xavier R, Hisamatsu T, Podolsky DK. Centaurin β1 down‐regulates nucleotide‐binding oligomerization domains 1‐ and 2‐dependent NF‐κB activation. J Biol Chem 2006; 281:36060–70. [DOI] [PubMed] [Google Scholar]