

Abstract
Helper-dependent adenoviral vectors mediate high efficiency gene editing in induced pluripotent stem cells without needing a designer nuclease thereby avoiding off-target cleavage. Because of their large cloning capacity of 37 kb, helper-dependent adenoviral vectors with long homology arms are used for gene editing. However, this makes vector construction and recombinant analysis difficult. Conversely, insufficient homology may compromise targeting efficiency. Thus, we investigated the effect of homology length on helper-dependent adenoviral vector targeting efficiency at the cystic fibrosis transmembrane conductance regulator locus in induced pluripotent stem cells and found a positive correlation. With 23.8 and 21.4 kb of homology, the frequencies of targeted recombinants were 50–64.6% after positive selection for vector integration, and 97.4–100% after negative selection against random integrations. With 14.8 kb, the frequencies were 26.9–57.1% after positive selection and 87.5–100% after negative selection. With 9.6 kb, the frequencies were 21.4 and 75% after positive and negative selection, respectively. With only 5.6 kb, the frequencies were 5.6–16.7% after positive selection and 50% after negative selection, but these were more than high enough for efficient identification and isolation of targeted clones. Furthermore, we demonstrate helper-dependent adenoviral vector-mediated footprintless correction of cystic fibrosis transmembrane conductance regulator mutations through piggyBac excision of the selectable marker. However, low frequencies (≤ 1 × 10−3) necessitated negative selection for piggyBac-excision product isolation.
Introduction
Recently, helper-dependent adenoviral vectors (HDAds) have been used to deliver donor DNA into human embryonic stem cells (ESCs), induced pluripotent stem cells (iPSCs), and adult stem cells to achieve high efficiency gene editing by homologous recombination to a wide variety of transcriptionally active and inactive loci, to achieve knockins, knockouts, and corrections.1,2,3,4,5,6,7,8,9,10,11 Collectively, these studies have all consistently demonstrated that HDAd-mediated gene editing of iPSCs and ESCs is not associated with ectopic random HDAd integrations, does not affect the undifferentiated state and pluripotency, and maintains genetic and epigenetic integrity. Indeed a recent study found that targeted gene correction in iPSCs by HDAd minimally impacts whole-genome mutational load as determined by whole genome sequencing.9 The major appeal of HDAd-mediated gene editing is that induction of an artificial double stranded break at the chromosomal target locus by a designer endonuclease is not required to achieve high targeting efficiency, thereby eliminating the potential for off-target cleavage.
HDAds possess many features that explain their effectiveness as a gene editing vector reviewed in ref. 12. They can very efficiently deliver foreign DNA into the nucleus of target cells. They are deleted of all viral-coding sequences thereby reducing their toxicity and increasing their cloning capacity to 37 kb. This tremendous cloning capacity permits inclusion of long homology arms capable of correcting multiple mutations, as well as inclusion of multiple selectable markers and promoters/enhancers/cis-acting elements, and other transgenes to enhance their gene editing efficiency. HDAds also rarely integrate randomly into the host genome and they offer the potential for in vivo gene editing.
As mentioned above, the large cloning capacity of HDAd permits inclusion of long homology arms to maximize targeting efficiency, a common feature in all of the aforementioned studies. However, the relationship between gene editing efficiency and the length of homology is not known. This is an important issue because inclusion of long homologies renders vector construction and manipulation difficult and also complicates Southern blot and polymerase chain reaction (PCR) analyses that are required to verify the genomic structure of targeted recombinants and in some cases inclusion of long homologies may not be possible. On the other hand, the efficiency of gene editing may be severely compromised by insufficient homology. Clearly, the need to determine the relationship between targeting efficiency and homology length is important for advancing this technology.
Another common feature of all HDAds used for gene editing is inclusion of a positive selectable marker flanked by either loxP or frt sites which permit selection for vector integration and subsequent removal of the positive selectable marker by transient expression of Cre or FLP recombinase, respectively. However, this strategy leaves behind a residual loxP or frt site at the target locus which is undesirable. Clearly, the ability to achieve footprintless genome modification is highly desirable, especially for potential clinic applications where introduction of only the minimally required genomic alteration would be desirable. Indeed it has been reported that the residual loxP site can interfere with expression of surrounding genes13 or potentially disrupt splicing elements.14
In this study, we address the important issues raised above by investigating the relationship between the efficiency of HDAd-mediated gene editing and the length of homology. We also demonstrate that HDAd can mediate footprintless gene editing by using piggyBac (PB)-mediated excision15 of the vector's positive selection marker.
Results
Effect of homology length on targeting
The objectives of this study were to determine the effect of homology length on the efficiency of HDAd-mediated gene editing and to achieve HDAd-mediated footprintless gene editing. As a model system, we targeted the cystic fibrosis transmembrane conductance regulator (CFTR) gene in a human iPS cell line (called CF17 (ref. 16)) that is compound heterozygous at the CFTR locus, with one allele ▵F508 and the other allele ▵I507 in exon 10 for correction with HDAds bearing the wild-type sequence in exon 10 (Figure 1a). As shown previously, targeted correction of either mutant allele restores expression of the mature CFTR glycoprotein and chloride channel function in epithelial cells derived from these iPSCs.16 To achieve the aforementioned objectives, a panel of HDAds bearing different total homology lengths to the CFTR gene was generated, all bearing the wild-type CFTR sequence (Figure 2). To minimize variability, each vector differs from the vector one size smaller and one size larger in only one of the two homology arms; HD-23.8-CFTR-neo contains a total of 23.8 kb of homology with 11.9 kb in the each of the two homology arms. HD-21.4-CFTR-neo contains a total of 21.4 kb of homology, having the same 11.9 kb right homology arm as HD-23.8-CFTR-neo but a shorter left homology arm of 9.5 kb. HD-14.8-CFTR-neo contains a total of 14.8 kb of homology, with the same 9.5 kb left homology arm as HD-21.4-CFTR-neo but a shorter right homology arm of 5.3 kb. HD-9.6-CFTR-neo contains a total of 9.6 kb of homology with the same 5.3 kb right homology arm as HD-14.8-CFTR-neo, but a shorter 4.3 kb left homology arm. HD-5.6-CFTR-neo contains a total of 5.6 kb of homology, with the same 4.3 kb left homology arm as HD-9.6-CFTR-neo but a shorter right homology arm of 1.3 kb. All HDAds possess a neomycin resistance marker (neo) to permit positive selection for HDAd integration which is flanked by PB inverted terminal repeats to permit its footprintless excision by PB transposase.15 All HDAds possess the herpes simplex virus thymidine kinase (HSV-TK) cassette outside the region of homology to permit negative selection against random vector integration.17
Figure 1.

Gene targeting at the cystic fibrosis transmembrane conductance regulator (CFTR) locus by helper-dependent adenoviral vector (HDAd). (a) A single reciprocal crossover in the right and left homology arms results in integration of the neomycin resistance marker (neo) into the CFTR gene rendering clones resistant to G418 (G418R). The herpes simplex virus thymidine kinase (HSV-TK) cassette in the HDAd permits negative selection against clones bearing random HDAd integrations rendering them sensitive to ganciclovir (GCVS) but the correctly targeted clones resistant to ganciclovir (GCVR). PiggyBac (PB) inverted terminal repeats (PB ITRs) flank the neo cassette to permit footprintless excision of neo in the presence of PB transposase. Sizes of the diagnostic ApaI fragments and the locations of the 5′ probe, 3′ probe and neo probe used for Southern analyses are shown. The positions of polymerase chain reaction (PCR) primers used to amplify the allele without targeted vector integration is shown. The ▵I507 and ▵F508 mutations are ~0.2 kb from the site of neo insertion. The position of the adenoviral packaging signal (Ψ) and adenoviral inverted terminal repeat (Ad ITR) are shown for the HDAd. Black rectangles represent wild-type exons 9 and 10, and the white rectangle represents mutant exon 10. (b) Southern blots of genomic DNA extracted from all 16 G418RGCVR colonies obtained from transduction of CF17 with HD-23.8-CFTR-neo at an multiplicity of infection (MOI) of 175 vp/cell analyzed with the 5′ probe, the 3′ probe and the neo probe showing targeted vector integration in all cases.
Figure 2.

The panel of helper-dependent adenoviral vector (HDAds) bearing different lengths of homology to the target cystic fibrosis transmembrane conductance regulator (CFTR) gene. The length of the homology arms are indicated for each vector. Each vector differs from the one above and below it in the length of only one homology arm. The restriction enzyme sites and corresponding vertical dotted lines indicate the identical positions between a given vector and one vector above and below it. See Figure 1 legend for abbreviations.
To determine the effect of homology length on the efficiency of gene targeting by HDAd, 2 × 106 CF17 cells were transduced with each of the HDAds shown in Figure 2 at a multiplicity of infection (MOI) of 175 vp/cell (Table 1, Transduction 1) or 350 vp/cell (Table 1, Transduction 2). Following selection, the total numbers of G418 resistant and ganciclovir resistant (G418RGCVR) colonies were enumerated (Table 1, column 3). DNA was extract from all G418RGCVR colonies, digested with ApaI and subject to Southern analysis to identify targeted recombinants. For all HDAds shown in Figure 2, targeted vector integration by a single crossover in each of the two homology arms results in the insertion of neo into the CFTR gene, converting the endogenous 30.1 kb ApaI fragment (revealed by the 5′probe and 3′ probe) into a 19.1 kb (revealed by the 5′ probe and neo probe) and a 14.2 kb ApaI fragment (revealed by the 3′ probe) (Figure 1a). The results of the Southern blot analyses for all G418RGCVR colonies are summarized in Table 1. Representative Southern blots are presented in Figure 1b, which show that all 16 G418RGCVR colonies obtained from transduction with HD-23.8-CFTR-neo at an MOI of 175 vp/cell were correctly targeted into one of the two CFTR alleles as evident by the presence of the 19.1 kb band when analyzed with the 5′ probe and the presence of the 14.2 kb band when analyzed with the 3′probe (the 31 kb band in both blots is the unmodified allele). This was further confirmed with the neo probe which hybridized to only the 19.1 kb band as expected.
Table 1. Gene targeting frequencies at the CFTR locus with HDAd.
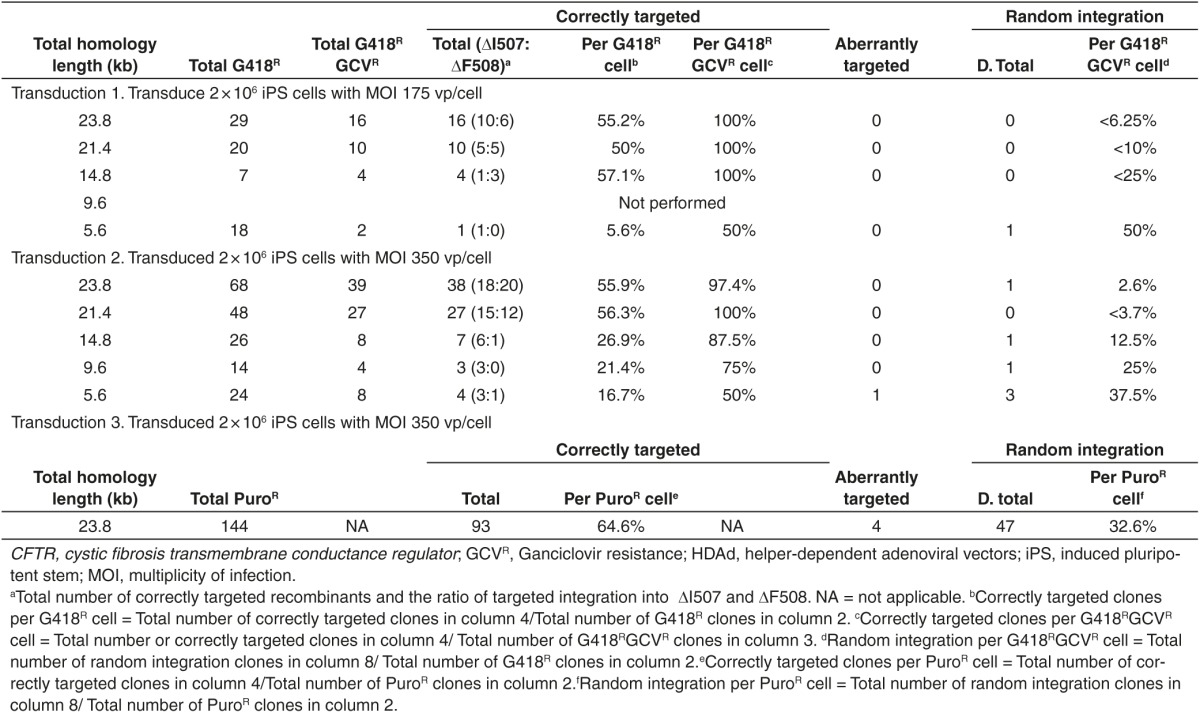
The results revealed that 23.8 kb of total homology yielded the highest number of targeted recombinants and reducing total homology to 21.4 kb resulted in a ~30% reduction in the number of targeted recombinants (Table 1, Transduction 1 and 2, column 4). However, both 23.8 kb and 21.4 kb of homology yielded comparable frequencies of targeted recombinants per G418R cell of 50–56.3% (Table 1, Transduction 1 and 2, column 5), and also comparable frequencies targeted recombinants per G418RGCVR of 97.4–100% (Table 1, Transduction 1 and 2, column 6). When total homology was reduced to 14.8 kb, the total number of target recombinant decreased to less than half that obtained with 21.4 kb (Table 1, Transduction 1 and 2, column 4). However, the frequencies of targeted recombinants per G418R cell remained relatively high at 26.9–57.1% (Table 1, Transduction 1 and 2, column 5) and the frequency of targeted recombinants per G418RGCVR cell also remained high at 87.5–100% (Table 1, Transduction 1 and 2, column 6). Reducing total homology to 9.6 kb resulted in a further reduction in the total number of targeted recombinants (Table 1, Transduction 2, column 4) as well as a reduction in the frequency of targeted recombinants per G418R cells to 21.4% (Table 1, Transduction 2, column 5) but the frequency of targeted recombinants per G418RGCVR cell remained relatively high at 75% (Table 1, Transduction 2, column 6). Further reducing total homology to only 5.6 kb did not appear to further reduce the total number of targeted recombinants compared with 9.6 kb total homology (Table 1, Transduction 1 and 2, column 4), but the frequency of targeted recombinants per G418R cells was reduced to 16.7% (Table 1, Transduction 1 and 2, column 5), and the frequency of targeted recombinants per G418RGCVR was reduced to 50% (Table 1, Transduction 1 and 2, column 6). Nevertheless, even with total homology as short as 5.6 kb, the frequency of targeted vector integration was still high enough to make identifying and isolating the targeted clones relatively easy and practical. As shown in Table 1, transduction with an MOI of 350 vp/cell yielded twofold to fourfold more targeted recombinants compared with an MOI of 175 vp/cell. Interestingly, we observed one clone from transduction with 350 vp/cell of HD-5.6-CFTR-neo, which appeared to have undergone aberrant targeting and this is addressed in more detail below.
For all 110 correctly targeted clones from transduction 1 and 2, Southern analyses with the 5′ and 3′ probe revealed the presence of the 31 kb and this indicated that the vector had integrated into only one of the two CFTR alleles in all cases (Figure 1b). To identify the targeted allele, PCR was used to amplify a 964 bp product specifically from the nontargeted allele and the product was sequenced to determine its identity. The results are summarized in Table 1, column 4 and suggest that there did not appear to be an obvious allelic preference for targeted vector integration when all HDAds were considered cumulatively; overall, 56.4% (62/110) of the targeted integration occurred at the ▵I507 allele while the remaining 43.6% (48/110) occurred at the ▵F508 allele. However, although the numbers are small, a preference for targeting into the ▵I507 allele might exist for the two vectors with the shortest homology lengths (HD-9.6-CFTR-neo and HD-5.6-CFTR-neo).
PB-mediated neo excision is inefficient
We selected clone 2–6, which had undergone targeted vector integration into the ▵F508 allele, for PB-mediated excision of the neo cassette. To accomplish this, 2 × 106 2–6 cells were transduced with HDAd-CAG-hyPB-VAI,18 a HDAd expressing the hyperactive PB transposase, at an MOI of 350 vp/cell. The transduced cells were plated at low density in the absence of G418 and 107 colonies were picked, and of these 34 were determined to be sensitive to G418 (G418S). PB-mediated excision of neo converts the 19.1 and 14.2 kb ApaI fragments back to a 31 kb ApaI fragment (Figure 1a). Unexpectedly, Southern analyses revealed that none of the 34 G418S clones had undergone PB-mediated excision of neo as evident by the continued presence of the 19.1 kb ApaI fragment revealed by the 5′ and neo probes and the 14.2 kb ApaI fragment revealed by the 3′ probe (Figure 3). In an independent repeated attempt, another 2 × 106 2–6 cells were transduced with HDAd-CAG-hyPB-VAI, and this time 112 colonies were picked, 26 of which were determined to be G418S. However, Southern analyses again revealed none had undergone PB-mediated neo excision (not shown). In a third attempt, we selected a different targeted clone, clone 1–4, that had undergone targeted vector integration into the ▵I507 allele. Following transduction of 2 × 106 1–4 cells with HDAd-CAG-hyPB-VAI at an MOI of 350 vp/cell, 240 colonies were picked and 59 were determined to be G418S. However, again none of the 59 clones had undergone PB-mediated neo excision as determined by Southern analyses (not shown). We do not understand why these clones were sensitive to G418 even though excision of the neo cassette had not occurred.
Figure 3.

Representative Southern blot analyses of genomic DNA extracted from G418S clones following transduction of targeted clone 2–6 with HDAd-CAG-hyPB-VAI showing no PB excision of the neo marker in all cases.
Negative selection needed for efficient isolation of PB-mediated excision products
That no PB-mediated neo excision could be identified after screening a total of 459 colonies suggested that this reaction occurs at a relatively low frequency and that negative selection would be needed to isolate these events. Therefore, to accomplish this, the neo cassette in HD-23.8-CFTR-neo was replaced with the positive/negative selectable PACTk cassette to generate HD-23.8-CFTR-PACtk (Figure 4a). PACTk is a fusion of the Puromycin N-acetyltransferase gene and the HSV-TK gene19 and therefore provides positive selection for vector integration by conferring puromycin resistance (puroR) and negative selection for loss of PACTk as a consequence of PB-mediated excision by conferring GCVR. Unfortunately, the use of PACTk meant that the HSV-TK cassette could no longer be used for negative selection against random vector integrations and thus was not included in HD-23.8-CFTR-PACTk. Fortunately, the relatively high frequencies of targeted integration after only positive selection (55.2–55.9%, Table 1, Transduction 1 and 2, column 5) should make identifying them relatively easy and practical even without negative selection against random integrations. A total of 144 puroR colonies were obtained following transduction of 2 × 106 CF17 cells with HD-23.8-CFTR-PACTk at an MOI of 350 vp/cell (Table 1, Transduction 3) and Southern analyses were performed on their extracted DNA. As shown in Figure 4a, targeted vector integration into the CFTR gene converts the endogenous 31 kb ApaI fragment (revealed by the 5′ and 3′ probes) into a 19 kb (revealed by the 5′ probe) and a 14.7 kb (revealed by the 3′ probe and PACTk probe) ApaI fragment. The results of the Southern analyses are summarized in Table 1 (Transduction 3) and a representative blot is shown in Figure 4b. The Southern analyses revealed that of the 144 puroR clones, 93 of these were correctly targeted into one of the two CFTR alleles (no biallelic targeting was observed), 47 had random vector integration, and four were aberrantly targeted (Table 1, Transduction 3). The four aberrantly targeted clones will be discussed in more detail below.
Figure 4.

Gene targeting at the CFTR locus with HD-23.8-CFTR-PACTk. (a) A single reciprocal crossover in the right and left homology arms results in integration of the PACTk marker to the CFTR gene rendering clones puroR. PB inverted terminal repeats (PB ITRs) flank the PACTk cassette to permit its footprintless excision in the presence of PB transposase. Sizes of the diagnostic ApaI fragments and the locations of the 5′ probe, 3′ probe and PACTk probe used for Southern analyses are shown. The positions of PCR primers used to amplify the allele without targeted vector integration is shown. The ▵I507 and ▵F508 mutations are ~0.2 kb from the site of PACTk insertion. The position of the adenoviral packaging signal (Ψ) and adenoviral inverted terminal repeat (Ad ITR) are shown for each HDAd. (b) Representative Southern blots of genomic DNA extracted from puroR clones analyzed with the 5′ external probe, the 3′ external probe and the PACTk probe showing targeted vector integration (clones 20, 64, 74, 96, 4, and 6), aberrant targeting (clones 21 and 57), and random vector integration (clones 1, 3, 11, 22, 43, 50, and 75). CFTR, cystic fibrosis transmembrane conductance regulator; HDAd, helper-dependent adenoviral vector, PB, piggyBac; PCR, polymerase chain reaction.
Next, we sought to achieve PB-mediated excision of the PACTk marker to accomplish footprintless correction of the CFTR mutation. Two correctly targeted clones, clone 7 and 27, were selected for PB-mediated excision. Clone 7 and 27 had targeted vector integration into the ▵F508 and ▵I507 allele, respectively, as determined by sequencing of the 964 bp PCR product from the unmodified allele (not shown). 2 × 106 clone 7 or clone 27 cells were transduced with HDAd-CAG-hyPB-VAI at an MOI of 350 vp/cell. Because the frequency of GCVR was unknown, the transduced cells were plated at a wide variety of cell densities in the absence of puromycin (Table 2), and 48 hours later, GCV was added to the culture media. The number of GCVR colonies were enumerated and presented in Table 2. To determine if PB-mediated PACTk excision was responsible for GCVR, DNA was extracted from 107 and 113 GCVR colonies originating from clone 7 and 27, respectively, and subjected to Southern analyses. As shown in Figure 4a, PB-mediated PACTk excision converts the 19 kb and the 14.7 kb ApaI fragments back to a 31 kb ApaI fragment. The results of the Southern analyses revealed that PB-mediated PACTk excision had occurred in all 220 GCVR colonies and a representative southern blot is presented in Figure 5. Table 2 shows that PB-mediated PACTk excision occurs at a frequency of ≤1 × 10−3. This explains why we were unable to identify PB-mediated excision of neo by simply screening 459 colonies. Thus the ability to perform negative selection afforded by PACTk is essential for efficient isolation of clones that had undergone PB-mediated PACTk excision. To confirm footprintless gene correction after PB-mediated PACTk excision, PCR was used to amplify a 964 bp region that encompassed both the site of ▵I507/▵F508 mutation and the location of downstream PACTk insertion from four GCVR clones, two derived from clone 7, and two from clone 27. The PCR product was then cloned into a bacterial plasmid (required because both alleles are amplified after PB excision of PACTk and this mixture cannot be directly sequenced) and sequenced and a representative result of this analysis is presented in Figure 6. The sequence analyses revealed that in all four cases, correction of the ▵I507 or ▵F508 mutation had occurred and that PB-mediated excision was indeed footprintless.
Table 2. Frequency of Ganciclovir resistance (GCVR) after transduction with HDAd-CAG-hyPB-VAI.
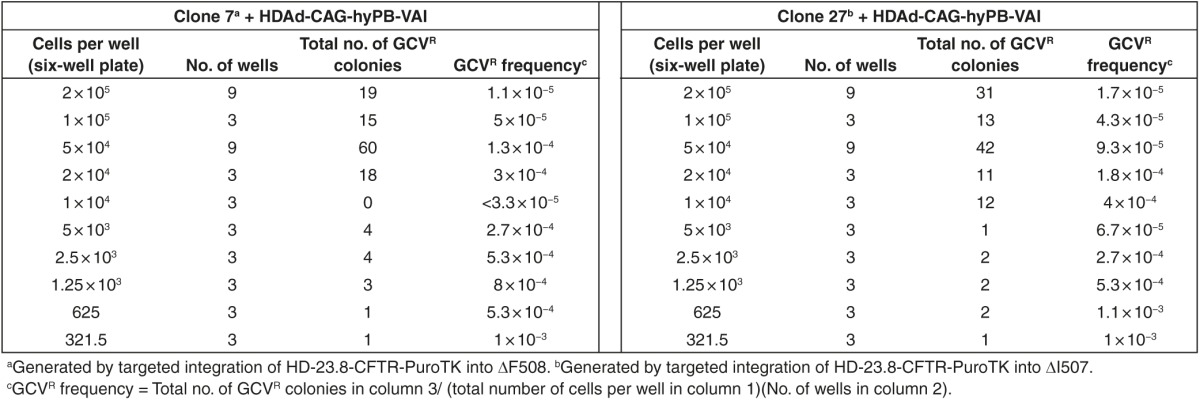
Figure 5.

Representative Southern blot analyses of genomic DNA extracted from G418S clones following transduction of targeted clone 7 with HDAd-CAG-hyPB-VAI showing PB excision of PACtk in all cases.
Figure 6.

Sequence analysis of a GCVR clone derived from clone 7 following PB-mediated excision of PACTk. Footprintless correction of the ▵F508 mutation is confirmed by the presence of the wild-type sequence (ATC, denoted wt) linked to the downstream piggyBac (PB) insertion site (denoted TTAA).
Aberrant gene targeting
As mentioned, four puroR clones from Transduction 3 appeared to have undergone aberrant targeting (clones 21, 57, 34, and 51), two of these, clone 21 and 57, are present in the Southern blot presented in Figure 4b. For clone 21, the expected 31 and 19 kb bands are revealed with the 5′ probe (Figure 4b) indicating that the left homology arm of the HDAd had integrated into one of the CFTR alleles by correct homologous recombination. However, the 3′ probe and the PACTk probe revealed a band that was larger than the expected 14.7 kb band (Figure 4b) indicating the right homology arm of the HDAd did not undergo correct homologous recombination with the target locus. Likewise, for clone 57, the expected 31 and 19 kb bands are revealed with the 5′ probe (Figure 4b) indicating that the left homology arm of the HDAd had integrated into one of the CFTR alleles by correct homologous recombination. However, the 3′ probe revealed only the 31 kb band and the PACTk probe revealed a band of high molecular weight band (Figure 4b) indicating the right homology arm of the HDAd did not undergo correct homologous recombination with the target locus. For clone 34, the 5′ probe revealed the expected 31 kb but not the expected 19 kb band, but instead an unexpected band between 31 kb and 19 kb was present (not shown), the 3′ probe revealed only the expected 31 kb band but not the expected 14.7 kb band, and the PACTk probe revealed a band of high molecular weight (not shown). For clone 51 the 5′ probe revealed the expected 31 and 19 kb bands, but the 3′ probe revealed only the expected 31 kb band but not the expected 14.7 kb band, and the PACTk probe did not reveal any band (not shown). As mentioned above, Transduction 2 with HD-5.6-CFTR-neo also generated one aberrantly targeted clone which possessed the expected 31 and 19 kb band as revealed by the 5′ probe, the expected 19 kb band revealed by the neo probe, but a band greater than the expected 14.1 kb band was revealed with the 3′ probe (not shown). In summary, in four of the five cases (expected for clone 34), the vector appears to have integrated into one of the CFTR alleles by correct homologous recombination with the left homology arm but not the right homology arm. In the case of clone 34, the vector appears to have integrated into one of the CFTR alleles (due to the presence of the expected 31 kb and a band of unexpected size with the 5′ probe), but this integration did not occur by correct homologous recombination in either the left or right homology arms. We made no further attempts to elucidate the genomic structure of these rare aberrantly targeted clones.
Discussion
We have systematically investigated the effects of homology length on gene targeting by HDAd and found that there is positive correlation between targeting efficiency and the total length of homology in HDAds. However, even with as little as 5.6 kb of total homology, the efficiency remains high enough to permit easy identification and isolation of the targeted recombinants. This high targeting frequency may be attributed to the relatively low frequency of random HDAd integration, a property that appears to be conferred by the adenoviral terminal protein that is covalently attached to the ends of the linear vector genome.20 Suzuki et al. compared the efficiency of targeting the Hprt locus in mouse ES cells with HDAds bearing different homology lengths.2 In that study, HPRT was nonfunctional due to an insertion of a 1.6 kb PGKneo cassette into exon 3 and HDAds bearing the wild-type exon 3 within 18.6 kb, 6.7 kb or 1.7 kb of homology were used for targeted correction. Suzuki et al. found that targeted correction was achieved only with 18.6 kb and not with 6.7 kb or 1.7 kb of homology, indicating that 6.7 kb of homology was not sufficient for targeting. This is in contrast to our study showing that 5.6 kb of homology was enough to target the CFTR locus. However, one major difference between our two studies is that the 3 bp deletion (▵I507 or ▵F508) we are trying to correct is significantly more subtle than the 1.6 kb insertion of PGKneo that they were trying to correct and this might influence the amount of homology required. Another major difference is that mouse ES cells might have different homology requirements than human iPSCs.
We also employed PB technology to achieve footprintless removal of the positive selectable marker, but our initial attempts to identify PB-mediated excision of the neo cassette by screening clones transduced with an HDAd expressing hyperactive PB transposase proved unsuccessful due to low efficiencies. To overcome this, we replaced the neo marker with the PACTk marker which permitted both positive selection for vector integration and negative selection for PB-mediated excision. Using PACTk, we were able to easily select for GCVR clones that had undergone PB-mediated excision despite the fact that we found PB-mediated PACTk excision to occur at a low frequency of ≤1 × 10−3 per transduced cell which explains why we were unable to identify these rare clones based on screening alone. Others have reported frequencies of 8 × 10−5 to 1.1 × 10−4for excision using hyperactive PB transposase in iPSCs.21 There also appeared to be an inverse correlation between the number of GCVR colonies and the number of cells plated per well (Table 2) and this is consistent with bystander killing, a known killing mechanism of the HSV-TK/GCV system in which cells that do no express HSV-TK are nevertheless killed by GCV because they are in gap-junction contact with cells that do express HSV-TK22. This suggests that a lower plate density is desirable during negative selection. While the use of PACTk permitted efficient isolation of PB-mediate PACTk excision, it precluded the use of the HSV-TK for negative selection against random vector integrations. Fortunately, this did not pose a significant problem because random vector integration with Ad-based vectors is rare due to the covalently attached Ad terminal proteins as discussed above. In the future, a diphtheria toxin23 or cytosine deaminase24 expression cassette could be added to the HDAd outside of the region of homology to serve as a negative selectable marker against random vector integrations if desired.
We have identified five clones that appeared to have undergone aberrant targeting. Of which four of these five clones appear to have undergone correct homologous recombination in the left homology arm but not in the right homology arm. We do not understand why homologous recombination appeared to occur with less fidelity in right arm in our targeting HDAds. Aberrantly targeted iPSCs have also been reported by others using HDAd, and these also appear to have undergone correct homologous recombination in the left arm but not in the right arm7.
Recently, Crane et al.16 reported zinc finger nuclease -mediated targeted correction of CFTR mutations in the same CF iPSCs used in this study. Using the same PACTk selectable marker used in this study, Crane et al. obtained an average of 29–32 puroR colonies per 2 × 106 iPSCs nucleofected with wild-type donor DNA and zinc finger nuclease expression plasmids, and found that 6.3% of the puroR colonies were correctly targeted into one CFTR allele. By comparison, HD-23.8-CFTR-PACTk yielded 144 puroR colonies per 2 × 106 cells transduced, 64.6% of which were correctly targeted into one CFTR allele. However, the two studies differ in so many variables that direct comparison of their targeting efficiencies is difficult. Nevertheless, an important advantage of HDAd is avoidance of off-target cleavage associated with using a designer nuclease (although none were found with the specific zinc finger nuclease used by Crane et al.). On the other hand, the nuclease approach described by Crane et al. used a total of only 1.6 kb homology which greatly simplifies vector construction. Crane et al. also reported a strong allelic bias in targeting which was attributed to a single base polymorphism 76 bp upstream of the zinc finger nuclease cleavage site. Specifically, the ▵I507 allele has an Adenine at this position while the ▵F508 allele has a Guanine, and strong preferential allelic targeting was observed depending on whether the donor DNA possessed an Adenine or Guanine at that position. The donor CFTR DNA in our HDAds possess an Adenine at that position and this could explain the apparent bias of targeting into the ▵I507 allele with the two HDAds bearing the shortest homologies. However, because the numbers are small, generating and analyzing more targeted recombinants with these two HDAds would be required to draw a definitive conclusion. The use of drug selection to isolate targeted recombinants could lead to genome instability. For example, ganciclovir which we used at the typical 2 µmol/l concentration for negative selection against PACtk, can induce chromosomal instability.25 This may be avoided by using 1-(2-deoxy-2-fluoro-β-D-arabinofuranosyl)-5-iodouracil which is effective for negative selection against PACtk at a 10-times lower concentration of 0.2 µmol/l and was not found to be associated with genomic instability.21,25
In summary, we have found that there is a direct relationship between the total length of homology and the efficiency of HDAd-mediated gene targeting in human iPSCs. However, even with as little as 5.6 kb total homology, targeting is achieved at practical efficiencies to permit easy isolation of targeted recombinants. We have also demonstrated for the first time, that HDAd can mediate footprintless correction of the CFTR mutation using PB-mediated excision of the selectable marker. However, PB-mediated excision is relatively inefficient requiring negative selection for efficient isolation of the excision products. Nevertheless, the benefits of footprintless excision outweigh the disadvantage of low efficiency for many applications.
Materials and methods
Helper-dependent adenoviral vectors. HD-23.8-CFTR-neo was constructed as follows: First, an HDAd plasmid containing the HSV-TK expression cassette was constructed from p▵28E4 (ref. 26). Next, a 23,786 bp fragment (positon 67,953–91,738 according to NCBI Reference Sequence: NC_000007.14) from BAC clone RP11-1152A23 (Sigma #RPCl11.C, St. Louis, MO) was cloned into this HDAd plasmid by recombineering.27 Next, the neomycin resistance marker flanked by PB inverted terminal repeats was inserted at position 79,857 (according to NCBI Reference Sequence: NC_000007.14) by recombineering. Finally, a LacZ expression cassette was inserted into the unique AscI site. The HDAds with shorter homology arms were derived from HD-23.8-CFTR-neo with the new borders of homology indicated by the restriction enzyme site shown in Figure 2. HD-23.8-CFTR-PACTk was derived from HD-23.8-CFTR-neo by replacing the neo expression cassette with the PACTk expression cassette.19 Further cloning details are available upon request. HDAd-CAG-hyPB-VAI is described elsewhere.18 HDAds were produced with 116 cells and AdNG163 helper virus as described elsewhere.28,29 HDAd titers were determined by absorbance at 260 nm as described elsewhere.29
Transduction of iPSCs. CF17, the feeder free human CF iPS cell line used in this study is described elsewhere16 and was maintained in mTeSR 1 (STEMCELL Technologies, Vancouver, Canada) on Matrigel (Corning, Tewksbury, MA) coated plates. The iPSCs were transduced with HDAd as described previously.18 Briefly, 2 × 106 cells were resuspended in 1 ml mTeSR 1 supplemented with Y27632 (Reagents Direct, Encinitas, CA) to 10 µmol/l in a 1.5 ml microfuge tube and infected with HDAd at an MOI of 175 or 350 vp/cell for 1 hour at 37oC with gentle rocking. Following infection, cells were washed twice with 1 ml mTeSR 1 supplemented with Y27632 to 10 µmol/l and plated into 10–12 Matrigel coated wells of six-well plates in mTeSR 1 supplemented with Y27632 to 10 µmol/l. For transductions 1 and 2 (Table 1), G418 was added to the media to a final concentration of 400 µg/ml 48 hours post-transduction, and 14 days post-transduction GCV was added to the media to a final concentration of 2 µmol/l and all surviving colonies were picked 21 days post-transduction. For transduction 3 (Table 1), puromycin was added to the media to a final concentration of 0.5 µg/ml 48 hours post-transduction and all surviving colonies were picked 13 days after transduction. DNA was extracted from all surviving colonies for Southern analysis.
PB excision. For PB excision, 2 × 106 iPSCs were transduced with HDAd-CAG-hyPB-VAI at an MOI of 350 vp/cell as described above. For targeted clones 2–6 and 1–4, generated with HD-23.8-CFTR-neo, cells were plated at 100, 250, 500, and 1,000 cells per well in nonselective media following transduction with HDAd-CAG-hyPB-VAI. A total of 459 well isolated colonies were picked from three independent transductions and these were screened for sensitivity to G418 and DNA was extracted from a total of 119 G418S clones for Southern analysis. For targeted clones 7 and 27 generated with HD-23.8-CFTR-PACTk, cells were plated in nonselective media following HDAd-CAG-hyPB-VAI transduction at the densities shown in Table 2. 48 hours post-transduction, GCV was added to the media to a final concentration of 2 µmol/l. 220 well isolated colonies were picked and DNA was extracted for Southern analysis.
DNA analysis. Genomic DNA from iPSCs was extracted from a single confluent well of a 24 or 12 well plate as follows: Cells were washed twice with 0.5 or 1 ml phosphate buffered saline (PBS), and 0.4 ml lysis buffer (10 mmol/l Tris-HCl pH 8.0, 10 mmol/l ethylenediaminetetraacetate (EDTA) pH 8.0, 100 mmol/l NaCl, 0.5% sodium dodecyl sulfate (SDS), 100 µg/ml proteinase K) was added to the well. The lysate was then transferred into a microfuge tube and incubated with gentle rocking overnight at 50 °C. The next day, the DNA was precipitated by the addition of 1 ml 95% ethanol, washed once with 1 ml 70% ethanol and resuspended in 10 mmol/l Tris-HCL pH 8.0.
Nonradioactive digoxigenin-based Southern blot hybridization was performed as recommended by the manufacturer (Roche, Indianapolis, IN). All Southern probes were digoxigenin-labelled PCR products generated according to manufacturer's recommendation (Roche). The 5′ probe was generated using the primers 5′-ATTTCAAGTGTCTTCGTCGG and 5′-GTAAGGTAAGTCCAGGTGC and the plasmid pLPBL-1-CFTR-ApaI-BamHI. The 3′ probe was generated using the primers 5′-TCATTGCCCTTTGTATGTGC and 5′-CATCCTCCACTGCCATTTC and the plasmid pLPBL-1-CFTR-BstBI-BstBI. The neo probe was generated using the primers 5′-GGATCGGCCATTGAACAAG and 5′-TGCGATGTTTCGCTTGGTG and the plasmid PL451. The PACTk probe was generated using the primers 5′-ATAGAGCCCACCGCATCC and 5′-AACGGCGACCTGTATAACG and the plasmid pLPBL-13-PACTk. Thermocycling conditions were as follows; 2 minutes at 95 °C, followed by 30 cycles of 95 °C for 30 seconds, 58 °C for 40 seconds and 72 °C for 40 seconds, and a final extension of 7 minutes at 72 °C.
Primers 5′-ATTGGAGGCAAGTGAATCCTGAGC and 5′-AGCAATAACTACTGAACCCACCATC were used to amplify the unmodified CFTR allele(s) encompassing the ▵I507/▵F508 mutation and the site of the selectable marker insertion from iPSC genomic DNA using HotStarPlus(Qiagen, Valencia, CA) according to the manufacturer's recommendations. Thermocycling conditions were as follows; 5 minutes at 95 °C, followed by 35 cycles of 94 °C for 1 minute; 68 °C for 1 minute and 72 °C for 1 minute, and a final extension of 10 minutes at 72 °C. For DNA extracted from the correctly targeted recombinants, the forward primer was used to sequence the PCR product to determine which allele vector integration had occur. For DNA extracted from clones that underwent PB-mediated excision of the PACTk expression cassette, the PCR products were first cloned into a plasmid (because both alleles are amplified) and then sequenced with the forward primer to verify footprintless gene correction.
Acknowledgments
This study was supported by the National Institutes of Health R01 DK067324 and by internal Baylor College of Medicine funds (PN), and the C. Harold and Lorine G. Wallace Distinguished University Chair (BRD). No competing financial interests exist.
References
- Ohbayashi, F, Balamotis, MA, Kishimoto, A, Aizawa, E, Diaz, A, Hasty, P et al. (2005). Correction of chromosomal mutation and random integration in embryonic stem cells with helper-dependent adenoviral vectors. Proc Natl Acad Sci USA 102: 13628–13633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki, K, Mitsui, K, Aizawa, E, Hasegawa, K, Kawase, E, Yamagishi, T et al. (2008). Highly efficient transient gene expression and gene targeting in primate embryonic stem cells with helper-dependent adenoviral vectors. Proc Natl Acad Sci USA 105: 13781–13786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, M, Suzuki, K, Qu, J, Saini, P, Dubova, I, Yi, F et al. (2011). Efficient correction of hemoglobinopathy-causing mutations by homologous recombination in integration-free patient iPSCs. Cell Res 21: 1740–1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, GH, Suzuki, K, Qu, J, Sancho-Martinez, I, Yi, F, Li, M et al. (2011). Targeted gene correction of laminopathy-associated LMNA mutations in patient-specific iPSCs. Cell Stem Cell 8: 688–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aizawa, E, Hirabayashi, Y, Iwanaga, Y, Suzuki, K, Sakurai, K, Shimoji, M et al. (2012). Efficient and accurate homologous recombination in hESCs and hiPSCs using helper-dependent adenoviral vectors. Mol Ther 20: 424–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umeda, K, Suzuki, K, Yamazoe, T, Shiraki, N, Higuchi, Y, Tokieda, K et al. (2013). Albumin gene targeting in human embryonic stem cells and induced pluripotent stem cells with helper-dependent adenoviral vector to monitor hepatic differentiation. Stem Cell Res 10: 179–194. [DOI] [PubMed] [Google Scholar]
- Liu, GH, Qu, J, Suzuki, K, Nivet, E, Li, M, Montserrat, N et al. (2012). Progressive degeneration of human neural stem cells caused by pathogenic LRRK2. Nature 491: 603–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, GH, Suzuki, K, Li, M, Qu, J, Montserrat, N, Tarantino, C et al. (2014). Modelling Fanconi anemia pathogenesis and therapeutics using integration-free patient-derived iPSCs. Nat Commun 5: 4330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki, K, Yu, C, Qu, J, Li, M, Yao, X, Yuan, T et al. (2014). Targeted gene correction minimally impacts whole-genome mutational load in human-disease-specific induced pluripotent stem cell clones. Cell Stem Cell 15: 31–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida, T, Ozawa, Y, Suzuki, K, Yuki, K, Ohyama, M, Akamatsu, W et al. (2014). The use of induced pluripotent stem cells to reveal pathogenic gene mutations and explore treatments for retinitis pigmentosa. Mol Brain 7: 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto, H, Ishimura, M, Ochiai, M, Takada, H, Kusuhara, K, Nakatsu, Y et al. (2016). BTK gene targeting by homologous recombination using a helper-dependent adenovirus/adeno-associated virus hybrid vector. Gene Ther 23: 205–213. [DOI] [PubMed] [Google Scholar]
- Brunetti-Pierri, N and Ng, P (2015). Helper-dependent adenoviral vectors for Gene Therapy. In: Templeton, Nancy Smyth (ed.). Gene and Cell Therapy: Therapeutic Mechanisms and Strategies, 4th edn. CRC Press: Boca Raton, FL, pp. 47–84 [Google Scholar]
- Meier, ID, Bernreuther, C, Tilling, T, Neidhardt, J, Wong, YW, Schulze, C et al. (2010). Short DNA sequences inserted for gene targeting can accidentally interfere with off-target gene expression. FASEB J 24: 1714–1724. [DOI] [PubMed] [Google Scholar]
- Friedel, RH, Wurst, W, Wefers, B and Kühn, R (2011). Generating conditional knockout mice. Methods Mol Biol 693: 205–231. [DOI] [PubMed] [Google Scholar]
- Elick, TA, Bauser, CA and Fraser, MJ (1996). Excision of the piggyBac transposable element in vitro is a precise event that is enhanced by the expression of its encoded transposase. Genetica 98: 33–41. [DOI] [PubMed] [Google Scholar]
- Crane, AM, Kramer, P, Bui, JH, Chung, WJ, Li, XS, Gonzalez-Garay, ML et al. (2015). Targeted correction and restored function of the CFTR gene in cystic fibrosis induced pluripotent stem cells. Stem Cell Reports 4: 569–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansour, SL, Thomas, KR and Capecchi, MR (1988). Disruption of the proto-oncogene int-2 in mouse embryo-derived stem cells: a general strategy for targeting mutations to non-selectable genes. Nature 336: 348–352. [DOI] [PubMed] [Google Scholar]
- Palmer, DJ, Grove, NC, Ng, P (2016). Helper virus-mediated downregulation of transgene expression permits production of recalcitrant helper-dependent adenoviral vector. Mol Ther Methods Clin Dev 3: 16039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karreman, C (1998). A new set of positive/negative selectable markers for mammalian cells. Gene 218: 57–61. [DOI] [PubMed] [Google Scholar]
- Holkers, M, Maggio, I, Henriques, SF, Janssen, JM, Cathomen, T and Gonçalves, MA (2014). Adenoviral vector DNA for accurate genome editing with engineered nucleases. Nat Methods 11: 1051–1057. [DOI] [PubMed] [Google Scholar]
- Yusa, K, Zhou, L, Li, MA, Bradley, A and Craig, NL (2011). A hyperactive piggyBac transposase for mammalian applications. Proc Natl Acad Sci USA 108: 1531–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholas, TW, Read, SB, Burrows, FJ and Kruse, CA (2003). Suicide gene therapy with Herpes simplex virus thymidine kinase and ganciclovir is enhanced with connexins to improve gap junctions and bystander effects. Histol Histopathol 18: 495–507. [DOI] [PubMed] [Google Scholar]
- Yagi, T, Ikawa, Y, Yoshida, K, Shigetani, Y, Takeda, N, Mabuchi, I et al. (1990). Homologous recombination at c-fyn locus of mouse embryonic stem cells with use of diphtheria toxin A-fragment gene in negative selection. Proc Natl Acad Sci USA 87: 9918–9922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milstone, DS, Bradwin, G and Mortensen, RM (1999). Simultaneous Cre catalyzed recombination of two alleles to restore neomycin sensitivity and facilitate homozygous mutations. Nucleic Acids Res 27: e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yusa, K (2013). Seamless genome editing in human pluripotent stem cells using custom endonuclease-based gene targeting and the piggyBac transposon. Nat Protoc 8: 2061–2078. [DOI] [PubMed] [Google Scholar]
- Toietta, G, Pastore, L, Cerullo, V, Finegold, M, Beaudet, AL and Lee, B (2002). Generation of helper-dependent adenoviral vectors by homologous recombination. Mol Ther 5: 204–210. [DOI] [PubMed] [Google Scholar]
- Liu, P, Jenkins, NA and Copeland, NG (2003). A highly efficient recombineering-based method for generating conditional knockout mutations. Genome Res 13: 476–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer, D and Ng, P (2003). Improved system for helper-dependent adenoviral vector production. Mol Ther 8: 846–852. [DOI] [PubMed] [Google Scholar]
- Palmer, DJ and Ng, P (2008). Methods for the production of helper-dependent adenoviral vectors. Methods Mol Biol 433: 33–53. [DOI] [PubMed] [Google Scholar]