

Abstract
Breast cancer (BC) is a heterogeneous disease, and different tumor characteristics and genetic variation may affect the clinical outcome. The FANCM c.5101C > T nonsense mutation in the Finnish population associates with increased risk of breast cancer, especially for triple‐negative breast cancer patients. To investigate the association of the mutation with disease prognosis, we studied tumor phenotype, treatment outcome, and patient survival in 3,933 invasive breast cancer patients, including 101 FANCM c.5101C > T mutation carriers and 3,832 non‐carriers. We also examined association of the mutation with nuclear immunohistochemical staining of DNA repair markers in 1,240 breast tumors. The FANCM c.5101C > T mutation associated with poor 10‐year breast cancer‐specific survival (hazard ratio (HR)=1.66, 95% confidence interval (CI) 1.09–2.52, p = 0.018), with a more pronounced survival effect among familial cases (HR = 2.93, 95% CI 1.5–5.76, p = 1.80 × 10−3). Poor disease outcome of the carriers was also found among the estrogen receptor (ER) positive subgroup of patients (HR = 1.8, 95% CI 1.09–2.98, p = 0.021). Reduced survival was seen especially among patients who had not received radiotherapy (HR = 3.43, 95% CI 1.6–7.34, p = 1.50 × 10−3) but not among radiotherapy treated patients (HR = 1.35, 95% CI 0.82–2.23, p = 0.237). Significant interaction was found between the mutation and radiotherapy (p = 0.040). Immunohistochemical analyses show that c.5101C > T carriers have reduced PAR‐activity. Our results suggest that FANCM c.5101C > T nonsense mutation carriers have a reduced breast cancer survival but postoperative radiotherapy may diminish this survival disadvantage.
Keywords: FANCM, breast cancer, survival, DNA repair, radiotherapy
Short abstract
What's new?
Variations in DNA repair genes can predispose individuals to breast cancer, with one example being FANCM c.5101C > T, a nonsense mutation in the Fanconi Anemia DNA repair pathway. In previous work, FANCM c.5101C > T was associated with increased breast cancer risk in the Finnish population. Here, the mutation is further shown to be associated with adverse breast cancer outcome. Mutation‐positive Finnish patients exhibited reduced long‐term survival and increased risk of disease recurrence. Survival was worse particularly for patients who were not treated with radiotherapy, indicating that FANCM c.5101C>T may interact with radiotherapy to improve disease outcome in mutation carriers.
Abbreviations
- BC
breast cancer
- BER
base excision repair
- CI
confidence interval
- CISH
chromogenic in situ hybridization
- DSB
double strand break
- ER
estrogen receptor
- FA
Fanconi anemia
- HR
hazard ratio
- HRM
high resolution melt
- IHC
immunohistochemistry
- KBCP
Kuopio breast cancer project
- NHEJ
non‐homologous end joining
- PARP
poly(ADP‐ribose) polymerase
- PR
progesterone receptor
- SSB
single strand break
- TNBC
triple‐negative breast cancer
Breast cancer is the most common cancer among women worldwide, and also the leading cause of female cancer death.1 Most breast cancer cases are sporadic, but around 15% have familial background. Hereditary predisposition to breast cancer is caused by variation in multiple genes commonly involved in DNA repair, especially with homologous recombination repair pathway.2 Recently, we identified a new breast cancer allele in the Finnish population in the FANCM gene, that functions in the Fanconi Anemia (FA) DNA repair pathway. The FANCM c.5101C > T (p.Q1701X, rs147021911) nonsense mutation increased the risk of breast cancer over twofold, and 3.5‐fold increased frequency was seen among the triple‐negative breast cancer (TNBC) cases.3
Predisposing mutations may associate with specific breast cancer phenotype or subgroup, as well as with patient prognosis and treatment outcome. CHEK2 and PALB2 truncating mutations, as well as FANCM c.5101C > T mutation, confer moderate risk for breast cancer, with a higher risk among patients with family history of breast cancer.3 – 7
CHEK2 c.1100delC and PALB2 c.1592delT mutations are associated also with an increased risk of breast cancer death or second breast cancer. Among patients with ER positive breast cancer, CHEK2 c.1100delC heterozygosity is associated with 1.6‐fold risk of breast cancer specific death and 3.5‐fold risk of a second breast cancer.8, 9 A significant proportion of PALB2 tumors are triple‐negative and the PALB2 mutation carriers have about 2‐fold increased risk of breast cancer death, independently of the triple‐negative status.7, 10
Here, we studied tumor characteristics, patient survival, and treatment outcome associated with the FANCM c.5101C > T mutation among 3,933 breast cancer patients in four breast cancer patient series from Finland. In addition, we examined the nuclear immunohistochemical staining of DNA repair markers in the mutation carrier and non‐carrier tumors from 1,240 invasive breast cancer cases.
Material and Methods
Subjects
Helsinki breast cancer series
The unselected breast cancer patient samples from Helsinki were collected at Helsinki University Central Hospital. From this cohort, 884 samples, including 79% of all consecutive, newly diagnosed breast cancer cases during the collection periods were collected at Department of Oncology in 1997–1998 and 2000.11, 12 In addition, 986 samples, including 87% of all consecutive, newly diagnosed breast cancer cases were collected at Department of Surgery in 2001–2004.13 Of these series, 397 cases had family history of breast cancer.
Additional familial breast cancer series was collected at Helsinki University Central Hospital Departments of Oncology and Clinical Genetics.13, 14 When combining the unselected and the additional familial samples, 524 patients had strong family history with at least three breast or ovarian cancers among first or second degree relatives (including the proband) and 568 patients had at least one first degree relative affected with breast or ovarian cancer. All the patients with strong family history were tested negative for BRCA1/2 mutations and the patients with one affected relative were tested negative for Finnish BRCA1/2 founder mutations as previously described.5, 15, 16 Only invasive cases were included in the analyses (N = 2,337).
All samples are genomic DNA isolated from peripheral blood. The patient genealogies were confirmed with population registries or hospital records and cancer diagnoses through the hospital records and the Finnish Cancer Registry. ER and progesterone hormone receptor (PR) status (positive when >10% of cells were stained) and tumor histology information were collected from pathology reports, HER2‐status is based on immunohistochemistry and gene amplification as described earlier.17, 18, 19 Information on breast cancer death was obtained from the Finnish Cancer Registry.
Tampere breast cancer series
The unselected breast cancer patient samples from Tampere area were collected in 1997–1999 and additional 336 incident cases in 1996–2004 at Tampere University Hospital as previously described.11, 13 Only invasive cases were included in the analysis (N = 650). All samples are genomic DNA isolated from peripheral blood. ER and PR hormone receptor status (positive when >10% of cells were stained), HER2‐status, and other clinicopathological information was obtained from patient and pathology reports and information on breast cancer death from the Finnish Cancer Registry.
Oulu breast cancer series
The unselected breast cancer patient samples from Northern Finland were collected at the Oulu University Hospital between the years 2000 and 2007. Only invasive cases were included in the analysis (N = 516). All samples are genomic DNA isolated from peripheral blood. HER2‐status was studied by means of immunohistochemistry (positivity defined as weak, moderate or strong levels of staining and negativity completely negative staining) and chromogenic in situ hybridization (CISH). ER and PR hormone receptor status (positive when >10% of cells were stained) and tumor histology information was collected from the pathology reports as described earlier.20, 21 Information on breast cancer death was obtained from the Oulu University Hospital.
Kuopio breast cancer series
For this study a sample set was used from The Kuopio Breast Cancer Project (KBCP), a prospective population‐based case‐control study conducted in 1990–1995. Women entering Kuopio University Hospital due to breast symptoms were invited to take part in the study at their first visit to the hospital. Altogether 516 women out of 1,919 were eventually diagnosed to have breast cancer. Hospital registries were used to collect information concerning clinicopathological features of the breast cancer, surgical and oncological treatments, and follow‐up.22, 23 ER and PR hormone receptors were classified as positive if the percentage of positive cells with nuclear staining was ≥ 10%. HER2 status assessment was conducted by immunohistochemistry (IHC). Samples with IHC score 2+ or 3+ were classified as HER2 positive (HER2+). Altogether, 430 female patients with invasive breast cancer were included in the survival analysis. All samples are genomic DNA isolated from peripheral blood.
This study was performed with informed consent from the patients and permission from the ethics committees of Helsinki University Hospital, Oulu University Hospital, Tampere University Hospital, University of Eastern Finland, and Kuopio University Hospital Board on Research Ethics.
Genotyping
FANCM c.5101C > T genotyping for the Helsinki and Tampere sample sets was performed with Sequenom MassARRAY system as previously described3 and for Oulu and Kuopio sample sets by using PCR‐based high resolution melt (HRM)—analysis and Sanger sequencing. The HRM PCR reactions were performed in 96 well plates using Type‐it HRM PCR Kit (Qiagen, Hilden, Germany) and CFX96 Real‐Time PCR Detection System (CFX96, Bio‐Rad, Hercules, CA). Primers used for the genotyping and sequencing FANCM c. 5101C > T mutation for Oulu and Kuopio cohorts were: F: 5'TCAAGTGAGGAGGAGAACAATG3', R: 5'TCAGCGATGTCTGTTTGCTC3'.
Statistical Analyses
All four datasets including altogether 3,933 invasive breast cancer patients from Helsinki, Tampere, Oulu, and Kuopio areas of Finland, were pooled for statistical analyses. All statistical analyses were performed using the R version 3.0.2 statistical software (http://www.r-project.org/). Kaplan–Meier survival curves and uni and multivariate Cox's proportional hazard models were used to estimate the hazard ratios and confidence intervals for survival and forest plots were drawn for visualization. All analyses were stratified by the study. The primary end point of the survival analyses was breast cancer death with 10‐year follow‐up time. In addition, 5‐year survival analysis with local recurrence as an endpoint was used for survival analyses in the radiotherapy‐based subgroups in the Helsinki data set (N = 2,337), where the information about local recurrence of the disease was available. Time‐to‐event was calculated from the date of the patient diagnosis and to account for the latency between diagnosis and recruitment into the study, all follow‐up times were left‐truncated. Cases with missing data were excluded from the analyses.
The multivariate analyses included the common clinically relevant factors (ER, grade, tumor size, nodal status) and/or cancer treatments (radiotherapy, endocrine therapy, and chemotherapy) as categorical co‐variates and were stratified by the study; inclusion of the study as a categorical co‐variate did not affect the result. In addition, the FANCM c.5101C > T genotype from the pooled data set was fitted into two Cox's proportional hazard models in order to test the interaction between the mutation and radiotherapy treatment. One model included the treatment and FANCM c.5101C > T genotype as individual covariates and the other included an interaction term between these two. Two‐way anova was used as a likelihood‐ratio test to compare the two models.24, 25
The p‐values for comparisons of histopathological features of mutation carriers and non‐carriers were calculated with Pearson's chi squared test or Fisher exact test (for n ≤ 5). Logistic regression was used for histopathological features with more than two categories. p‐values <0.05 were considered statistically significant.
To test whether FANCM mutation status correlates with immunohistochemical expression of markers involved in DNA damage response and repair, we analyzed a number of markers that have been stained and scored as described in our previous studies: BRCA1, FANCD2, RAD51, XPF, PAR26; ATM,18 gamma‐HA2X,27 and TP53.28 For the continuously scored markers (BRCA1, FANCD2, RAD51, XPF, and PAR; % positive nuclei and staining intensity score as determined by automated analysis), association with FANCM mutation status was tested using a Kruskal–Wallis test. All other markers used categorical scoring and a χ 2 test was employed as the test for association. Further information is available in Supporting Information Appendix.
Results
All survival analysis results are based on the 3,933 invasive breast cancer cases in the pooled data set with 581 breast cancer deaths, except the survival analysis among radiotherapy‐based subgroups with local recurrence as an endpoint is based on the Helsinki data set with 2,337 invasive samples, including 344 breast cancer deaths. The pooled data set includes 101 FANCM c.5101C > T mutation carriers and 3,832 non‐carriers, Helsinki data set includes 61 mutation carriers and 2,276 non‐carriers. The tumor characteristics of the patients and detailed description of all the datasets used are presented in Table 1.
Table 1.
Description of the patient data sets used in this study
| Helsinki | Tampere | Oulu | Kuopio | |
|---|---|---|---|---|
| No. of cases | 2,337 | 650 | 516 | 430 |
| No. of mutation carriers | 61 (2.6%) | 26 (4%) | 5 (1%) | 9 (2%) |
| Vital status | ||||
| Alive | 1,482 (64%) | 448 (69%) | 362 (70%) | 176 (41%) |
| Deceased: all‐cause | 511 (21%) | 118 (18%) | 94 (18%) | 161 (37%) |
| Deceased: breast cancer | 344 (15%) | 84 (13%) | 60 (12%) | 93 (22%) |
| Follow‐up mean ±SD (years) | 8.16 ± 2.4 | 7.44 ± 2.13 | 5.17 ± 2.92 | 7.78 ± 3.08 |
| Age at diagnosis, mean [range] | 56.3 [21–95] | 58.9 [30–88] | 57.4 [28–92] | 58.1 [23–91] |
| Estrogen receptor | ||||
| Negative | 430 (18%) | 128 (20%) | 96 (19%) | 101 (23%) |
| Positive | 1,803 (77%) | 508 (78%) | 385 (75%) | 300 (70%) |
| Missing data | 104 (5%) | 14 (2%) | 35 (7%) | 29 (7%) |
| Grade | ||||
| 1 | 580 (25%) | 197 (30%) | 76 (15%) | 115 (27%) |
| 2 | 980 (42%) | 226 (35%) | 212 (41%) | 196 (46%) |
| 3 | 651 (28%) | 133 (20%) | 177 (34%) | 115 (27%) |
| Missing data | 126 (5%) | 94 (14%) | 51 (10%) | 4 (1%) |
| T/tumor size category | ||||
| 1 | 1,409 (60%) | 401 (62%) | 238 (46%) | 229 (53%) |
| 2 | 743 (32%) | 213 (33%) | 226 (44%) | 161 (37%) |
| 3 | 69 (3%) | 24 (4%) | 15 (3%) | 23 (5%) |
| 4 | 82 (4%) | – | – | 17 (4%) |
| Missing data | 34 (1%) | 12 (2%) | 37 (7%) | – |
| N (nodal metastasis) | ||||
| Negative | 1,263 (54%) | 390 (69%) | 265 (51%) | 251 (58%) |
| Positive | 1,036 (44%) | 260 (40%) | 216 (42%) | 171 (40%) |
| Missing data | 38 (2%) | – | 35 (7%) | 8 (2%) |
| M (distant metastasis) | ||||
| Negative | 2,253 (96.5%) | 630 (97%) | 492 (95%) | 419 (97%) |
| Positive | 73 (3%) | 12 (2%) | 24 (5%) | 11 (3%) |
| Missing data | 11 (0.5%) | 8 (1%) | – | – |
| Histological type | ||||
| Ductal | 1,597 (68%) | 537 (83%) | 371 (71%) | 281 (65%) |
| Lobular | 470 (20%) | 86 (13%) | 78 (15%) | 73 (17%) |
| Medullar | 29 (1%) | – | 2 (1%) | 8 (2%) |
| Other | 240 (10%) | 18 (3%) | 30 (6%) | 68 (16%) |
| NA | 1 | 9 (1%) | 35 (7%) | – |
| Radiotherapy | ||||
| Yes | 1,829 (78%) | 493 (76%) | 423 (82%) | 251 (58%) |
| No | 443 (19%) | 155 (24%) | 87 (17%) | 179 (42%) |
| Missing data | 65 (3%) | 2 | 6 (1%) | – |
| Chemotherapy | ||||
| Yes | 870 (37%) | 131 (20%) | 215 (42%) | 83 (19%) |
| No | 1,405(60%) | 511 (79%) | 297 (58%) | 347 (81%) |
| Missing data | 62 (3%) | 8 (1%) | 4 (1%) | – |
| Endocrine therapy | ||||
| Yes | 1,055 (45%) | 204 (32%) | 243 (47%) | 105 (24%) |
| No | 1,207 (52%) | 444 (68%) | 268 (52%) | 325 (76%) |
| Missing data | 65 (3%) | 2 | 5 (1%) | – |
Histopathological Features of the FANCM C.5101C > T Positive Tumors
The association of the FANCM c.5101C > T mutation with histopathological features of the tumors was studied in the pooled data among all cases and separately among ER positive cases (Table 2). The mutation did not associate with any common clinical feature, however the breast tumors from the c.5101C > T mutation carriers were more often of triple negative phenotype (p = 0.060, compared with tumors from non‐carriers).
Table 2.
Histopathological features of FANCM c.5101C > T‐mutation carriers and wild type tumors
| Category | FANCM c.5101C>T | % | FANCM wt | % | p | Model |
|---|---|---|---|---|---|---|
| All breast cancer cases | ||||||
| Grade | 0.263 | Logistic regression | ||||
| 1 | 25 | 26.00% | 943 | 26.00% | ||
| 2 | 36 | 37.00% | 1,578 | 44.00% | ||
| 3 | 36 | 37.00% | 1,040 | 30.00% | ||
| T | 0.255 | Logistic regression | ||||
| 1 | 53 | 53.00% | 2,224 | 59.00% | ||
| 2 | 41 | 41.00% | 1,302 | 34.50% | ||
| 3 | 2 | 2.00% | 129 | 3.00% | ||
| 4 | 4 | 4.00% | 95 | 2.50% | ||
| N | 0.380 | Pearson chisq. | ||||
| neg | 52 | 52.00% | 2,117 | 56.20% | ||
| pos | 48 | 48.00% | 1,653 | 43.80% | ||
| M | 0.770 | Fisher | ||||
| neg | 99 | 99.00% | 3,695 | 96.90% | ||
| pos | 2 | 1.00% | 118 | 3.10% | ||
| ER | 0.432 | Pearson chisq. | ||||
| neg | 23 | 23.00% | 726 | 19.80% | ||
| pos | 77 | 77.00% | 2,936 | 80.20% | ||
| PR | 0.380 | Pearson chisq. | ||||
| neg | 39 | 39.00% | 1,271 | 34.80% | ||
| pos | 61 | 61.00% | 2,386 | 65.20% | ||
| Her2 | 0.167 | Pearson chisq. | ||||
| neg | 67 | 90.50% | 2,336 | 91.50% | ||
| pos | 7 | 9.50% | 422 | 8.50% | ||
| TN | 0.060 | Pearson chisq. | ||||
| TN | 13 | 14.00% | 297 | 8.50% | ||
| NOT TN | 80 | 86.00% | 3,215 | 91.50% | ||
| Morphology | 0.366 | Logistic regression | ||||
| Ductal | 78 | 77.00% | 2,708 | 71.50% | ||
| Lobular | 14 | 14.00% | 693 | 18.30% | ||
| Medullar | 1 | 1.00% | 38 | 1.00% | ||
| Other | 8 | 8.00% | 348 | 9.20% | ||
| ER‐positive breast cancer cases | ||||||
| Grade | 0.813 | Logistic regression | ||||
| 1 | 25 | 33.50% | 874 | 31.50% | ||
| 2 | 33 | 44.00% | 1,379 | 49.50% | ||
| 3 | 17 | 22.50% | 524 | 19.00% | ||
| T | 0.279 | Logistic regression | ||||
| 1 | 45 | 57.00% | 1,827 | 63.00% | ||
| 2 | 27 | 35.00% | 934 | 32.00% | ||
| 3 | 1 | 3.00% | 87 | 3.00% | ||
| 4 | 4 | 5.00% | 67 | 2.00% | ||
| N | 0.500 | Pearson chisq. | ||||
| neg | 40 | 52.50% | 1,265 | 43.50% | ||
| pos | 36 | 47.50% | 1,644 | 46.50% | ||
| M | 0.775 | Fisher | ||||
| neg | 76 | 98.70% | 2,841 | 98.00% | ||
| pos | 1 | 1.30% | 65 | 2.00% | ||
| PR | 0.754 | Pearson chisq. | ||||
| neg | 17 | 22.00% | 604 | 20.00% | ||
| pos | 60 | 88.00% | 2,326 | 80.00% | ||
| Her2 | 1 | Fisher | ||||
| neg | 52 | 91.00% | 1,943 | 90.00% | ||
| pos | 5 | 9.00% | 219 | 10.00% | ||
| Morphology | 0.587 | Logistic regression | ||||
| Ductal | 56 | 73.00% | 2,007 | 68.00% | ||
| Lobular | 14 | 18.00% | 635 | 22.00% | ||
| Medullar | 0 | 0.00% | 4 | 0.00% | ||
| Other | 7 | 9.00% | 286 | 10.00% | ||
Abbreviations: T: tumor size class; M: distant metastasis; ER: estrogen receptor; PR: progesterone receptor
FANCM C.5101C > T Mutation Associates with Breast Cancer Survival
To evaluate the association of the FANCM c.5101C > T mutation with the disease outcome, we examined 10‐year breast cancer specific survival by Cox's univariate proportional hazard analysis in 3,933 invasive breast cancer patients from Helsinki, Tampere, Oulu, and Kuopio data sets. The mutation was associated with poor breast cancer‐specific survival in the pooled data set stratified for study (HR = 1.66, 95% CI 1.09–2.52, p = 0.018). Absolute uncorrected survival rates are illustrated in Figure 1a. However, in the multivariate survival analysis including the common clinical features (ER, grade, tumor size, nodal status) and the conventional cancer treatments (radiotherapy, chemotherapy, endocrine treatment) the mutation was not significantly and independently prognostic in the pooled data set (HR = 1.44, 95% CI 0.91–2.26, p = 0.133) (Supporting Information Table 1).
Figure 1.
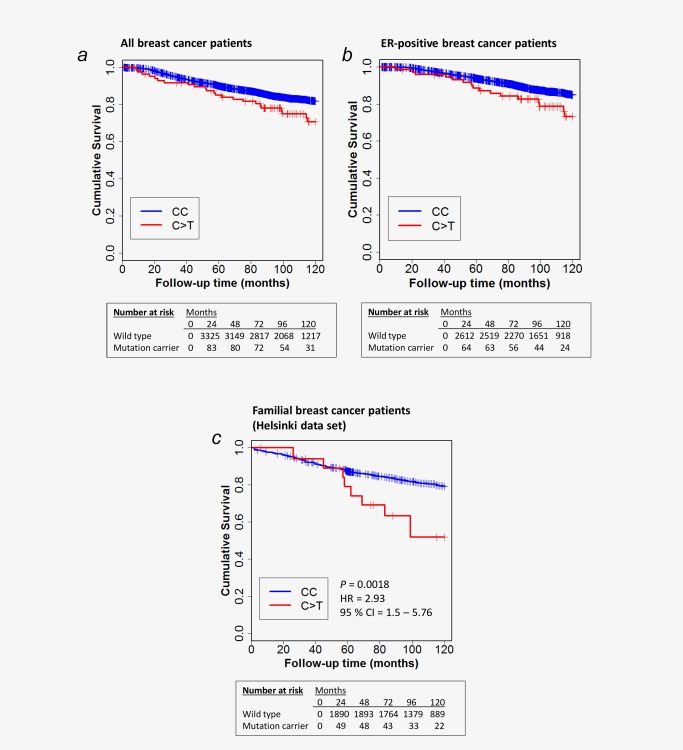
Kaplan–Meier plots of cumulative survival for breast cancer death in 10 years. Absolute uncorrected survival rates are presented among the pooled data set (HR = 1.62, 95% CI 1.07–2.46, p Cox's regression = 0.023; (a) and among ER‐positive patients (HR = 1.8, 95% CI 1.09–2.98, p Cox's regression = 0.021; (b) Results for survival analysis among familial cases (C) from Helsinki.
As the mutation associates with triple‐negative phenotype with poor survival as such, we analyzed the survival specifically also among ER positive cases. The mutation associated with reduced survival also in the ER‐positive group of patients in the pooled data set stratified for study (HR = 1.8, 95% CI 1.09–2.98, p = 0.021). Absolute uncorrected survival rates are illustrated in Figure 1b. Furthermore, as the FANCM c.5101C > T mutation associates with familial breast cancer risk, we performed the survival analysis for the invasive familial cases (N = 1,006) among the Helsinki dataset in which familial status was available for the samples. The breast cancer specific survival was worse for mutation carriers among patients with family history of the disease (HR = 2.93, 95% CI 1.5–5.76, p = 1.80 × 10−3; Fig. 1c).
Survival in Subgroups Defined by Tumor Phenotype and Treatment
To examine the survival effect of the FANCM c.5101C > T mutation in more detail, we performed univariate Cox's proportional hazard analysis (endpoint: breast cancer death in 10 years) in subgroups based on the tumor phenotype (ER, PR, TN, nodal status, tumor size, grade) among the pooled data set (N = 3,933). In addition, we performed univariate Cox's proportional hazard analysis by the conventional cancer treatment options (endocrine treatment, radiotherapy, and/or chemotherapy) to examine the treatment outcome of the FANCM c.5101C > T mutation carriers. Forest plot was drawn for visualizing hazard ratios and confidence intervals (Fig. 2). As the worse survival was also seen among the ER‐positive patients, we performed similar subgroup analyses (PR, TN, nodal status, tumor size, grade, and the anticancer treatments) among ER‐positive patients (N = 3,013) (Supporting Information Fig. 1). Heterogeneity in the survival effect was seen for the c.5101C > T mutation carriers related to radiotherapy treatment, with significantly reduced survival especially among patients who had not received radiotherapy (HR = 3.43, 95% CI 1.6–7.34, p = 1.50 × 10−3) but not among radiotherapy treated patients (HR = 1.35, 95% CI 0.82–2.23, p = 0.237).
Figure 2.
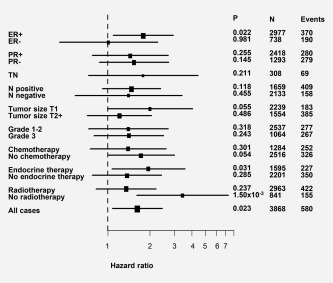
Forest plot of hazard ratios and their confidence intervals for the FANCM c.5101 C > T mutation in the pooled data set and in different subgroups including the clinical factors and conventional cancer treatments. The Cox proportional hazard model was used for 10‐year breast‐cancer specific survival. Horizontal lines represent 95% confidence intervals. ER = estrogen receptor, PR = progesterone receptor, TN =triple negative, N = nodal metastasis status. All cases = all cases after samples with missing data are excluded.
To further examine the radiotherapy outcome among the c.5101C > T carriers, we performed survival analysis with local recurrence (within 5 years) as an endpoint in the Helsinki data set where the recurrence information was available (N = 2,337). Increased risk for local recurrence was observed for mutation carriers who had not received radiotherapy (HR = 6.19, 95% CI 1.46–26.2, p = 0.013, Supporting Information Table 2) but not among radiotherapy treated patients (HR = 0.98, 95% CI = 0.24–4.00, p = 0.979). In the multivariate model, the FANCM c.5101C > T mutation is only borderline significant (p = 0.086), however the hazard ratios remain consistent.
Next, we tested interaction between FANCM c.5101C > T genotype and radiotherapy treatment with Cox's proportional hazard model stratified with study among pooled data set, including 2,996 patients who had received radiotherapy and 864 who had not (Table 3A). A significant interaction was seen between the mutation and radiotherapy treatment (p = 0.032), with a protective hazard ratio (HR = 0.37, 95% CI 0.15–0.92). A likelihood‐ratio test comparing models with interaction term and model with independent covariates displayed an interactive effect between the covariates (p (interaction)=0.040). These results suggest that FANCM‐mutation positive breast cancer patients may benefit from radiotherapy more than non‐carriers, an issue that should be further investigated to clarify the absolute benefits from radiotherapy to such patients.
Table 3.
A) Cox's proportional hazard model to test the interaction between radiotherapy treatment and FANCM c.5101C > T mutation with breast cancer death as an endpoint; B) Local recurrence as an endpoint
| Covariate | HR | p | 95% CI | Endpoint |
|---|---|---|---|---|
| A | ||||
| Model 1: no interaction | Breast cancer death (10 yrs) | |||
| RS147021911 | 1.71 | 0.011 | 1.13–2.60 | |
| Radiotherapy | 0.70 | 1.0 × 10−4 | 0.58‐0.84 | |
| Covariate | HR | p | 95% CI | Endpoint |
| Model 2: interaction | Breast cancer death (10 yrs) | |||
| RS147021911 | 3.72 | 7.00 × 10−4 | 1.74‐7.95 | |
| Radiotherapy | 0.72 | 8.40 × 10−4 | 0.59–0.87 | |
| RS147021911:Radiotherapy | 0.37 | 0.032 | 0.15–0.92 | |
| Likelihood ratio test p values | 0.040 | |||
| B | ||||
| Covariate | HR | p | 95% CI | Endpoint |
| Model 1: no interaction | Local recurrence (5 yrs) | |||
| RS147021911 | 1.71 | 0.298 | 0.62–4.64 | |
| Radiotherapy | 0.48 | 1.05 × 10−3 | 0.31–0.75 | |
| Covariate | HR | p | 95% CI | Endpoint |
| Model 2: interaction | Local recurrence (5 yrs) | |||
| RS147021911 | 5.96 | 1.50 × 10−3 | 1.42–25.11 | |
| Radiotherapy | 0.52 | 4.05 × 10−3 | 0.33–0.81 | |
| RS147021911:Radiotherapy | 0.16 | 0.080 | 0.02–1.23 | |
| Likelihood ratio test p values | 0.090 | |||
We further studied the survival interaction of FANCM mutation with radiotherapy using similar interaction model with local recurrence (within 5 years) as an endpoint in the Helsinki data set (N = 2,069) (Table 3B). Due to the smaller sample size and thus loss of statistical power, the significance of the interactive effect is not apparent (likelihood‐ratio test p values 0.090). However, even more pronounced protective hazard ratio was seen for FANCM c.5101C > T mutation and radiotherapy interaction (HR = 0.16), compared to significantly increased hazard ratio for mutation alone (HR = 5.96).
Immunohistochemical Analyses
In the association analysis between FANCM c.5101C > T mutation status and DNA repair related immunohistochemical markers, a statistically significant association was detected between nuclear poly‐ADP‐ribose (PAR; a measurement of PARP activity) staining and mutated FANCM. PAR staining was reduced in FANCM c.5101C > T mutation carrier tumors, both in terms of the proportion of positively stained tumor nuclei (p = 0.016, Kruskal‐Wallis test) and staining intensity (p = 0.011, Kruskal–Wallis test) (Supporting Information Fig. 2). No other immunohistochemical markers were associated with mutated FANCM (Supporting Information Table 3).
Discussion
This study evaluated the survival association, tumor characteristics, and treatment outcome for Finnish breast cancer patients carrying the FANCM c.5101C > T mutation. We detected an association between the FANCM c.5101C > T mutation and adverse breast cancer outcome (HR = 1.66, 95% CI 1.09 – 2.52, p = 0.018, N = 3,832 [non‐carriers], N = 101 [mutation carriers]). The breast cancer specific survival was worse among familial cases (HR = 2.93, 95% CI 1.5–5.76, p = 1.80 × 10−3, N = 981 [non‐carriers], N = 25 [mutation carriers]).
When examining the tumors of the FANCM c.5101C > T mutation carriers, a borderline significant association of the mutation was seen with triple‐negative tumors (p = 0.060, compared with tumors from non‐carriers). This is in line with the previous risk analysis, in which the FANCM c.5101C > T mutation was found to be associated with 3.6‐fold increased risk for triple‐negative subtype of breast cancer.3 This type of breast cancer is generally aggressive with poor prognosis and no effective therapies available.29 However, our survival analysis indicates that the poor prognosis associated with FANCM c.5101C > T mutation is not only a result of the higher incidence of the triple‐negative tumors, as the mutation also associates with worse survival among the ER‐positive subgroup of patients. Yet in the multivariate survival analysis including conventional prognostic markers and treatments, the FANCM c.5101C > T mutation was not independently prognostic (HR = 1.44, 95% CI 0.91‐2.26, p = 0.133).
The comprehensive survival analyses revealed an association with FANCM c.5101C > T mutation and radiotherapy outcome. Interaction analyses with a hazard ratio of 0.37 (95% CI 0.15–0.95, p = 0.032) for the mutation:radiotherapy interaction compared to the HR of 3.72 for the mutation alone (95% CI 1.74–7.95, p = 7.00 × 10−4) in the interaction model indicate that the mutation carriers may benefit from radiotherapy. To this end, we performed the interaction analyses also with local recurrence in five years as an endpoint, as radiotherapy is commonly used to prevent such events. While this interaction model is not statistically significant in the smaller sample set, the more pronounced protective hazard ratio of 0.16 for the radiotherapy and FANCM c.5101C > T interaction further supports our observations that carrying the FANCM c. 5101C > T mutation increases the risk for local recurrence and subsequently also death from breast cancer, however the mutation carriers seem to benefit from postoperative radiotherapy. From the pathobiological point of view, we propose that the increased risk of local recurrence and death may reflect enhanced genomic instability and hence aggressiveness due to impaired DNA repair in the tumors with the FANCM c. 5101C > T mutation. On the positive side, such enhanced genetic instability and suboptimal repair capacity seem to represent a specific vulnerability of such tumors, manifest particularly after an extra burden of difficult‐to repair DNA damage caused by ionizing radiation treatment. Overall, these results are especially interesting, as markers associated with radiotherapy treatment outcome for cancer patients have not been previously described. However, further studies in larger datasets are needed to validate the radiotherapy outcome for FANCM mutation carriers.
FANCM is a multifunctional protein, acting as an anchor protein for both Fanconi Anemia and Bloom syndrome complexes, two molecular pathways that functionally overlap in these genetic disorders.30, 31, 32 As a part of the FA pathway, FANCM operates in the interstrand crosslink repair to facilitate various DNA repair processes, such as homologous recombination and non‐homologous end‐joining (NHEJ) pathway.30, 33 Inactivation of the FA pathway leads to hypersensitivity to DNA crosslinking agents, and in the absence of FANCM, the formation of the FA and Bloom's complexes is unsuccessful and this may explain the tumorigenetic characteristics of defective FANCM protein.32 Interestingly, in addition to BRCA‐genes, recent studies link several Fanconi anemia pathway genes also with sensitivity to PARP inhibition, including PALB2, RAD51C, and SLX4, 34, 35, 36 as well as FANCM.35 Mutations in FANCM were found to cause hypersensitivity to PARP inhibitors, indicating that FANCM actually has a role in the cellular defense against PARP inhibition.37 This may reflect the several roles FANCM has in cells also outside the Fanconi Anemia pathway, including replisome stability and cell cycle checkpoint activation when DNA repair is needed.38, 39, 40
Taking the DNA repair functions of FANCM in consideration, we examined nuclear immunohistochemical staining profiles of DNA repair markers of the FANCM c.5101C > T mutation carriers. Among eight examined markers, the mutation was associated with low expression of poly (ADP‐ribose) marker (PAR), which measures the activity of the PARP enzymes participating in DNA repair processes in cells,41 indicating that the mutation carriers have decreased PARP‐activity. It must be noted that our immunohistochemical method measures the overall poly(ADP‐ribosyl)ation levels in tumor nuclei, and is therefore not specific to any particular PARP enzyme or biological process. The best known example of PARylation occurs in response to DNA damage, where the binding and activity of PARP promotes DNA repair through the single‐strand break (SSB), double‐strand break (DSB), or base excision repair (BER) pathways.42 In the case of excessive DNA damage, hyper‐PARylation may also be a signal for cell death.43 PARylation has additionally been reported to play a role in mitosis, chromatin remodeling, regulation of transcription, and the organization of genomic regulatory regions via insulator elements.44 We can therefore only speculate on the specific functional significance of the FANCM‐associated reduction in PARylation observed in our breast tumor samples. We did not detect a change in gamma‐H2AX staining, suggesting that a major quantitative change in overall DNA damage is not the case here. Since both FANCM and PARP are involved in resolving replication stress,45, 46, 47 it is possible that the FANCM c.5101C > T mutation‐associated reduction in PAR staining indicates a replication stress sensitive phenotype that would respond strongly to the extreme replication stress caused by radiation therapy. While the causal relationship of FANCM with reduced PARylation levels remains unclear, our data may have therapeutic implications.27, 48 Given the role of FANCM in resolving replication stress, the FANCM‐mutant tumors may be especially sensitive to drugs that further exacerbate the extent of replication stress, such as PARP inhibitors. Based on our present results and the emerging knowledge in the field, we suggest that the subset of FANCM‐mutant tumors may be particularly vulnerable to PARP inhibitors, used either as a monotherapy or, as our data indicate, combined with radiotherapy. Future preclinical and clinical studies should test the feasibility of these conceptually plausible options.
Conclusions
Our findings indicate that the FANCM c.5101C > T mutation in Fanconi Anemia pathway associates with the disease outcome of breast cancer. Based on the large series of Finnish breast cancer patients, we have shown here that the mutation carriers have worse long‐term survival and increased risk for local recurrence, however the survival may be improved with radiotherapy. Further analyses in larger datasets are warranted to clarify the survival effects and functional mechanisms associated with the mutation, especially on the efficacy of radiotherapy. Such studies may eventually help to understand the biological mechanisms affecting tumor progression and further support efforts for creating more targeted treatment combinations and risk estimation.
Author Contributions
J.I.K., R.F., C.B. and H.N. designed the study and drafted the manuscript.
J.I.K. analyzed and pooled the data.
J.I.K. and A.T. carried out the molecular genetic studies.
J.I.K. performed the statistical analyses with R.F., S.K., M.J. and L.M.P.
J.B. and J.I.B. performed and evaluated some of the immunohistochemical analyses and J.B. contributed to conceptual discussions and manuscript writing.
T.M., K.P., A.M., M.T., V.‐M.K., R.W., A.K. and K.A. contributed samples and patient information. All authors read and approved the final manuscript.
Supporting information
Supporting Information
Figure 1
Figure 2
Table 1
Table 2
Table 3
Acknowledgements
Authors would like to thank all the volunteered patients who participate in this study. Helsinki breast cancer study thanks research nurses Irja Erkkilä and Virpi Palola from Helsinki University Hospital for their help with collecting patient data and samples, and the staff at the Technology Centre, Institute for Molecular Medicine Finland (FIMM) for SNV marker genotyping of the Helsinki sample set. The Finnish Cancer Registry is gratefully acknowledged for the cancer diagnostic data and Drs. David Weaver and Kam Sprott for the help with immunohistochemistry data. Oulu breast cancer study would like to thank Leena Keskitalo and Annika Väntänen for their technical assistance. Kuopio breast cancer study thanks Eija Myöhänen for skillful technical assistance.
The Helsinki breast cancer study has been supported by the Helsinki University Central Hospital Research Fund, the Academy of Finland (132473), the Sigrid Juselius Foundation, and the Cancer Society of Finland, and by the Finnish Cultural Foundation and the Paulo Foundation for L.M.P., and Biomedicum Helsinki Foundation for J.K. The Oulu breast cancer study was supported by the Academy of Finland (284605), the Cancer Society of Finland, the Sigrid Juselius Foundation, the University of Oulu, the University of Oulu Support Foundation, the special Governmental EVO funds for Oulu University Hospital‐based research activities for R.W., and the Academy of Finland (250083) for K.P. The Kuopio breast cancer study was supported by the special Government Funding of Kuopio University Hospital Grants, The Cancer Society of Finland, and the strategic fund of the University of Eastern Finland. J.B. and J.I.B. are supported by the Novo Nordisk Foundation and the Danish Cancer Society, the Danish National Research Foundation (DNRF125, Center of Excellence CARD), the Swedish Research Council and CancerFonden.
This article was published online on 19 September 2016. An error was subsequently identified. This notice is included in the online and print versions to indicate that both have been corrected on 26 September 2016.
Disclosure: The authors have declared no conflicts of interest.
References
- 1. Torre LA, Bray F, Siegel RL, et al. Global cancer statistics, 2012. CA Cancer J Clin 2015;65:87–108. [DOI] [PubMed] [Google Scholar]
- 2. Foulkes WD. Inherited susceptibility to common cancers. N Engl J Med 2008;359:2143–53. [DOI] [PubMed] [Google Scholar]
- 3. Kiiski JI, Pelttari LM, Khan S, et al. Exome sequencing identifies FANCM as a susceptibility gene for triple‐negative breast cancer. Proc Natl Acad Sci USA 2014;111:15172–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. CHEK2 Breast Cancer Case‐Control Consortium CHEK2*1100delC and susceptibility to breast cancer: a collaborative analysis involving 10,860 breast cancer cases and 9,065 controls from 10 studies. Am J Hum Genet 2004;74:1175–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Vahteristo P, Bartkova J, Eerola H, et al. A CHEK2 genetic variant contributing to a substantial fraction of familial breast cancer. Am J Hum Genet 2002;71:432–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Antoniou AC, Casadei S, Heikkinen T, et al. Breast‐cancer risk in families with mutations in PALB2. N Engl J Med 2014;371:497–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Heikkinen T, Kärkkäinen H, Aaltonen K, et al. The breast cancer susceptibility mutation PALB2 1592delT is associated with an aggressive tumor phenotype. Clin Cancer Res 2009;15:3214–22. [DOI] [PubMed] [Google Scholar]
- 8. Weischer M, Nordestgaard BG, Pharoah P, et al. CHEK2*1100delC heterozygosity in women with breast cancer associated with early death, breast cancer‐specific death, and increased risk of a second breast cancer. J Clin Oncol 2012;30:4308–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Schmidt MK, Tollenaar RA, de Kemp SR, et al. Breast cancer survival and tumor characteristics in premenopausal women carrying the CHEK2*1100delC germline mutation. J Clin Oncol 2007;25:64–9. [DOI] [PubMed] [Google Scholar]
- 10. Cybulski C, Kluźniak W, Huzarski T, et al. Clinical outcomes in women with breast cancer and a PALB2 mutation: a prospective cohort analysis. Lancet Oncol 2015;6:638–44. [DOI] [PubMed] [Google Scholar]
- 11. Syrjäkoski K, Vahteristo P, Eerola H, et al. Population‐based study of BRCA1 and BRCA2 mutations in 1035 unselected Finnish breast cancer patients. J Natl Cancer Inst 2000;92:1529–31. [DOI] [PubMed] [Google Scholar]
- 12. Kilpivaara O, Bartkova J, Eerola H, et al. Correlation of CHEK2 protein expression and c.1100delC mutation status with tumor characteristics among unselected breast cancer patients. Int J Cancer 2005;113:575–80. [DOI] [PubMed] [Google Scholar]
- 13. Fagerholm R, Hofstetter B, Tommiska J, et al. NAD(P)H:quinone oxidoreductase 1 NQO1*2 genotype (P187S) is a strong prognostic and predictive factor in breast cancer. Nat Genet 2008;40:844–53. [DOI] [PubMed] [Google Scholar]
- 14. Eerola H, Blomqvist C, Pukkala E, et al. Familial breast cancer in southern Finland: how prevalent are breast cancer families and can we trust the family history reported by patients? Eur J Cancer 2000;36:1143–8. [DOI] [PubMed] [Google Scholar]
- 15. Vehmanen P, Friedman LS, Eerola H, et al. Low proportion of BRCA1 and BRCA2 mutations in Finnish breast cancer families: evidence for additional susceptibility genes. Hum Mol Genet 1997;6:2309–15. [DOI] [PubMed] [Google Scholar]
- 16. Vahteristo P, Eerola H, Tamminen A, et al. A probability model for predicting BRCA1 and BRCA2 mutations in breast and breast‐ovarian cancer families. Br J Cancer 2001;84:704–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Eerola H, Heikkilä P, Tamminen A, Aittomäki K, Blomqvist C, Nevanlinna H. Histopathological features of breast tumours in BRCA1, BRCA2 and mutation‐negative breast cancer families. Breast Cancer Res 2005;7:R93–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tommiska J, Bartkova J, Heinonen M, et al. The DNA damage signalling kinase ATM is aberrantly reduced or lost in BRCA1/BRCA2‐deficient and ER/PR/ERBB2‐triple‐negative breast cancer. Oncogene 2008;27:2501–6. [DOI] [PubMed] [Google Scholar]
- 19. Sarantaus L, Vahteristo P, Bloom E, et al. BRCA1 and BRCA2 mutations among 233 unselected Finnish ovarian carcinoma patients. Eur J Hum Genet 2001;9:424–30. [DOI] [PubMed] [Google Scholar]
- 20. Tervasmäki A, Winqvist R, Pylkäs K. Recurrent CYP2C19 deletion allele is associated with triple‐negative breast cancer. BMC Cancer 2014;14:902–2407. 14‐902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Vuorela M, Pylkäs K, Hartikainen JM, et al. Further evidence for the contribution of the RAD51C gene in hereditary breast and ovarian cancer susceptibility. Breast Cancer Res Treat 2011;130:1003–10. [DOI] [PubMed] [Google Scholar]
- 22. Hartikainen JM, Tuhkanen H, Kataja V, et al. An autosome‐wide scan for linkage disequilibrium‐based association in sporadic breast cancer cases in eastern Finland: three candidate regions found. Cancer Epidemiol Biomarkers Prev 2005;14:75–80. [PubMed] [Google Scholar]
- 23. Kauppinen JM, Kosma VM, Soini Y, et al. ST14 gene variant and decreased matriptase protein expression predict poor breast cancer survival. Cancer Epidemiol Biomarkers Prev 2010;19:2133–42. [DOI] [PubMed] [Google Scholar]
- 24. Yates F. The analysis of multiple classifications with unequal numbers in the different classes. J Am Stat Assoc 1934;29:51–66. [Google Scholar]
- 25. Chambers JM, Hastie TJ. Statistical Models in S, 1992, Wadsworth & Brooks/Cole. [Google Scholar]
- 26. Fagerholm R, Sprott K, Heikkinen T, et al. Overabundant FANCD2, alone and combined with NQO1, is a sensitive marker of adverse prognosis in breast cancer. Ann Oncol 2013;24:2780–5. [DOI] [PubMed] [Google Scholar]
- 27. Bartkova J, Horejsi Z, Koed K, et al. DNA damage response as a candidate anti‐cancer barrier in early human tumorigenesis. Nature 2005;434:864–70. [DOI] [PubMed] [Google Scholar]
- 28. Tommiska J, Eerola H, Heinonen M, et al. Breast cancer patients with p53 Pro72 homozygous genotype have a poorer survival. Clin Cancer Res 2005;11:5098–103. [DOI] [PubMed] [Google Scholar]
- 29. Stevens KN, Vachon CM, Couch FJ. Genetic susceptibility to triple‐negative breast cancer. Cancer Res 2013;73:2025–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kottemann MC, Smogorzewska A. Fanconi anaemia and the repair of Watson and Crick DNA crosslinks. Nature 2013;493:356–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Meetei AR, Sechi S,M, Wallisch M, et al. A multiprotein nuclear complex connects Fanconi anemia and Bloom syndrome. Mol Cell Biol 2003;23:3417–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Deans AJ, West SC. FANCM connects the genome instability disorders Bloom's Syndrome and Fanconi Anemia. Mol Cell 2009;36:943–53. [DOI] [PubMed] [Google Scholar]
- 33. Castella M, Jacquemont C, Thompson EL, et al. FANCI Regulates Recruitment of the FA Core Complex at Sites of DNA Damage Independently of FANCD2. PLoS Genet 2015;11:e1005563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Buisson R, Dion‐Côté AM, Coulombe Y, et al. Cooperation of breast cancer proteins PALB2 and piccolo BRCA2 in stimulating homologous recombination. Nat Struct Mol Biol 2010;17:1247–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Min A, Im SA, Yoon YK, et al. RAD51C‐deficient cancer cells are highly sensitive to the PARP inhibitor olaparib. Mol Cancer Ther 2013;12:865–77. [DOI] [PubMed] [Google Scholar]
- 36. Kim Y, Spitz GS, Veturi U, et al. Regulation of multiple DNA repair pathways by the Fanconi anemia protein SLX4. Blood 2013;121:54–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Stoepker C, Faramarz A, Rooimans MA, et al. DNA helicases FANCM and DDX11 are determinants of PARP inhibitor sensitivity. DNA Repair (Amst) 2015;26:54–64. [DOI] [PubMed] [Google Scholar]
- 38. Schwab RA, Blackford AN, Niedzwiedz W. ATR activation and replication fork restart are defective in FANCM‐deficient cells. Embo J 2010;29:806–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Collis SJ, Ciccia A, Deans AJ, et al. FANCM and FAAP24 function in ATR‐mediated checkpoint signaling independently of the Fanconi anemia core complex. Mol Cell 2008;32:313–24. [DOI] [PubMed] [Google Scholar]
- 40. Luke‐Glaser S, Luke B, Grossi S, et al. FANCM regulates DNA chain elongation and is stabilized by S‐phase checkpoint signalling. Embo J 2010;29:795–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. De Vos M, Schreiber V, Dantzer F. The diverse roles and clinical relevance of PARPs in DNA damage repair: current state of the art. Biochem Pharmacol 2012;84:137–46. [DOI] [PubMed] [Google Scholar]
- 42. D'Amours D, Desnoyers S, D'Silva I, et al. Poly(ADP‐ribosyl)ation reactions in the regulation of nuclear functions. Biochem J 1999;342:249–68. [PMC free article] [PubMed] [Google Scholar]
- 43. Bürkle A. PARP‐1: a regulator of genomic stability linked with mammalian longevity. Chembiochem 2001;2:725–8. [DOI] [PubMed] [Google Scholar]
- 44. Kim MY, Zhang T, Kraus WL. Poly(ADP‐ribosyl)ation by PARP‐1: 'PAR‐laying' NAD+ into a nuclear signal. Genes Dev 2005;19:1951–67. [DOI] [PubMed] [Google Scholar]
- 45. Yang YG, Cortes U, Patnaik S, et al. Ablation of PARP‐1 does not interfere with the repair of DNA double‐strand breaks, but compromises the reactivation of stalled replication forks. Oncogene 2004;23:3872–82. [DOI] [PubMed] [Google Scholar]
- 46. Bryant HE, Petermann E, Schultz N, et al. PARP is activated at stalled forks to mediate Mre11‐dependent replication restart and recombination. Embo J 2009;28:2601–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Blackford AN, Schwab RA, Nieminuszczy J, et al. The DNA translocase activity of FANCM protects stalled replication forks. Hum Mol Genet 2012;21:2005–16. [DOI] [PubMed] [Google Scholar]
- 48. Halazonetis TD, Gorgoulis VG, Bartek J. An oncogene‐induced DNA damage model for cancer development. Science 2008;319:1352–5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Figure 1
Figure 2
Table 1
Table 2
Table 3