

Abstract
Osteoarthritis is a serious disease of articular cartilage. The pathogenic factors contributing to this disorder are inflammation, extracellular matrix degradation and failure to rebuild the articular cartilage. Preclinical studies suggest that microRNA-140 may play a protective role in osteoarthritis development, but little is known about the mechanism by which this occurs. Here we present the results of forced expression of microRNA-140 in an in vitro model of osteoarthritis, evaluated by global proteomics analysis. We show that inflammation was reduced through the altered levels of multiple proteins involved in the nuclear factor of kappa light polypeptide gene enhancer in B-cells 1 pathway. microRNA-140 upregulated many of the components involved in the synthesis of hyaline extracellular matrix and reduced the levels of aggrecanases and syndecan 4, thus potentially both increasing cartilage repair and reducing cartilage breakdown. These results show how forced expression of microRNA-140 is likely to counteract all three pathogenic processes, and support the idea that intra-articular injection of microRNA-140 may benefit patients suffering from early osteoarthritis.
Introduction
Articular cartilage covers the end of opposing bones in synovial joints. It provides smooth and almost frictionless movement of the bones against each other, and protection from damage by load and tensile forces. These properties are provided by a unique combination of extracellular matrix (ECM) molecules, of which the most important are type 2 collagen (COL2), the proteoglycan aggrecan (ACAN) and the glycosaminoglycan chondroitin sulfate (CS).1 Osteoarthritis (OA) is a disease that leads to gradual degradation of the articular cartilage ECM resulting in pain, stiffness and swelling of affected joints. OA is the most common form of arthritis and one of the major causes of disability in the western world, and it constitutes a huge economic burden for the society.2 Currently, no treatment has been shown to stop or reverse the progression of OA. In many patients, the end result will be joint replacement. There are several known risk factors associated with OA such as joint injury, inflammation, age, obesity, genetics and gender, but the molecular mechanisms behind OA are not fully understood. However, several of these pathogenic factors are thought to act through the increased secretion of interleukin 1β (IL1β) into the joint space.3 IL1B is an inflammatory mediator that acts through the nuclear factor of kappa light polypeptide gene enhancer in B-cells 1 (NFKB) pathway to induce expression of many genes that are upregulated in OA cartilage such as IL1B, IL6, IL8, and the matrix degrading enzymes matrix metalloprotein 13 (MMP13) and a disintegrin-like and metallopeptidase with thrombospondin type 1 motif 5 (ADAMTS5), responsible for degradation of COL2 and ACAN, respectively.4
microRNAs (miRNAs) are also dysregulated in OA cartilage. miRNAs are small double stranded RNA molecules that regulate gene expression by binding to complementary sequences in mRNA molecules, leading to either degradation of the mRNA or repression of translation.5,6 This makes miRNA therapeutics a promising treatment option for several diseases.7,8 microRNA-140 (miR-140) is regarded as a cartilage specific miRNA because it is predominantly expressed in cartilaginous tissues during development.9,10 Knockout studies have revealed miR-140 to be an important factor in OA development, as miR-140-/- mice showed OA-like changes such as accelerated proteoglycan loss and ECM degradation, while mice overexpressing miR-140 were protected against degradation of proteoglycans and COL2 in a model of antigen-induced arthritis.11 In the same study the aggrecanase ADAMTS5 was shown to be a target of miR-140, and it was suggested that this could explain the protective role of miR-140 in OA.
In this study we investigated the role of miR-140-5p (hereafter referred to as miR-140) using an in vitro model of OA. Human articular chondrocytes (ACs) derived from OA cartilage overexpressing miR-140 were cultured in pathophysiologically relevant concentrations of recombinant human IL1B (rhIL1B) and analyzed using real-time quantitative polymerase chain reaction (RT-qPCR), western blot and global proteomics. Mass spectrometry-based proteomics was used because miRNAs can regulate gene expression at the protein level without affecting mRNA levels. We show that that forced expression of miR-140 increased protein levels of SOX9, ACAN and chondroitin sulfate N-acetylgalactosaminyltransferase 1 (CSGALNACT1), an enzyme involved in the synthesis of CS. Decreased expression was found for a range of inflammation mediators, including IL1B, IL6 and IL8. Likewise, signaling through the NFKB pathway is likely to be inhibited by increased expression of the NFKB inhibitor IKBA. Finally, cartilage degradation may be reduced by the possible downregulation of ADAMTS5 and the increase in syndecan 4 (SDC4), an inhibitor of ADAMTS5 activation. Thus, miR-140 may act through many pathways to counteract the development of OA.
Results
Establishing an in vitro model of IL1B-induced OA
To investigate the protective role of miR-140 in OA, the in vitro model of adding rhIL1B to cell cultures was first optimized and validated by dose-response and time course analysis. Figure 1a,b shows that 0.1 ng/ml of rhIL1B induced near maximal transcription levels of IL1B, IL8, and MMP13 in ACs (i) and bone-marrow-derived mesenchymal stem cells (ii), often used in OA and cartilage research because of their chondrogenic potential. This concentration of rIL1B was used for the rest of the experiments.
Figure 1.

Upregulation of OA-associated genes in response to rhIL1B. (a) RT-qPCR analysis of IL1B, IL8, and MMP13 mRNA levels after stimulating ACs with increasing doses of rhIL1B. (b) RT-qPCR analysis of IL1B, IL8, and MMP13 mRNA levels after stimulating bone-marrow-derived mesenchymal stem cells (BM-MSCs) with increasing doses of rhIL1B. (c) Expression of IL1B, IL6, IL8, and MMP13 during 48 hours of stimulation with rhIL1B 0.1 ng/ml. (d) Expression of IL1B, IL6, IL8, and MMP13 during 7 days of stimulation with rhIL1B 0.1 ng/ml. Cells used in a, b, c, and d are from four different donors. Numbers in parentheses indicate relative fold change values relative to time zero or the first detectable value. N.D, not detected; OA, osteoarthritis; IL1B, interleukin 1β; RT-qPCR, real time-quantitative polymerase chain reaction; rhIL1B, recombinant human IL1B; MMP13, matrix metalloprotein 13.
In a time course experiment, rhIL1B induced maximum expression level for IL1B, IL6, and IL8 at 5–8hours and for MMP13 at 20 hours in ACs (Figure 1c). IL1B, IL6, and IL8 mRNA levels were gradually decreased over 1 week in response to continuous stimulation of rhIL1B, but were still upregulated compared with unstimulated cells (Figure 1d). In contrast, MMP13 mRNA levels decreased to baseline values between days 2 and 5.
Proteome changes following rhILB1 stimulation
IL1B activates the NFKB pathway which subsequently induces synthesis of IL1B, IL6, IL8, MMP13, and ADAMTS5.4 To validate the upregulation of these proteins, and also look for other proteins that might change following rhIL1B stimulation, mass spectrometry-based proteomics was performed on ACs from three donors. The cells were stimulated with rhIL1B for 20 hours before harvesting for analysis. IL1B, IL6, and MMP13 were confirmed to be upregulated at the protein level (Supplementary dataset 1, sheet 1). There was also a 56-, 12- and 4-fold upregulation of IL8 for the three donors, but this was not found to be statistically significant. As for all the previous experiments the mRNA for the respective proteins was upregulated in the three donors, including IL8 (see Supplementary Figure S1). In total, 91 proteins were upregulated. Several of them were involved in the NFKB pathway such as TNF receptor associated factor 1 and the NFKB transcription factors v-rel avian reticuloendotheliosis viral oncogene homolog (REL) and RELB. A total of 78 proteins were downregulated (see Supplementary dataset 1, sheet 2). The inhibitor of NFKB, NFKBIB (IKB-β), was also downregulated (see Supplementary dataset 1 for all differently expressed proteins). A gene ontology (GO) term analysis was performed, and showed that the upregulated proteins were involved in inflammatory responses, regulation of signaling, cell adhesion, carbohydrate and unsaturated fatty acid metabolism, and cartilage development while the downregulated proteins were mainly involved in metabolic processes, cell death, and cell cycle (Supplementary dataset 2, sheet 1 and 2). These results showed that addition of 0.1 ng/ml of rhIL1B had a dramatic effect on the ACs and confirmed translation of IL1B, IL6, and MMP13 mRNAs into proteins.
miR-140 inhibits mediators of inflammation and cartilage degradation and upregulates chondrogenic proteins
We proceeded to test the effect of forced expression of miR-140 in this system by electroporation of a synthetic miR-140 mimic into monolayer expanded ACs on day 23 of culture. Previously we showed that liposomal transfection induced immune responses in MSCs and ACs. In contrast, no immune response was observed after electroporation.12 Electroporation was therefore used in this study. Two days after electroporation miR-140 increased ~ 120–140 fold in the miR-140 electroporated cells compared with controls for all three donors (Figure 2a). The levels of miR-140 after electroporation corresponded perfectly to the endogenous levels of miR-140 in native cartilage (Figure 2a, donors 1 and 3). We and others have recently shown RALA, ADAMTS5, and BMP2 to be targets of miR-140. These were all reduced by miR-140 (Figure 2b), showing that the electroporation of miR-140 gave functionally appropriate results. One day after electroporation rhIL1B was added to the cells for 20 hours and RT-qPCR was used to analyze the expression of IL1B, IL6, IL8, OA-relevant chemokine (C-C motif) ligand 5 (CCL5), and ADAMTS4 mRNA.13,14 All genes except MMP13 were downregulated by miR-140 (Figure 3a). The synovial membrane is also known to be inflamed in OA patients, and synovial fibroblasts (SFs) are thought to contribute to OA development by secreting inflammatory cytokines. It is therefore desirable that miR-140 reduce inflammation also in the SFs. Figure 3b shows that miR-140 also inhibited IL1B, IL6, and IL8 in rhIL1B-stimulated SFs. Again MMP13 was not affected. This is consistent with previous findings where treatment with anti-miR-140 did not affect MMP13 protein15
Figure 2.

Functional analysis of miR-140 2 days after electroporation. (a) RT-qPCR analysis of miR-140 levels in ACs electroporated with a negative control sequence (Control), miR-140 and in native articular cartilage. (b) Relative expression of RALA, BMP2, and ADAMTS5 in control cells and miR-140 electroporated cells. Cells from three donors were used in a and b. U6 and GAPDH were used as a reference genes in a and b, respectively. In a and b the cells were stimulated for 20 hours before analysis. In c the cells were stimulated for 0, 30 minutes, 1, 3, 5, 8, 20, and 48 hours. In d the cells were stimulated for 1, 2, 5, and 7 days. Min, minutes; h, hours. RT-qPCR, real time-quantitative polymerase chain reaction; ACs, articular chondrocytes.
Figure 3.

Results of miR-140 overexpression in rhIL1B stimulated ACs. The cells were electroporated with a negative control sequence or miR-140. The following day the cells were stimulated with rhIL1B for 20 hours before analysis. RT-qPCR analysis of IL1B, IL6, IL8, CCL5, ADAMTS4, and MMP13 in ACs (a) and synovial fibroblasts (SFs) (b). Numbers in parentheses indicate fold change values relative to results for control transfection plus IL1B stimulation. Differentially shaded bars indicate the different genes. IL1B, interleukin 1β; RT-qPCR, real time-quantitative polymerase chain reaction; rhIL1B, recombinant human IL1B; MMP13, matrix metalloprotein 13; ACs, articular chondrocytes.
We proceeded with proteomics analysis to look for differentially expressed proteins in the miR-140 transfected ACs. Mass spectrometry analysis showed that miR-140 decreased 43 and increased 114 proteins (Supplementary dataset 1, sheet 3, and 4). Selected proteins are listed in Table 1. IL1B and IL6 were downregulated, consistent with the RT-qPCR results. IL8 was downregulated 1.9-, 3.2-, and 17.5-fold in the three donors, but this was not statistically significant. IL8 is therefore not included in the lists. Several other proteins involved in inflammation, NFKB signaling and OA were downregulated (Table 1 and Figure 4). In particular, annexin 6 (ANXA6), E3 ubiquitin-protein ligase ZFP91 (ZFP91), MAGE family member D1 (MAGED1), tripartite motif containing 4 (TRIM4), and DEAD-box helicase 41 (DDX41) are known to be involved in the activation of the NFKB pathway. This suggested that miR-140 inhibited inflammation by inhibiting the NFKB pathway. IKB is known to be the most potent inhibitor of the NFKB pathway. IKBA was found to be upregulated in donor 2 and 3 only, and thus is not on the list of significantly upregulated proteins. As activation of the NFKB pathway happens within minutes after stimulation, we performed western blotting to look at the IKBA levels at early time points. Two AC donors were used, including donor 1 that was not significantly upregulated according to the proteomics (Figure 5a). In both donors IKBA was increased in miR-140-electroporated unstimulated cells and after 30 minutes of rIL1B stimulation. IKBA was also increased after 20 hours, but with minor differences in donor 2. This shows that miR-140 inhibited activation of NFKB also by increasing IKBA. Finally, integrin alpha 2 (ITGA2), known to facilitate inflammatory degradation of articular cartilage, was greatly reduced (Table 1).16 The GO term analysis showed that the downregulated proteins were associated with regulation of response to stimulus, biological adhesion, cell activation, regulation of vascular endothelial growth factor production, and hyaluronan metabolic process (Supplementary dataset 2, sheet 3).
Table 1. Differentially expressed proteins after miR-140 transfection and rhIL1B stimulation.
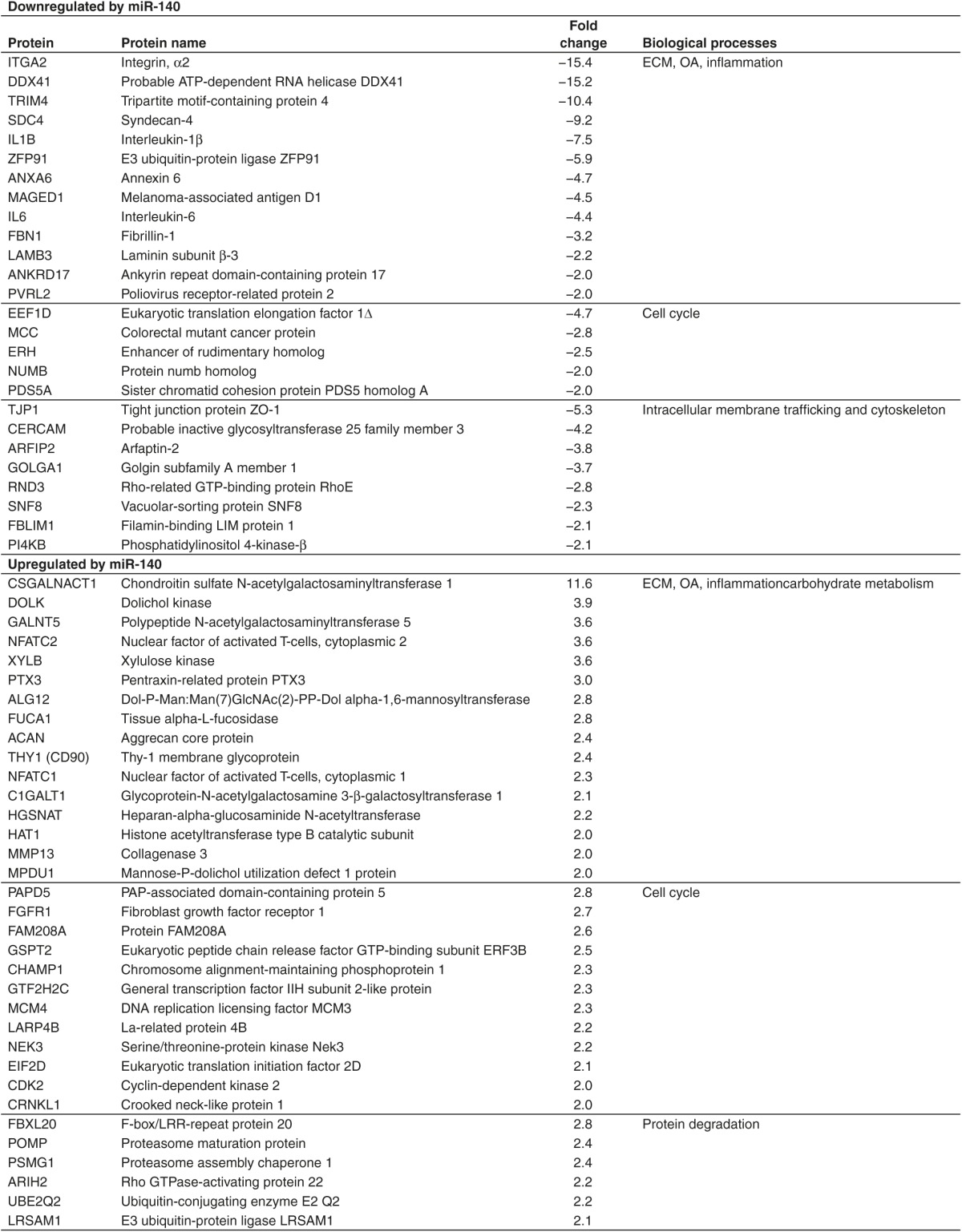
Figure 4.

Impact of miR-140 overexpression on proteins involved in the NFKB signaling pathway. NFKB activators (upper right corner) and target genes (in the nucleus) of NFKB were downregulated by miR-140, while two inhibitors were upregulated (upper left corner). IL1B binds to IL1R1. This activates IRAK kinases and the IKK complex which phosphorylates the NFKB inhibitor IKB. Phosphorylated IKB is then ubiquitinated and degraded in the proteasome. This releases NFKB. NFKB1 or -2 heterodimerizes with REL, RELA or RELB, translocates into the nucleus and turns on transcription of NFKB target genes. IL1B, interleukin 1β.
Figure 5.

Western blot analysis of inflammation and chondrogenesis proteins. (a) IKB protein levels were analyzed in unstimulated cells, after 30 minutes (min) and 20 hours (h) following rhIL1B stimulation. (b) IL1B, IL6, IL8, ACAN, SOX9, and CSGALNACT1 in cells electroporated with control or miR-140 for 1 day and subsequently stimulated with rhIL1B for 20 hours. ACTB was used as loading control in a and b. (c) RT-qPCR analysis of ACAN, SOX9, and CSGALNACT mRNA levels after electroporation with miR-140 and stimulation with rhIL1B. D1, Donor 1; D2, Donor 2; D3, Donor 3. IL1B, interleukin 1β; RT-qPCR, real time-quantitative polymerase chain reaction; rhIL1B, recombinant human IL1B.
Of the upregulated proteins, many are known to play important roles in ECM and carbohydrate metabolism, OA, inflammation, cell cycle, and protein degradation (Table 1). Among the most interesting upregulated proteins were ACAN, CSGALNACT1, nuclear factor of activated T-cells (NFAT)C1, NFATC2, MMP13, and histone acetyltransferase type B catalytic subunit (HAT1). ACAN is the most important proteoglycan in articular cartilage while CSGALNACT1 is very important for synthesis of CS and normal development of articular cartilage in mice.17,18,19 In mice, loss of NFATC1 and NFATC2 leads to OA.20,21 HAT1 is an inhibitor of NFKB signaling.22
Previously, we showed that inhibition of miR-140 reduced both ACAN and SOX9 protein levels.23 However, SOX9 was not differently expressed in the proteomic analysis. In addition, IL8 was not significantly downregulated. SOX9 and IL8 were therefore included as a part of the western blot validation experiments for the proteomic analysis (Figure 5b). The results confirmed downregulation of IL1B, IL6, and IL8; and upregulation of ACAN, SOX9, and CSGALNACT1 (two known isoforms) in all the three donors. Interestingly, the upregulation of ACAN, SOX9, and CSGALNACT1 protein occurred in the absence of similar changes in mRNA levels (Figure 5c). ADAMTS5 mRNA was reduced following miR-140 transfection (Figure 2b), but ADAMTS5 was not detected in the proteomics analysis, presumably for technical reasons. Western blotting of ADAMTS5 showed a number of bands, of which only the smallest band (25 kDa) was downregulated in two of the three donors (Figure 5b). However, SDC4 binds to ADAMTS5 and has been shown to be essential for the activation of ADAMTS5.24 SDC4 was greatly reduced following miR-140 transfection (Table 1), which could lead to reduced ADAMTS5 activity in the absence of greatly reduced ADAMTS5 protein. According to GO term analysis the upregulated proteins were mainly associated with different metabolic processes, protein modification, cellular component organization, and development process (Supplementary dataset 2, sheet 4).
Several of the downregulated proteins are predicted targets of miR-140 (Table 2), which suggests that downregulation of these proteins is mediated through the canonical pathways for miRNA bioactivity. Three such predicted targets are the mRNAs transcribed from the IL1B, IL6, SDC4 genes. To validate these mRNAs as targets for miR-140, luciferase reporter constructs containing the 3′-untranslated region (UTR) of IL1B, IL6, SDC4 or three control constructs containing the 3′UTR of ACTB, a random 3′UTR and a vector without 3′UTR (EMPTY), were cotransfected into HEK293 cells together with a negative control miR or miR-140 mimic (Figure 6). A lower luciferase signal was detected for IL1B, IL6, and SDC4 in the miR-140 transfected cells, but not in the three control vectors. This shows that IL1B, IL6, and SDC4 are targets of miR-140. We also performed a search in the PITA database to assess the binding strength (represented as an energetic score-ddG) of the 3′UTR of IL1B, IL6, and SDC4 to the miR-140 seed sequence. The binding strength scores were moderate and comparable to values from other validated targets of miR-140 (Supplementary dataset 3).
Table 2. Proteins that were downreguated by miR-140 and are predicted targets of miR-140.
Figure 6.

microRNA luciferase reporter assays. Luciferase reporter constructs containing the indicated 3′UTR and negative control miR (control) and miR-140 mimic were cotransfected into Lenti-X 293T cells. After 24 hours, the luciferase signal from the cells was detected by photon counting. Results were calculated from technical triplicates (n = 3) and shown as mean ± SD. 3′-UTR, 3′-untranslated region.
Discussion
OA is a serious disease that impacts on the life of a considerable proportion of the adult human population. The pathogenic factors contributing to this disorder are inflammation, ECM degradation and failure to rebuild the articular cartilage. miR-140 has been shown in animal models to protect against arthritis, in part through the downregulation of ADAMTS5. Using an in vitro model of OA we now extend this insight by showing that forced expression of miR-140 inhibits the NFKB signaling pathway and downstream inflammatory cytokines, possibly inhibits ECM degradation proteins and enhances levels of proteins essential for the regeneration of new hyaline cartilage. Recently it was shown that intra-articular injection of miR-140 ameliorated rheumatoid arthritis in mice.25 The current results support the notion that intra-articular injection of miR-140 in patients with asymptomatic or early stage OA may prevent the onset of full-blown disease.
The in vitro model of adding rhIL1B to ACs is a well-established model. In this study, the concentration of rhIL1B added was 1/100 of the concentration frequently used by others. Where 0.1 ng/ml corresponds to the concentration of IL1B found in the synovial fluid of OA patients,26 and it is therefore a pathophysiologically relevant concentration. It is also similar to the concentration of IL1B found to be secreted following sterile trauma to other tissues.27
To simulate the effect of intraarticular injection, we transfected miR-140 into ACs in the rhIL1B induced OA model. The rationale behind this was several fold: loss of miR-140 has been shown to induce OA in knockout mice, overexpression of miR-140 in transgenic mice has been shown to protect against collagen-induced arthritis and miR-140 expression has been found to be reduced in cartilage and synovial fluid of human OA patients.11,28,29,30 These observations all suggest that overexpression of miR-140 in cells within the joints could benefit patients with OA, but they have not contributed much to our understanding of how this might work. In this study, we used global proteomics to look at the effect of miR-140. This is likely to be particularly important, since miRNAs can affect protein levels in the absence of mRNA changes.
IL1B activates the NFKB pathway. Our data suggest that miR-140 inhibits this pathway because the NFKB target genes IL1B, IL6, IL8, CCL5, ADAMTS4, and ADAMTS5 were inhibited at the mRNA level, whereas IL1B, IL6, and IL8 were also downregulated at the protein level. IL1B, IL6, and SDC4 were also formally identified and validated as targets of miR-140 by luciferase reporter assays. The most obvious mechanisms for inhibition of the NFKB pathway would be through direct degradation of IL1B, IL6, and SDC4 mRNA by miR-140 and the demonstrated upregulation of IKBA, known to bind NFKB and inhibit downstream signaling. However, our current results suggest that also other mechanisms may be involved. HAT1 was upregulated by miR-140. This protein forms a repressor complex that inhibits NFKB signaling.22 In addition, miR-140 downregulated a number of other proteins involved in the NFKB pathway. ANXA6 is increased in human OA cartilage. This protein interacts with RELA to stimulate NFKB activity. Loss of ANXA6 leads to reduced cartilage destruction in mouse OA.31,32,33 ZFP91 is involved in the activation of NFKB2,34 while MAGED1 and TRIM4 have both been shown to activate NFKB.35,36 Knocking down DDX41 inhibits activation of NFKB37 (Table 1 and Figure 4). These data suggest that miR-140 inhibits the NFKB pathway at several different points of attack. Finally, miR-140 transfection led to greatly reduced levels of ITGA2. This may impact on the inflammatory component of OA pathogenesis, because ITGA2 knockout mice showed reduced inflammatory cartilage degradation and clinical symptoms in two models of rheumatoid arthritis.16
Forced expression of miR-140 also led to upregulation of proteins of essential importance for neochondrogenesis (Table 1 and Figure 5b). The transcription factor SOX9 is often called the master regulator of chondrogenesis, because it regulates the transcription of the essential cartilage ECM molecules COL2 and ACAN. Inhibition of miR-140 during in vitro chondrogenesis of MSCs inhibited both SOX9 and ACAN expression.23 This was confirmed in this study, where overexpression of miR-140 led to increased synthesis of SOX9. This could explain the increased ACAN levels. However, the minor increase in ACAN mRNA compared with the huge increase in ACAN protein suggests that post-transcriptional mechanisms may also be involved. Another important chondrogenesis molecule upregulated by miR-140 was CSGALNACT1. This enzyme is involved in the synthesis of CS in hyaline cartilage.17 The importance of CSGALNACT1 is perhaps best demonstrated in studies of endemic degenerative OA and primary OA, two disorders where OA occurs without any known predisposing factor. In both these conditions CSGALNACT1 was considerably down-regulated.19 In another study, CSGALNACT1 knockout mice were found to have thinner articular cartilage with more degraded ACAN compared with wildtype mice.18 This suggests that upregulation of CSGALNACT1 following miR-140 transfection will be an important mechanisms for regeneration of the cartilage. CSGALNACT1 mRNA was not changed, again indicating post-transcriptional mechanisms for the increase in CSGALNACT1 protein. Two other molecules, upregulated following miR-140 transfection, are known to be extremely important for cartilage health: NFATC1 and NFATC2. Originally described to be transcription factors involved in immune responses in T-cells, these molecules have recently been implicated in OA pathogenesis. First, Nfatc2-/- mice were shown to exhibit normal skeletal development but to display loss of COL2 and ACAN, overexpression of MMP1A, MMP13, and ADAMTS5 and inflammatory cytokines IL1B, IL6, and IL17. These mice showed gradual development of OA changes between 12 and 24 months.21 When mice with cartilage specific conditional deletion of Nfatc1 were bred onto the Nfatc2-/- strain, OA developed much earlier with COL2 and ACAN degradation and osteophyte formation.20 Based on these data it seems reasonable to assume that upregulation of NFATC1 and NFATC2 will counteract the development of human OA.
It is possible that overexpression of miR-140 also inhibits cartilage ECM degradation. Both ADAMTS4 and ADAMTS5 were downregulated at the mRNA level. Neither ADAMTS4 nor ADAMTS5 were in the list of detectable proteins from the proteomics analysis, presumably for technical reasons. ADAMTS5 western blot showed slightly reduced intensity of a 25 kD band in two of three donors, consistent with the size of the catalytic domain, but no reduced intensity of other detectable bands. However, SDC4 was greatly reduced by miR-140. SDC4 has been shown to control the activation and aggrecanase activity of ADAMTS5. Loss of SDC4 protects against cartilage degradation in vivo.24 Thus, miR-140 overexpression may lead to reduced aggrecanase activity through downregulation of SDC4 without greatly affecting ADAMTS5 protein levels. There was a twofold upregulation of MMP13 by miR-140. This may suggest increased degradation of COL2 following miR-140 electroporation. However, MMPs seem unable to degrade COL2 in the presence of an intact proteoglycan network.38,39,40 Thus, the collagen network may be protected against MMP13 degradation by miR-140 via upregulation of ACAN and synthesis of CS by CSGALNACT1. Li et al. recently reported that miR-140 downregulates MMP13 protein in murine chondrocytes.41 This contrasts with our results. The explanation may be differences between species, but it could also be due to the use of different reagents and transfection methods. In our experiments we have used electroporation and synthetic miRNA mimics, while Li et al. used lipofection and DNA plasmids. Previously, we showed that lipofection alone can induce immune repsonses in human ACs and MSCs.12 Further studies are needed to investigate the effect of miR-140 on the synthesis and function of cartilage degrading enzymes.
In this study, we have mainly looked into individual proteins that are known to be involved in inflammation, OA, and cartilage biology. GO term analysis showed that proteins downregulated as a result of forced expression of miR-140 resulted in terms associated with immune and inflammatory processes such as regulation of response to stimulus, biological adhesion, and cell activation while the upregulated proteins were associated with metabolic processes, protein modification, cellular component organization, and cell cycle. This is in accordance with the proteins we manually picked to make Table 1.
One final aspect of these results deserves consideration: the mechanism of action of miR-140 overexpression. The canonical hypothesis of miRNA bioactivity says that nucleotides 2–8, termed the seed sequence of the miRNA binds by base-pairing to a target sequence, frequently located in the 3′-UTR of the mRNA. The results will be either sequestration or degradation of the mRNA or repression of protein translation, both leading to reduced levels of the target protein. Based on the seed sequence of miR-140, target mRNAs can be predicted and validated. Of the 43 proteins downregulated following miR-140 overexpression in this study, only 14 were predicted to be targets for miR-140, and several of these satisfied only some of the algorithms. We validated IL1B, IL6, and SDC4 to be targets of miR-140, but the mechanism for the downregulation of the rest of the 40 proteins, and for the upregulation of all the 114 upregulated proteins remains to be explained. One possibility is that one or more of the predicted and/or validated targets, impacts on the synthesis of all the other differentially expressed proteins described here. This is possible; some of the validated and predicted targets listed in Table 2 are involved in signal transduction or protein/protein interactions and the NFKB pathway. However, a surprisingly large proportion of the differentially expressed proteins following miR-140 overexpression were likely to have a beneficial effect on OA pathogenesis through several mechanisms and many different points of action. This opens up to the possibility that miR-140 overexpression also exerts its bioactivity in an organ- and disease specific manner.
Materials and methods
All reagents used in this study are listed in Supplementary Table S1.
Isolation and culture of cells. ACs, bone-marrow-derived mesenchymal stem cells and SFs were isolated and cultured in monolayer as previously described.23,42 The Regional Committee for Medical Research Ethics, Southern Norway approved the study and all donors gave written consent.
Electroporation and stimulation with recombinant IL1B. 1 × 106 monolayer expanded ACs and SFs were electroporated with 5 μmol/l of synthetic Pre-miR miRNA precursor negative control and Pre-miR miRNA-140-5p precursor (Thermo Fisher Scientific, Waltham, MA) in a total volume of 100 μl using the Amaxa nucleofection system according to the Human Chondrocyte Nucleofector Kit (Lonza, Walkersville, MD). The Pre-miR miRNA precursors are small (21 nucleotides) double-stranded RNA molecules designed to mimic mature miRNAs. The Pre-miR miRNA precursors are chemically modified to ensure that only one of the strands is functional. Thus, miR-140-3p is not functional in the assay used in this study. The dose of the transfected miR-140-5p was based on dose-response experiments using the positive control miR-1 and its target PTK9. One day after electroporation, cells were stimulated with 0.1 ng/ml rhIL1B (R&D systems, Minneapolis, MN) for 20 hours before analysis. Nonelectroporated cells were stimulated with rhIL1B for 20 hours.
Total RNA/miRNA isolation, cDNA synthesis, and RT-qPCR. Total RNA containing miRNAs were isolated using the miRNeasy mini kit according to the protocol from the manufacturer (Qiagen, Germantown, MD). RNA purity and quality was checked by Nanodrop and agarose gel electrophoresis, respectively. cDNA synthesis and RT-qPCR were performed following protocols from the manufacturer using the High Capacity cDNA Reverse Transcription Kit and the Taqman MicroRNA Reverse Transcription Kit (Thermo Fisher Scientific). For mRNA and miRNA analysis 200 ng in a total volume of 20 µl and 10 ng in a total volume of 15 µl of total RNA, respectively, was reverse transcribed into cDNA. All samples were run in technical triplicates. For mRNA analysis, each replicate contained 1.0 µl cDNA in a total volume of 25 µl, while 1.33 cDNA in a total volume of 20 µl was used for miRNA analysis. The thermocycling parameters were 95°C for 10 minutes followed by 40 cycles of 95°C for 15 seconds and 60°C for 1 minute. GAPDH was used as endogenous control for mRNA analysis while U18 were used for miRNA analysis. RT-qPCR results are a shown as relative fold changes using mean values from technical triplicates with a 95% confidence interval. All donors are shown separately in the figures.
Western blotting. Cell lysates corresponding to 250,000 cells were loaded onto a 4–20% gradient polyacrylamide gel (Biorad, Hercules, CA). Proteins were separated be gel electrophoresis, transferred to PVDF membranes and incubated with appropriate antibodies before visualizing the bands using the myECL imager (Thermo Fisher Scientific). ACTB was used as loading control for all western blotting experiments. In Figure 5b all proteins were developed on the same membrane except for ADAMTS5 which was developed on a separate membrane. The membrane was stripped and reprobed using the Restore stripping buffer from Thermo Scientific. The loading control in Figure 5b is representative for both membranes.
Mass spectrometry. All experiments were performed on an Easy nLC1000 nano-LC system connected to a quadrupole–Orbitrap (QExactive) mass spectrometer (ThermoElectron, Bremen, Germany) equipped with a nanoelectrospray ion source (EasySpray/Thermo). For liquid chromatography separation we used an EasySpray column (C18, 2 µm beads, 100 Å, 75 μm inner diameter) (Thermo) capillary of 25 cm bed length. The flow rate used was 0.3 μl/min, and the solvent gradient was 5% B to 30% B in 240 minutes, then 90% B wash in 20 minutes. Solvent A was aqueous 0.1% formic acid, whereas solvent B was 100% acetonitrile in 0.1% formic acid. Column temperature was kept at 60°C. The mass spectrometer was operated in the data-dependent mode to automatically switch between MS and MS/MS acquisition. Survey full scan MS spectra (from m/z 400 to 1,200) were acquired in the Orbitrap with resolution R = 70,000 at m/z 200 (after accumulation to a target of 3,000,000 ions in the quadruple). The method used allowed sequential isolation of the most intense multiply-charged ions, up to 10, depending on signal intensity, for fragmentation on the HCD cell using high-energy collision dissociation at a target value of 100,000 charges or maximum acquisition time of 100 ms. MS/MS scans were collected at 17,500 resolution at the Orbitrap cell. Target ions already selected for MS/MS were dynamically excluded for 30 seconds. General mass spectrometry conditions were: electrospray voltage, 2.0 kV; no sheath and auxiliary gas flow, heated capillary temperature of 250°C, normalized HCD collision energy 25%. Ion selection threshold was set to 1e4 counts. Isolation width of 3.0 Da was used.
Protein identification and label-free quantitation. MS raw files were submitted to MaxQuant software version 1.4.0.8 (Max-Planck Institute for Biochemistry, Am Klopferspitz 18, D-82152 Martinsried, Germany) for protein identification.43 Parameters were set as follow: protein N-acetylation, methionine oxidation and pyrogluatamte conversion of Glu and Gln as variable modifications. First search error window of 20 ppm and mains search error of 6 ppm. Trypsin without proline restriction enzyme option was used, with two allowed miscleavages. Minimal unique peptides were set to 1, and false discovery rate (FDR) allowed was 0.01 (1%) for peptide and protein identification. Label-free quantitation was set with a retention time alignment window of 3 minutes. The Uniprot human database was used (download from December 2013). Generation of reversed sequences was selected to assign FDR rates. Normalized label-free quantification (LFQ) intensity values were imported into the R software and paired t-tests were used to identify differently expressed proteins. P < 0.05 was considered significant. All proteins that did not change in the same direction in all donors were excluded. Multiple testing removed most of the differently expressed proteins, including all the proteins that were validated to be differently expressed on all three donors using western blotting and RT-qPCR. Thus, correction for multiple testing was not performed, as this would have introduced a number of false negative results. As this approach runs the risk of including false positive results, the most important and interesting protein changes were validated by western blotting. The mass spectrometry data are accessible in the ProteomeXchange database with accession number PXD003362. Reviewer account details: Username: reviewer64130@ebi.ac.uk, Password: ZmPxZSLu.
GO analysis. GO analysis was performed using the GO enrichment analysis at the Gene Ontology Consortium homepage (http://geneontology.org/). “Biological processes” was used as ontology.
Prediction of miR-140 targets. Evaluation of predicted targets of miR-140 was performed using the miRwalk-search engine by searching in five different databases: DIANAmt, miRanda, miRWalk, PITA, and Targetscan. The results in Supplementary dataset 3 are from the PITA database.
microRNA luciferase reporter assays. Luciferase reporter assays (GoClone Reporter, SwitchGear Genomics, Menlo Park, CA) for 3′UTRs from selected possible targets and appropriate controls were performed in a 96-well format (50 ng of reporter/well) using Lenti-X 293T cells and the Lightswitch Luciferase Assay System. Luciferase measurements in cells were performed 24 hours after transfection by photon counting using Spectrum IVIS µCT instrument and LivingImage Software version 4.3.1 from Perkin Elmer (Waltham, MA). Cells were exposed for 10 seconds without filters.
SUPPLEMENTARY MATERIAL Figure S1. Response to addition of rhIL1B. RT-qPCR of IL1B, IL6, IL8, and MMP13 in cells from three donors, (A), (B), and (C), respectively. Table S1. List of reagents used. Supplementary Data.
Acknowledgments
We acknowledge Manuela Zucknick for help with the statistical analysis of the mass spectrometry data and Jan Øyvind Moskaug for help with the microscope and software for detection of luciferase signal. All MS data were processed at the Proteomics Core Facility, Oslo University Hospital Rikshospitalet. This study was funded by a post doc grant for TAK from South-Eastern Norway Regional Health Authority. None of the authors have competing or financial interests which could create a potential conflicts of interest or the appearance of a conflict of interest with regard to the work. All authors have either drafted or revised the manuscript and approved the final manuscript.
Supplementary Material
References
- Seibel, MJ, Robins, SP, and Bilezikian, JP (2006). Dynamics of Bone and Cartilage Metabolism, Academic Press: San Diego. [Google Scholar]
- Centers for Disease Control and Prevention (CDC) (2007). National and state medical expenditures and lost earnings attributable to arthritis and other rheumatic conditions–United States, 2003. MMWR Morbidity and mortality weekly report 56: 4–7. [PubMed] [Google Scholar]
- Daheshia, M and Yao, JQ (2008). The interleukin 1beta pathway in the pathogenesis of osteoarthritis. J Rheumatol 35: 2306–2312. [DOI] [PubMed] [Google Scholar]
- Goldring, MB (2012). Chondrogenesis, chondrocyte differentiation, and articular cartilage metabolism in health and osteoarthritis. Ther Adv Musculoskelet Dis 4: 269–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutvágner, G and Zamore, PD (2002). A microRNA in a multiple-turnover RNAi enzyme complex. Science 297: 2056–2060. [DOI] [PubMed] [Google Scholar]
- Lee, RC, Feinbaum, RL and Ambros, V (1993). The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 75: 843–854. [DOI] [PubMed] [Google Scholar]
- Bader, AG (2012). miR-34 - a microRNA replacement therapy is headed to the clinic. Front Genet 3: 120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanford, RE, Hildebrandt-Eriksen, ES, Petri, A, Persson, R, Lindow, M, Munk, ME et al. (2010). Therapeutic silencing of microRNA-122 in primates with chronic hepatitis C virus infection. Science 327: 198–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuddenham, L, Wheeler, G, Ntounia-Fousara, S, Waters, J, Hajihosseini, MK, Clark, I et al. (2006). The cartilage specific microRNA-140 targets histone deacetylase 4 in mouse cells. FEBS Lett 580: 4214–4217. [DOI] [PubMed] [Google Scholar]
- Wienholds, E, Kloosterman, WP, Miska, E, Alvarez-Saavedra, E, Berezikov, E, de Bruijn, E et al. (2005). MicroRNA expression in zebrafish embryonic development. Science 309: 310–311. [DOI] [PubMed] [Google Scholar]
- Miyaki, S, Sato, T, Inoue, A, Otsuki, S, Ito, Y, Yokoyama, S et al. (2010). MicroRNA-140 plays dual roles in both cartilage development and homeostasis. Genes Dev 24: 1173–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsen, TA and Brinchmann, JE (2013). Liposome delivery of microRNA-145 to mesenchymal stem cells leads to immunological off-target effects mediated by RIG-I. Mol Ther 21: 1169–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alaaeddine, N, Olee, T, Hashimoto, S, Creighton-Achermann, L and Lotz, M (2001). Production of the chemokine RANTES by articular chondrocytes and role in cartilage degradation. Arthritis Rheum 44: 1633–1643. [DOI] [PubMed] [Google Scholar]
- Verma, P and Dalal, K (2011). ADAMTS-4 and ADAMTS-5: key enzymes in osteoarthritis. J Cell Biochem 112: 3507–3514. [DOI] [PubMed] [Google Scholar]
- Tardif, G, Hum, D, Pelletier, JP, Duval, N and Martel-Pelletier, J (2009). Regulation of the IGFBP-5 and MMP-13 genes by the microRNAs miR-140 and miR-27a in human osteoarthritic chondrocytes. BMC Musculoskelet Disord 10: 148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters, MA, Wendholt, D, Strietholt, S, Frank, S, Pundt, N, Korb-Pap, A et al. (2012). The loss of α2β1 integrin suppresses joint inflammation and cartilage destruction in mouse models of rheumatoid arthritis. Arthritis Rheum 64: 1359–1368. [DOI] [PubMed] [Google Scholar]
- Sakai, K, Kimata, K, Sato, T, Gotoh, M, Narimatsu, H, Shinomiya, K et al. (2007). Chondroitin sulfate N-acetylgalactosaminyltransferase-1 plays a critical role in chondroitin sulfate synthesis in cartilage. J Biol Chem 282: 4152–4161. [DOI] [PubMed] [Google Scholar]
- Sato, T, Kudo, T, Ikehara, Y, Ogawa, H, Hirano, T, Kiyohara, K et al. (2011). Chondroitin sulfate N-acetylgalactosaminyltransferase 1 is necessary for normal endochondral ossification and aggrecan metabolism. J Biol Chem 286: 5803–5812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng, J, Wu, C, Ma, W, Zhang, Y, Hou, T, Xu, H et al. (2013). Abnormal expression of chondroitin sulphate N-acetylgalactosaminyltransferase 1 and Hapln-1 in cartilage with Kashin-Beck disease and primary osteoarthritis. Int Orthop 37: 2051–2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenblatt, MB, Ritter, SY, Wright, J, Tsang, K, Hu, D, Glimcher, LH et al. (2013). NFATc1 and NFATc2 repress spontaneous osteoarthritis. Proc Natl Acad Sci USA 110: 19914–19919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, J, Gardner, BM, Lu, Q, Rodova, M, Woodbury, BG, Yost, JG et al. (2009). Transcription factor Nfat1 deficiency causes osteoarthritis through dysfunction of adult articular chondrocytes. J Pathol 219: 163–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadler, AJ, Suliman, BA, Yu, L, Yuan, X, Wang, D, Irving, AT et al. (2015). The acetyltransferase HAT1 moderates the NF-κB response by regulating the transcription factor PLZF. Nat Commun 6: 6795. [DOI] [PubMed] [Google Scholar]
- Karlsen, TA, Jakobsen, RB, Mikkelsen, TS and Brinchmann, JE (2014). microRNA-140 targets RALA and regulates chondrogenic differentiation of human mesenchymal stem cells by translational enhancement of SOX9 and ACAN. Stem Cells Dev 23: 290–304. [DOI] [PubMed] [Google Scholar]
- Echtermeyer, F, Bertrand, J, Dreier, R, Meinecke, I, Neugebauer, K, Fuerst, M et al. (2009). Syndecan-4 regulates ADAMTS-5 activation and cartilage breakdown in osteoarthritis. Nat Med 15: 1072–1076. [DOI] [PubMed] [Google Scholar]
- Peng, JS, Chen, SY, Wu, CL, Chong, HE, Ding, YC, Shiau, AL et al. (2016). Amelioration of experimental autoimmune arthritis through targeting of synovial fibroblasts by intra-articular delivery of microRNAs 140-3p and 140-5p. Arthritis Rheumatol 68: 370–381. [DOI] [PubMed] [Google Scholar]
- Vangsness, CT Jr, Burke, WS, Narvy, SJ, MacPhee, RD and Fedenko, AN (2011). Human knee synovial fluid cytokines correlated with grade of knee osteoarthritis–a pilot study. Bull NYU Hosp Jt Dis 69: 122–127. [PubMed] [Google Scholar]
- Sjögren, F and Anderson, C (2009). Sterile trauma to normal human dermis invariably induces IL1beta, IL6 and IL8 in an innate response to “danger”. Acta Derm Venereol 89: 459–465. [DOI] [PubMed] [Google Scholar]
- Iliopoulos, D, Malizos, KN, Oikonomou, P and Tsezou, A (2008). Integrative microRNA and proteomic approaches identify novel osteoarthritis genes and their collaborative metabolic and inflammatory networks. PLoS One 3: e3740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyaki, S, Nakasa, T, Otsuki, S, Grogan, SP, Higashiyama, R, Inoue, A et al. (2009). MicroRNA-140 is expressed in differentiated human articular chondrocytes and modulates interleukin-1 responses. Arthritis Rheum 60: 2723–2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, M, Liu, L, Xiao, T and Guo, W (2012). [Detection of the expression level of miR-140 using realtime fluorescent quantitative PCR in knee synovial fluid of osteoarthritis patients]. Zhong Nan Da Xue Xue Bao Yi Xue Ban 37: 1210–1214. [DOI] [PubMed] [Google Scholar]
- Campbell, KA, Minashima, T, Zhang, Y, Hadley, S, Lee, YJ, Giovinazzo, J et al. (2013). Annexin A6 interacts with p65 and stimulates NF-κB activity and catabolic events in articular chondrocytes. Arthritis Rheum 65: 3120–3129. [DOI] [PubMed] [Google Scholar]
- Minashima, T, Campbell, K and Kirsch, T (2013). Annexins: novel therapeutic targets for the treatment of osteoarthritis? J Am Acad Orthop Surg 21: 256–257. [DOI] [PubMed] [Google Scholar]
- Pfander, D, Swoboda, B and Kirsch, T (2001). Expression of early and late differentiation markers (proliferating cell nuclear antigen, syndecan-3, annexin VI, and alkaline phosphatase) by human osteoarthritic chondrocytes. Am J Pathol 159: 1777–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin, X, Jin, HR, Jung, HS, Lee, SJ, Lee, JH and Lee, JJ (2010). An atypical E3 ligase zinc finger protein 91 stabilizes and activates NF-kappaB-inducing kinase via Lys63-linked ubiquitination. J Biol Chem 285: 30539–30547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matluk, N, Rochira, JA, Karaczyn, A, Adams, T and Verdi, JM (2010). A role for NRAGE in NF-kappaB activation through the non-canonical BMP pathway. BMC Biol 8: 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan, J, Li, Q, Mao, AP, Hu, MM and Shu, HB (2014). TRIM4 modulates type I interferon induction and cellular antiviral response by targeting RIG-I for K63-linked ubiquitination. J Mol Cell Biol 6: 154–163. [DOI] [PubMed] [Google Scholar]
- Zhang, Z, Yuan, B, Bao, M, Lu, N, Kim, T and Liu, YJ (2011). The helicase DDX41 senses intracellular DNA mediated by the adaptor STING in dendritic cells. Nat Immunol 12: 959–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little, CB, Barai, A, Burkhardt, D, Smith, SM, Fosang, AJ, Werb, Z et al. (2009). Matrix metalloproteinase 13-deficient mice are resistant to osteoarthritic cartilage erosion but not chondrocyte hypertrophy or osteophyte development. Arthritis Rheum 60: 3723–3733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little, CB and Fosang, AJ (2010). Is cartilage matrix breakdown an appropriate therapeutic target in osteoarthritis–insights from studies of aggrecan and collagen proteolysis? Curr Drug Targets 11: 561–575. [DOI] [PubMed] [Google Scholar]
- Pratta, MA, Yao, W, Decicco, C, Tortorella, MD, Liu, RQ, Copeland, RA et al. (2003). Aggrecan protects cartilage collagen from proteolytic cleavage. J Biol Chem 278: 45539–45545. [DOI] [PubMed] [Google Scholar]
- Li, X, Zhen, Z, Tang, G, Zheng, C and Yang, G (2016). MiR-29a and MiR-140 protect chondrocytes against the anti-proliferation and cell matrix signaling changes by IL-1β. Mol Cells 39: 103–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuno, M, Katano, H, Otabe, K, Komori, K, Matsumoto, Y, Fujii, S et al. (2015). Platelet-derived growth factor (PDGF)-AA/AB in human serum are potential indicators of the proliferative capacity of human synovial mesenchymal stem cells. Stem Cell Res Ther 6: 243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox, J and Mann, M (2008). MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol 26: 1367–1372. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.