

Summary
Zika virus (ZikV) has emerged as a potential threat to human health worldwide. A member of the Flaviviridae, ZikV is transmitted to humans by mosquitoes. It is related to other pathogenic vector‐borne flaviviruses including dengue, West Nile and Japanese encephalitis viruses, but produces a comparatively mild disease in humans. As a result of its epidemic outbreak and the lack of potential medication, there is a need for improved vaccine/drugs. Computational techniques will provide further information about this virus. Comparative analysis of ZikV genomes should lead to the identification of the core characteristics that define a virus family, as well as its unique properties, while phylogenetic analysis will show the evolutionary relationships and provide clues about the protein's ancestry. Envelope glycoprotein of ZikV was obtained from a protein database and the most immunogenic epitope for T cells and B cells involved in cell‐mediated immunity, whereas B cells are primarily responsible for humoral immunity. We mainly focused on MHC class I potential peptides. YRIMLSVHG,VLIFLSTAV and MMLELDPPF,GLDFSDLYY are the most potent peptides predicted as epitopes for CD4+ and CD8+ T cells, respectively, whereas MMLELDPPF and GLDFSDLYY had the highest pMHC‐I immunogenicity score and these are further tested for interaction against the HLA molecules, using in silico docking techniques to verify the binding cleft epitope. However, this is an introductory approach to design an epitope‐based peptide vaccine against ZikV; we hope that this model will be helpful in designing and predicting novel vaccine candidates.
Keywords: artificial neural network, epitopes, Immune Epitope Database, immunogenomics, MHC class, Zika virus
Introduction
Zika virus (ZikV) has emerged as a mosquito‐borne virus that was first identified in the Zika forest of Uganda in 1947 in rhesus monkeys through a monitoring network of sylvatic yellow fever. It was subsequently identified in humans in 1952 at the same place (Uganda) followed by the United Republic of Tanzania. Outbreaks of ZikV disease have been recorded in Africa, the Americas, Asia and the Pacific1. In 2007, the first documented epidemicity of ZikV occurred in the Federated States of Micronesia where 185 suspected cases were reported, of which 49 were confirmed and 59 were considered probable.2 The world is now mobilizing to tackle the latest threat to global health security – ZikV, which is now spreading speedily in other parts of the world (Fig. 1) as mentioned in a CDC report of 9 March 2016 (http://www.cdc.gov/zika/about/overview.html). ZikV belongs to the Flaviviridae and the genus Flavivirus, and is therefore a relative of Dengue, Japanese encephalitis, West Nile and yellow fever viruses. Like other flaviviruses, ZikV is enveloped and icosahedral with a non‐segmented, single‐stranded, positive‐sense RNA genome 10 794 kb in length with two flanking non‐coding regions (5′ and 3′ NCR) and a single long open reading frame encoding a polyprotein: 5′‐C‐prM‐E‐NS1‐NS2A‐NS2BNS3‐NS4A‐NS4B‐NS5‐3′, that is cleaved into capsids (C), precursor of membrane (prM), envelope (E) and seven non‐structural (NS) proteins.3, 4 It is most closely related to the Spondweni virus and is one of the two viruses in the Spondweni virus clade.3, 5 People with ZikV infection usually have symptoms that can include mild fever, skin rashes, conjunctivitis, muscle and joint pain, malaise or headache. These symptoms are normally mild and last for 2–7 days. The incubation period of ZikV is not clear, but is likely to be a few days (WHO February 2016). Currently, there is no treatment/medicine or vaccine to cure ZikV infection and there are no good diagnostic tests, so the development of new therapeutic agents, vaccines or anti‐viral drugs against ZikV is very important. This development has not yet been reported, but a group of scientists from Bharat Biotech International Limited at Hyderabad in India have applied to patent a ZikV vaccine that is not yet approved by WHO. Integration of computational techniques provides a novel approach for integrating immunogenetics and immunogenomics with bioinformatics for the development of vaccines. This is known as vaccinomics.6 These approaches had previously been used to address the development of new vaccines against diseases such as multiple sclerosis,7 Dengue,8 malaria,9 influenza10 and tumours.11 However, these methods of vaccine development usually work through the identification of HLA ligands and T‐cell epitopes,12 which specify the selection of the potent vaccine candidates associated with the Transporter of Antigen Presentation (TAP) molecules.13, 14
Figure 1.

Depicts the most infected countries and territories with active transmission of the Zika virus (CDC_report).
The purpose of our present study is to promote the designing of a vaccine against ZikV using in silico methods, taking envelope glycoprotein of ZikV into consideration. The reason for choosing envelope glycoprotein is because of its function. These proteins are important for viral attachment to the host cell surface, and are also responsible for facilitating an immune response in the host cell. Therefore, we designed an epitope‐based peptide vaccine against ZikV using the vaccinomics approach, with an expectation that wet laboratory research will validate our prediction.
Materials and methods
The flow chart (see Supplementary material, Fig. S1) summarizes the steps followed to predict the most probable epitopes in the envelope glycoprotein of ZikV.
Sequence retrieval
The ZikV envelope glycoprotein is important for viral attachment and facilitating the immune response on the host cell surface. Determination of protein/peptide sequences is a basic requirement for biomedical research, so ZikV envelope glycoprotein sequences were obtained from UniProt (www.uniprot.org/) in fasta format.15
Sequence analysis
Sequence analysis is the process of subjecting a DNA, RNA or peptide sequence to any of a wide range of analytical methods to understand its features, function, structure or evolution. The BLASTp16 program screens homologous sequences from its database and selects those sequences that are more similar to our ZikV envelope glycoprotein; we also performed multiple sequence alignment, analysing the evolutionary divergence in the envelope glycoprotein of ZikV from other close species using MUSCLE (http://www.ebi.ac.uk/Tools/msa/muscle/), a multiple sequence alignment tool.17
Protein antigenicity prediction
To determine the most potent antigenic protein of the ZikV envelope glycoproteins, we used an online server vaxijen_v2.0,18 with a default threshold value. All the antigenic proteins of ZikV with their respective scores were obtained then sorted in excel. A single antigenic protein with maximum antigenicity scores was selected for further evaluation.
Protein secondary and tertiary structure prediction and validation
We predicted the secondary structure of the envelope glycoprotein of ZikV using an online server cfssp (Chou & Fasman Secondary Structure Prediction)19, 20 because the antigenic part of the protein is more likely to belong to the β‐sheet region.21 To confirm the predicted three‐dimensional (3D) structures of the selected peptides the pep‐fold Peptide Structure Prediction server22 was used but they were not all accessible for interaction. So, we predicted the 3D structure of our protein using I‐Tasser (Iterative Threading ASSEmbly Refinement).23 I‐Tasser introduced an ab initio modelling method using at least 10 templates to build a 3D structure of our protein. This structure was validated on the basis of basic parameters such as z‐score, which should lie between 1 and 5, C‐score is the confidence score and should be high, root‐mean‐square deviation (RMSD) is always considered ≤ 4 Å as appropriate, and in the Ramachandran plot maximum residues should be in the allowed region except for some like glycine (85–98% residues can be considered).24 Each generated 3D structure is then validated by procheck servers (http://services.mbi.ucla.edu/SAVES/), which check the stereo‐chemical properties of a protein 3D structure, resulting in a number of Post‐Script plots giving a detailed analysis of its respective 3D structure overall and of its residue‐by‐residue geometry. After analysing these plots we can infer the availability of the best structure generated by these tool(s). Once we have built the model, minimization of the energy level of this model is done using the swiss_pdb viewer tool.25 Pymol graphics26 was used to superimpose the predicted structure of peptides with the 3D structure of the ZikV envelope glycoprotein to check whether the peptide was within the accessible range for interaction with HLA molecules.
T‐cell epitope identification
CD8+ T‐cell epitope identification
For the identification of the T‐cell epitope, we used the netctl_1.2 online tool27 for epitope identification using a 0·95 threshold to maintain sensitivity and specificity of 0·90 and 0·95, respectively. The tool expands the prediction for 12 MHC‐I supertypes and integrates the prediction of peptide MHC‐I binding, proteasomal C‐terminal cleavage with TAP transport efficiency. These predictions were performed by an artificial neural network, weighted TAP transport efficiency matrix and a combined algorithm for MHC‐I binding and proteasomal cleavage efficiency was then used to determine the overall scores and translated into sensitivity/specificity. On the basis of this overall score, seven8 best peptides (epitopes) were selected for further evaluation.
For the prediction of peptides binding to MHC‐I, we used a tool from the Immune Epitope Database (IEDB) and calculate IC50 values for peptides binding to specific MHC‐I molecules.28 For the binding analysis, all the alleles were selected with word length of nine residues and binding affinity < 200 nm for further analysis. Another tool (named as MHC‐NP) provided by the IEDB server was used to assess the probability that a given peptide was naturally processed and bound to a given MHC molecule.29
Validation of approach
We repeated the analysis for validating the strategy (see Supplementary material, Fig. S2) on the well known haemagglutinin protein of a common influenza strain for the identification of CD8+ T‐cell epitopes. This analysis supported our approach of prediction and gave us reliability for considering the predicted peptides as potential candidate epitopes (see Supplementary material, Table S1). We independently searched each predicted epitope in the IEDB and found that many of them were the exact match whereas others had some variation in the position (start position and end position).
Epitope conservancy and immunogenicity prediction
Epitope conservancy tries to elucidate the degree of similarity between the epitope and the target (i.e. given) sequence. This property of epitope gives us the promise of its availability in a range of different strains. Hence for the analysis of the epitope conservancy, the web‐based tool from IEDB30 analysis resources was used. Immunogenicity prediction can uncover the degree of influence (or efficiency) of the respective epitope to produce an immunogenic response. The T‐cell class I pMHC immunogenicity predictor at IEDB, which uses amino acid properties as well as their position within the peptide to predict the immunogenicity of a class I peptide MHC (pMHC) complex.31
Allergenicity assessment
The prediction of allergenicity for our epitope is important for vaccine development. Allergenicity assessment aims to predict allergens and non‐allergens with high sensitivity and specificity, without compromising efficiency in classification of proteins with similar sequences to known allergens.32 We used the online tool AllerHunter server.33 This server predicts allergenicity through a combinational prediction, by using both incorporation of the Food and Agriculture Organization/WHO allergenicity evaluation system and Support Vector Machines pairwise sequence similarity. AllerHunter predicts allergens as well as non‐allergens with high specificity.
CD4+ T‐cell epitope identification
CD4+ T‐cell epitopes have an important task in eliciting strong protective immune responses during peptide (epitope)‐based vaccination. They also play a key role in humoral immunity by providing help to B cells, enabling effective antibody class switching and affinity maturation.34 The prediction of these epitopes focuses on the peptide‐binding process by MHC class II proteins.35 We predicted that all 129 possible epitopes with an IC50 value < 100 were considered as potential T‐cell epitopes. We used an online tool predivac (http://predivac.biosci.uq.edu.au) to predict these possible epitopes.
HLA and epitope interaction analysis using molecular docking studies
Epitope model generation
From the computationally predicted epitopes from the group of 110 different epitopes from the ZikV envelope glycoprotein sequence according to all MHC (A1‐B62) supertypes, only seven epochal peptide (epitopes) co‐ordinates were extracted from a modelled 3D structure of ZikV envelope glycoprotein and their energy levels were minimized using a swiss pdb viewer 25 then labelled using pymol. Those epitopes showing the higher probable score for further docking analysis were selected.
Retrieval of HLA allele molecules
Probable 3D structure of HLA alleles was retrieved from the Protein DataBank (PDB).36
Molecular docking and analysis
The predicted peptides (i.e. candidate epitopes) were found to bind in the groove of their respective HLA alleles. The docking study was performed using autodock 4.0,37 using an implementation of the Lamarckian genetic algorithm (GA), to model the peptide binding to HLA molecules.38 The output from 10 independent GA runs for each peptide was processed and that with the lowest binding affinity was considered, taking into consideration weighted terms for van der Waal's dispersion/repulsion, hydrogen bonding, electrostatics and dissolution interactions. The interactions were visualized with pymol version 1.7.4.4, a molecular graphics system (Schrödinger, LLC, Portland, OR). Molecular docking studies were required to show whether the target was binding to the specific region. In our case it validated results obtained from the epitope prediction workflow, i.e. its availability for binding (or its presence on the surface), and the predicted epitope was valid in inducing immune response.
B‐cell epitope identification
B lymphocytes are the cells that are differentiated into antibody‐secreting plasma cells and memory cells. The prediction of B‐cell epitopes was performed to find the potential antigen that provides an assurance of humoral immunity. IEDB were used to identify the B‐cell antigenicity including the classical propensity scale methods such as the Kolaskar and Tongaonkar antigenicity scales,39 Parker hydrophilicity prediction,40 Emini surface accessibility prediction,41 Karplus and Schulz flexibility prediction42 and the Chou and Fasman β‐turn prediction tool43 as the antigenic parts of a protein belong to the β‐turn regions.44 Parker hydrophilicity prediction was applied to all the hydrophilicity parameters extensively used in all of the algorithms to predict which amino acid residues were antigenic.40 The Bepipred linear epitope prediction45 tool uses a combinatorial algorithm comprising both hidden Markov model and propensity scale methods for antigenic propensity and so performs significantly better than any of the other methods.46
Results
Sequence retrieval and analysis
We retrieved the envelope glycoprotein of ZikV from the UniProt (ID:‐A0A060H177) database, as the glycoprotein was considered, and also predicted, to be the most immunogenic protein.47 Then we performed BLASTp for the envelope glycoprotein of ZikV, we found that 11 other viruses (Ilheus_virus, Rocio_virus, Alfuy_virus, Naranjal_virus, Aroa_virus, Bainyik_virus, Saint_Louis_encephalitis_virus, Japanese_encephalitis_virus, Murray_encephalitis_virus, West Nile viruses and Spondweni_virus) have similar homologues with > 55% identical sequences. Multiple sequence alignment was performed to identify regions of similarity that may be a consequence of functional, structural, or evolutionary relationships among their sequences. We divided all viruses into two groups for good alignment results. In the first group, we noted that the residues C 3, G 5, R 9, D 10, F 11, E 13, G 14, S 16, G 17, T 19, W 20, D 22, V 24, L 25, E 26, C 30, T 32, M 34, D 37, K 38, P 39, T 40, D 42, Y 61, C 75, P 76, T 77, G 79, E 80, K 85, D 87, C 92, D 98, R 99, G 100, W 101, G 102, N 103, G 104, C 105, G 106, L 107, F 108, G 109, K 110, G 111, S 123, T 115, C 116, A 117, K 118, R 119,C 212, G 127, I 130, E 133, Y 137, V 143, H 144, P 175, G 182, G 185, C 191, E 192, R 194, G 196, Y 203, T 206, K 210, L 213, V 214, H 215, W 218, F 219, D 221, L 224, P 225, W 226, W 237, E 241, L 243, E 245, F 246, H 250, A 251, Q 254, L 259, G 260, S 261, Q 262, E 263, G 264, H 267, A 269, L 270, A 271, G 272, A 273, S 287, G 288, H 287, L 290, K 291, C 292, R 293, K 295, K 298, K 302, G 303, Y 306, C 309, F 313, F 315, P 319, T 322, H 324, G 325, T 326, E 330, Y 333, G 334, G 337, P 338, C 339, P 342, G 357, R 358, T 361, N 363, P 364, N 372, K 374, E 378, P 381, P 382, F 383, G 384, D 385, S 386, Y 387, I 388, G 391, G 393, E 397, H 399, H 400, H 401, W 402, H 403 were totally conserved and residues 1, 4, 12, 21, 23, 31, 33, 35, 37, 41, 45, 51, 52, 53, 65, 68, 69, 70, 72, 80, 81, 86, 90, 91, 113, 120, 134, 135, 136, 139, 141, 155, 159, 164, 169, 171, 172, 175, 184, 189, 190, 195, 197, 198, 205, 207, 212, 215, 217, 222, 228, 232, 244, 244, 248,255, 257, 267, 274, 285, 296, 297, 299, 301, 305, 320, 321, 329, 331, 334, 337, 342, 353,354, 360,368, 366, 367, 375, 377, 380, 390, 391,398, 401, and 406 were strongly similar in ZikV, Spondweni_virus, St_L_Enc_Virus, West_Nile_viruses, Japanese_encephalitis_virus and Murray_encephalitis_virus. In a second group, residues C 121, I 130, E 133, N 134, Y 137, K 167, P 174, G 181, G 184, C 190, E 191, P 192, R 193, L 196, D 197, Y 202,V 213, W 217, D 220 L 223, P 224, W 236, N 238 E 244, F 245, H 249,A 250, Q 253, V 255, L 258, Q 261, E 262, G 263, A 269, L 270, G 272, A 273, G 290, H 291, L 292, K 293, C 294, R 295, K 300, K 304, G 305 were totally conserved and residues 124, 135, 139, 143, 154, 156, 169, 170, 180, 184, 189, 190, 195, 205, 206, 209, 211, 212, 217, 218, 220, 224, 242, 243, 247, 256, 264, 265, 266, 273, 287, 289, 296, 298, 299, 301 and 303 were strongly similar in ZikV, Naranjal_virus, Aroa_virus, Bainyik_virus, Alfuy_virus, Ilheus_virus and Rocio_virus. On analysing multiple sequence alignment carefully with respect to epitopic conservancy and comparing it with the epitope prediction, we found 15 highly conserved information blocks (shown as yellow rectangles) that inhabit almost all epitopes (see Fig. 2).
Figure 2.

Multiple sequence alignment, showing the fully conserved region highlighted in yellow blocks; identical and similar regions are indicated by ‘*’ and ‘:’, respectively, including all species. Moreover, the rectangles designate the predicted T‐cell epitope regions in two colours: red (CD8+) and green (CD4+).
Antigenic prediction
We predicted the most potent antigenic protein of ZikV envelope glycoprotein using an online server vaxijen_v2.0, which is based on auto‐cross covariance transformation of protein sequences into uniform vectors of principal amino acid properties.18 The overall antigenic prediction score was 0·6178 (probable antigen) at 0·4 threshold value.
Protein structure prediction and validation
The secondary structure of protein from its amino acid sequence describes the α‐helix, β‐sheets and random coil. Our ZikV envelope glycoprotein is 504 residues long, of which 180 residues(35·7%) form sheet, 61 residues form turn and 305 residues (60·5%) form helix regions of the protein (Fig. 3). In the secondary structure, 35·7% region of the target protein remains as β‐sheet. In several experiments, it was shown that the antigenic part of the protein was more likely to belong to the β‐sheet region.21 For the docking analysis, the 3D structures of the selected peptides were designed (see Fig. 4) using the pep‐fold Peptide Structure Prediction server that searches for known 3D protein structures from PDB that are homologous to the epitope source sequence. Keeping this in mind we carried out homology modelling of full ZikV glycoprotein instead of peptide. The 3D structure built by the I‐tasser used the top three templates 3J65A, 3J27A and 4CCTA of 10 PDB templates. The average confidence score of the predicted model was 1·50; the z‐scores were 2·96, 4·11 and 3·55 for the top three templates, respectively; and the average RMSD of our predicted structure was 0·481 from the top three templates. The Ramachandran plot for our model showing residues in the allowed region was > 85% (Fig. 5). Verified 3D48 predicted that 87·50% of the residues had an average 3D score of 0·2 for the best predicted model and at least 80% of the amino acids should have scored ≥ 0·2 in the 3D/1D profile. We superimposed predicted peptides with the our modelled 3D structure and found that mostly epitopes were in the accessible area of proteins, which means that interactions occur easily.
Figure 3.
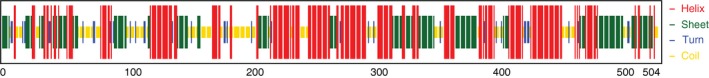
Represents the composition of secondary structure from amino acid residues of Zika virus envelope glycoprotein. Only 35·7% residues form sheet, 60·5% form helices and 3·8% residues form the turn region.
Figure 4.
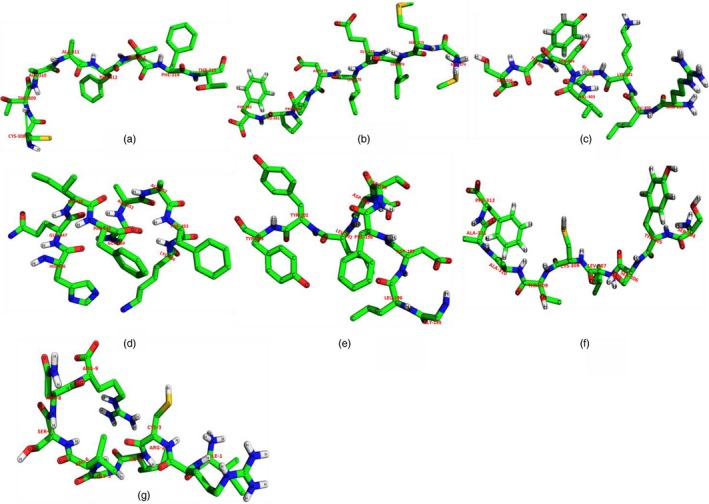
Modelled peptide structure of seven CD8+ T‐cell epitopes represented by sequence as (a) CTAAFTFTK, (b) MMLELDPPF, (c) RLKGVSYSL, (d) HQIFGAAFK, (e) GLDFSDLY, (f) SYSLCTAAF, (g) IRCIGVSNR, respectively.
Figure 5.

(a) Modelled three‐dimenasional structure of Zika virus envelope glycoprotein; (b) Top three templates superimposed with our modelled protein with an average of RMSD = 0·481; (c) Ramachandran plot of our modelled protein showing the residues in allowed the region.
T‐cell epitope identification
CD8+ T‐cell epitope identification
The NetCTL server predicted 110 different epitopes in ZikV envelope glycoprotein but only seven most potential peptides were selected, based on their high combinatorial score, for further analysis. Using the MHC‐I binding prediction tool, which is based on SMM, we chose those MHC‐I alleles for which the epitopes showed higher affinity (IC50 < 200 nm). For cleaving of the peptide bonds, so converting the protein into peptide, proteasomes played a key role. The peptide molecule allied with class I MHC molecules and the peptide–MHC molecule after proteasomal cleavage were transported to the cell membrane where they were presented to T helper cells. The total score of each epitope–HLA interaction was considered and a higher score meant higher processing efficiency. The two peptides E1 and E2 (MMLELDPPF and GLDFSDLYY) among seven were found to interact with most of the MHC‐I alleles, including HLA‐A*32:01, HLA‐B*53:01, HLA‐A*02:06, HLA‐B*35:01, HLA‐A*02:01, HLA‐A*29:02, HLA‐A*23:01, HLA‐C*05:01, HLA‐C*14:02, HLA‐B*15:01, HLA‐C*53:01, HLA‐A*01:00, HLA‐C*12:03 and HLA‐C*03:03. The MHC‐NP prediction tool29 was used to find the highest probable score of our predicted peptides – both E1 and E2 had highest probable scores of 0·7516 and 0·6756, respectively, for HLA‐B*53:01. All the predicted peptides had maximum identity for conservancy hit and 100% maximum identity was found, except for one epitope which had identity of 44·44%. I‐pMHC immunogenicity prediction49 analysis of epitopes E1 and E2 had the highest I‐pMHC immunogenicity scores of 0·7356 and 0·96115, respectively (Table 1).
Table 1.
The seven potential CD8+ T‐cell epitopes along with their interacting MHC class I alleles and total processing score, epitopes conservancy_hits and pMHC‐I immunogenicity score
| Epitopes | Position | NetCTL Combined score | Epitope_Conservancy_Hit (MAX. Identity %) | MCH‐I interaction with an affinity of IC50 < 200 and the total score (proteasome score, TAP score, MHC‐I score, processing score) | pMHC‐I immunogenicity score |
|---|---|---|---|---|---|
| CTAAFTFTK | 308–316 |
0·8605; B62 0·9834; A3 |
100 | HLA‐A*68:01; 7·79 (0·2) | 0·08405 |
| HLA‐A*11:01; 10.00(0·2) | |||||
| HLA‐C*12:03; 43·15 (−6·7) | |||||
| HLA‐C*03:03; 67·08 (0·42) | |||||
| HLA‐A*03:01; 68·99 (0·4) | |||||
| HLA‐A*31:01; 196·55 (2·4) | |||||
| MMLELDPPF | 374–382 |
0·9556; A2 0·9841; A24 |
100 | HLA‐B*35:01; 12·11 (0·3) | 0·7356 |
| HLA‐C*14:02; 18·31 (2·8) | |||||
| HLA‐A*32:01; 19·54 (0·3) | |||||
| HLA‐B*15:01; 29·58 (0·2) | |||||
| HLA‐A*02:01; 112·49 (1·8) | |||||
| HLA‐B*53:01; 96·43 (0·4) | |||||
| HLA‐A*29:02; 126·49 (0·8) | |||||
| HLA‐C*03:03; 67·08 (0·42) | |||||
| HLA‐A*23:01; 164·22 (0·6) | |||||
| HLA‐B*15:02; 7·76 (1·17) | |||||
| HLA‐A*02:06; 24·19 (0·3) | |||||
| RLKGVSYSL | 299–307 |
1·1578; A2 1·5559; B8 |
44·44 | HLA‐C*12:03; 21·15 (27) | 0·07322 |
| HLA‐A*32:01; 36·55 (0·3) | |||||
| HLA‐C*03:03; 111·84 (56) | |||||
| HLA‐A*30:01; 156·66 (2·4) | |||||
| HLA‐B*08:01; 166·17 (0·6) | |||||
| HQIFGAAFK | 446–456 |
1·2774; A3 0·9725; B62 |
100 | HLA‐C*03:03; 29·55 (21) | 0·5754 |
| HLA‐C*12:03; 29·87 (43) | |||||
| HLA‐A*30:01; 97·27 (1·6) | |||||
| HLA‐A*68:01; 138·19 (2·1) | |||||
| HLA‐A*11:01; 180·43 (1·4) | |||||
| GLDFSDLYY | 195–203 |
3·844; A1 0·9025; A3 |
100 | HLA‐A*29:02; 35·54 (0·3) | 0·96115 |
| HLA‐C*14:02; 57·76 (11) | |||||
| HLA‐A*01:01; 110·8 (0·2) | |||||
| HLA‐C*05:01; 4·25 (0·5 | |||||
| HLA‐C*12:03; 10·67 (12) | |||||
| HLA‐B*53:01; 43·76 (0·51) | |||||
| SYSLCTAAF | 304–312 |
1·4265; A24 0·8502; B62 |
100 | HLA‐C*14:02; 7·76 (1·2) | 0·08405 |
| HLA‐C*03:03; 59·1 (39) | |||||
| HLA‐A*24:02; 85·83 (0·3) | |||||
| HLA‐C*07:02; 92·29 (3·4) | |||||
| HLA‐C*12:03; 93·16 (96) | |||||
| HLA‐A*23:01; 104·58 (0·4) | |||||
| IRCIGVSNR | 1–9 |
1·7210; A1 0·657; B27 |
100 | HLA‐C*12:03; 38·92 (61) | 0·5675 |
| HLA‐C*07:02; 124·5 (5·2) | |||||
| HLA‐C*14:02; 128·71 (29) | |||||
| HLA‐C*07:01; 199·6 (9·4) |
Allergenicity assessment
The allergenicity prediction was precisely calculated using the AllerHunter tool and predicted the query sequence as non‐allergen with a score of 0·01 with 91·67% sensitivity and 89·3% specificity.
CD4+ T‐cell epitope identification
For the identification of MHC class II potential peptide epitopes, we used the MHC class II binding prediction tool predivac, which predicted 129 epitopes for HLA‐DRB_1 with IC50 < 100. After analysis of all the possible epitopes, we found that of 129 epitopes only two – YRIMLSVHG and VLIFLSTAV – interacted with mostly HLA_DRB_1 and could act as potential CD4+ T‐cell epitopes (Table 2).
Table 2.
Presents two most potential CD4+ T‐cell epitopes along with their interacting MHC class II alleles with affinity IC50 < 100 and PREDIVAC scores
| Position | Epitopes | Interacting MHC class II allele with an affinity of IC50 < 100 (IC50 values; PREDIVAC score) |
|---|---|---|
| 314–322 | FTKIPAETL |
HLA‐DRB1*15:01; 14 (89·35), HLA‐DRB1*01:01; 16 (83·76) HLA‐DRB1*04:04; 37 (87·34), HLA‐DRB1*07:01; 80 (82·86) HLA‐DRB1*04:01; 81 (87·34) |
| 137–145 | YRIMLSVHG |
HLA‐DRB1*01:01; 14 (94·08), HLA‐DRB1*15:01; 40 (81·01) HLA‐DRB1*04:01; 60 (87·88), HLA‐DRB1*04:04; 61 (91·01) |
Molecular docking studies for HLA and epitopes interaction analysis
To ensure the interaction between HLA molecules and our predicted potential epitopes, we performed molecular docking using autodock 4.0. Among all the MHC class 1 alelles, only HLA‐A*53:01 had a maximum probable score for our most potent epitopes E1 and E2. The crystal structure of HLA‐A*53:01 molecules retrieved by PDB. As a result we found that both the epitopes E1 and E2 interacted with HLA‐A*53:01 with strong binding affinities of −8·34 kcal/mol and −8·1 kcal/mol, respectively (Fig. 6 and 7). The epitope E1 is bound in the groove of the HLA‐B*53:01 molecules with residues ASN‐63, GLN‐70, ASN‐80, TYR‐84 and THR‐143 forming regular hydrogen bonds whereas residues TYR‐7, TYR‐9, ILE‐66, THR‐73, SER‐77, TYR‐99 and GLU‐163 form bonds as a result of sharing electrons (which may happen as a result of charge distribution) between oxygen atoms with covalent characters. E2 interact with the residues GLN‐70, SER‐77 and ASN‐80 forming regular hydrogen bonds while residues ILE‐66, THR‐73, ASP‐74, TYR‐84, SER‐143, TYR‐116, ASN‐114 and GLU‐163 form bonds as a result of sharing electrons (which may happen due to charge distribution) between oxygen atoms with covalent characters.
Figure 6.

Represents the epitope MMLELDPPF, which binds in the groove of the HLA‐B*53:01 molecules. Residues ASN‐63, GLN‐70, ASN‐80, TYR‐84 and THR‐143 form regular hydrogen bonds while residues TYR‐7, TYR‐9, ILE‐66, THR‐73, SER‐77, TYR‐99 and GLU‐163 form bonds as a result of electron sharing (which may happen due to charge distribution) between oxygen atoms with covalent characters.
Figure 7.

Epitope MMLELDPPF binding in the groove of the HLA‐B*53:01 molecules. Residues GLN‐70, SER‐77 and ASN‐80 form regular hydrogen bonds while residues ILE‐66, THR‐73, ASP‐74, TYR‐84, SER‐143, TYR‐ 116, ASN‐114 and GLU‐163 form bonds as a result of sharing electrons (may happen due to charge distribution) between oxygen atoms with covalent characters.
B‐cell epitope prediction
We predict B‐cell epitope identification, here using an amino acid scale‐based method. We used different analysis methods for the prediction of a continuous B‐cell epitope.
The Emini surface accessibility prediction method analysed the surface accessibility. The average surface accessibility was 1·0106 and minimum 0·074, and region 159–164 was found to have the maximum surface accessibility with a score of 6·347.
For antigenicity prediction we used the the Kolaskar and Tongaonkar antigenicity prediction method, which evaluated the analysed antigenicity on the basis of the physicochemical properties of amino acids and their abundances in experimentally known epitopes. The average antigenic propensity of our ZikV envelope glycoprotein was 1·034 with a maximum of 1·05 and minimum 0·966. The set threshold value for determination of antigenicity was 1. Region 163–172 residues were found to have more propensity for antigenicity.
The Chaus and Fasman β‐turn prediction method predicts the β‐turn. The β‐turns are frequently accessible and significantly hydrophilic in nature. The properties possessed by the β‐turns are also applicable to the antigenic regions of the protein.50 The region 151–161 residues were considered as a β‐turn region. It has been proved experimentally that the flexibility of the peptide is correlated to antigenicity. The Karplus and Schulz flexibility prediction method predicts an average flexibility of 0·9537 and minimum of 0·883 and the region 159–167 was found to be the most flexible with a maximum score of 1·140. The Parker Hydrophilicity Prediction tool predicts the hydrophilicity of ZikV envelope glycoprotein with an average score of 1·46, minimum 0·074 and the region 154–160 was found to be 7·23 with maximum hydrophilicity.
Finally, Bepipred linear epitope prediction was used, which is based on a Hidden Markov model, the best single method for predicting linear B‐cell epitopes. We predicted that the peptide sequences from 157 to 166 amino acids were capable of inducing the desired immune response as B‐cell epitopes. After the comparative and cross reference analysis, we found that the region between 150 and 175 amino acid residues in the ZikV envelope glycoprotein was capable of inducing the desired immune response as B‐cell epitopes.(Table 3) (Fig. 8).
Table 3.
Combined B‐cell linear epitope prediction showed the region from 150 to 170 amino acid residues have the highest antigenic propensity for B‐cell linear epitopes in Zika virus envelope glycoprotein
| Method | Region | Residues | Score | ||
|---|---|---|---|---|---|
| Max. | Avg. | Min. | |||
| Chou & Fasman Beta‐Turn Prediction | 152–161 | IVNDTGHETD | 1·205 | 1·098 | 1·0055 |
| Emini Surface Accessibility Prediction | 159–164 | ETDENR | 6·347 | 1·015 | 0·074 |
| Karplus & Schulz Flexibility Prediction | 159–167 | ETDENRAK | 1·104 | 0·955 | 0·875 |
| Kolaskar & Tongaonkar Antigenicity | 163–170 | NRAKVEI | 1·054 | 1·003 | 0·966 |
| Parker Hydrophilicity Prediction | 154–160 | NDTGHET | 7·234 | 1·464 | ‐6·100 |
| Bepipred Linear Epitope Prediction | 157–166 | GHETDENRAK | 1·884 | 0·027 | ‐2·366 |
Figure 8.

Combined B‐cell linear epitope prediction showed the region from 150 to 170 amino acid residues had the highest antigenic propensity for B‐cell linear epitopes. Surrounded by six differently coloured lines, which cover the region 150–170 amino acid residues in Zika virus envelope glycoprotein, each line indicating different analysis methods with the maximum scores.
Discussion
Vaccine development is a long, complex process, often lasting 10–15 years and requiring a combination of public and private involvement. With the advancement of sequence‐based technology, we have gained information about the proteomics and genomics of the different viruses. Vaccine development for ZikV is based on screening of multiple epitopes with the most antigenic properties that direct the immune system to protect human beings from ZikV infection The purpose of our study was to screen new, high‐potential immunogenic epitopes for T cells because vaccines against T‐cell epitopes are more promising as they evoke a long‐lasting immune response, and because with antigenic drift, an antigen can easily escape the memory response of antibody.51 We have designed a peptide‐based vaccine using various bioinformatics tools and a vaccinomics approach. This in silico approach has already been used to address the development of new vaccines for combating diseases like multiple sclerosis, Dengue, malaria, influenza and tumours. In the present study we focused on MHC class I potential peptide epitopes only, and we also identified MHC class II and B‐cell epitopes in ZikV envelope glycoprotein. There are many criteria that need to be fulfilled by a vaccine candidate epitope, and the predicted seven most potent peptides fulfilled all the criteria. The initial criterion is the conservancy of the epitopes, which was measured by the IEDB conservancy analysis tool and we found 100% maximum identity of all except one epitope, which had only 44·44% identical. Of these seven peptides, only MMLELDPPF and GLDFSDLYY were found to interact with most of the MHC class I alleles, but HLA‐B*53:0 had the highest probable score for both peptides (Table 4).
Table 4.
Results obtained after docking of epitopes (MMLELDPPF and GLDFSDLYY) with HLA‐B*53:01 molecules
| Potential epitopes | ∆Gb | H‐bonds | Other Forces involved | Contact residues |
|---|---|---|---|---|
| MMLELDPPF (E1) | −8·34 kcal/mole | 5 | Hydrogen bonds, Hydrophobic and π‐π interactions | TYR‐7, TYR‐9, ASN‐63, ILE‐66, GLN‐70, THR‐73, SER‐77, ASN‐80, TYR‐84, TYR‐99, THR‐143, GLU‐163 |
| GLDFSDLYY (E2) | −8·1 kcal/mole | 3 | ILE‐66, GLN‐70, THR‐73, ASP‐74, SER‐77, ASN‐80, TYR‐84, SER‐143, TYR‐116, ASN‐114 |
However, in vaccine development, allergenicity is a prominent hurdle. Today, most vaccines stimulate the immune system into an ‘allergic’ reaction, through induction of type 2 T helper cells and immunoglobulin E. Here, our proposed peptide has allergenicity scores of 0·01 for both, and so it was considered as a non‐allergen. Molecular docking studies are required to confirm that the target is binding to the specific region. In our case it validated our results obtained from the epitope prediction workflow, i.e. its availability for binding (meaning its presence on the surface), to show if the predicted epitope is valid in inducing an immune response. We have found promising binding of ≤ −8·0 kcal/mol affinity of binding with HLA antigen, which verifies the binding cleft epitope interaction with the HLA molecule when it is applied in vivo. T‐cells work to transfer information from the inflammation site by recognizing pathogens using signatures (often called epitope), so providing cell‐mediated innate immunity. So, the identification of CD4+ T‐cell epitopes is important and we found 129 epitopes for HLA‐DRB_1 with an IC50 < 100. After filtration criterion is applied, only two epitopes (YRIMLSVHG and VLIFLSTAV) were found to interact with most of the HLA_DRB_1 molecules and could be considered as potential CD4+ T‐cell epitopes. The identified B‐cell epitopes in the ZikV envelope glycoprotein were from region 150–170 amino acid residues. This region is identified as being most capable of inducing the desired immune response as B‐cell epitopes and also important for developing peptide vaccine. This region has the capability to induce the desired immune response and so can be used by immunologists as a B‐cell epitope.
At present, vaccines are mostly based on B‐cell immunity. But recently, vaccine based on the T‐cell epitope has been encouraged because the host can generate a strong immune response by CD8+ T cells against the infected cell. Further, our results (similarity analysis and epitope prediction) showed that the epitopes we predicted were found conserved in all the selected viruses during the course of evolution (presented in multiple sequence alignment) and noticed a high degree of similarity with their results.52 This implies that the predicted candidate epitopes (peptide) will be considered as a broad‐spectrum potential vaccine if developed.
This type of in silico study has recently received experimental validation,53 they identified a multi‐epitope cluster secretory protein of Mycobacterium tuberculosis (Ag85B) that bound to 15 HLA class I and three class II molecules and later their prediction was experimentally validated in vitro. Comparing this with our study, we found that we have followed the same in silico approach but in some cases were more specific in the selection of MHC class I and class II molecules. Khan et al. had chosen MHC‐I and MHC‐II alleles for which the epitopes showed higher affinity (IC50 < 500 nm) but in our case we selected those peptides that showed higher affinity (IC50 < 200 nm and IC50 < 100 nm) for MHC‐I and MHC‐II alleles, respectively, because the immunogenic property of each predicted T‐cell epitope was characterized by its IC50 value, which indicates the peptide's binding affinity to HLA molecules and the number of corresponding restricting HLA alleles. Peptides with lower IC50 values showed good inhibition.54 We also predicted immunogenicity and allergenicity assessment of the peptides. All predictions were that high specificity would be maintained, which we can define by a stringent threshold value. Additionally, we identified that B‐cell epitopes can potentially guide experimental epitope mapping and may also be valuable for the interpretation of results from experiments based on antibody affinity binding such as ELISA, radioimmunoassay and Western blotting. We have suggested that the proposed epitopes would be able to trigger an efficacious immune response as a peptide vaccine in vivo.
Conclusion
Immunoinformatics has emerged as a promising field for predicting epitope agents. Viruses (such as ZikV, human immunodeficiency virus and Ebola virus) elicit both the humoral and T‐cell immunity. Hence, our analysis infers the epitopes (we found say E1, E2) that help to promote immunity against ZikV. This is done in a sense that as soon as the virus tries to attach to the host cell, the peptide (as vaccine) will recognize it and present this information to a broad spectrum of protector cells (T and B cells). As our epitopes elicit the sense of interaction to both CD8 and CD4 they can mimic antigen presentation and so help antibody formation inside the host. In this way these computational approaches save both the expenditure and the time needed to screen a large number of possible epitopes compared with experimental techniques and also guide the experimental work with high confidence of finding the desired results.
Disclosure
The authors report no conflicts of interests in this work.
Supporting information
Figure S1. Flow chart showing the complete Epitope prediction protocol.
{kind=link}
Figure S2. Flow chart showing repeated analysis for validation.
{kind=link}
Table S1. Predicted immunogenic sequences of hemagglutinin protein of a common influenza strain.
Acknowledgements
The authors are grateful to the Centre for Interdisciplinary Research in Basic Sciences (CIRBSc), Jamia Millia Islamia‐110025 for providing the research infrastructure and we gratefully acknowledge the IEDB team. Shahnwaz Ali and Md Zubbair Malik are supported by ICMR‐SRF PhD fellowship.
References
- 1. WHO , Zika situation report. URL http://www.who.int/emergencies/zika-virus/situation-report/10-march-2016/en/ 2016.
- 2. Samarasekera U, Triunfol M. Concern over Zika virus grips the world. Lancet 2016; 387:521–4. [DOI] [PubMed] [Google Scholar]
- 3. Faye O, Freire CCM, Iamarino A, Faye O, de Oliveira JVC, Diallo M et al Molecular evolution of Zika Virus during its emergence in the 20th century. Bird B, editor. PLoS Negl Trop Dis 2014; 8:e2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kuno G, Chang G‐JJ. Full‐length sequencing and genomic characterization of Bagaza, Kedougou, and Zika viruses. Arch Virol 2007; 152:687–96. [DOI] [PubMed] [Google Scholar]
- 5. Fields BN, Knipe DM, Howley PM. Fields' virology, 5th edn Philadelphia: Wolters Kluwer Health/Lippincott Williams & Wilkins, 2007:2 [Google Scholar]
- 6. Poland GA, Ovsyannikova IG, Jacobson RM. Application of pharmacogenomics to vaccines. Pharmacogenomics 2009; 10:837–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bourdette DN, Edmonds E, Smith C, Bowen JD, Guttmann CRG, Nagy ZP et al A highly immunogenic trivalent T cell receptor peptide vaccine for multiple sclerosis. Mult Scler 2005; 11:552–61. [DOI] [PubMed] [Google Scholar]
- 8. Tambunan USF. In silico analysis of envelope dengue virus‐2 envelope dengue Virus‐3 Protein as the backbone of dengue virus tetravalent vaccine by using homology modeling method. OnLine J Biol Sci 2009; 9:6–16. [Google Scholar]
- 9. López JA, Weilenman C, Audran R, Roggero MA, Bonelo A, Tiercy JM et al A synthetic malaria vaccine elicits a potent CD8(+) and CD4(+) T lymphocyte immune response in humans. Implications for vaccination strategies. Eur J Immunol 2001; 31:1989–98. [DOI] [PubMed] [Google Scholar]
- 10. Shahsavandi S, Ebrahimi MM, Sadeghi K, Mahravani H. Design of a heterosubtypic epitope‐based peptide vaccine fused with hemokinin‐1 against influenza viruses. Virol Sin 2015; 30:200–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Knutson KL, Schiffman K, Disis ML. Immunization with a HER‐2/neu helper peptide vaccine generates HER‐2/neu CD8 T‐cell immunity in cancer patients. J Clin Invest 2001; 107:477–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Petrovsky N, Brusic V. Computational immunology: The coming of age. Immunol Cell Biol 2002; 80:248–54. [DOI] [PubMed] [Google Scholar]
- 13. Brusic V, Bajic VB, Petrovsky N. Computational methods for prediction of T‐cell epitopes–a framework for modelling, testing, and applications. Methods 2004; 34:436–43. [DOI] [PubMed] [Google Scholar]
- 14. Nielsen M, Lundegaard C, Lund O, Keşmir C. The role of the proteasome in generating cytotoxic T‐cell epitopes: insights obtained from improved predictions of proteasomal cleavage. Immunogenetics 2005; 57:33–41. [DOI] [PubMed] [Google Scholar]
- 15. Apweiler R. UniProt: the universal protein knowledgebase. Nucleic Acids Res 2004; 32:D115–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol 1990; 215:403–10. [DOI] [PubMed] [Google Scholar]
- 17. Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 2004; 32:1792–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Doytchinova IA, Flower DR. VaxiJen: a server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinformatics 2007; 8:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chou PY, Fasman GD. Prediction of protein conformation. Biochemistry (Mosc) 1974; 13:222–45. [DOI] [PubMed] [Google Scholar]
- 20. Chou PY, Fasman GD. Conformational parameters for amino acids in helical, β‐sheet, and random coil regions calculated from proteins. Biochemistry (Mosc) 1974; 13:211–22. [DOI] [PubMed] [Google Scholar]
- 21. Hasan A, Hossain M, Alam J. A computational assay to design an epitope‐based peptide vaccine against Saint Louis encephalitis virus. Bioinforma Biol Insights 2013; 7:347–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Thévenet P, Shen Y, Maupetit J, Guyon F, Derreumaux P, Tufféry P. PEP‐FOLD: an updated de novo structure prediction server for both linear and disulfide bonded cyclic peptides. Nucleic Acids Res. 2012; 40:W288–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yang J, Yan R, Roy A, Xu D, Poisson J, Zhang Y. The I‐TASSER Suite: protein structure and function prediction. Nat Methods 2014; 12:7–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Aftab ALAM. In Silico analysis of mmpl gene family of mycobacterium tuberculosis: a novel target for anti–TB drugs. Glob J 2016; 5:212–9. [Google Scholar]
- 25. Johansson MU, Zoete V, Michielin O, Guex N. Defining and searching for structural motifs using DeepView/Swiss‐PdbViewer. BMC Bioinformatics 2012; 13:173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schr″odinger LLC. (2015). The PyMOL molecular graphics system. URL http://www.schrodinger.com/pymol/ February 2016.
- 27. Larsen MV, Lundegaard C, Lamberth K, Buus S, Lund O, Nielsen M. Large‐scale validation of methods for cytotoxic T‐lymphocyte epitope prediction. BMC Bioinformatics 2007; 8:424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Buus S, Lauemøller SL, Worning P, Kesmir C, Frimurer T, Corbet S et al Sensitive quantitative predictions of peptide‐MHC binding by a “Query by Committee” artificial neural network approach. Tissue Antigens 2003; 62:378–84. [DOI] [PubMed] [Google Scholar]
- 29. Giguère S, Drouin A, Lacoste A, Marchand M, Corbeil J, Laviolette F. MHC‐NP: predicting peptides naturally processed by the MHC. J Immunol Methods 2013; 400–401:30–6. [DOI] [PubMed] [Google Scholar]
- 30. Bui H‐H, Sidney J, Li W, Fusseder N, Sette A. Development of an epitope conservancy analysis tool to facilitate the design of epitope‐based diagnostics and vaccines. BMC Bioinformatics 2007; 8:361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Moutaftsi M, Peters B, Pasquetto V, Tscharke DC, Sidney J, Bui H‐H et al A consensus epitope prediction approach identifies the breadth of murine T(CD8+)‐cell responses to vaccinia virus. Nat Biotechnol 2006; 24:817–9. [DOI] [PubMed] [Google Scholar]
- 32. Muh HC, Tong JC, Tammi MT. AllerHunter: A SVM‐pairwise system for assessment of allergenicity and allergic cross‐reactivity in proteins. Rapallo F, editor. PLoS One 2009; 6:e5861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Muh HC, Tong JC, Tammi MT. AllerHunter: A SVM‐pairwise system for assessment of allergenicity and allergic cross‐reactivity in proteins. Rapallo F, editor. PLoS One 2009; 4:e5861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fraser CC, H. Altreuter D, Ilyinskii P, Pittet L, LaMothe RA, Keegan M et al Generation of a universal CD4 memory T cell recall peptide effective in humans, mice and non‐human primates. Vaccine 2014; 32:2896–903. [DOI] [PubMed] [Google Scholar]
- 35. Oyarzún P, Ellis JJ, Bodén M, Kobe B. PREDIVAC: CD4+ T‐cell epitope prediction for vaccine design that covers 95% of HLA class II DR protein diversity. BMC Bioinformatics 2013; 14:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Berman HM. The protein data bank. Nucleic Acids Res 2000; 28:235–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS et al AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J Comput Chem 2009; 30:2785–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Patronov A, Dimitrov I, Flower DR, Doytchinova I. Peptide binding prediction for the human class II MHC allele HLA‐DP2: a molecular docking approach. BMC Struct Biol 2011; 11:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kolaskar AS, Tongaonkar PC. A semi‐empirical method for prediction of antigenic determinants on protein antigens. FEBS Lett 1990; 276:172–4. [DOI] [PubMed] [Google Scholar]
- 40. Parker JM, Guo D, Hodges RS. New hydrophilicity scale derived from high‐performance liquid chromatography peptide retention data: correlation of predicted surface residues with antigenicity and X‐ray‐derived accessible sites. Biochemistry (Mosc) 1986; 25:5425–32. [DOI] [PubMed] [Google Scholar]
- 41. Emini EA, Hughes JV, Perlow DS, Boger J. Induction of hepatitis A virus‐neutralizing antibody by a virus‐specific synthetic peptide. J Virol 1985; 55:836–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Karplus PA, Schulz GE. Prediction of chain flexibility in proteins: A tool for the selection of peptide antigens. Naturwissenschaften 1985; 72:212–3. [Google Scholar]
- 43. Chou PY, Fasman GD. Empirical predictions of protein conformation. Annu Rev Biochem 1978; 47:251–76. [DOI] [PubMed] [Google Scholar]
- 44. Rini J, Schulze‐Gahmen U, Wilson I. Structural evidence for induced fit as a mechanism for antibody‐antigen recognition. Science 1992; 255:959–65. [DOI] [PubMed] [Google Scholar]
- 45. Larsen JEP, Lund O, Nielsen M. Improved method for predicting linear B‐cell epitopes. Immunome Res 2006; 2:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Haste Andersen P, Nielsen M, Lund O. Prediction of residues in discontinuous B‐cell epitopes using protein 3D structures. Protein Sci 2006; 15:2558–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Roehrig JT, Mathews JH, Trent DW. Identification of epitopes on the E glycoprotein of Saint Louis encephalitis virus using monoclonal antibodies. Virology 1983; 128:118–26. [DOI] [PubMed] [Google Scholar]
- 48. Eisenberg D, Lüthy R, Bowie JU. VERIFY3D: assessment of protein models with three‐dimensional profiles. Methods Enzymol 1997; 277:396–404. [DOI] [PubMed] [Google Scholar]
- 49. Calis JJA, Maybeno M, Greenbaum JA, Weiskopf D, De Silva AD, Sette A et al Properties of MHC class I presented peptides that enhance immunogenicity. PLoS Comput Biol 2013; 9:e1003266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Rose GD, Gierasch LM, Smith JA. Turns in peptides and proteins. Adv Protein Chem 1985; 37:1–109. [DOI] [PubMed] [Google Scholar]
- 51. Chiou S‐S, Fan Y‐C, Crill WD, Chang R‐Y, Chang G‐JJ. Mutation analysis of the cross‐reactive epitopes of Japanese encephalitis virus envelope glycoprotein. J Gen Virol 2012; 6:1185–92. [DOI] [PubMed] [Google Scholar]
- 52. Xu X, Sette A, Peters B. Computational analysis of Zika virus: Flavivirus antibody epitope data mapped onto the Zika virus proteome suggest potential shared and unique epitopes. URL www.iedb.org/downloader.php?file_name=doc/ZIKV_report_1.2.pdf 2016.
- 53. Khan MK, Zaman S, Chakraborty S, Chakravorty R, Alam MM, Bhuiyan TR et al In silico predicted mycobacterial epitope elicits in vitro T‐cell responses. Mol Immunol 2014; 61:16–22. [DOI] [PubMed] [Google Scholar]
- 54. Lundegaard C, Lamberth K, Harndahl M, Buus S, Lund O, Nielsen M. NetMHC‐3.0: accurate web accessible predictions of human, mouse and monkey MHC class I affinities for peptides of length 8‐11. Nucleic Acids Res 2008; 36:W509–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Flow chart showing the complete Epitope prediction protocol.
Figure S2. Flow chart showing repeated analysis for validation.
Table S1. Predicted immunogenic sequences of hemagglutinin protein of a common influenza strain.