

Abstract
8-Amino-3,8-dideoxy-D-manno-octulosonic acid (Kdo8N) is a unique amino sugar that has thus far only been observed on the lipopolysaccharides of marine bacteria belonging to the genus Shewanella. Although its biological function is still unclear, it is thought that the sugar is important for the integrity of the bacterial cell outer membrane. A three-gene cluster required for the biosynthesis of Kdo8N was first identified in Shewanella oneidensis. Here we describe the three-dimensional structures of two of the enzymes required for Kdo8N biosynthesis in S. oneidensis, namely, KdnB and KdnA. The structure of KdnB was solved to 1.85-Å resolution, and its overall three-dimensional architecture places it into the Group III alcohol dehydrogenase superfamily. A previous study suggested that KdnB did not require NAD(P) for activity. Strikingly, although the protein was crystallized in the absence of any cofactors, the electron density map clearly revealed the presence of a tightly bound NAD(H). In addition, a bound metal was observed, which was shown via X-ray fluorescence to be a zinc ion. Unlike other members of the Group III alcohol dehydrogenases, the dinucleotide cofactor in KdnB is tightly bound and cannot be removed without leading to protein precipitation. With respect to KdnA, it is a pyridoxal 5′-phosphate or (PLP)-dependent aminotransferase. For this analysis, the structure of KdnA, trapped in the presence of the external aldimine with PLP and glutamate, was determined to 2.15-Å resolution. The model of KdnA represents the first structure of a sugar aminotransferase that functions on an 8-oxo sugar. Taken together the results reported herein provide new molecular insight into the biosynthesis of Kdo8N.
Graphical Abstract
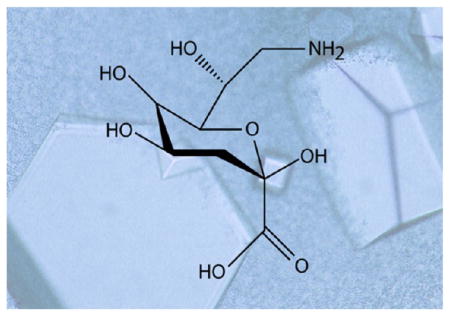
The lipopolysaccharide or LPS is a complex glycoconjugate that decorates the outermost layer of most Gram-negative bacteria.1,2 It is composed of a lipid A component, a core polysaccharide, and an O-specific polysaccharide (or O-antigen). It is the lipid A portion that anchors the complex glycoconjugate into the bacterial cell outer membrane. In most Gram-negative bacteria, the sugar bridging the lipid A component to the core polysaccharide is 3-deoxy-D-manno-octulosonic acid or Kdo. Interestingly, in marine bacteria of the genus Shewanella, Kdo is further modified to 8-amino-3,8-dideoxy-D-manno-octulosonic acid or Kdo8N.3–6 As of 2007, 40 bacterial species have been assigned to the genus Shewanella, all of which have been found in marine habitats.7 Due to the ability of these organisms to survive in harsh environments, they display fascinating physiological and metabolic characteristics. As a consequence, there has been considerable interest in employing them, for example, in bioremediation applications, energy-generating processes, and the biosynthesis of omega-3 polyunsaturated fatty acids.7
Recently, the laboratory of the late Professor Christian R. H. Raetz described the gene cluster in Shewanella oneidensis required for the biosynthesis of Kdo8N.8 In this elegant investigation, three enzymes, namely, KdnB, KdnA, and KdsB, were biochemically characterized. Additionally, a ΔkdnAkdnB knockout strain of S. oneidensis was constructed. Although the biological role of Kdo8N is not well understood, the knockout strain demonstrated increased susceptibility to polymyxin B, an antimicrobial agent that functions by permeabilizing bacterial cell outer membranes.9 As hypothesized by the Raetz laboratory, the presence of Kdo8N could play a role in increasing the integrity of the Shewanella outer membrane.
In the proposed pathway depicted in Scheme 1, the first step in the biosynthesis of Kdo8N is the oxidation of the C-8 carbon of Kdo to yield an aldehyde moiety. This reaction is catalyzed by KdnB, which requires a divalent metal ion for activity. In the subsequent step, the aldehyde-containing sugar is aminated by KdnA, a PLP-dependent enzyme that utilizes L-glutamate as its nitrogen source. Finally, KdsB catalyzes the coupling of Kdo8N to CMP to produce the nucleotide-linked sugar required for its attachment to lipid A. We were especially intrigued by KdnB, which based on the data presented in the paper, appeared to be an oxidase using a metal ion and molecular oxygen rather than NAD+ for activity. Given our long-standing interest in unusual sugars found in the lipopolysaccharides of Gram-negative bacteria and, in particular, the enzymes required for their biosynthesis, we initiated a structural analysis of both KdnB and KdnA. Herein we present the high-resolution structures of these proteins. The molecular architectures of both KdnB and KdnA provide new insight into the biosynthesis of this intriguing Kdo variant found, thus far, exclusively in the genus Shewanella.
Scheme 1.

MATERIALS AND METHODS
Protein Expression and Purification
The genes encoding KdnA and KdnB (S. oneidensis ATCC 700550) were cloned using Platinum Pfx DNA polymerase (Invitrogen). Primers were designed that incorporated NdeI and XhoI restrictions sites. For KdnA, the PCR product was digested with NdeI and XhoI and ligated into pET31. The gene was constructed such that the final protein contained a C-terminal histidine tag with the sequence LEHHHHHH. The KdnB PCR product was similarly digested and ligated into pET28T, a laboratory pET28(b+) derived vector that had been previously modified to incorporate a TEV protease cleavage site after the N-terminal polyhistidine tag.10
The pET31-kdnA plasmid was used to transform Rosetta2-(DE3) Escherichia coli cells (Novagen). The cultures were grown with shaking in lysogeny broth supplemented with 100 mg/L ampicillin and 50 mg/L chloramphenicol at 37 °C until an optical density of approximately 0.8 was reached at 600 nm. The flasks were cooled in an ice bath, and the cells were induced with 1 mM isopropyl β-D-1-thiogalactopyranoside and allowed to express protein at 16 °C for 24 h.
The cells were harvested by centrifugation and disrupted by sonication on ice. The lysate was cleared by centrifugation, and KdnA was purified utilizing Ni-NTA resin (Qiagen) according to the manufacturer’s instructions. The protein was dialyzed against 10 mM Tris-HCl (pH 8.0) and 200 mM NaCl and concentrated to 22 mg/mL based on an extinction coefficient of 1.08 (mg/mL)−1 cm−1.
The pET28T-kdnB plasmid was used to transform Rosetta2-(DE3) E. coli cells (Novagen). The cultures were grown with shaking in lysogeny broth supplemented with 50 mg/L kanamycin and 50 mg/L chloramphenicol at 37 °C and grown and induced in a similar manner to that described for KdnA. KdnB was purified utilizing Ni-NTA resin (Qiagen) according to the manufacturer’s instructions. TEV protease was added to the pool of purified protein (approximately 1:20 molar ratio), and the mixture was allowed to incubate for 36 h at 4 °C, after which time the protease and uncleaved KdnB were removed by passage over Ni-NTA resin. The cleaved protein was dialyzed against 10 mM Tris-HCl (pH 8.0) and 200 mM NaCl and concentrated to 18 mg/mL based on an extinction coefficient of 0.85 (mg/mL) −1 cm−1.
Crystallizations
Crystallization conditions for KdnA were surveyed by the hanging drop method of vapor diffusion using a laboratory-based sparse matrix screen. The enzyme was screened in the presence of 1 mM PLP and 50 mM monosodium L-glutamate. Initial crystals were observed in trials conducted at 20 °C using 20% poly(ethylene glycol) 3350 buffered with 100 mM MES (pH 6.0) as the precipitant. X-ray diffraction quality crystals of the protein were subsequently grown from precipitant solutions composed of 16–21% poly(ethylene glycol) 3350, 100 mM MES (pH 6.0), 200 mM KCl, and 150 mM MgCl2. These crystals belonged to the monoclinic space group C2 with unit cell dimensions of a = 266.6 Å, b = 61.7 Å, c = 99.6 Å and β = 102.0°. There were two dimers in the asymmetric unit. The crystals were prepared for X-ray data collection by serially transferring them to a cryoprotectant solution composed of 30% poly(ethylene glycol) 3350, 150 mM MgCl2, 250 mM KCl, 250 mM NaCl, 1 mM PLP, 50 mM monosodium L-glutamate, 10% ethylene glycol, and 100 mM MES (pH 6.0).
Crystallization conditions for KdnB were surveyed by the hanging drop method of vapor diffusion using a laboratory-based sparse matrix screen in the presence and absence of Kdo. A well diffracting crystal form was identified from the crystallization experiments conducted at 4 °C and using as the precipitant a poly(ethylene glycol) 8000 solution containing tetramethylammonium chloride and homo-PIPES (pH 5.0). The same crystal form appeared in the presence or absence of Kdo. X-ray diffraction quality crystals of the protein were subsequently grown from precipitant solutions composed of 7–9% poly-(ethylene glycol) 8000, 100 mM homo-PIPES (pH 5.0), and 1.0 M tetramethylammonium chloride. These crystals belonged to the hexagonal space group P6322 with unit cell dimensions of a = b = 107.6 Å, and c = 149.9 Å, and they contained one subunit in the asymmetric unit. The crystals were prepared for X-ray data collection by serially transferring them to a cryoprotectant solution composed of 20% poly(ethylene glycol) 8000, 1.5 M tetramethylammonium chloride, 200 mM NaCl, 5% ethylene glycol, and 100 mM homo-PIPES (pH 5.0).
X-ray Data Collection, Processing and Structural Analyses
X-ray data sets were collected in house using a Bruker AXS Platinum 135 CCD detector controlled with the Proteum software suite (Bruker AXS Inc.) The X-ray source was Cu Kα radiation from a Rigaku RU200 X-ray generator equipped with Montel optics and operated at 50 kV and 90 mA. The X-ray data sets were processed with SAINT and scaled with SADABS (Bruker AXS Inc.). Relevant X-ray data collection statistics are listed in Table 1.
Table 1.
X-ray Data Collection Statistics
| KdnB | KdnA | |
|---|---|---|
| resolution limits (Å) | 50–1.85 (1.95–1.85)b | 50–2.15 (2.25–2.15)b |
| number of independent reflections | 44036 (6274) | 83041 (9801) |
| completeness (%) | 99.3 (99.0) | 95.8 (89.4) |
| redundancy | 7.6 (4.7) | 2.9 (1.8) |
| avg I/avg σ(I) | 11.2 (3.1) | 7.8 (2.4) |
| Rsym (%)a | 8.3 (39.9) | 7.9 (25.4) |
Rsym = (Σ|I − Ī|/Σ I) × 100.
Statistics for the highest resolution bin.
The structure of KdnA was determined via molecular replacement using the software package PHASER,11 with the coordinates of QdtB serving as the search model.12 4-fold averaging and solvent flattening yielded an electron density map that allowed for essentially a complete tracing of the KdnA polypeptide chain. The model was subjected to alternate cycles of refinement with REFMAC13 and manual model building with COOT.14,15 KdnB was also solved using PHASER with the search model being that of PDB entry 3RF7. The KdnB model was refined in a similar manner as that for KdnA. Model refinement statistics are listed in Table 2.
Table 2.
Refinement Statistics
| KdnB | KdnA | |
|---|---|---|
| resolution limits (Å) | 50–1.85 | 50–2.15 |
| R-factora (overall)%/no. reflections | 18.1/44036 | 17.4/83041 |
| R-factor (working)%/no. reflections | 17.9/41818 | 17.1/78941 |
| R-factor (free)%/no. reflections | 21.8/2218 | 22.6/4100 |
| number of protein atoms | 2793 | 12215 |
| number of heteroatoms | 500 | 816 |
| average B values | ||
| protein atoms (Å2) | 18.3 | 22.9 |
| ligand (Å2) | 17.6 | 17.8 |
| solvent (Å2) | 28.9 | 21.0 |
| weighted RMS deviations from ideality | ||
| bond lengths (Å) | 0.011 | 0.011 |
| bond angles (deg) | 2.0 | 2.1 |
| planar groups (Å) | 0.007 | 0.01 |
| Ramachandran regions (%)b | ||
| most favored | 92.9 | 88.3 |
| additionally allowed | 7.1 | 11.7 |
R-factor = (Σ|Fo − Fc|/Σ|Fo|) × 100 where F0 is the observed structure-factor amplitude and Fc is the calculated structure-factor amplitude.
Distribution of Ramachandran angles according to PROCHECK.28
Determination of Metal Identity
A fluorescence emission spectrum was recorded on a single KdnB crystal using the insertion-device beamline 19-ID at the Structural Biology Center (Argonne National Laboratory).
Size Exclusion Chromatography
The quaternary structures of purified KdnA and KdnB were analyzed using a Superdex 200 10/300 (GE Healthcare) gel filtration column and an ÄKTA HPLC system. The samples were loaded and run in 10 mM Tris (pH 8.0) and 200 mM NaCl. The column was run at a speed of 0.5 mL/min at ambient temperature. The standards utilized were alcohol dehydrogenase (molecular weight ~150 000), bovine serum albumin (molecular weight ~66 000), and carbonic anhydrase (molecular weight ~29 000).
Removal of Tightly Bound NAD(H) from KdnB
KdnB (20 mg/mL) was mixed 1:1 with 8 M urea and subsequently dialyzed against the same. Over a period of 4 days the protein/urea solution was dialyzed stepwise against lower and lower urea concentrations. When the urea concentration reached 2 M, 20 mM Tris (pH 8) and 200 mM NaCl were included in the dialysis solutions. Final dialysis solutions (1 M and below) also contained 2 mM TCEP. The protein solution became turbid when the urea concentration was reduced to 0.5 M. After complete removal of the urea, less than 5% of the starting protein remained in solution based on absorption readings at 280 nm.
RESULTS
Structure of KdnB
Size exclusion chromatography was used to determine the quaternary structure of KdnB, and based on the results shown in Figure 1, it can be concluded that the enzyme functions as a dimer. The crystals utilized in this investigation belonged to the space group P6322 with the dimer 2-fold axis coincident to a crystallographic dyad, thereby reducing the contents of the asymmetric unit to one monomer. The model was refined to 1.85-Å resolution with an overall R-factor of 18.1%. The electron density for the polypeptide chain was continuous from the N- to the C-terminus (356 amino acid residues). In addition, three amino acid residues belonging to the N-terminal purification tag were visible (Gly-His-Gly). Shown in Figure 2a is a ribbon representation of the KdnB dimer, which has overall dimensions of ~70 Å × 70 Å × 90 Å and a total buried surface area of ~3500 Å2. Secondary structural elements involved in the formation of the subunit/subunit interface include the first two β-strands (Asn 5 to Val 8 and Lys 11 to Phe 14) and α-helices six and seven (Glu 210 to Val 226 and Lys 234 to Ala 250) from each subunit.
Figure 1.

Size exclusion gel filtration chromatography. Shown in the red and blue traces are the retention times for KdnA and KdnB, respectively, versus absorbance. The black line shows the retention times for alcohol dehydrogenase (molecular weight ~150 000), bovine serum albumin or BSA (molecular weight ~66 000), and carbonic anhydrase (molecular weight ~29 000).
Figure 2.

Structure of KdnB from S. oneidensis. A ribbon representation of the KdnB dimer is shown in (a) with Subunits 1 and 2 displayed in violet and blue, respectively. The subunit/subunit interface is highlighted in green. The 2-fold rotational axis of the dimer is perpendicular to the plane of the paper. A close-up view of a single subunit is presented in (b). The dinucleotide binding motif is depicted in blue and green, whereas the C-terminal helical domain is colored in violet. The active site is wedged between the two domains. The tightly bound NAD(H) is drawn in sticks, and the position of the bound zinc ion is indicated by the green sphere. The topology of the dinucleotide binding motif observed in KdnB is decidedly different from that observed in the canonical Rossmann fold as shown in (c). The topology drawings were prepared with Pro-Origami29 and modified in Adobe Illustrator. Parts (a) and (b) were prepared with PyMOL.30
The subunit of the dimer adopts a two-domain architecture with the N-terminal region defined by Ser 2 to Glu 184 and the C-terminal portion delineated by Thr 185–Ile 356 (Figure 2b). The N-terminal domain is dominated by a six-stranded parallel β-sheet flanked on either side by two α-helices, whereas the C-terminal domain is composed entirely of α-helices. The β-sheet found in the N-terminal domain is reminiscent of the classical Rossmann fold, but the topology is different when compared to that observed, for example, in horse liver alcohol dehydrogenase (Figure 2c).
In light of a previous report suggesting that KdnB was not dependent upon NAD+ for activity, the crystallization conditions did not include any additional cofactors.8 As shown in Figure 3a, there was clear electron density corresponding to a bound NAD(H) molecule (the oxidation state of the nicotinamide ring is unknown). Both the nicotinamide and adenine rings of the NAD(H) cofactor are in the anti-conformation, and the riboses adopt C2′-endo puckers. In addition to the dinucleotide ligand, there was clear electron density for a bound metal ion. As described in Materials and Methods, a fluorescence scan conducted at Argonne National Laboratories on a KdnB crystal was indicative of a zinc ion (Figure 3b). The zinc ion, which was modeled at full occupancy, is ligated by Asp 194 (2.2 Å), His 258 (2.4 Å), His 272 (2.2 Å), and a water molecule (2.4 Å) in a distorted tetrahedral coordination sphere. The metal/ligand bond distances are well within those typically observed in protein structures.16
Figure 3.

KdnB active site. Electron density corresponding to the bound dinucleotide is shown in (a). The difference map, contoured at 3σ and calculated with F0 − Fc coefficients, shows the position of the bound nucleotide. There is no model bias in this omit map. A fluorescence scan on the KdnB crystal is displayed in (b) and shows that the bound metal observed in the active site is zinc. A close-up view of the active site is depicted in (c). Ordered water molecules are represented as red spheres. The zinc ion is displayed in green. Possible hydrogen bonding interactions are indicated by the dashed lines.
Shown in Figure 3c is a close-up view of the active site. The dinucleotide is anchored into the active site by extensive interactions with protein side chains and ordered water molecules. A glycine rich loop (Gly 98, Gly 99, Gly 100, Ser 101), which connects β-strand five to α-helix four, envelops the pyrophosphate moiety of the dinucleotide where it provides hydrogen-bonding interactions. The zinc is positioned within 5 Å of the C-4 carbon of the nicotinamide ring. The overall molecular architecture of KdnB places it into the Group III alcohol dehydrogenase superfamily as will be discussed further below.17
Structure of KdnA
On the basis of size exclusion chromatography, KdnA also functions as a dimer (Figure 1). Crystals employed in the structural analysis of KdnA belonged to the space group C2 with two dimers in the asymmetric unit. The model was refined to 2.15-Å resolution with an overall R-factor of 17.4%. The electron density for each polypeptide chain backbone in the asymmetric unit was continuous from Pro 2 to Gln 395. Given that the α-carbons for the four subunits in the asymmetric unit superimpose with root-mean-square deviations of between 0.2 and 0.3 Å, the following discussion refers only to the first dimer in the X-ray coordinate file.
Shown in Figure 4a is a ribbon representation of the KdnA dimer, which has overall dimensions of ~75 Å × 63 Å × 82 Å and an extensive total buried surface area of 6000 Å2. KdnA adopts the classical aspartate aminotransferase fold.18 Each subunit contains 13 α-helices and 13 β-strands. The major tertiary structural element is a seven-stranded mixed β-sheet surrounded by α-helices. There is an additional four-stranded antiparallel β-sheet located near the C-terminus and a short β-hairpin motif delineated by Gly 164 to Leu 171. The extended loop (Ser 211 to Ser 237) that connects α-helices seven and eight projects into the active site of the second subunit of the dimer.
Figure 4.

Structure of KdnA from S. oneidensis. A ribbon representation of the KdnA dimer is shown in (a) with the individual subunits of the dimer colored in violet and blue. The β-strands forming the seven-stranded mixed β-sheet are highlighted in green, whereas those forming the four-stranded antiparallel β-sheet and the β-hairpin motif are colored in yellow. The 2-fold rotational axis of the dimer lies in the plane as indicated by the black arrow. The observed difference electron density for the external aldimine is shown in (b). The map, contoured at 3σ and calculated with F0 − Fc coefficients, shows the position of the ligand before it was ever included in the X-ray coordinate file. A close-up view of the binding region around the external aldimine is presented in (c). Ordered water molecules are represented by the red spheres, and possible hydrogen bonding interactions are indicated by the dashed lines. Asterisks on the amino acid labels indicate that the residues are provided by the second subunit in the dimer.
KdnA was crystallized in the presence of PLP and L-glutamate. Shown in Figure 4b is the electron density observed for the ligand. At this resolution it was not possible to determine whether the external aldimine or the ketimine intermediate had been trapped in the active site. Regardless, the position of the intermediate defines the active site, a close-up view of which is presented in Figure 4c. The PLP-derived intermediate is nestled at the end of the large β-sheet with the positive end of a helix dipole moment projecting toward the phosphoryl group of the cofactor (third α-helix of the subunit formed by Gly 61–Ala 71). The phosphoryl moiety of the cofactor is anchored into the active site by the side chains of Thr 62, Ser 181, and Asp 183 from Subunit 1 and Asn 233 from Subunit 2. Given the close distance between the carboxylate oxygen of Asp 183 and the phosphate group of the PLP intermediate (2.7 Å), one of the groups must be protonated. In addition to the side chain interactions, the phosphoryl group lies within hydrogen bonding distance to the backbone amide groups of Gly 61 and Thr 62 and two water molecules. One of the side chain oxygens of Asp 157 lies 2.5 Å from the ring nitrogen of the cofactor. Gln 160 bridges the ring hydroxyl and the α-carboxylate groups of the intermediate. The imidazole side chain nitrogen of His 325 and a water molecule also lie within 3.2 Å of the α-carboxylate group. Finally, the carboxylate of the glutamate side chain forms a striking salt bridge with the guanidinium group of Arg 222 from Subunit 2. An ordered water molecule bridges the phosphoryl group of the intermediate and the glutamate side chain carboxylate. In the aspartate aminotransferase family there is typically a conserved lysine residue that forms an internal aldimine with the PLP cofactor. In KdnA, this residue is Lys 186 (Figure 4c).
DISCUSSION
In the initial paper describing the characterization of the enzymes required for the biosynthesis of Kdo8N (Scheme 1), it was postulated that KdnB might represent a novel class of alcohol oxidases and that KdnB and KdnA work in concert to produce the modified sugar.8 This conclusion was based on the fact that product formation was not stimulated by the addition of NAD+, that incubation of Kdo with only KdnB never led to the formation of a detectable intermediate, and that H2O2 was produced when Mn-KdnB and PLP-KdnA were incubated with Kdo and L-glutamate. It was thus unexpected that when the structure of KdnB was determined in our laboratory, tightly bound NAD(H) was observed. Indeed, treatment of KdnB with urea followed by slow dialysis resulted in precipitated protein, suggesting that the cofactor is important for the structural integrity of the enzyme. This phenomenon has been observed in other enzymes including UDP-galactose 4-epimerase from E. coli.19
From the structural analysis of KdnB described here, it is now known that the enzyme binds zinc and belongs to the Group III superfamily of alcohol dehydrogenases.20 Although enzymes belonging to this superfamily were first postulated to exist in 1994 on the basis of bioinformatics approaches, it was not until 2001 that the three-dimensional structure of a family member, glycerol dehydrogenase from Bacillus stearothermophilus, was revealed by the laboratory of Professor David Rice.17 For this X-ray analysis, three different structures were determined (all in the presence of zinc): the unliganded enzyme, the binary complex of the protein with NAD+, and the binary complex with the glycerol substrate. On the basis of these structural models, a catalytic mechanism was proposed whereby the C-2 hydroxyl oxygen of glycerol interacts with the Zn2+ ion. This interaction would lower the pKa of the hydroxyl group such that the attached proton could be easily removed via a water-mediated proton shuttle. Upon collapse of the resultant alkoxide intermediate, the C-2 hydrogen of the glycerol molecule would be transferred as a hydride to C-4 of the NAD+ cofactor to produce NADH. Thus, in the case of the Group III enzymes there is no need for an enzymatic base to assist the removal of the proton from the substrate hydroxyl group.
A similar mechanism can be envisioned for KdnB. Specifically the hydroxyl group at C-8 could coordinate to the bound zinc ion in a manner that orients the C-8 hydrogen of the Kdo substrate toward C-4 of the dinucleotide cofactor. A possible model of this is presented in Figure 5. Importantly, there is a rather large open cleft in the KdnB/NAD(H) complex that is filled with bound solvent molecules. This region could clearly accommodate the rest of the sugar moiety. All attempts to produce a KdnB/NADH/Kdo ternary complex were unsuccessful, presumably because of the low pH (5.0) at which the crystals were obtained. A better understanding of substrate binding will require a crystal form more amenable to complex formation. This work is in progress.
Figure 5.

Model of the Michaelis complex for KdnB. The structure of Kdo was positioned into the active site such that its C-8 hydroxyl coordinates to the zinc ion and the hydrogen that is transferred as a hydride is directed toward C-4 of the nicotinamide ring. The hydrogens, colored in white, were added using the program PyMOL.
The fact that the dinucleotide cofactor can be easily removed from glycerol dehydrogenase begs the question as to why this is not the case for KdnB. There are several differences between the two enzymes with respect to the hydrogen bonding patterns surrounding the cofactors, albeit they are quite minor. For example, in KdnB the adenine ring of the cofactor is anchored into the binding pocket by the side chain of Thr 140 and the carbonyl groups of Thr 140 and Leu 179 (Figure 3). In glycerol dehydrogenase there are only two interactions between the NAD+ cofactor and Thr 118 (side chain and carbonyl group). Indeed, it appears that the loop connecting β-strand nine to α-helix five in KdnB (Ser 177–Glu 184) is located closer to the cofactor than that observed for glycerol dehydrogenase. Calculation of the NAD(H) buried surface area between the two enzymes is somewhat more informative, however. According to calculations made using “areaimol” in CCP4, the buried NAD(H) surface area in KdnB is 70%, whereas that for glycerol dehydrogenase is about 35%.21,22 This difference in buried surface area can be seen in Figure 6. Given that X-ray models are static in nature, however, it is difficult to define precisely what factors are involved in allowing NAD(H) to function as a cofactor or as a cosubstrate in the Group III alcohol dehydrogenases. Nevertheless, the model of KdnB presented here emphasizes the need to consider whether an isolated enzyme, predicted to be dinucleotide-dependent, exchanges its cofactor or not. It is thus not surprising that in the original analysis of KdnB the addition of NAD+ did not stimulate product formation because NADH was already tightly bound and could not be exchanged.
Figure 6.

Space-filling representations. Shown in (a) is a space-filling representation of the KdnB subunit colored in light blue with the bound dinucleotide displayed in standard atom colors. The same view for glycerol dehydrogenase is shown in (b), and as can be seen the dinucleotide is considerably more solvent exposed.
The structure of KdnA reported here represents the first molecular model of a sugar aminotransferase that functions on an 8-oxo group. Until this analysis, the only known models for sugar aminotransferases were those enzymes that catalyze amination reactions on either 3-oxo or 4-oxo sugars. Unlike KdnA, these PLP-dependent aminotransferases typically require nucleotide-linked sugars as their substrates.23,24 In 2013, the structure of one aminotransferase that functions on a free sugar was reported, however, namely, NtdA from Bacillus subtilis.25 This enzyme is involved in the biosynthesis of kanosamine, a sugar antibiotic. The sequence identities and similarities between NtdA and KdnA are 38% and 56%, respectively over ~200 structurally equivalent α-carbons. The insightful structural analysis of NtdA revealed an additional N-terminal domain not observed in previous sugar aminotransferase structures including KdnA and a different active site sequence motif surrounding the catalytic lysine. Specifically, in NtdA, there is a tyrosine residue positioned on the N-terminal side of the catalytic lysine (Tyr 246, Lys 247). This tyrosine residue precludes the ability of NtdA to bind a nucleotide-linked sugar because it projects into the pocket that would normally be occupied by the nucleobase of the substrate.
A search of the Protein Data Bank using PDBefold26 reveals that the overall architecture of KdnA matches closely to that observed for QdtB from Thermoanaerobacterium thermosaccharolyticum, a sugar aminotransferase that functions on a dTDP-linked sugar.12 The α-carbons for the subunits of KdnA and QdtB correspond with a root-mean-square deviation of 1.8 Å. Shown in Figure 7 is a superposition of the active sites of KdnA and QdtB highlighting the key features that preclude the former enzyme from binding a nucleotide-linked sugar. In QdtB there is an alanine residue preceding the active site lysine (Lys 185), which in KdnA is a valine residue. The side chain of Val 185 fills a pocket that would normally be occupied by the C-5 carbon and the C-5 oxygen of the nucleotide ribose. Additionally, in QdtB the aromatic side chains of Trp 31 and Phe 32 from the second subunit form a cradle for the thymine ring of the substrate. In KdnA, the positions of the side chains of Phe 23 and Phe 29 from the second subunit would sterically hinder the binding of a nucleotide base.
Figure 7.

Superposition of the active site regions of KdnA and QdtB. Those ligands and amino acid residues associated with KdnA are displayed in violet and blue, and the external aldimine is colored in green bonds. Those residues and the external aldimine associated with QdtB are displayed in gray bonds. Asterisks on the amino acid labels indicate that the residues are provided by the second subunit of the dimer.
In summary, the three-dimensional architectures of two key enzymes, KdnB and KdnA, involved in the biosynthesis of Kdo8N have been revealed at high resolution. Kdo8N is an unusual sugar that has thus far only been observed on the lipopolysaccharides of the genus Shewanella. Importantly some strains, such as S. putrefaciens and S. algae, are opportunistic human and marine animal pathogens.27 Given the evidence that the presence of Kdo8N may increase the integrity of the Shewanella outer membrane, the models presented here may prove useful in the future design of antibiotics.
Acknowledgments
Funding
This research was supported in part by an NIH grant (GM115921 to H.M.H.).
We thank Professor Peter Tipton for helpful comments. A portion of the research described in this paper was performed at Argonne National Laboratory, Structural Biology Center at the Advanced Photon Source (U.S. Department of Energy, Office of Biological and Environmental Research, under Contract DE-AC02-06CH11357). We gratefully acknowledge Dr. Norma E. C. Duke for collecting the fluorescence scan on the crystal at Argonne National Laboratories. This paper is dedicated in memory of Professor Christian R. H. Raetz and in appreciation of the authors who continued their investigations on KdnA and KdnB.
ABBREVIATIONS
- CMP
cytidine monophosphate
- homo-PIPES
homopiperazine-1,4-bis(2-ethanesulfonic acid)
- HPLC
high-performance liquid chromatography
- MES
2-(N-morpholino)ethanesulfonic acid
- MOPS
3-(N-morpholino)propanesulfonic acid
- NAD+
nicotinamide adenine dinucleotide (oxidized)
- NADH
nicotinamide adenine dinucleotide (reduced)
- Ni-NTA
nickel nitrilotriacetic acid
- PLP
pyridoxal 5′-phosphate
- PCR
polymerase chain reaction
- TEV
tobacco etch virus
- TCEP
tris(2-carboxyethyl)-phosphine
- Tris
tris-(hydroxymethyl)aminomethane
Footnotes
Accession Codes
X-ray coordinates have been deposited in the Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ. (accession nos. 5K8B and 5K8C).
The authors declare no competing financial interest.
References
- 1.Raetz CR, Whitfield C. Lipopolysaccharide endotoxins. Annu Rev Biochem. 2002;71:635–700. doi: 10.1146/annurev.biochem.71.110601.135414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Raetz CR, Reynolds CM, Trent MS, Bishop RE. Lipid A modification systems in gram-negative bacteria. Annu Rev Biochem. 2007;76:295–329. doi: 10.1146/annurev.biochem.76.010307.145803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vinogradov E, Korenevsky A, Beveridge TJ. The structure of the rough-type lipopolysaccharide from Shewanella oneidensis MR-1, containing 8-amino-8-deoxy-Kdo and an open-chain form of 2-acetamido-2-deoxy-D-galactose. Carbohydr Res. 2003;338:1991–1997. doi: 10.1016/s0008-6215(03)00325-2. [DOI] [PubMed] [Google Scholar]
- 4.Vinogradov E, Korenevsky A, Beveridge TJ. The structure of the core region of the lipopolysaccharide from Shewanella algae BrY, containing 8-amino-3, 8-dideoxy-D-manno-oct-2-ulosonic acid. Carbohydr Res. 2004;339:737–740. doi: 10.1016/j.carres.2003.12.010. [DOI] [PubMed] [Google Scholar]
- 5.Vinogradov E, Kubler-Kielb J, Korenevsky A. The structure of the carbohydrate backbone of the LPS from Shewanella spp. MR-4. Carbohydr Res. 2008;343:2701–2705. doi: 10.1016/j.carres.2008.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Leone S, Molinaro A, De Castro C, Baier A, Nazarenko EL, Lanzetta R, Parrilli M. Absolute configuration of 8-amino-3,8-dideoxyoct-2-ulosonic acid, the chemical hallmark of lipopolysaccharides of the genus Shewanella. J Nat Prod. 2007;70:1624–1627. doi: 10.1021/np0702988. [DOI] [PubMed] [Google Scholar]
- 7.Hau HH, Gralnick JA. Ecology and biotechnology of the genus Shewanella. Annu Rev Microbiol. 2007;61:237–258. doi: 10.1146/annurev.micro.61.080706.093257. [DOI] [PubMed] [Google Scholar]
- 8.Gattis SG, Chung HS, Trent MS, Raetz CR. The origin of 8-amino-3,8-dideoxy-D-manno-octulosonic acid (Kdo8N) in the lipopolysaccharide of Shewanella oneidensis. J Biol Chem. 2013;288:9216–9225. doi: 10.1074/jbc.M113.453324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vaara M. Polymyxins and their novel derivatives. Curr Opin Microbiol. 2010;13:574–581. doi: 10.1016/j.mib.2010.09.002. [DOI] [PubMed] [Google Scholar]
- 10.Thoden JB, Holden HM. The molecular architecture of human N-acetylgalactosamine kinase. J Biol Chem. 2005;280:32784–32791. doi: 10.1074/jbc.M505730200. [DOI] [PubMed] [Google Scholar]
- 11.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thoden JB, Schaffer C, Messner P, Holden HM. Structural analysis of QdtB, an aminotransferase required for the biosynthesis of dTDP-3-acetamido-3,6-dideoxy-alpha-D-glucose. Biochemistry. 2009;48:1553–1561. doi: 10.1021/bi8022015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr, Sect D: Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 14.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr, Sect D: Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 15.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr, Sect D: Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Laitaoja M, Valjakka J, Janis J. Zinc coordination spheres in protein structures. Inorg Chem. 2013;52:10983–10991. doi: 10.1021/ic401072d. [DOI] [PubMed] [Google Scholar]
- 17.Ruzheinikov SN, Burke J, Sedelnikova S, Baker PJ, Taylor R, Bullough PA, Muir NM, Gore MG, Rice DW. Glycerol dehydrogenase. structure, specificity, and mechanism of a family III polyol dehydrogenase. Structure. 2001;9:789–802. doi: 10.1016/s0969-2126(01)00645-1. [DOI] [PubMed] [Google Scholar]
- 18.Schneider G, Kack H, Lindqvist Y. The manifold of vitamin B6 dependent enzymes. Structure. 2000;8:R1–6. doi: 10.1016/s0969-2126(00)00085-x. [DOI] [PubMed] [Google Scholar]
- 19.Frey PA, Hegeman AD. Chemical and stereochemical actions of UDP-galactose 4-epimerase. Acc Chem Res. 2013;46:1417–1426. doi: 10.1021/ar300246k. [DOI] [PubMed] [Google Scholar]
- 20.Reid MF, Fewson CA. Molecular characterization of microbial alcohol dehydrogenases. Crit Rev Microbiol. 1994;20:13–56. doi: 10.3109/10408419409113545. [DOI] [PubMed] [Google Scholar]
- 21.Lee B, Richards FM. The interpretation of protein structures: estimation of static accessibility. J Mol Biol. 1971;55:379–400. doi: 10.1016/0022-2836(71)90324-x. [DOI] [PubMed] [Google Scholar]
- 22.Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, Wilson KS. Overview of the CCP4 suite and current developments. Acta Crystallogr, Sect D: Biol Crystallogr. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Holden HM, Cook PD, Thoden JB. Biosynthetic enzymes of unusual microbial sugars. Curr Opin Struct Biol. 2010;20:543–550. doi: 10.1016/j.sbi.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Romo AJ, Liu HW. Mechanisms and structures of vitamin B(6)-dependent enzymes involved in deoxy sugar biosynthesis. Biochim Biophys Acta, Proteins Proteomics. 2011;1814:1534–1547. doi: 10.1016/j.bbapap.2011.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van Straaten KE, Ko JB, Jagdhane R, Anjum S, Palmer DR, Sanders DA. The structure of NtdA, a sugar aminotransferase involved in the kanosamine biosynthetic pathway in Bacillus subtilis, reveals a new subclass of aminotransferases. J Biol Chem. 2013;288:34121–34130. doi: 10.1074/jbc.M113.500637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Krissinel E, Henrick K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr, Sect D: Biol Crystallogr. 2004;60:2256–2268. doi: 10.1107/S0907444904026460. [DOI] [PubMed] [Google Scholar]
- 27.Khashe S, Janda JM. Biochemical and pathogenic properties of Shewanella alga and Shewanella putrefaciens. J Clin Microbiol. 1998;36:783–787. doi: 10.1128/jcm.36.3.783-787.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Laskowski RA, Moss DS, Thornton JM. Main-chain bond lengths and bond angles in protein structures. J Mol Biol. 1993;231:1049–1067. doi: 10.1006/jmbi.1993.1351. [DOI] [PubMed] [Google Scholar]
- 29.Stivala A, Wybrow M, Wirth A, Whisstock JC, Stuckey PJ. Automatic generation of protein structure cartoons with Pro-origami. Bioinformatics. 2011;27:3315–3316. doi: 10.1093/bioinformatics/btr575. [DOI] [PubMed] [Google Scholar]
- 30.DeLano WL. The PyMOL Molecular Graphics System. DeLano Scientific; San Carlos, CA, USA: 2002. [Google Scholar]