

Abstract
Abstract
Synapses are essential components of neurons and allow information to travel coordinately throughout the nervous system to adjust behavior to environmental stimuli and to control body functions, memories, and emotions. Thus, optimal synaptic communication is required for proper brain physiology, and slight perturbations of synapse function can lead to brain disorders. In fact, increasing evidence has demonstrated the relevance of synapse dysfunction as a major determinant of many neurological diseases. This notion has led to the concept of synaptopathies as brain diseases with synapse defects as shared pathogenic features. In this review, which was initiated at the 13th International Society for Neurochemistry Advanced School, we discuss basic concepts of synapse structure and function, and provide a critical view of how aberrant synapse physiology may contribute to neurodevelopmental disorders (autism, Down syndrome, startle disease, and epilepsy) as well as neurodegenerative disorders (Alzheimer and Parkinson disease). We finally discuss the appropriateness and potential implications of gathering synapse diseases under a single term. Understanding common causes and intrinsic differences in disease‐associated synaptic dysfunction could offer novel clues toward synapse‐based therapeutic intervention for neurological and neuropsychiatric disorders.
In this Review, which was initiated at the 13th International Society for Neurochemistry (ISN) Advanced School, we discuss basic concepts of synapse structure and function, and provide a critical view of how aberrant synapse physiology may contribute to neurodevelopmental (autism, Down syndrome, startle disease, and epilepsy) as well as neurodegenerative disorders (Alzheimer's and Parkinson's diseases), gathered together under the term of synaptopathies.
Read the Editorial Highlight for this article on page 783.
Keywords: Alzheimer disease, autism, Down syndrome, epilepsy, hyperekplexia, synapses
Abbreviations used
- AD
Alzheimer disease
- ASD
autism spectrum disorders
- CAMs
cell‐adhesion molecules
- DSCAM
down syndrome cell‐adhesion molecule
- ECM
extracellular matrix
- FMRP
fragile X mental retardation protein
- FXS
fragile X syndrome
- GIRK2
2G‐protein‐activated inwardly rectifying potassium channel
- GlyRs
glycine receptors
- LRRK2
leucine‐rich repeat kinase 2
- LTD
long‐term depression
- mGluRs
metabotropic glutamate receptors
- MMPs
matrix metalloproteinases
- NFTs
neurofibrillary tangles
- PD
Parkinson disease
- PSD
post‐synaptic density
- RIM
Rab3a interacting molecule
- SE
status epilepticus
- TLE
temporal lobe epilepsy
Synapses are integral components of neurons and allow an organized flux of information in the brain. The emergence, diversification, and specialization of synapses played a central role in the evolution of higher brain functions and cognition in vertebrates (reviewed in Ryan and Grant 2009). On the one hand, modulation of synapse activity constitutes a major strategy to control brain homeostasis. On the other hand, slight but persistent perturbations in synapse physiology can result in major defects that may manifest as brain disorders.
The increasingly used term ‘synaptopathy’ refers to brain disorders that have arisen from synaptic dysfunction. The term goes back to a review by the Brundin laboratory discussing Huntington's disease as a result of synaptic failure (Li et al. 2003). In its broadest definition, it summarizes any perturbation in which aberrant mechanisms correlate with synaptic dysfunction regardless of its pathophysiological origin. This may result in a perplexing use of the term ‘synaptopathy’, which may mask whether a certain synaptic malfunction is the cause or the consequence of a given pathophysiological process.
Increasing evidence demonstrates the importance of synapse dysfunction as a major determinant of several neurodevelopmental diseases [autism spectrum disorders (ASD), Down syndrome, startle disease, and epilepsy] and neurodegenerative diseases (Alzheimer and Parkinson disease). Here, we present a critical overview of synapse dysfunction in the aforementioned disorders and discuss possible causes for synapse failure as putative common denominators. Understanding the molecular underpinnings leading to synaptic dysfunction will aid in the development of tailored synapse‐targeted therapies for neurological disorders.
Synapse structure and function
Synapses constitute the basic information transfer units in the nervous system and can be divided into two groups: electrical and chemical synapses. Electrical synapses allow for the direct transfer of charged ions and small molecules through pores known as gap junctions, mostly found in glial cells (reviewed in Rouach et al. 2002). Within the chemical synapse, electrical activity is unidirectionally transferred from one neuron (pre‐synaptic terminal) to another (post‐synaptic terminal) through chemical mediators. Action potentials travel along axons to induce release of neurotransmitters from vesicles in the pre‐synaptic bouton into the synaptic cleft; the activation of specific ionotropic receptors by the neurotransmitters is then again transduced into an electrical signal that depolarizes the post‐synaptic cell and is transmitted downstream.
Synapse function involves highly specialized molecular machineries at the pre‐ and post‐synapses. However, its homeostasis and plasticity also require the contribution of surrounding glia end feets and the extracellular matrix (ECM). Therefore, the classical concept of a synapse, as consisting of the neuronal pre‐ and a post‐synapse, needs to be extended to engulf glial cells and the ECM, thereby creating the term of the quadripartite synapse (reviewed in Sykova and Nicholson 2008).
Chemical synapses transduce either excitatory or inhibitory signals that increase or decrease the likelihood of firing action potentials in target cells, respectively. The most abundant excitatory neurotransmitter in the central nervous system is glutamate. GABA is the main inhibitory neurotransmitter in the adult forebrain, whereas glycine mediates inhibitory neurotransmission mainly in the brainstem and spinal cord (Fogarty et al. 2016).
Pre‐synaptic specializations
The structure of the pre‐synaptic terminal is essentially the same at excitatory and inhibitory synapses. The main difference, however, lies in neurotransmitter‐synthesizing enzymes and transporters. Basically, the signal to release neurotransmitter vesicles consists of a steep rise in intracellular Ca2+ levels as a result of pre‐synaptic voltage‐dependent calcium channels, mainly CaV2.1 and CaV2.2, which are activated upon action potential‐dependent depolarization of the pre‐synaptic membrane (reviewed in Catterall and Few 2008). Increased Ca2+ levels are sensed by the synaptic vesicle protein synaptotagmin I, which leads to conformational changes in the Soluble NSF Attachment Protein Receptor (SNARE) protein complex to mediate fusion of the vesicle with the plasma membrane. The pre‐synaptic scaffold, more specifically the cytomatrix at the active zone, consists of a protein meshwork ensuring fast but adjustable release of neurotransmitters. Bassoon, piccolo, munc‐13, Rab3a interacting molecule (RIM), and RIM‐binding proteins are prominent components of the cytomatrix at the active zone and anchor pre‐synaptic membrane proteins, such as ion channels and cell‐adhesion molecules (Ackermann et al. 2015). The efficacy of neurotransmitter release is regulated upon overall changes in network activity to maintain neuronal activity in a physiological range, in a process termed homeostatic plasticity, which modulates the expression of pre‐synaptic scaffold proteins (Lazarevic et al. 2011, 2013).
Post‐synaptic specializations
As compared with pre‐synapses, the molecular composition of post‐synaptic elements of excitatory and inhibitory synapses comprises more intrinsic differences. Excitatory post‐synapses are mostly formed on dendritic spines, which are protrusions from dendrites, and contain the post‐synaptic density (PSD), an electron‐dense structure at the post‐synaptic site (Gray 1959). The presence of a visible PSD allows one to morphologically distinguish between asymmetric (mainly excitatory) and symmetric (inhibitory) synapses (Colonnier 1968). The PSD is formed by a multilayered scaffold of densely organized proteins underneath the post‐synaptic membrane that anchors cell surface proteins, neurotransmitter receptors, and cell‐adhesion molecules. Furthermore, the PSD harbors intracellular signaling molecules and cytoskeletal filaments. Classes of scaffold proteins comprise homer (homer 1–3), the ProSAP/Shank family (Shank 1, 2, 3), and the membrane‐associated guanylate kinase family, including PSD95 and SAP102. PSD95 anchors ionotropic glutamate receptors (iGluRs) at synaptic sites, whereas homer which interacts with metabotropic glutamate receptors (mGluRs), along with the ProSAP/shank family build a scaffold in the deeper layer of the PSD, which is linked to actin filaments and is involved in spine growth (reviewed in Sheng and Kim 2011).
Glutamate activates a number of different cognate receptors within the post‐synaptic membrane of excitatory synapses. The group of iGluRs, which are ligand‐gated heterotetrameric ion channels, consists of the pharmacologically defined α‐amino‐3‐hydroxy‐5‐methylisoxazole‐4‐propionate (AMPA)‐, NMDA‐, and kainate‐type receptors. The subunit composition of these receptors is cell type and brain region specific, and in part developmentally regulated (Traynelis et al. 2010). AMPA receptors (AMPARs) mediate the fast depolarizing currents, whereas NMDA receptors (NMDARs) are more important in modulating synaptic responses as a result of their voltage‐dependent opening mechanism, their higher permeability for Ca2+, and their coupling to intracellular signaling effectors, such as CaMKII (Strack and Colbran 1998). mGluRs are subdivided into three classes, depending on their coupled G‐protein (reviewed in Niswender and Conn 2010). The subunit composition of glutamate receptors defines their functionality and, hence, receptor composition at individual synapses is crucial for synaptic transmission properties.
Conversely, inhibitory post‐synapses are usually formed directly at the soma or dendritic shaft. The scaffold of inhibitory post‐synapses contains gephyrin, which anchors neurotransmitter receptors, and collybistin, that regulates the clustering of gephyrin at GABAergic synapses. There are two described ionotropic GABA receptors: GABAA‐ and GABAC‐Rs, which form chloride channels to mediate fast hyperpolarization of post‐synaptic membranes (Lujan et al. 2005). In contrast, the metabotropic GABABRs are coupled via G‐proteins to potassium channels, leading to slow inhibitory post‐synaptic currents (reviewed in Gassmann and Bettler 2012). As GABABRs mediate their inhibitory effect via potassium channels, they always act as inhibitory receptors, in contrast to the GABAA – and GABACRs, which depend on the equilibrium potential of chloride.
Glycine receptors (GlyRs) are heteropentameric ionotropic receptors that are permeable to chloride and mediate inhibitory neutransmission. GABA and glycine can be co‐released in the brainstem and in the spinal cord, and a subset of hippocampal interneurons and pyramidal neurons present glycine receptor clusters at GABAergic synapses (Jonas et al. 1998; Levi et al. 2004). It is worth mentioning that glycine can also act as a co‐agonist to NMDARs at excitatory synapses in the forebrain (Johnson and Ascher 1987).
Integration of synapses across the cleft
Cell‐adhesion molecules (CAMs) physically connect pre‐ and post‐synaptic elements across the synaptic cleft. Synaptic CAMs include cadherins, Ig‐CAMs, neurexins, neuroligins (NLGNs), ephrins (Eph), and Eph receptors (reviewed in Benson and Huntley 2012). CAMs not only maintain the structure of mature synapses but also play roles in synapse development. As interaction between CAMs is not merely structural but provides indispensable molecular clues for the formation of new synapses, CAM mutual recognition is required for synaptogenesis (reviewed in Bukalo and Dityatev 2012).
The best‐studied pair of synaptic CAMs is constituted by the pre‐synaptic neurexin and its post‐synaptic partner neuroligin (Dean et al. 2003). This heterophilic interaction is essential for synapse structure and trans‐synaptic signaling (Varoqueaux et al. 2006). Over‐expression or suppression of neuroligins leads to an increase or decrease in the total amount of synapses in neurons, respectively (Dahlhaus et al. 2010; Shipman et al. 2011), highlighting their importance for synaptogenesis. CaMKII‐dependent phosphorylation of neuroligins is necessary for their synaptogenic properties (Bemben et al. 2014), which offers a putative activity‐dependent regulation of synapse enhancement in a neuroligin‐dependent manner (Chubykin et al. 2007).
The tetrapartite synapse
In addition to the pre‐ and post‐synapse, the model of a tetrapartite synapse includes glial end feet and the ECM. Astrocytes are closely associated with synapses and actively control synaptic transmission. They monitor neuronal activity by increasing intracellular levels of calcium and respond by secretion of a variety of molecules (Chung et al. 2015b; reviewed in Bezzi and Volterra 2001). Secretion of D‐serine, for example, is essential for glutamatergic transmission, as it acts as a co‐agonist for NMDARs (Schell et al. 1995). Furthermore, astrocytes play an important role in synaptogenesis, by secreting thrombospondin, a major factor for synapse formation (Christopherson et al. 2005).
The ECM enwraps synapses and matures postnatally to form dense, net‐like structures on a subset of cells, mainly parvalbumin‐positive interneurons. Hyaluronic acid, which acts as a backbone to coordinate proteoglycans and glycoproteins, is a major component of ECM (Frischknecht and Seidenbecher 2008). Furthermore, ECM‐modifying enzymes, such as matrix metalloproteinases (MMPs), and a disintegrin and metalloproteinase with thrombospondin motifs proteins are present in the brain (Porter et al. 2005; Sbai et al. 2008). Substantial evidence has indeed suggested significant roles for MMPs, especially MMP‐9, in neurotransmitter receptor availability and synapse function (reviewed in Michaluk et al. 2009; Lepeta and Kaczmarek 2015; Vafadari et al. 2015). Remodeling ECM influences plasticity processes in functional and structural levels and affects memory (Frischknecht et al. 2009; Gogolla et al. 2009; de Vivo et al. 2013; Valenzuela et al. 2014). In addition to the hyaluronic acid‐based ECM, there are several other extracellular molecules including laminins, reelin, agrin, thrombospondins, and many more that influence synaptic function (Heikkinen et al. 2014).
Synaptic plasticity
Synaptic plasticity refers to the ability of synapses to modify their structure and tonus after persistent electrical activity and/or signaling. This property can occur in both pre‐ or post‐synaptic compartments, allowing synapses to adapt to different contexts and, importantly, enabling learning and memory processes (reviewed in Bailey et al. 2015; Kandel et al. 2014; Citri and Malenka 2008).
As a prototypic form of synaptic plasticity, long‐term potentiation (LTP) induction by repeated synaptic activity promotes the activation of NMDARs and subsequent Ca2+ influx. NMDARs function as coincidence detectors in synaptic activity, as they require both pre‐synaptic release of glutamate and basal depolarization of the post‐synaptic membrane initiated by AMPARs. Such mechanisms instigate changes in the kinase/phosphatase balance and gene expression that are important for the persistent modification of synapse structure and function (reviewed in Citri and Malenka 2008; Kandel et al. 2014; Rosenberg et al. 2014). In addition to LTP, there is evidence for additional forms of long‐term synapse plasticity. Long‐term depression (LTD) can be induced by low‐frequency stimulation and is thought to contribute to refining memory engrams in the brain (reviewed in Dietz and Manahan‐Vaughan 2016). Synaptic scaling relates to a homeostatic plasticity that globally adjusts synaptic strength in a given neuron in response to prolonged stimuli (reviewed in Tatavarty et al. 2013; Turrigiano 2012). Metaplasticity is another form of adaptation that utilizes the strength of synaptic activity as a mechanism of calibration for the threshold of a synapse to express its plasticity (Lee et al. 2010).
Synaptic dysfunction as risk for brain disorders
The complexity of human brain functions is linked to the accelerated evolution of synapses in primates. Synapses can operate with a certain degree of autonomy from the cell body (reviewed in Brose et al. 2010) and their outstanding complexity results in a great variety of possible synaptic alterations. Synaptic dysfunction can result either from alterations in their intrinsic synaptic molecular mechanisms or be secondary to changes in additional biochemical processes in the surrounding environment.
Mutations in genes encoding synaptic proteins have been shown to result in altered protein levels/function in a number of neurological disorders. In addition, impaired synapse function is a feature of several neurological disorders not fully explained by genetic mutations, such as Alzheimer and Parkinson disease. Moreover, decreased synapse density and function often occur prior to neuronal death. Thus, it is tempting to speculate that molecular elements of synapses are key candidates to contribute to or even cause brain malfunction. In the following sections, we will review central aspects of synapse dysfunction in prevalent neurodevelopmental and neurodegenerative disorders.
Synapse dysfunction in neurodevelopmental disorders
Autism spectrum disorders
ASD form a group of diverse neurodevelopmental conditions defined by two core symptoms: social deficits that include communication and interaction impairments; and stereotypical, repetitive, and restricted behaviors. One of the main features of ASD is the high level of heterogeneity resulting in complexity both when considering symptoms and causative factors. It is in fact likely that every single disorder within the group has its unique mechanisms and consequences, with environmental and genetic factors playing roles in the etiology of ASDs. Around 600 genetic variations, many of which affect synaptic protein genes, have been related to ASD and, despite a wide diversity in these genes, some of them belong to a common pathway, suggesting that core mechanisms may exist (reviewed in Huguet et al. 2013). The imbalance between excitation and inhibition in neocortical areas has been proposed as a key process underlying ASD pathogenesis (reviewed in Baudouin et al. 2012). Here, we will focus on changes in synaptic protein synthesis and plasticity caused by alterations of glutamate receptors.
mGluRs
It has been recently demonstrated that alterations in signaling, expression, and function of group I mGluRs are related to neurodevelopmental disorders (reviewed in D'Antoni et al. 2014). Group I mGluRs comprising mGluR1 and mGluR5 have been proposed as key regulators of syndromic and non‐syndromic forms of ASD, making them possible therapeutic targets.
mGluR5 signaling was shown to be affected in opposing directions in both Fragile X syndrome (FXS) and tuberous sclerosis syndrome (TSC): FXS is the most common monogenic form of ASD and inherited intellectual disability (ID). It is caused by a mutation in the FMR1 gene that encodes the Fragile X mental retardation protein (FMRP), which regulates trafficking, stability, and translation of many RNAs coding for proteins implicated in synaptic plasticity (reviewed in Budimirovic and Kaufmann 2011). Mark Bear and colleagues proposed that FMRP suppresses translation downstream of mGluR5 and that absence of FMRP lead to exaggerated synthesis of proteins required for mGluR‐dependent LTD, thereby enhancing its magnitude (Bear et al. 2004). Following this hypothesis, different approaches were used to rescue disease phenotypes in Fmr1‐deficient mice, including allosteric inhibition and genetic ablation of mGluR5 (Auerbach et al. 2011).
TSC is another genetic syndrome associated with ASD and ID. The syndrome is caused by heterozygous mutations in genes of the TSC1/2 complex involved in an mTOR‐mediated signaling pathway that couples cell surface receptors to protein synthesis (Auerbach et al. 2011). Several mouse models of TSC exhibit decreased mGluR5‐mediated LTD caused by impaired protein synthesis. Consistently, treatment with an allosteric mGluR5 agonist was able to restore mGluR5‐mediated LTD (Auerbach et al. 2011).
NMDARs
The implication of NMDARs in the etiology of ASD has been supported by both clinical and non‐clinical studies. Clinical studies have identified genetic variants in the GRIN2A and GRIN2B genes encoding the GluN2A and GluN2B subunits of the NMDAR, respectively (O'Roak et al. 2012; Kenny et al. 2014). It is highly plausible that differences in subunit composition affect functional properties of NMDARs and/or NMDAR‐dependent plasticity (reviewed in Paoletti et al. 2013) This notion is supported by the fact that mice with constitutively reduced expression of the GluN1 subunit present core symptoms of ASD, such as defective communication and sociability in addition to repetitive behavior. Furthermore, disruption of NMDARs causes auditory‐evoked response endophenotypes that are often observed in individuals with ASD (Gandal et al. 2012).
A role for NMDARs in ASD is further supported by the fact that social withdrawal and repetitive behavior in individuals with ASD can be alleviated by the NMDAR co‐agonist D‐cycloserine (Posey et al. 2004; Urbano et al. 2014). By contrast, NMDAR antagonists memantine and amantadine improve ASD‐related symptoms including social deficits, inappropriate language, stereotypy, cognitive impairments, lethargy, irritability, inattention, and hyperactivity in humans (reviewed in Hosenbocus and Chahal 2013). Thus, ASD could result, at least in part, from deviations in hormetic NMDAR responses.
Shank
Several genetic studies have reported alterations in the gene encoding of the post‐synaptic scaffold protein shank in ASD. Shank2 −/− mice, lacking exons 6 and 7, show reduced hippocampal NMDAR function, whereas mice lacking only exon 7 show up‐regulation of NMDARs in synaptosomes, increased NMDAR/AMPAR ratio, and enhanced NMDAR‐dependent LTP (Schmeisser et al. 2012; Won et al. 2012). However, both Shank2 −/− models described show very similar social deficits, which support the notion that deviation in NMDAR function in either direction can result in ASD‐like phenotypes. Interestingly, aberrant NMDAR function and behavioral deficits observed in those mice could be normalized with systemic D‐cycloserine and administration of the positive modulator of mGluR5 3‐Cyano‐N‐1,3‐diphenyl‐1H‐pyrazol‐5‐ylbenzamide (CDPPB) (Won et al. 2012). Shank1 has similarly been associated with autism‐like behavioral changes. Shank1 −/− mice display increased anxiety‐like behavior and deficits in contextual fear learning but improvements in spatial learning, which corresponds well with the cognitive changes observed in many individuals with ASD. In addition, Shank1 −/− mice show smaller dendritic spines in CA1 pyramidal hippocampal neurons and weaker synaptic transmission (Hung et al. 2008), supporting recent evidence that dendritic spine abnormalities are associated with ASDs (Hung et al. 2008; Fang et al. 2014).
Neuroligin
Several mutations in genes encoding NLGNs have been associated with ASD (Jamain et al. 2003; Laumonnier et al. 2004). Knocking in the ASD‐associated R451C substitution into the endogenous NLGN3 locus caused a prominent decrease in NLGN3 levels that resulted in impaired social behaviors, enhanced spatial learning, and increased synaptic inhibition in the mouse somatosensory cortex. In the hippocampus, this mutation caused an increase in AMPAR‐mediated excitatory synaptic transmission, up‐regulation of GluN2B, and strengthened LTP (Etherton et al. 2011). Another ASD‐associated mutation in NLGN3, R704C, did not alter synapse formation but caused a selective decrease in AMPAR‐mediated synaptic transmission in mice, whereas NMDAR‐driven synaptic transmission and pre‐synaptic neurotransmitter release were not affected (Chanda et al. 2013). These results suggest NLGN3 as a key player in synaptic transmission by modulating the recruitment of AMPARs to post‐synaptic sites at excitatory synapses (Etherton et al. 2011). Similarly, mice lacking NLGN1 showed ASD‐like behavior and reductions in NMDAR function that could be reversed with D‐cycloserine (Blundell et al. 2010) or memantine (Chung et al. 2015a). Interestingly, reduced LTD is a shared feature between a syndromic form of autism (e.g. FXS) and a non‐syndromic form of autism partly caused by mutations in Nlgn genes in mice (Baudouin et al. 2012).
Toward shared pathways in ASD
ASD is a very complex group of disorders associated with aberrant synaptic transmission and plasticity. A range of animal models has demonstrated that scaffolding and adhesion proteins such as Shank and NLGNs play a key role in the regulation of NMDARs and AMPARs. Altered expression, trafficking, and function of these two groups of receptors are postulated to play a crucial role in the development of ASD. Moreover, evidence suggests mGluR signaling as a shared pathway in disorders associated with ID and ASD. However, the molecular mechanism leading to alterations in group I mGluRs in ASD remains to be elucidated.
Overall, the different animal models have been very useful for the understanding of the neurobiological pathways affecting mGluRs and iGluRs in ASD; however, the models have their limitations as they cannot reproduce all characteristics of the human disorder and only allow studies of ASD at reduced genetic complexity. Most animal models represent alterations in only one single gene, whereas individuals with ASD often have several gene alterations. Understanding how synapse dysfunction develops in ASD, including the associated molecular machinery, remains a challenge that needs to be overcome in search for effective and safe pharmacological treatment for this diverse group of disorders.
Down syndrome
Down syndrome (DS) is the most common genetic form of ID and occurs in ~ 10 of 10 000 live births (Khoshnood et al. 2011). Individuals with DS show deficits in learning and memory, language and executive functions since early childhood, resulting from the presence of an extra copy of chromosome 21 (Hsa21). DS patients exhibit an age‐dependent reduction in dendritic branching and spine density that likely involves impaired reorganization of the actin cytoskeleton in neurons (Marin‐Padilla 1976; Takashima et al. 1981). Several cytoskeleton‐related genes are located in Hsa21, including DSCR1, DYRK1A, and S100B, and have been proposed as candidates to contribute to the neuronal architecture abnormalities observed in DS (Dierssen et al. 2009). In addition, DS patients show early onset Alzheimer‐like neurodegeneration (reviewed in Lott and Dierssen 2010).
Multiple studies support that an over‐inhibition of synapses by increased GABAergic circuitry may be the main force for synaptic dysfunctions in DS as it affects the excitation–inhibition balance in the brain (Kurt et al. 2004; reviewed in Fernandez and Garner 2007; Hanson et al. 2007), which may alter synaptic connectivity and cognitive function. Thus, it is plausible to speculate that altered synapse structure and function induced by aberrant expression of Hsa21 and some non‐Hsa21 genes underlie cognitive symptoms in DS. One such gene recently emerged as modulating the excessive inhibitory tone. It encodes the type 2 G‐protein‐activated inwardly rectifying potassium channel (GIRK2). Elevated expression of GIRK2 has been found in the hippocampus of theTs65Dn mouse, which is the most widely used DS model. Enhanced GIRK2 levels result in a more hyperpolarized resting potential, cognitive deficits, reduced reversal of potentiation, and enhanced LTD in the Ts65Dn mice (reviewed in Cramer and Galdzicki 2012). Augmented coupling between GABAB receptors and GIRK2 channels in Ts65Dn hippocampus may potentially account for the impaired expression of LTP in the hippocampus (Kleschevnikov et al. 2004; Siarey et al. 2006). Several mechanisms are thought to mediate the excitatory–inhibitory imbalance, namely, the decreased numbers of excitatory synapses, excessive GABA drive as a result of higher synaptic vesicle release probability, and/or increased number of inhibitory interneurons in the neocortex and hippocampus (Chowdhury et al. 2010).
Ts65Dn mice represent the only model that has been used in the pre‐clinical evaluation of drugs for improvement in learning and memory. It has been criticized as ~ 45% of Hsa21 orthologous genes do not show trisomy in those mice. Furthermore, this model bears an extra trisomic Mmu17 region, which carries genes that are not orthologs of Hsa21 genes. Despite these limitations, such mice survive until adulthood and present cognitive deficits, structural, and functional alterations that recapitulate the ones observed in DS individuals (Reeves et al. 1995; Escorihuela et al. 1998). Ts65Dn mice present age‐associated shifts in the excitation–inhibition balance and showed a 30% reduction in the numbers of asymmetric synapses in the temporal cortex, whereas both symmetric and asymmetric synapses appear significantly larger (Kurt et al. 2000). In addition, Ts65Dn synaptic structures show reduced activity‐dependent synaptic plasticity, as they are less responsive to stimulation (Dierssen et al. 2003). Electrophysiological abnormalities include deficits in GABAergic and glutamatergic transmission in the hippocampus and cerebellum; among them are age‐ and cell type‐specific deficits in hippocampal LTP and LTD (Siarey et al. 1999; Belichenko et al. 2009).
One of the key molecules governing neural circuitry formation in the developing brain is Down syndrome cell‐adhesion molecule (DSCAM), a member of the immunoglobulin superfamily that has been linked to neurodevelopmental deficits in DS. It is expressed in various brain regions during development and its levels decrease after birth (Yamakawa et al. 1998; Agarwala et al. 2001). DSCAM is necessary for axon guidance and proper refinement of neuronal networks (Blank et al. 2011) and its different variants mark neurons with individual identity, thus fostering self‐recognition during establishment of neuronal networks (Hattori et al. 2007; Fuerst et al. 2009). Increased levels of DSCAM in DS lead to abnormally large pre‐synaptic arbors (Kim et al. 2013) and impair precise synaptic targeting and neural circuit function (Cvetkovska et al. 2013).
Recent studies point to endocytic dysfunction as an early pathological change in DS (Cataldo et al. 2000, 2008). Altered levels of two Hsa21‐encoded proteins – intersectin and synaptojanin (encoded by ITSN1 and SYNJ1, respectively) – impair the recycling of synaptic proteins, interfering with the pre‐synaptic vesicle machinery in DS. In addition, DSCR1 and DYRK1A provide an upstream control for ITSN1 and SYNJ1 function in regulation of vesicle endocytosis; thus, in DS vesicle trafficking is deregulated at multiple levels (reviewed in Garner and Wetmore 2012). Another key gene triplicated in DS encodes amyloid‐β precursor protein (APP). The extra APP copy results in elevated β‐amyloid production, likely explaining the accelerated Alzheimer‐like phenotype observed in DS patients and perhaps limiting synapse function and cognition during adulthood (reviewed in Head et al. 2016). Alterations in the endocytic pathway in DS contribute to amyloidogenesis and further promote β‐amyloid deposits and tau pathology in DS brains (Cataldo et al. 2008).
Approaches targeting altered neurotransmitter systems and synaptic function, including the excitation–inhibition imbalance, have been proposed as therapeutic interventions to ameliorate cognitive deficits in DS. Chronic administration of non‐competitive GABAAR antagonists picrotoxin and pentylenetetrazole separately rescued compromised LTP and improved learning and memory in DS mice (Fernandez et al. 2007). Because of patient safety concerns regarding the seizure‐inducing potential of pentylenetetrazole and higher susceptibility of DS individuals to seizures (Lujic et al. 2011), other compounds with similar blocking capabilities but reduced seizure‐inducing features have now been under investigation, among which are molecules targeting specific subunits of GABAA receptors (Braudeau et al. 2011). These actions of GABAA antagonists support the over‐inhibition hypothesis by GABAergic circuitry in DS.
As a result of the high prevalence of early onset Alzheimer disease in DS, pharmacotherapies for DS based on drugs approved for Alzheimer disease have been proposed and tested in both mice and humans. Memantine, an uncompetitive antagonist of NMDA receptors, has also been proposed as a therapeutic approach to the management of DS because of the interaction of NMDAR with proteins encoded by Hsa21 genes (Siddiqui et al. 2008). Recent studies have shown that epigallocatechin gallate, a green tea catechin that promotes neuroprotection and neuroplasticity inhibits the kinase activity of DYRK1A, up‐regulated in DS. Furthermore, epigallocatechin gallate promotes the non‐amyloidogenic processing of APP (Fernandez et al. 2010), which would also ameliorate the amyloid burden in DS. In addition to pharmacological therapies, a number of non‐pharmacological approaches have been tested in the attempt to alleviate DS structural and cognitive alterations. The enhancement of physiological neuroplasticity by early intervention programs, special education and cognitive stimulation, have been proposed for DS individuals as environmental enrichment is able to profoundly affect the brain structure and function at the molecular level resulting in the improvement of cognitive function in euploid people (reviewed in Baroncelli et al. 2010; Sale et al. 2009).
Hyperekplexia (Startle Disease)
Hyperekplexia, also termed startle disease, is a rare hereditary neurological disorder caused by dysfunction of glycinergic transmission. Hyperekplexia is characterized by the exaggeration of the normal physiological startle reflex to tactile, auditory, or other stimuli (Suhren et al. 1966). The cases of hyperekplexia can be divided into two groups: major and minor. Patients belonging to the ‘major’ group exhibit three hallmarks: stiffness at birth, exaggerated startling response, and temporary generalized stiffness following startle. ‘Minor’ patients demonstrate only the startling response without stiffness (Suhren et al. 1966; Andermann et al. 1980; Dooley and Andermann 1989; Brown et al. 1991; Pascotto and Coppola 1992; Tijssen et al. 2002). Babies generally present generalized stiffness shortly after birth which increases following handling but is not present during sleep (Koning‐Tijssen and Brouwer 2000). Although it usually persists throughout life, proper treatment and aging can alleviate symptoms. Albeit rare, hyperekplexia may be associated with brain damage and sudden infant death related to breath‐holding episodes. Stiffness of legs and wide gait are generally present in adult patients which often results in patients suffering regular falls (Kirstein and Silfverskiold 1958).
Mutations in glycinergic signaling
Defective glycinergic signaling is the major cause of hyperekplexia. GlyRs are composed of five homologous subunits; α1, α2, α3, α4, and β, which differ in both their patterns of distribution in the CNS and expression windows (reviewed in Lynch 2004). To date, mutations in five genes involved in glycinergic signaling have been associated with hyperekplexia. Mutations in the gene for the α1 subunit of the GlyR, GLRA1, form the most common cause of hyperekplexia (Shiang et al. 1993; Buckwalter et al. 1994; Rees et al. 1994; Saul et al. 1999; Chung et al. 2010). Mutations generally result in defective subcellular localization, trafficking, ligand sensitivity, or opening of the ligand‐gated ion channel. Furthermore, decreased agonist affinity and GlyR‐mediated chloride currents ultimately lead to impaired glycinergic signaling upon startle response and, hence, increases in muscle tone.
There are two mouse models harboring mutations within the α1 subunit of the GlyR: cincinnati mice and oscillator (spd ot), both unfortunately showing lethal phenotypes (Buckwalter et al. 1994; Kling et al. 1997; Holland et al. 2006; Villmann et al. 2009). Homozygous spd ot mice are lethal because of a premature stop codon in Glra1 (Buckwalter et al. 1994). Surprisingly, the same mutation (R316X) does not lead to lethality in humans, although it also results in a premature stop codon (Tsai et al. 2004; Schaefer et al. 2015). The spasmodic (spd) mouse, which also carries a mutation within the α1 subunit of GlyR, exhibits milder symptoms with normal lifespan (Lane et al. 1987).
Surprisingly, except for GLRA1, no mutations have been found in any of the other GLRA genes coding for α subunits, but mutations in GLRB, coding for the β subunit, have been identified (Al‐Owain et al. 2012). Mutations in GLRB give rise to more severe phenotypes than those seen in patients with GLRA1 mutations, although the underlying mechanisms remain unclear. The spastic mouse (spa) harbors a mutation within the GlyR β subunit, developing a mild or severe phenotype depending on the genetic background strain (Kingsmore et al. 1994; Mulhardt et al. 1994; Becker et al. 2012).
Mutations in the glycine transporter GlyT2 (SLC6A5) are postulated as the second major cause of hyperekplexia (Rees et al. 2006). Mice with mutated GlyT2 also show a lethal phenotype (Rees et al. 2006; Harvey et al. 2008). In addition, mutations in genes encoding the GlyR‐ and GABAAR‐interacting protein gephyrin (GPHN) (Rees et al. 2003) and the exchange factor collybistin (ARHGEF9) (Harvey et al. 2004) have also been identified in hyperekplexia patients.
Autoantibodies
In 2008, a phenotype similar to classical hyperekplexia was described after discovery of autoantibodies against GlyR subunits in patients from 14 months to 65 years old (Damasio et al. 2013; Carvajal‐Gonzalez et al. 2014). In addition to loss‐of‐function mutations, autoimmune antibodies could thus represent a novel mechanism of defective glycinergic signaling in hyperekplexia. Although the underlying pathways are not clearly understood thus far, several case reports have been published favoring enhanced internalization of GlyRs as one of the likely pathomechanisms (Carvajal‐Gonzalez et al. 2014). In line with this, patients with the stiff‐person syndrome, closely related to hyperekplexia (Khasani et al. 2004), may produce autoantibodies against amphiphysin and show compromised central inhibition. Amphiphysin is involved in clathrin‐mediated endocytosis. As GABAergic interneurons show fast neurotransmission, they need compensatory endocytosis, which is impaired by the autoantibodies against amphiphysin, leading to network disinhibition (Werner et al. 2016). Clonazepam, an allosteric potentiator of GABAARs, is commonly used as effective therapy for hyperekplexia patients with different types of mutations (Andermann et al. 1980; Ryan et al. 1992; Bakker et al. 2009; James et al. 2013; Thomas et al. 2013). Thus, enhancing GABA‐mediated inhibitory neurotransmission successfully counteracts deficits in the glycinergic neurotransmission, although the molecular mechanisms behind such effects remain unclear.
Epilepsy
Epilepsy comprises a group of common neurological disorders characterized by the occurrence of epileptic seizures, which are defined as uncontrolled and excessive synchronized electrical activity of central neurons. Several conditions, including infection, stroke, and traumatic brain injury, have been reported to increase the susceptibility of developing epilepsy, although a mechanistic understanding of neural events driving epileptogenesis is still limited (reviewed in Pitkänen and Lukasiuk 2009; Zelano et al. 2015; Pitkänen et al. 2016). Temporal lobe epilepsy (TLE) is the most frequent form of epilepsy, with the hippocampal formation and surrounding cortical regions being regarded as the most prevalently affected areas in the human brain (reviewed in Pitkanen et al. 2015). Epileptogenesis is an ongoing process, with substantial progress after the first seizure. In support of this notion, the first seizure episode acutely increases the risk of sequential events, and some studies have reported that human TLE can be progressive (reviewed in Pitkanen et al. 2015; Cardoso et al. 2006).
In addition, epilepsy can be triggered by genetic mutations affecting key modulators of neuronal function, including sodium channels, neurotransmitter transporters, and synaptic proteins (reviewed in Noebels 2015). The growing list of epilepsy‐related mutations poses new questions as to how single gene mutations and acquired conditions, such as traumatic brain injury, could converge to similar epileptic phenotypes (reviewed in Staley 2015). It is worth mentioning that, although epileptogenesis usually initiates at early stages of life, epilepsy has a neurodegenerative component, facilitating aberrant synapse morphology and function that could result in cognitive failure and increased risk for dementia (Wittner et al. 2002; Leite et al. 2005; Keezer et al. 2016).
While the underlying cause of epilepsy remains largely unknown, current evidence supports a hypothesis in that an altered balance of excitatory and inhibitory circuits facilitates the occurrence of the first seizure episode, which in turn elicits cellular and molecular changes that may lower thresholds for sequential episodes. It is thought that, in TLE models, glutamatergic neurotransmission is enhanced (Peret et al. 2014; Soukupova et al. 2015), whereas GABA release is decreased (Soukupova et al. 2014), leading to aberrant signaling and an overall increase in excitatory neuronal transmission. Furthermore, alterations in neuronal morphology, glial cells, and ECM have been related to the onset and development of epilepsy (Wetherington et al. 2008; Quirico‐Santos et al. 2013; Singh et al. 2013).
As a disorder affecting neurotransmission, it is reasonable to speculate that synapse function is compromised by epilepsy. Indeed, animal models of status epilepticus (SE) and epileptic patients present reduced dendritic branching, dendritic spine loss (von Campe et al. 1997; Isokawa 2000; Singh et al. 2013), and altered neurotransmitter receptor composition (Lopes et al. 2015; Zhou et al. 2015) at remaining synapses. Alterations in the subunit composition and distribution of AMPARs and NMDARs likely contribute to abnormal excitatory tonus during seizures (Lopes et al. 2015; Zhou et al. 2015). Likewise, it is conceivable that reduced inhibitory tonus arising from malfunctioning hippocampal interneurons may further exacerbate SE (Staley 2015).
Accumulating evidence indicates that molecules that act as neuromodulators could also participate in the pathogenesis of epilepsy. Indeed, neuropeptides and small molecule co‐agonists known to regulate network excitability and transmission tonus have been investigated for their potential to enhance seizure threshold, and gene therapy has been proposed as a treatment approach (Riban et al. 2009). Among neuropeptides, brain‐derived neurotrophic factor appears to promote epileptogenesis via TrkB activation (Liu et al. 2013; Gu et al. 2015), but neuropeptide Y was shown to suppress abnormal excitatory currents in human epileptic tissue (Ledri et al. 2015). In addition, disruption of adenosine and D‐serine signaling mechanisms associates with post‐synaptic molecular changes and lower seizure threshold (Klatte et al. 2013; Boison 2016). Finally, very recent evidence has shown a role for the endocannabinoid system in regulating susceptibility to epilepsy (reviewed in Soltesz et al. 2015). In fact, experimental observations have demonstrated that components of endocannabinoid signaling pathways can be deregulated in epilepsy, thereby dampening protective effects of tonic inhibition against seizures (reviewed in Soltesz et al. 2015).
Studying a very common disorder, that affects around 50 million people worldwide (as determined by the World Health Organization; http://www.who.int/mediacentre/factsheets/fs999/en/), the epilepsy research community is in dire need of animal models that fully recapitulate human disease progression. This is mainly because of the underlying causes being largely unknown. Most in vivo experimental observations to date have been obtained from chemically induced SE in mice (reviewed in Pitkanen et al. 2015), in which drugs such as kainic acid or other neurotoxic agents are used to induce epileptic seizures. Nevertheless, such models have revealed several features that may contribute to the understanding of how normal networks start to seize.
Current therapeutics for epilepsy include anti‐convulsants and anti‐epileptics, such as valproate, which increases the amount of the inhibitory GABA neurotransmitter, and phenytoin, which prolongs inactivation of Na+ channels. Levetiracetam is another commonly used anti‐epileptic agent thought to act by binding to the pre‐synaptic protein SV2a and reducing neurotransmitter release at glutamatergic synapses (reviewed in Lynch et al. 2004). The multivariate nature of epilepsy highlights the need to establish the full range of synapse/network modifications that develop in epileptic brains. Dissecting the awry components that mediate epileptogenesis might foster the amplification of available treatments, and may possibly reduce the current fraction of intractable cases.
Synapses and neurodegenerative diseases
Alzheimer disease
Alzheimer disease (AD) is a neurodegenerative disorder that accounts for around 60% of all dementia cases (Barker et al. 2002; Ferri et al. 2005). AD is a highly sporadic and idiopathic disease, with only a small fraction of cases (1–5%) clearly attributable to familial mutations. The most common AD‐linked mutations are located in the amyloid‐β precursor protein (APP) gene and in gene regions encoding the catalytic cores of the gamma‐secretases presenilin 1 (PSEN1) or presenilin 2 (PSEN2) (Goate et al. 1991; Levy‐Lahad et al. 1995; Sherrington et al. 1995). For sporadic AD, many genetic susceptibility factors have been described to date, the most important being apolipoprotein E (APOE) allele 4 (APOE4) (Corder et al. 1993).
AD brains contain extracellular deposits of the amyloid beta (Aβ) peptide, termed amyloid plaques, and intracellular neurofibrillary tangles (NFTs), mostly composed of aggregated tau protein (Selkoe et al. 2012). Notably, NFTs appear to precede amyloid plaque deposition during AD progression in humans (Braak and Braak 1997). Several etiopathogenic hypotheses have been put forward in AD, the most common being the amyloid cascade hypothesis (reviewed in Hardy and Higgins 1992; Hardy and Selkoe 2002) and the tau hypothesis (Gray et al. 1987), linking the histopathological hallmarks to AD initiation. These paradigms have been substantially improved over the years after findings that neither Aβ plaques nor NFT volume correlate well with the severity of dementia, whereas the loss of nerve endings and synaptic dysfunction more closely associates with cognitive impairment (Terry et al. 1991; Nagy et al. 1995; Kril et al. 2002).
Conversely, soluble Aβ oligomers, instead of deposited plaques, are now thought to underlie widespread neurotoxicity and synaptic loss (reviewed in Ferreira et al. 2015) and, indeed, the degree of dementia in patients correlates well with the levels of oligomers in post mortem brains (Lue et al. 1999; McLean et al. 1999; Gong et al. 2003; Bilousova et al. 2016). Levels of Aβ oligomers are increased in the brains of AD patients over control brains (Gong et al. 2003; Xia et al. 2009), and their synaptic presence is linked to poorer cognition regardless of classical neuropathology (Bjorklund et al. 2012; Bilousova et al. 2016). Studies in transgenic animal models of AD have shown that synaptic and cognitive impairments are associated with the elevation in soluble oligomeric Aβ species prior to the appearance of plaques and tangles (Mucke et al. 2000; Billings et al. 2005; Tu et al. 2014), and studies in rodents suggest that soluble Aβ oligomers, but not monomers, can disrupt hippocampal LTP and can also impair memory (Walsh et al. 2002).
The precise mechanisms of Aβ toxicity are not fully understood, but it is believed that oligomers can interact with a number of synaptic proteins including mGluRs and iGluRs (De Felice et al. 2007; Renner et al. 2010; Um et al. 2013), α7‐nicotinic acetylcholine receptors (Wang et al. 2000), p75 neurotrophin receptor (Yaar et al. 1997; Sotthibundhu et al. 2008), NLGN‐1 (Dinamarca et al. 2011), cellular prion protein (Lauren et al. 2009), and PSD95 (Pham et al. 2010). The synaptic action of Aβ triggers aberrant activation of NMDARs (Wu and Hou 2010), Ca2+ deregulation (Lazzari et al. 2015), and cellular stress, which in turn leads to synaptic dysfunction and neuronal loss (reviewed in Ferreira et al. 2015). Soluble Aβ further exacerbates neurotransmitter release at excitatory synapses, including glutamate and the NMDAR co‐agonist D‐serine, thereby triggering post‐synaptic neurotoxicity and synapse failure (Paula‐Lima et al. 2013; Talantova et al. 2013; Madeira et al. 2015). The fact that cognitive decline appears in patients before extensive neuronal loss is detected reinforces the notion that synapse dysfunction is likely to be an early cause for memory loss in AD (reviewed in Selkoe 2002; Ferreira et al. 2015).
Accumulating evidence indicates that mechanisms of cellular stress and apoptosis could be overactive in AD (reviewed in D'Amelio et al. 2012; Lourenco et al. 2015) and some reports have postulated that such processes could also be locally activated to prune synapses or limit their function (Li et al. 2010; Jo et al. 2011; Lourenco et al. 2013; Erturk et al. 2014). For example, the activation of stress‐related kinases has been recently implicated in synapse and cognitive dysfunction in AD (reviewed in Lourenco et al. 2015), and this might relate to impaired local translation at synapses (Lourenco et al. 2013; Ma et al. 2013). Another example arises from the non‐apoptotic role of caspases in limiting synapse function (Li et al. 2010; Jo et al. 2011; Erturk et al. 2014). Notably, impairments in synapse plasticity initiated by Aβ are thought to require caspase 3 activation (Jo et al. 2011).
Recent data has also implicated epigenetic alterations as mediators of AD‐related synapse and memory defects. AD brains present altered epigenetic/epigenomic signatures (Gräff et al. 2012; Gjoneska et al. 2015; Shen et al. 2016), and increased activity of histone deacetylases contribute to disturbances in brain transcriptional profiles and memory in animal models of AD (Gräff et al. 2012; Benito et al. 2015). Consistently, histone deacetylases inhibitors were shown to attenuate memory impairment in AD mice (Ricobaraza et al. 2009, 2010; Kilgore et al. 2010). Additional evidence has further suggested a role for neuronal cell cycle re‐entry in AD‐linked pathological changes (McShea et al. 2007; Varvel et al. 2008; Bonda et al. 2009; Bhaskar et al. 2014; Shen et al. 2016).
In addition to Aβ, PSEN function was also shown to be important for synaptic transmission, as conditional knock out of presenilin was shown to impair NMDA receptor function and LTP, and PSEN mutations were demonstrated to cause abnormal calcium handling in neurons (Zhang et al. 2010). Several other protein defects have also been described in AD pathology, including protein phosphatase 2A family of proteins and their regulators, which have been linked to amyloidogenesis, tau hyperphosphorylation, and synapse deficits (Sontag and Sontag 2014). Furthermore, post‐transcriptional regulation by regulatory microRNAs (miRNAs) is altered in AD patients. For example, miR‐132, a regulator of tau (Lau et al. 2013) and acetylcholinesterase (Shaked et al. 2009), is drastically down‐regulated in AD patients (Lau et al. 2013) leading to elevated tau aggregation (Smith et al. 2015). Importantly, tau pathology appears to be itself a substantial contributor of synapse dysfunction in AD, and has been recently proposed to propagate across brain structures, in a manner similar to Aβ (reviewed in Spires‐Jones and Hyman 2014).
For many years, molecular explanations of AD progression have relied on isolated and competing hypotheses that would not intersect (as evidenced in disputes between the so‐termed BAPtist and TAUist groups, and reviewed in Selkoe et al. 2012). Nevertheless, a novel molecular understanding of AD has integrated these opposing views in a converging scenario toward synapse failure as underlying cause of cognitive decline. As expected for a complex disease, Aβ, tau, and APOE4 features appear to interact to promote cognitive decline and neurodegeneration in AD, whereas they may independently contribute to synapse malfunction and cognitive impairment (Shankar et al. 2008; Zhu et al. 2012; Min et al. 2015). APOE4 is known to stimulate brain Aβ deposition (Reiman et al. 2009) and, in turn, Aβ promotes abnormal tau phosphorylation (De Felice et al. 2008; Jin et al. 2011) to mediate synapse and memory defects in mouse models of AD (Roberson et al. 2007, 2011; Ittner et al. 2010). Notably, APOE4 appears to impair synapses, at least in part, through the action of Aβ oligomers and tau (Andrews‐Zwilling et al. 2010; Koffie et al. 2012). Thus, multiple factors could act as initiators of common signaling pathways that ultimately lead to synapse impairment and AD neuropathology.
Current pharmacological treatments, largely based on enhancing cholinergic signaling or attenuating NMDAR function at glutamatergic synapses, only provide modest symptomatic relief and do not treat the underlying cause or delay disease progression (reviewed in Selkoe 2011). Observations arising from two Phase III anti‐amyloid clinical trials have been somewhat disappointing, and have strengthened the notion that combating deposited amyloid would not represent the most effective strategy toward AD therapy (Doody et al. 2014; Salloway et al. 2014). Although synapses do not appear to be the starting point for AD pathology, the notion that several upstream pathogenic mechanisms act synergistically to promote synapse dysfunction/loss indicate that strategies aimed at preventing synapse failure could provide greater therapeutic benefit for cognitive decline in AD.
Parkinson disease
Parkinson disease (PD) is characterized by progressive degeneration of nigrostriatal dopaminergic neurons, leading to loss of motor function, rigidity, postural instability, tremor, and bradykinesia. Both familial (5–10% of all cases) and acquired (90–95% of all cases) forms of Parkinsonism are usually caused by defects in dopamine (DA) metabolism. PD leads to profound immobility and disability that usually affects people over the age of 60. Decreases in DA inputs from basal ganglia result in impaired control of motor circuits that ultimately leads to clinical symptoms. Deposition of Lewy bodies (LB), protein aggregates‐containing alpha‐synuclein (α‐syn) often found in remaining dopaminergic neurons, is regarded as central to the pathogenic mechanisms underlying PD (Spillantini et al. 1997). Notably, synapse dysfunction contributes to the progression of PD, with impairments in protein trafficking, autophagy, and mitochondrial function (Lynch‐Day et al. 2012; Hunn et al. 2015; Pickrell and Youle 2015). Although most PD cases are idiopathic, some autosomal‐dominant genetic causes have been identified, enabling the investigation of possible pathogenic mechanisms, including PARK1 – PARK8 mutations.
Dopaminergic neurons within the substantia nigra (SN) preferentially degenerate within PD pathogenesis. A neuroanatomical analysis of SN indicated that susceptible neurons have 10–100 times the number of synapses of neighboring neurons, with relatively large axonal lengths (~ 50 mm in rats) (reviewed in Bolam and Pissadaki 2012). This poses a large demand for efficient trafficking of organelles and vesicles within these neurons and may explain the selective vulnerability of these cells in PD (Giehm et al. 2011).
Synaptic dystrophy characterizes the early stages of PD neuropathology (Janezic et al. 2013). SNARE protein complexes, which mediate the membrane fusion of exocytosis vesicles, are prone to misfolding and non‐specific interactions, thus depending on efficient chaperones to remove them from the synapse (Chandra et al. 2005). α‐syn is one such oligomerization‐prone chaperones that cause mislocalization of SNARE complexes associated with decreased DA release and synaptic dystrophy (Tokuda et al. 2010; Winner et al. 2011; Rockenstein et al. 2014). It is thus possible that dysfunctional dopaminergic synapses contribute to disease progression even before evident neurodegeneration takes place.
Leucine‐rich repeat kinase 2 (LRRK2) is a large multidomain protein, whose mutations relate to autosomal‐dominant forms of PD, and variations in LRRK2 have been associated with up to 40% of sporadic PD cases in defined cohorts (Piccoli et al. 2011). The function of LRRK2 has not been completely understood, although growing evidence implicates roles in intracellular trafficking, microtubule stability, vesicular recycling, and autophagy (reviewed in Berwick and Harvey 2013; Law et al. 2014; Berwick and Harvey 2011). As expected, mutated LRRK2 correlates with decreases in synaptic plasticity and neurogenesis (Matta et al. 2012; Berwick and Harvey 2013). LRRK2 has also been shown to interact with other genes associated with sporadic PD susceptibility, including rab‐7‐like protein 1 (RAB7L1) and cyclin‐G‐associated kinase (MacLeod et al. 2013; Beilina et al. 2014), proteins that promote the removal of vesicles through the autophagy–lysosome pathway. LRRK2‐positive membrane compartments are involved in pre‐synaptic vesicle trafficking and autophagy, as demonstrated by localization of LRRK2 to synaptic vesicles (Xiong et al. 2010; Piccoli et al. 2011), electrophysiological defects following knock‐down or over‐expression of LRRK2 in cultured neurons (Shin et al. 2008; Piccoli et al. 2011), and biochemical interaction between LRRK2 and the early endosomal marker Rab5b (Shin et al. 2008). Defective autophagy appears to play a significant role in synaptic dysfunction in the PD neurodegenerative cascade (reviewed in Plowey and Chu 2011). Functional loss of autophagy induces PD‐linked features and modifies synapse structure and function (Ahmed et al. 2012). In addition to LRRK2 (Nakamura et al. 2011), a number of PD‐associated proteins, including α‐syn (Lynch‐Day et al. 2012) and PINK1 (Li et al. 2005), have been linked to autophagy.
Research in the field of PD pathogenesis has mostly focused on dopaminergic defects in the SN, ultimately corresponding to PD motor outcomes. However, recent evidence has indicated that other neurotransmitter systems become affected in PD, and may respond to comorbid alteration in patients. For instance, patients and experimental models of PD present altered serotonergic and adrenergic transmission (Buddhala et al. 2015; Deusser et al. 2015). Deficits in both dopamine and serotonin‐dependent mechanisms might further explain the depressive behavior frequently reported in PD. In addition, PD mice present impaired synaptic plasticity in the hippocampus, which might correlate with cognitive deficits (Sweet et al. 2015). Hence, rather than being restricted to SN, synapse dysfunction appears to be present at several brain regions and in different neurotransmitter systems, likely explaining the complex phenotype in PD.
Significant advances have been made in discovering therapeutic options to minimize motor impairment in PD. In fact, administration of dopamine precursors, such as L‐DOPA, or agents that stimulate dopamine synthesis have been clinically in use and effectively rescue motor impairment in humans. Still, etiological components, as well as the molecular points of convergence in PD, remain to be established to optimize therapeutic approaches. Cellular mechanisms that directly impact synaptic function, including autophagy, endosomal trafficking, and mitochondrial homeostasis, likely constitute effective future interventional targets in PD therapy. In this regard, a deeper understanding of the common bases of neurotransmission and synapse dysfunction triggered in both sporadic and familial forms of PD might offer new possibilities for treating this debilitating disorder.
Concluding remarks
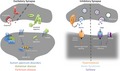
The intricate assembly of molecular machineries at the synapse works coordinately to mediate proper synaptic activity in the brain. Impairing the function of a single component in this convoluted system can compromise synapse function, ultimately leading to a group of brain diseases termed synaptopathies. The discovery of several synapse‐linked gene mutations associated with different brain disorders has opened the possibility that synapse‐targeted therapeutics could be of assistance in such disorders. More recently, the notion of synaptopathies has been extended to include not only diseases originated at the synapse but also all those that present synapse defects as a leading route to clinical symptoms. Thus, genetic or sporadic conditions that lead to abnormal excitatory (summarized in Fig. 1) or inhibitory (Fig. 2) synapse function could pave the way for neurodevelopmental or age‐related disorders to manifest.
Figure 1.

Schematic overview of excitatory synapse‐linked disease mechanisms. Functional overview of the excitatory synapse with main proteins and pathways affected in autism spectrum disorders (ASD), Alzheimer disease (AD), and Parkinson disease (PD). Proteins and pathways are color coded for each disorder (ASD – blue; AD – green; PD – red). Dashed line divides the pre‐synaptic part into dopaminergic and glutamatergic terminals. ROS – reactive oxygen species. Altered (either increased or decreased) function of NMDA and AMPA receptors, disrupted mGluR signaling and consequently changes in long‐term depression (LTD) are postulated to play a crucial role in the development of ASD. Such mechanisms likely culminate in altered protein synthesis at the post‐synapse. Notably, FMRP deficiency results in elevated translation and autism‐like behaviors in FXS. In AD, the post‐synaptic action of Aβ oligomers and tau aggregates result in deregulation of NMDA receptors (NMDARs), cellular stress, and decreased protein synthesis. Aβ oligomers further act through interactions with neuroligin‐1 and mGluR5 to impair neuronal homeostasis. Pre‐synaptic disturbances, including impaired BDNF transport and excessive D‐serine production, have also been associated with AD. Such abnormal molecular cascades ultimately result in defective LTP and memory. In PD, defective autophagy and α‐synuclein‐mediated mislocalization of SNARE complexes associate with decreased DA release and mitochondrial function. This leads to increases in oxidative stress, likely driving impaired dopaminergic tonus and neuronal death that are characteristic of PD.
Figure 2.

Schematic overview of inhibitory synapse‐linked disease mechanisms. Functional overview on the inhibitory synapse with main proteins and pathways affected in hyperekplexia, Down syndrome (DS), and epilepsy. Proteins and pathways are color coded for each disorder (hyperekplexia – orange; DS – blue; epilepsy – purple). Dashed line divides the scheme into glycinergic and GABAergic synapse. The major cause of hyperekplexia is defective glycinergic signaling resulting from mutations in genes involving subunits of GlyRs, glycine transporter, or GlyR‐interacting proteins. Excitation–inhibition imbalance underlies deficient synapse function in epilepsy and DS. In epilepsy, abnormal excitatory tonus results from decreased GABA release and concurrent enhanced glutamatergic neurotransmission. In DS, an over‐inhibition of synapses by increased GABAergic circuitry and endocytic dysfunction is considered the main synaptic hallmark of the disease.
Whereas traditional therapeutic options for synapse‐linked diseases have mostly focused on pharmacological approaches to correct neurotransmitter signaling, recent paradigm shifts in the field of synaptopathies could offer novel translational perspectives that consider additional components of synapses. In this regard, large‐scale projects have been devoted to understand how alterations in single synaptic components could result in major symptoms of neurological diseases (Bayés et al. 2010; Grant 2012). Thus, understanding these neurological diseases through ‘synaptic glasses’ could result in perspectives that likely yield novel therapeutic approaches for neurodevelopmental and/or neurodegenerative disorders.
Acknowledgments and conflict of interest disclosure
This review was initiated at the 13th International Society for Neurochemistry (ISN) Advanced School held in Mission Beach, Australia, in August 2015. We thank the ISN for the support provided, as well as all ISN School faculty members, who devoted their time to share their views on synapse biology and dysfunction and to encourage young scientists. We specially thank Elizabeth J. Coulson (University of Queensland, Australia) for her assistance and support during ISN School. Constanze Seidenbecher is an editor for Journal of Neurochemistry. Barbara Schweitzer is currently working in Journal of Neurochemistry's Editorial Office. All authors wish to acknowledge ISN for financial and educational support, and declare no conflict of interest.
Read the Editorial Highlight for this article on page 783.
References
- Ackermann F., Waites C. L. and Garner C. C. (2015) Presynaptic active zones in invertebrates and vertebrates. EMBO Rep. 16, 923–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwala K. L., Ganesh S., Suzuki T. et al (2001) Dscam is associated with axonal and dendritic features of neuronal cells. J. Neurosci. Res. 66, 337–346. [DOI] [PubMed] [Google Scholar]
- Ahmed I., Liang Y., Schools S., Dawson V. L., Dawson T. M. and Savitt J. M. (2012) Development and characterization of a new Parkinson's disease model resulting from impaired autophagy. J. Neurosci. 32, 16503–16509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al‐Owain M., Colak D., Al‐Bakheet A. et al (2012) Novel mutation in GLRB in a large family with hereditary hyperekplexia. Clin. Genet. 81, 479–484. [DOI] [PubMed] [Google Scholar]
- Andermann F., Keene D., Andermann E. and Quesney L. (1980) Startle disease or hyperekplexia: further delineation of the syndrome. Brain 103, 985–997. [DOI] [PubMed] [Google Scholar]
- Andrews‐Zwilling Y., Bien‐Ly N., Xu Q. et al (2010) Apolipoprotein E4 causes age‐ and Tau‐dependent impairment of GABAergic interneurons, leading to learning and memory deficits in mice. J. Neurosci. 30, 13707–13717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auerbach B. D., Osterwell E. K. and Bear M. F. (2011) Mutations causing syndromic autism define an axis of synaptic pathophysiology. Nature 480, 63–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey C. H., Kandel E. R. and Harris K. M. (2015) Structural components of synaptic plasticity and memory consolidation. Cold Spring Harb. Perspect. Biol. 7, a021758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakker M. J., Peeters E. A. and Tijssen M. A. (2009) Clonazepam is an effective treatment for hyperekplexia due to a SLC6A5 (GlyT2) mutation. Mov. Disord. 24, 1852–1854. [DOI] [PubMed] [Google Scholar]
- Barker W. W., Luis C. A., Kashuba A. et al (2002) Relative frequencies of Alzheimer disease, Lewy body, vascular and frontotemporal dementia, and hippocampal sclerosis in the State of Florida Brain Bank. Alzheimer Dis. Assoc. Disord. 16, 203–212. [DOI] [PubMed] [Google Scholar]
- Baroncelli L., Braschi C., Spolidoro M., Begenisic T., Sale A. and Maffei L. (2010) Nurturing brain plasticity: impact of environmental enrichment. Cell Death Differ. 17, 1092–1103. [DOI] [PubMed] [Google Scholar]
- Baudouin S. J., Gaudias J., Gerharz S. et al (2012) Shared synaptic pathophysiology in syndromic and nonsyndromic rodent models of autism. Science 338, 128–132. [DOI] [PubMed] [Google Scholar]
- Bayés À., devan Lagemaat L. N. , Collins M. O., Croning M. D. R., Whittle I. R., Choudhary J. S. and Grant S. G. N. (2010) Characterization of the proteome, diseases and evolution of the human postsynaptic density. Nat. Neurosci. 14, 19–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bear M. F., Huber K. M. and Warren S. T. (2004) The mGluR theory of fragile X mental retardation. Trends Neurosci. 27, 370–377. [DOI] [PubMed] [Google Scholar]
- Becker K., Braune M., Benderska N., Buratti E., Baralle F., Villmann C., Stamm S., Eulenburg V. and Becker C. M. (2012) A retroelement modifies pre‐mRNA splicing: the murine Glrbspa allele is a splicing signal polymorphism amplified by long interspersed nuclear element insertion. J. Biol. Chem. 287, 31185–31194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beilina A., Rudenko I. N., Kaganovich A. et al (2014) Unbiased screen for interactors of leucine‐rich repeat kinase 2 supports a common pathway for sporadic and familial Parkinson disease. Proc. Natl Acad. Sci. USA, 111, 2626–2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belichenko N. P., Belichenko P. V., Kleschevnikov A. M., Salehi A., Reeves R. H. and Mobley W. C. (2009) The “Down syndrome critical region” is sufficient in the mouse model to confer behavioral, neurophysiological, and synaptic phenotypes characteristic of Down syndrome. J. Neurosci. 29, 5938–5948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bemben M. A., Shipman S. L., Hirai T., Herring B. E., Li Y., Badger J. D., 2nd , Nicoll R. A., Diamond J. S. and Roche K. W. (2014) CaMKII phosphorylation of neuroligin‐1 regulates excitatory synapses. Nat. Neurosci. 17, 56–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benito E., Urbanke H., Ramachandran B. et al (2015) HDAC inhibitor‐dependent transcriptome and memory reinstatement in cognitive decline models. J. Clin. Investig. 125, 3572–3584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson D. L. and Huntley G. W. (2012) Synapse adhesion: a dynamic equilibrium conferring stability and flexibility. Curr. Opin. Neurobiol. 22, 397–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berwick D. C. and Harvey K. (2011) LRRK2 signaling pathways: the key to unlocking neurodegeneration? Trends Cell Biol. 21, 257–265. [DOI] [PubMed] [Google Scholar]
- Berwick D. C. and Harvey K. (2013) LRRK2: an éminence grise of Wnt‐mediated neurogenesis? Front. Cell Neurosci. 7, 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezzi P. and Volterra A. (2001) A neuron‐glia signalling network in the active brain. Curr. Opin. Neurobiol. 11, 387–394. [DOI] [PubMed] [Google Scholar]
- Bhaskar K., Maphis N., Xu G. et al (2014) Microglial derived tumor necrosis factor‐alpha drives Alzheimer's disease‐related neuronal cell cycle events. Neurobiol. Dis. 62, 273–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billings L. M., Oddo S., Green K. N., McGaugh J. L. and LaFerla F. M. (2005) Intraneuronal Abeta causes the onset of early Alzheimer's disease‐related cognitive deficits in transgenic mice. Neuron 45, 675–688. [DOI] [PubMed] [Google Scholar]
- Bilousova T., Miller C. A., Poon W. W. et al (2016) Synaptic amyloid‐beta oligomers precede p‐tau and differentiate high pathology control cases. Am. J. Pathol. 186, 185–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjorklund N. L., Reese L. C., Sadagoparamanujam V. M., Ghirardi V., Woltjer R. L. and Taglialatela G. (2012) Absence of amyloid beta oligomers at the postsynapse and regulated synaptic Zn2+ in cognitively intact aged individuals with Alzheimer's disease neuropathology. Mol. Neurodegener. 7, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blank M., Fuerst P. G., Stevens B. et al (2011) The Down syndrome critical region regulates retinogeniculate refinement. J. Neurosci. 31, 5764–5776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blundell J., Blaiss C. A., Etherton M. R., Espinosa F., Tabuchi K., Walz C., Bolliger M. F., Sudhof T. C. and Powell C. M. (2010) Neuroligin‐1 deletion results in impaired spatial memory and increased repetitive behavior. J. Neurosci. 30, 2115–2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boison D. (2016) Adenosinergic signaling in epilepsy. Neuropharmacology. 104, 131–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolam J. P. and Pissadaki E. K. (2012) Living on the edge with too many mouths to feed: why dopamine neurons die. Mov. Disord. 27, 1478–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonda D. J., Evans T. A., Santocanale C. et al (2009) Evidence for the progression through S‐phase in the ectopic cell cycle re‐entry of neurons in Alzheimer disease. Aging 1, 382–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H. and Braak E. (1997) Frequency of stages of Alzheimer‐related lesions in different age categories. Neurobiol. Aging 18, 351–357. [DOI] [PubMed] [Google Scholar]
- Braudeau J., Dauphinot L. and Duchon A. (2011) Chronic treatment with a promnesiant GABA‐A ‐selective inverse agonist increases immediate early genes expression during memory processing in mice and rectifies their expression levels in a down syndrome mouse model. Adv. Pharmacol. Sci. 2011, 153218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brose N., O'Connor V. and Skehel P. (2010) Synaptopathy: dysfunction of synaptic function? Biochem. Soc. Trans. 38, 443–444. [DOI] [PubMed] [Google Scholar]
- Brown P., Rothwell J., Thompson P., Britton T., Day B. and Marsden C. (1991) The hyperekplexias and their relationship to the normal startle reflex. Brain, 114 (Pt 4), 1903–1928. [DOI] [PubMed] [Google Scholar]
- Buckwalter M. S., Cook S. A., Davisson M. T., White W. F. and Camper S. A. (1994) A frameshift mutation in the mouse alpha 1 glycine receptor gene (Glra1) results in progressive neurological symptoms and juvenile death. Hum. Mol. Genet. 3, 2025–2030. [DOI] [PubMed] [Google Scholar]
- Buddhala C., Loftin S. K., Kuley B. M., Cairns N. J., Campbell M. C., Perlmutter J. S. and Kotzbauer P. T. (2015) Dopaminergic, serotonergic, and noradrenergic deficits in Parkinson disease. Ann. Clin. Transl. Neurol. 2, 949–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budimirovic D. B. and Kaufmann W. E. (2011) What can we learn about autism from studying fragile X syndrome? Dev. Neurosci. 33, 379–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bukalo O. and Dityatev A. (2012) Synaptic cell adhesion molecules. Adv. Exp. Med. Biol. 970, 97–128. [DOI] [PubMed] [Google Scholar]
- von Campe G., Spencer D. D. and de Lanerolle N. C. (1997) Morphology of dentate granule cels in the human epileptogenic hippocampus. Hippocampus 7, 472–488. [DOI] [PubMed] [Google Scholar]
- Cardoso T. A., Coan A. C., Kobayashi E., Guerreiro C. A., Li L. M. and Cendes F. (2006) Hippocampal abnormalities and seizure recurrence after antiepilpetic drug withdrawal. Neurology 67, 134–136. [DOI] [PubMed] [Google Scholar]
- Carvajal‐Gonzalez A., Leite M. I., Waters P. et al (2014) Glycine receptor antibodies in PERM and related syndromes: characteristics, clinical features and outcomes. Brain 137, 2178–2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cataldo A. M., Peterhoff C. M., Troncoso J. C., Gomez‐Isla T., Hyman B. T. and Nixon R. A. (2000) Endocytic pathway abnormalities precede amyloid beta deposition in sporadic Alzheimer's disease and Down syndrome: differential effects of APOE genotype and presenilin mutations. Am. J. Pathol. 157, 277–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cataldo A. M., Mathews P. M., Boiteau A. B. et al (2008) Down syndrome fibroblast model of Alzheimer‐related endosome pathology: accelerated endocytosis promotes late endocytic defects. Am. J. Pathol. 173, 370–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catterall W. A. and Few A. P. (2008) Calcium channel regulation and presynaptic plasticity. Neuron 59, 882–901. [DOI] [PubMed] [Google Scholar]
- Chanda S., Marro S., Wernig M. and Sudhof T. C. (2013) Neurons generated by direct conversion of fibroblasts reproduce synaptic phenotype caused by autism‐associated neuroligin‐3 mutation. Proc. Natl Acad. Sci. USA 110, 16622–16627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra S., Gallardo G., Fernandez‐Chacon R., Schluter O. M. and Sudhof T. C. (2005) a‐Synuclein cooperates with CSPa in preventing neurodegeneration. Cell 123, 383–396. [DOI] [PubMed] [Google Scholar]
- Chowdhury L., Chakraborty S., Chakrabarti J., Bhattacharya D. and Mandals M. (2010) Analgesic effect of low gynaecological surgery under general anaesthesia. J. Indian Med. Assoc. 108, 734–737. [PubMed] [Google Scholar]
- Christopherson K. S., Ullian E. M., Stokes C. C. et al (2005) Thrombospondins are astrocyte‐secreted proteins that promote CNS synaptogenesis. Cell 120, 421–433. [DOI] [PubMed] [Google Scholar]
- Chubykin A. A., Atasoy D., Etherton M. R., Brose N., Kavalali E. T., Gibson J. R. and Sudhof T. C. (2007) Activity‐dependent validation of excitatory versus inhibitory synapses by neuroligin‐1 versus neuroligin‐2. Neuron 54, 919–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung S. K., Vanbellinghen J. F., Mullins J. G. et al (2010) Pathophysiological mechanisms of dominant and recessive GLRA1 mutations in hyperekplexia. J. Neurosci. 30, 9612–9620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung W., Choi S. Y., Lee E. et al (2015a) Social deficits in IRSp53 mutant mice improved by NMDAR and mGluR5 suppression. Nat. Neurosci. 18, 435–443. [DOI] [PubMed] [Google Scholar]
- Chung W. S., Allen N. J. and Eroglu C. (2015b) Astrocytes control synapse formation, function, and elimination. Cold Spring Harb. Perspect. Biol. 7, a020370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Citri A. and Malenka R. C. (2008) Synaptic plasticity: multiple forms, functions, and mechanisms. Neuropsychopharmacology, 33, 18–41. [DOI] [PubMed] [Google Scholar]
- Colonnier M. (1968) Synaptic patterns on different cell types in the different laminae of the cat visual cortex. An electron microscope study. Brain Res. 9, 268–287. [DOI] [PubMed] [Google Scholar]
- Corder E. H., Saunders A. M., Strittmatter W. J., Schmechel D. E., Gaskell P. C., Small G. W., Roses A. D., Haines J. L. and Pericak‐Vance M. A. (1993) Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science 261, 921–923. [DOI] [PubMed] [Google Scholar]
- Cramer N. and Galdzicki Z. (2012) From abnormal hippocampal synaptic plasticity in down syndrome mouse models to cognitive disability in down syndrome. Neural. Plast. 2012, 101542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cvetkovska V., Hibbert A. D., Emran F. and Chen B. E. (2013) Overexpression of Down syndrome cell adhesion molecule impairs precise synaptic targeting. Nat. Neurosci. 16, 677–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlhaus R., Hines R. M., Eadie B. D., Kannangara T. S., Hines D. J., Brown C. E., Christie B. R. and El‐Husseini A. (2010) Overexpression of the cell adhesion protein neuroligin‐1 induces learning deficits and impairs synaptic plasticity by altering the ratio of excitation to inhibition in the hippocampus. Hippocampus 20, 305–322. [DOI] [PubMed] [Google Scholar]
- Damasio J., Leite M. I., Coutinho E., Waters P., Woodhall M., Santos M. A., Carrilho I. and Vincent A. (2013) Progressive encephalomyelitis with rigidity and myoclonus: the first pediatric case with glycine receptor antibodies. JAMA Neurol. 70, 498–501. [DOI] [PubMed] [Google Scholar]
- D'Amelio M., Sheng M. and Cecconi F. (2012) Caspase‐3 in the central nervous system: beyond apoptosis. Trends Neurosci. 35, 700–709. [DOI] [PubMed] [Google Scholar]
- D'Antoni S., Spatuzza M., Bonaccorso C. M., Musumeci S. A., Ciranna L., Nicoletti F., Huber K. M. and Catania M. V. (2014) Dysregulation of group‐I metabotropic glutamate (mGlu) receptor mediated signalling in disorders associated with Intellectual Disability and Autism. Neurosci. Biobehav. Rev. 46(Pt 2), 228–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Felice F. G., Velasco P. T., Lambert M. P., Viola K., Fernandez S. J., Ferreira S. T. and Klein W. L. (2007) Abeta oligomers induce neuronal oxidative stress through an N‐methyl‐D‐aspartate receptor‐dependent mechanism that is blocked by the Alzheimer drug memantine. J. Biol. Chem. 282, 11590–11601. [DOI] [PubMed] [Google Scholar]
- De Felice F. G., Wu D., Lambert M. P. et al (2008) Alzheimer's disease‐type neuronal tau hyperphosphorylation induced by A beta oligomers. Neurobiol. Aging 29, 1334–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean C., Scholl F. G., Choih J., DeMaria S., Berger J., Isacoff E. and Scheiffele P. (2003) Neurexin mediates the assembly of presynaptic terminals. Nat. Neurosci. 6, 708–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deusser J., Schmidt S., Ettle B., Plotz S., Huber S., Muller C. P., Masliah E., Winkler J. and Kohl Z. (2015) Serotonergic dysfunction in the A53T alpha‐synuclein mouse model of Parkinson's disease. J. Neurochem. 135, 589–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dierssen M., Benavides‐Piccione R., Martinez‐Cue C., Estivill X., Florez J., Elston G. N. and DeFelipe J. (2003) Alterations of neocortical pyramidal cell phenotype in the Ts65Dn mouse model of Down syndrome: effects of environmental enrichment. Cereb. Cortex 13, 758–764. [DOI] [PubMed] [Google Scholar]
- Dierssen M., Herault Y. and Estivill X. (2009) Aneuploidy: from a physiological mechanism of variance to Down syndrome. Physiol. Rev. 89, 887–920. [DOI] [PubMed] [Google Scholar]
- Dietz B. and Manahan‐Vaughan D. (2016) Hippocampal long‐term depression is facilitated by the acquisition and updating of memory of spatial auditory content and requires mGlu5 activation. Neuropharmacology pii: S0028‐3908(16)30061‐2. doi: 10.1016/j.neuropharm.2016.02.026. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Dinamarca M. C., Weinstein D., Monasterio O. and Inestrosa N. C. (2011) The synaptic protein neuroligin‐1 interacts with the amyloid beta‐peptide. Is there a role in Alzheimer's disease? Biochemistry 50, 8127–8137. [DOI] [PubMed] [Google Scholar]
- Doody R. S., Thomas R. G., Farlow M. et al (2014) Phase 3 trials of solanezumab for mild‐to‐moderate Alzheimer's disease. N. Engl. J. Med. 370, 311–321. [DOI] [PubMed] [Google Scholar]
- Dooley J. and Andermann F. (1989) Startle disease or hyperekplexia: adolescent onset and response to valproate. Pediatr. Neurol. 5, 126–127. [DOI] [PubMed] [Google Scholar]
- Erturk A., Wang Y. and Sheng M. (2014) Local pruning of dendrites and spines by caspase‐3‐dependent and proteasome‐limited mechanisms. J. Neurosci. 34, 1672–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escorihuela R. M., Vallina I. F., Martinez‐Cue C., Baamonde C., Dierssen M., Tobena A., Florez J. and Fernandez‐Teruel A. (1998) Impaired short‐ and long‐term memory in Ts65Dn mice, a model for Down syndrome. Neurosci. Lett. 247, 171–174. [DOI] [PubMed] [Google Scholar]
- Etherton M. R., Tabuchi K., Sharma M., Ko J. and Sudhof T. C. (2011) An autism‐associated point mutation in the neuroligin cytoplasmic tail selectively impairs AMPA receptor‐mediated synaptic transmission in hippocampus. EMBO J. 30, 2908–2919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang W. Q., Chen W. W., Jiang L., Liu K., Yung W. H., Fu A. K. and Ip N. Y. (2014) Overproduction of upper‐layer neurons in the neocortex leads to autism‐like features in mice. Cell Rep. 9, 1635–1643. [DOI] [PubMed] [Google Scholar]
- Fernandez F. and Garner C. C. (2007) Over‐inhibition: a model for developmental intellectual disability. Trends Neurosci. 30, 497–503. [DOI] [PubMed] [Google Scholar]
- Fernandez F., Morishita W., Zuniga E., Nguyen J., Blank M., Malenka R. C. and Garner C. C. (2007) Pharmacotherapy for cognitive impairment in a mouse model of Down syndrome. Nat. Neurosci. 10, 411–413. [DOI] [PubMed] [Google Scholar]
- Fernandez J. W., Rezai‐Zadeh K., Obregon D. and Tan J. (2010) EGCG functions through estrogen receptor‐mediated activation of ADAM10 in the promotion of non‐amyloidogenic processing of APP. FEBS Lett. 584, 4259–4267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira S. T., Lourenco M. V., Oliveira M. M. and De Felice F. G. (2015) Soluble amyloid‐beta oligomers as synaptotoxins leading to cognitive impairment in Alzheimer's disease. Front. Cell Neurosci. 9, 191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferri C. P., Prince M., Brayne C. et al (2005) Global prevalence of dementia: a Delphi consensus study. Lancet 366, 2112–2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogarty M. J., Kanjhan R., Bellingham M. C. and Noakes P. G. (2016) Glycinergic neurotransmission: a potent regulator of embryonic motor neuron dendritic morphology and synaptic plasticity. J. Neurosci. 36, 80–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frischknecht R. and Seidenbecher C. I. (2008) The crosstalk of hyaluronan‐based extracellular matrix and synapses. Neuron. Glia. Biol. 4, 249–257. [DOI] [PubMed] [Google Scholar]
- Frischknecht R., Heine M., Perrais D., Seidenbecher C. I., Choquet D. and Gundelfinger E. D. (2009) Brain extracellular matrix affects AMPA receptor lateral mobility and short‐term synaptic plasticity. Nat. Neurosci. 12, 897–904. [DOI] [PubMed] [Google Scholar]
- Fuerst P. G., Bruce F., Tian M., Wei W., Elstrott J., Feller M. B., Erskine L., Singer J. H. and Burgess R. W. (2009) DSCAM and DSCAML1 function in self‐avoidance in multiple cell types in the developing mouse retina. Neuron 64, 484–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandal M. J., Anderson R. L., Billingslea E. N., Carlson G. C., Roberts T. P. and Siegel S. J. (2012) Mice with reduced NMDA receptor expression: more consistent with autism than schizophrenia? Genes Brain Behav. 11, 740–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garner C. C. and Wetmore D. Z. (2012) Synaptic pathology of Down syndrome. Adv. Exp. Med. Biol. 970, 451–468. [DOI] [PubMed] [Google Scholar]
- Gassmann M. and Bettler B. (2012) Regulation of neuronal GABA(B) receptor functions by subunit composition. Nat. Rev. Neurosci. 13, 380–394. [DOI] [PubMed] [Google Scholar]
- Giehm L., Svergun D. I., Otzen D. E. and Vestergaard B. (2011) Low‐resolution structure of a vesicle disrupting [alpha]‐synuclein oligomer that accumulates during fibrillation. Proc. Natl Acad. Sci. USA, 108, 3246–3251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gjoneska E., Pfenning A. R., Mathys H., Quon G., Kundaje A., Tsai L. H. and Kellis M. (2015) Conserved epigenomic signals in mice and humans reveal immune basis of Alzheimer's disease. Nature 518, 365–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goate A., Chartier‐Harlin M. C., Mullan M. et al (1991) Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer's disease. Nature 349, 704–706. [DOI] [PubMed] [Google Scholar]
- Gogolla N., Caroni P., Luthi A. and Herry C. (2009) Perineuronal nets protect fear memories from erasure. Science 325, 1258–1261. [DOI] [PubMed] [Google Scholar]
- Gong Y., Chang L., Viola K. L., Lacor P. N., Lambert M. P., Finch C. E., Krafft G. A. and Klein W. L. (2003) Alzheimer's disease‐affected brain: presence of oligomeric A beta ligands (ADDLs) suggests a molecular basis for reversible memory loss. Proc. Natl Acad. Sci. USA 100, 10417–10422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gräff J., Rei D., Guan J.‐S. et al (2012) An epigenetic blockade of cognitive functions in the neurodegenerating brain. Nature 483, 222–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant S. G. (2012) Synaptopathies: diseases of the synaptome. Curr. Opin. Neurobiol. 22, 522–529. [DOI] [PubMed] [Google Scholar]
- Gray E. G. (1959) Axo‐somatic and axo‐dendritic synapses of the cerebral cortex: an electron microscope study. J. Anat. 93, 420–433. [PMC free article] [PubMed] [Google Scholar]
- Gray E. G., Paula‐Barbosa M. and Roher A. (1987) Alzheimer's disease: paired helical filaments and cytomembranes. Neuropathol. Appl. Neurobiol., 13, 91–110. [DOI] [PubMed] [Google Scholar]
- Gu B., Huang Y. Z., He X. P., Joshi R. B., Jang W. and McNamara J. O. (2015) A peptide uncoupling BDNF receptor TrkB from phospholipase Cgamma1 prevents epilepsy induced by status epilepticus. Neuron 88, 484–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson J. E., Blank M., Valenzuela R. A., Garner C. C. and Madison D. V. (2007) The functional nature of synaptic circuitry is altered in area CA3 of the hippocampus in a mouse model of Down's syndrome. J. Physiol. 579, 53–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J. A. and Higgins G. A. (1992) Alzheimer's disease: the amyloid cascade hypothesis. Science 256, 184–185. [DOI] [PubMed] [Google Scholar]
- Hardy J. and Selkoe D. J. (2002) The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297, 353–356. [DOI] [PubMed] [Google Scholar]
- Harvey K., Duguid I. C., Alldred M. J. et al (2004) The GDP‐GTP exchange factor collybistin: an essential determinant of neuronal gephyrin clustering. J. Neurosci. 24, 5816–5826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey R. J., Carta E., Pearce B. R., Chung S. K., Supplisson S., Rees M. I. and Harvey K. (2008) A critical role for glycine transporters in hyperexcitability disorders. Fron. Mol. Neurosci. 1, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattori D., Demir E., Kim H. W., Viragh E., Zipursky S. L. and Dickson B. J. (2007) Dscam diversity is essential for neuronal wiring and self‐recognition. Nature 449, 223–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Head E., Lott I. T., Wilcock D. M. and Lemere C. A. (2016) Aging in down syndrome and the development of Alzheimer's disease neuropathology. Curr. Alzheimer Res. 13, 18–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heikkinen A., Pihlajaniemi T., Faissner A. and Yuzaki M. (2014) Neural ECM and synaptogenesis. Prog. Brain Res. 214, 29–51. [DOI] [PubMed] [Google Scholar]
- Holland K. D., Fleming M. T., Cheek S., Moran J. L., Beier D. R. and Meisler M. H. (2006) De novo exon duplication in a new allele of mouse Glra1 (spasmodic). Genetics 174, 2245–2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosenbocus S. and Chahal R. (2013) Memantine: a review of possible uses in child and adolescent psychiatry. J. Can. Acad. Child Adolesc. Psychiatry. 22, 166–171. [PMC free article] [PubMed] [Google Scholar]
- Huguet G., Ey E. and Bourgeron T. (2013) The genetic landscapes of autism spectrum disorders. Annu. Rev. Genomics Hum. Genet. 14, 191–213. [DOI] [PubMed] [Google Scholar]
- Hung A. Y., Futai K., Sala C. et al (2008) Smaller dendritic spines, weaker synaptic transmission, but enhanced spatial learning in mice lacking Shank1. J. Neurosci. 28, 1697–1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunn B. H., Cragg S. J., Bolam J. P., Spillantini M. G. and Wade‐Martins R. (2015) Impaired intracellular trafficking defines early Parkinson's disease. Trends Neurosci. 38, 178–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isokawa M. (2000) Remodeling dendritic spines of dentate granule cells in temporal lobe epilepsy patients and the rat pilocarpine model. Epilepsia 41, S14–S17. [DOI] [PubMed] [Google Scholar]
- Ittner L. M., Ke Y. D., Delerue F. et al (2010) Dendritic Function of Tau Mediates Amyloid‐β Toxicity in Alzheimer's Disease Mouse Models. Cell 142, 387–397. [DOI] [PubMed] [Google Scholar]
- Jamain S., Quach H., Betancur C. et al (2003) Mutations of the X‐linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat. Genet. 34, 27–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James V. M., Bode A., Chung S. K. et al (2013) Novel missense mutations in the glycine receptor beta subunit gene (GLRB) in startle disease. Neurobiol. Dis. 52, 137–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janezic S., Threlfell S., Dodson P. D. et al (2013) Deficits in dopaminergic transmission precede neuron loss and dysfunction in a new Parkinson model. Proc. Natl Acad. Sci. USA 110, E4016–E4025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin M., Shepardson N., Yang T., Chen G., Walsh D. and Selkoe D. J. (2011) Soluble amyloid beta‐protein dimers isolated from Alzheimer cortex directly induce Tau hyperphosphorylation and neuritic degeneration. Proc. Natl Acad. Sci. USA 108, 5819–5824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo J., Whitcomb D. J., Olsen K. M. et al (2011) Abeta(1‐42) inhibition of LTP is mediated by a signaling pathway involving caspase‐3, Akt1 and GSK‐3beta. Nat. Neurosci. 14, 545–547. [DOI] [PubMed] [Google Scholar]
- Johnson J. W. and Ascher P. (1987) Glycine potentiates the NMDA response in cultured mouse brain neurons. Nature 325, 529–531. [DOI] [PubMed] [Google Scholar]
- Jonas P., Bischofberger J. and Sandkuhler J. (1998) Corelease of two fast neurotransmitters at a central synapse. Science 281, 419–424. [DOI] [PubMed] [Google Scholar]
- Kandel E. R., Dudai Y. and Mayford M. R. (2014) The molecular and systems biology of memory. Cell 157, 163–186. [DOI] [PubMed] [Google Scholar]
- Keezer M. R., Sisodiya S. M. and Sander J. W. (2016) Comorbidities of epilepsy: current concepts and future perspectives. Lancet Neurol. 15, 106–115. [DOI] [PubMed] [Google Scholar]
- Kenny E. M., Cormican P., Furlong S. et al (2014) Excess of rare novel loss‐of‐function variants in synaptic genes in schizophrenia and autism spectrum disorders. Mol. Psychiatry 19, 872–879. [DOI] [PubMed] [Google Scholar]
- Khasani S., Becker K. and Meinck H. M. (2004) Hyperekplexia and stiff‐man syndrome: abnormal brainstem reflexes suggest a physiological relationship. J. Neurol. Neurosurg. Psychiatry 75, 1265–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoshnood B., Greenlees R., Loane M., Dolk H., Committee E. P. M. and Group E. W. (2011) Paper 2: EUROCAT public health indicators for congenital anomalies in Europe. Birth Defects Res. A Clin. Mol. Teratol. 91 Suppl 1, S16–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilgore M., Miller C. A., Fass D. M., Hennig K. M., Haggarty S. J., Sweatt J. D. and Rumbaugh G. (2010) Inhibitors of class 1 histone deacetylases reverse contextual memory deficits in a mouse model of Alzheimer's disease. Neuropsychopharmacology, 35, 870–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J. H., Wang X., Coolon R. and Ye B. (2013) Dscam expression levels determine presynaptic arbor sizes in Drosophila sensory neurons. Neuron 78, 827–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingsmore S. F., Giros B., Suh D., Bieniarz M., Caron M. G. and Seldin M. F. (1994) Glycine receptor beta‐subunit gene mutation in spastic mouse associated with LINE‐1 element insertion. Nat. Genet. 7, 136–141. [DOI] [PubMed] [Google Scholar]
- Kirstein L. and Silfverskiold B. P. (1958) A family with emotionally precipitated drop seizures. Acta Psychiatr. Neurol. Scand. 33, 471–476. [DOI] [PubMed] [Google Scholar]
- Klatte K., Kirschstein T., Otte D. et al (2013) Impaired D‐serine‐mediated cotransmission mediates cognitive dysfunction in epilepsy. J. Neurosci. 33, 13066–13080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleschevnikov A. M., Belichenko P. V., Villar A. J., Epstein C. J., Malenka R. C. and Mobley W. C. (2004) Hippocampal long‐term potentiation suppressed by increased inhibition in the Ts65Dn mouse, a genetic model of Down syndrome. J. Neurosci. 24, 8153–8160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kling C., Koch M., Saul B. and Becker C. M. (1997) The frameshift mutation oscillator (Glra1(spd‐ot)) produces a complete loss of glycine receptor alpha1‐polypeptide in mouse central nervous system. Neuroscience 78, 411–417. [DOI] [PubMed] [Google Scholar]
- Koffie R. M., Hashimoto T., Tai H. C. et al (2012) Apolipoprotein E4 effects in Alzheimer's disease are mediated by synaptotoxic oligomeric amyloid‐beta. Brain 135, 2155–2168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koning‐Tijssen M. and Brouwer O. (2000) Hyperekplexia in the first year of life. Mov. Disord. 15, 1293–1296. [DOI] [PubMed] [Google Scholar]
- Kril J. J., Patel S., Harding A. J. and Halliday G. M. (2002) Neuron loss from the hippocampus of Alzheimer's disease exceeds extracellular neurofibrillary tangle formation. Acta Neuropathol. 103, 370–376. [DOI] [PubMed] [Google Scholar]
- Kurt M. A., Davies D. C., Kidd M., Dierssen M. and Florez J. (2000) Synaptic deficit in the temporal cortex of partial trisomy 16 (Ts65Dn) mice. Brain Res. 858, 191–197. [DOI] [PubMed] [Google Scholar]
- Kurt M. A., Kafa M. I., Dierssen M. and Davies D. C. (2004) Deficits of neuronal density in CA1 and synaptic density in the dentate gyrus, CA3 and CA1, in a mouse model of Down syndrome. Brain Res. 1022, 101–109. [DOI] [PubMed] [Google Scholar]
- Lane P. W., Ganser A. L., Kerner A. L. and White W. F. (1987) Spasmodic, a mutation on chromosome 11 in the mouse. J. Hered. 78, 353–356. [DOI] [PubMed] [Google Scholar]
- Lau P., Bossers K., Janky R. et al (2013) Alteration of the microRNA network during the progression of Alzheimer's disease. EMBO Mol. Med. 5, 1613–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laumonnier F., Bonnet‐Brilhault F., Gomot M. et al (2004) X‐linked mental retardation and autism are associated with a mutation in the NLGN4 gene, a member of the neuroligin family. Am. J. Hum. Genet. 74, 552–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauren J., Gimbel D. A., Nygaard H. B., Gilbert J. W. and Strittmatter S. M. (2009) Cellular prion protein mediates impairment of synaptic plasticity by amyloid‐beta oligomers. Nature 457, 1128–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law B. M., Spain V. A., Leinster V. H. et al (2014) A direct interaction between leucine‐rich repeat kinase 2 and specific beta‐tubulin isoforms regulates tubulin acetylation. J. Biol. Chem. 289, 895–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarevic V., Schone C., Heine M., Gundelfinger E. D. and Fejtova A. (2011) Extensive remodeling of the presynaptic cytomatrix upon homeostatic adaptation to network activity silencing. J. Neurosci. 31, 10189–10200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarevic V., Pothula S., Andres‐Alonso M. and Fejtova A. (2013) Molecular mechanisms driving homeostatic plasticity of neurotransmitter release. Front. Cell Neurosci. 7, 244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazzari C., Kipanyula M. J., Agostini M., Pozzan T. and Fasolato C. (2015) Abeta42 oligomers selectively disrupt neuronal calcium release. Neurobiol. Aging 36, 877–885. [DOI] [PubMed] [Google Scholar]
- Ledri M., Sorensen A. T., Madsen M. G. et al (2015) Differential effect of neuropeptides on excitatory synaptic transmission in human epileptic hippocampus. J. Neurosci. 35, 9622–9631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M. C., Yasuda R. and Ehlers M. D. (2010) Metaplasticity at single glutamatergic synapses. Neuron 66, 859–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leite J. P., Neder L., Arisi G. M., Carlotti C. G., Jr , Assirati J. A. and Moreira J. E. (2005) Plasticity, synaptic strength, and epilepsy: what can we learn from ultrastructural data? Epilepsia 46(Suppl 5), 134–141. [DOI] [PubMed] [Google Scholar]
- Lepeta K. and Kaczmarek L. (2015) Matrix metalloproteinase‐9 as a novel player in synaptic plasticity and schizophrenia. Schizophr. Bull. 41, 1003–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levi S., Logan S. M., Tovar K. R. and Craig A. M. (2004) Gephyrin is critical for glycine receptor clustering but not for the formation of functional GABAergic synapses in hippocampal neurons. J. Neurosci. 24, 207–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy‐Lahad E., Wasco W., Poorkaj P. et al (1995) Candidate gene for the chromosome 1 familial Alzheimer's disease locus. Science 269, 973–977. [DOI] [PubMed] [Google Scholar]
- Li J.‐Y., Plomann M. and Brundin P. (2003) Huntington's disease: a synaptopathy? Trends Mol. Med. 9, 414–420. [DOI] [PubMed] [Google Scholar]
- Li Y., Tomiyama H., Sato K. et al (2005) Clinicogenetic study of PINK1 mutations in autosomal recessive early‐onset parkinsonism. Neurology 64, 1955–1957. [DOI] [PubMed] [Google Scholar]
- Li Z., Jo J., Jia J. M., Lo S. C., Whitcomb D. J., Jiao S., Cho K. and Sheng M. (2010) Caspase‐3 activation via mitochondria is required for long‐term depression and AMPA receptor internalization. Cell 141, 859–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G., Gu B., He X. P., Joshi R. B., Wackerle H. D., Rodriguiz R. M., Wetsel W. C. and McNamara J. O. (2013) Transient inhibition of TrkB kinase after status epilepticus prevents development of temporal lobe epilepsy. Neuron 79, 31–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes M. W., Lopes S. C., Costa A. P. et al (2015) Region‐specific alterations of AMPA receptor phosphorylation and signaling pathways in the pilocarpine model of epilepsy. Neurochem. Int. 87, 22–33. [DOI] [PubMed] [Google Scholar]
- Lott I. T. and Dierssen M. (2010) Cognitive deficits and associated neurological complications in individuals with Down's syndrome. Lancet Neurol. 9, 623–633. [DOI] [PubMed] [Google Scholar]
- Lourenco M. V., Clarke J. R., Frozza R. L. et al (2013) TNF‐alpha mediates PKR‐dependent memory impairment and brain IRS‐1 inhibition induced by Alzheimer's beta‐amyloid oligomers in mice and monkeys. Cell Metab. 18, 831–843. [DOI] [PubMed] [Google Scholar]
- Lourenco M. V., Ferreira S. T. and De Felice F. G. (2015) Neuronal stress signaling and eIF2alpha phosphorylation as molecular links between Alzheimer's disease and diabetes. Prog. Neurobiol. 129, 37–57. [DOI] [PubMed] [Google Scholar]
- Lue L. F., Kuo Y. M., Roher A. E. et al (1999) Soluble amyloid beta peptide concentration as a predictor of synaptic change in Alzheimer's disease. Am. J. Pathol. 155, 853–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lujan R., Shigemoto R. and Lopez‐Bendito G. (2005) Glutamate and GABA receptor signalling in the developing brain. Neuroscience 130, 567–580. [DOI] [PubMed] [Google Scholar]
- Lujic L., Bosnjak V. M., Delin S., Duranovic V. and Krakar G. (2011) Infantile spasms in children with Down syndrome. Coll Antropol. 35(Suppl 1), 213–218. [PubMed] [Google Scholar]
- Lynch J. W. (2004) Molecular structure and function of the glycine receptor chloride channel. Physiol. Rev. 84, 1051–1095. [DOI] [PubMed] [Google Scholar]
- Lynch B. A., Lambeng N., Nocka K., Kensel‐Hammes P., Bajjalieh S. M., Matagne A. and Fuks B. (2004) The synaptic vesicle protein SV2A is the binding site for the antiepileptic drug levetiracetam. Proc. Natl Acad. Sci. USA 101, 9861–9866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch‐Day M. A., Mao K., Wang K., Zhao M. and Klionsky D. J. (2012) The role of autophagy in Parkinson's disease. Cold Spring Harb. Perspect Med. 2, a009357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma T., Trinh M. A., Wexler A. J., Bourbon C., Gatti E., Pierre P., Cavener D. R. and Klann E. (2013) Suppression of eIF2α kinases alleviates Alzheimer's disease–related plasticity and memory deficits. Nat. Neurosci. 16, 1299–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLeod David. A., Rhinn H., Kuwahara T. et al (2013) Article: RAB7L1 interacts with LRRK2 to modify intraneuronal protein sorting and Parkinson's disease risk. Neuron 77, 425–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madeira C., Lourenco M. V., Vargas‐Lopes C. et al (2015) d‐serine levels in Alzheimer's disease: implications for novel biomarker development. Transl. Psychiatry. 5, e561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin‐Padilla M. (1976) Pyramidal cell abnormalities in the motor cortex of a child with Down's syndrome. A Golgi study. J. Comp. Neurol. 167, 63–81. [DOI] [PubMed] [Google Scholar]
- Matta S., Van Kolen K., da Cunha R. et al (2012) Article: LRRK2 controls an EndoA phosphorylation cycle in synaptic endocytosis. Neuron 75, 1008–1021. [DOI] [PubMed] [Google Scholar]
- McLean C. A., Cherny R. A., Fraser F. W., Fuller S. J., Smith M. J., Beyreuther K., Bush A. I. and Masters C. L. (1999) Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer's disease. Ann. Neurol. 46, 860–866. [DOI] [PubMed] [Google Scholar]
- McShea A., Lee H. G., Petersen R. B., Casadesus G., Vincent I., Linford N. J., Funk J. O., Shapiro R. A. and Smith M. A. (2007) Neuronal cell cycle re‐entry mediates Alzheimer disease‐type changes. Biochim. Biophys. Acta 1772, 467–472. [DOI] [PubMed] [Google Scholar]
- Michaluk P., Mikasova L., Groc L., Frischknecht R., Choquet D. and Kaczmarek L. (2009) Matrix metalloproteinase‐9 controls NMDA receptor surface diffusion through integrin beta1 signaling. J. Neurosci. 29, 6007–6012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min S. W., Chen X., Tracy T. E. et al (2015) Critical role of acetylation in tau‐mediated neurodegeneration and cognitive deficits. Nat. Med. 21, 1154–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mucke L., Masliah E., Yu G. Q. et al (2000) High‐level neuronal expression of abeta 1‐42 in wild‐type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J. Neurosci. 20, 4050–4058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulhardt C., Fischer M., Gass P., Simon‐Chazottes D., Guenet J. L., Kuhse J., Betz H. and Becker C. M. (1994) The spastic mouse: aberrant splicing of glycine receptor beta subunit mRNA caused by intronic insertion of L1 element. Neuron 13, 1003–1015. [DOI] [PubMed] [Google Scholar]
- Nagy Z., Esiri M. M., Jobst K. A. et al (1995) Relative roles of plaques and tangles in the dementia of Alzheimer's disease: correlations using three sets of neuropathological criteria. Dementia 6, 21–31. [DOI] [PubMed] [Google Scholar]
- Nakamura K., Nemani V. M., Azarbal F. et al (2011) Direct membrane association drives mitochondrial fission by the Parkinson disease‐associated protein alpha‐synuclein. J. Biol. Chem. 286, 20710–20726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niswender C. M. and Conn P. J. (2010) Metabotropic glutamate receptors: physiology, pharmacology, and disease. Annu. Rev. Pharmacol. Toxicol. 50, 295–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noebels J. (2015) Pathway‐driven discovery of epilepsy genes. Nat. Neurosci. 18, 344–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Roak B. J., Vives L., Fu W. et al (2012) Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science 338, 1619–1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paoletti P., Bellone C. and Zhou Q. (2013) NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nat. Rev. Neurosci. 14, 383–400. [DOI] [PubMed] [Google Scholar]
- Pascotto A. and Coppola G. (1992) Neonatal hyperekplexia: a case report. Epilepsia 33, 817–820. [DOI] [PubMed] [Google Scholar]
- Paula‐Lima A. C., Brito‐Moreira J. and Ferreira S. T. (2013) Deregulation of excitatory neurotransmission underlying synapse failure in Alzheimer's disease. J. Neurochem. 126, 191–202. [DOI] [PubMed] [Google Scholar]
- Peret A., Christie L. A., Ouedraogo D. W., Gorlewicz A., Epsztein J., Mulle C. and Crepel V. (2014) Contribution of aberrant GluK2‐containing kainate receptors to chronic seizures in temporal lobe epilepsy. Cell Rep. 8, 347–354. [DOI] [PubMed] [Google Scholar]
- Pham E., Crews L., Ubhi K. et al (2010) Progressive accumulation of amyloid‐beta oligomers in Alzheimer's disease and in amyloid precursor protein transgenic mice is accompanied by selective alterations in synaptic scaffold proteins. FEBS J. 277, 3051–3067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccoli G., Condliffe S. B., Bauer M. et al (2011) LRRK2 controls synaptic vesicle storage and mobilization within the recycling pool. J. Neurosci. 31, 2225–2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickrell Alicia M. and Youle Richard J. (2015) The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson's disease. Neuron 85, 257–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitkänen A. and Lukasiuk K. (2009) Molecular and cellular basis of epileptogenesis in symptomatic epilepsy. Epilepsy Behav. 14, 16–25. [DOI] [PubMed] [Google Scholar]
- Pitkanen A., Lukasiuk K., Dudek F. E. and Staley K. J. (2015) Epileptogenesis. Cold Spring Harb. Perspect Med. 5, pii: a022822. doi: 10.1101/cshperspect.a022822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitkänen A., Roivainen R. and Lukasiuk K. (2016) Development of epilepsy after ischaemic stroke. Lancet Neurol. 15, 185–197. [DOI] [PubMed] [Google Scholar]
- Plowey E. D. and Chu C. T. (2011) Synaptic dysfunction in genetic models of Parkinson's disease: a role for autophagy? Neurobiol. Dis. 43, 60–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter S., Clark I. M., Kevorkian L. and Edwards D. R. (2005) The ADAMTS metalloproteinases. Biochem. J. 386, 15–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posey D. J., Kem D. L., Swiezy N. B., Sweeten T. L., Wiegand R. E. and McDougle C. J. (2004) A pilot study of D‐cycloserine in subjects with autistic disorder. Am. J. Psychiatry 161, 2115–2117. [DOI] [PubMed] [Google Scholar]
- Quirico‐Santos T., Nascimento Mello A., Casimiro Gomes A., de Carvalho L. P., de Souza J. M. and Alves‐Leon S. (2013) Increased metalloprotease activity in the epileptogenic lesion–Lobectomy reduces metalloprotease activity and urokinase‐type uPAR circulating levels. Brain Res. 1538, 172–181. [DOI] [PubMed] [Google Scholar]
- Rees M. I., Andrew M., Jawad S. and Owen M. J. (1994) Evidence for recessive as well as dominant forms of startle disease (hyperekplexia) caused by mutations in the alpha 1 subunit of the inhibitory glycine receptor. Hum. Mol. Genet. 3, 2175–2179. [DOI] [PubMed] [Google Scholar]
- Rees M. I., Harvey K., Ward H. et al (2003) Isoform heterogeneity of the human gephyrin gene (GPHN), binding domains to the glycine receptor, and mutation analysis in hyperekplexia. J. Biol. Chem. 278, 24688–24696. [DOI] [PubMed] [Google Scholar]
- Rees M. I., Harvey K., Pearce B. R. et al (2006) Mutations in the gene encoding GlyT2 (SLC6A5) define a presynaptic component of human startle disease. Nat. Genet. 38, 801–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves R. H., Irving N. G., Moran T. H., Wohn A., Kitt C., Sisodia S. S., Schmidt C., Bronson R. T. and Davisson M. T. (1995) A mouse model for Down syndrome exhibits learning and behaviour deficits. Nat. Genet. 11, 177–184. [DOI] [PubMed] [Google Scholar]
- Reiman E. M., Chen K., Liu X. et al (2009) Fibrillar amyloid‐beta burden in cognitively normal people at 3 levels of genetic risk for Alzheimer's disease. Proc. Natl Acad. Sci. USA 106, 6820–6825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renner M., Lacor P. N., Velasco P. T., Xu J., Contractor A., Klein W. L. and Triller A. (2010) Deleterious effects of amyloid beta oligomers acting as an extracellular scaffold for mGluR5. Neuron 66, 739–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riban V., Fitzsimons H. L. and During M. J. (2009) Gene therapy in epilepsy. Epilepsia 50, 24–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricobaraza A., Cuadrado‐Tejedor M., Perez‐Mediavilla A., Frechilla D., Del Rio J. and Garcia‐Osta A. (2009) Phenylbutyrate ameliorates cognitive deficit and reduces tau pathology in an Alzheimer's disease mouse model. Neuropsychopharmacology, 34, 1721–1732. [DOI] [PubMed] [Google Scholar]
- Ricobaraza A., Cuadrado‐Tejedor M., Marco S., Pérez‐Otaño I. and García‐Osta A. (2010) Phenylbutyrate rescues dendritic spine loss associated with memory deficits in a mouse model of Alzheimer disease. Hippocampus 22, 1040–1050. [DOI] [PubMed] [Google Scholar]
- Roberson E. D., Scearce‐Levie K., Palop J. J., Yan F., Cheng I. H., Wu T., Gerstein H., Yu G. Q. and Mucke L. (2007) Reducing endogenous tau ameliorates amyloid beta‐induced deficits in an Alzheimer's disease mouse model. Science 316, 750–754. [DOI] [PubMed] [Google Scholar]
- Roberson E. D., Halabisky B., Yoo J. W. et al (2011) Amyloid‐beta/Fyn‐induced synaptic, network, and cognitive impairments depend on tau levels in multiple mouse models of Alzheimer's disease. J. Neurosci. 31, 700–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockenstein E., Nuber S., Overk C. R. et al (2014) Accumulation of oligomer‐prone a‐synuclein exacerbates synaptic and neuronal degeneration in vivo. Brain, 137, 1496–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg T., Gal‐Ben‐Ari S., Dieterich D. C., Kreutz M. R., Ziv N. E., Gundelfinger E. D. and Rosenblum K. (2014) The roles of protein expression in synaptic plasticity and memory consolidation. Fron. Mol. Neurosci. 7, 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouach N., Avignone E., Meme W., Koulakoff A., Venance L., Blomstrand F. and Giaume C. (2002) Gap junctions and connexin expression in the normal and pathological central nervous system. Biol. Cell 94, 457–475. [DOI] [PubMed] [Google Scholar]
- Ryan T. J. and Grant S. G. N. (2009) The origin and evolution of synapses. Nat. Rev. Neurosci. 10, 701–712. [DOI] [PubMed] [Google Scholar]
- Ryan S. G., Sherman S. L., Terry J. C., Sparkes R. S., Torres M. C. and Mackey R. W. (1992) Startle disease, or hyperekplexia: response to clonazepam and assignment of the gene (STHE) to chromosome 5q by linkage analysis. Ann. Neurol. 31, 663–668. [DOI] [PubMed] [Google Scholar]
- Sale A., Berardi N. and Maffei L. (2009) Enrich the environment to empower the brain. Trends Neurosci. 32, 233–239. [DOI] [PubMed] [Google Scholar]
- Salloway S., Sperling R., Fox N. C. et al (2014) Two phase 3 trials of bapineuzumab in mild‐to‐moderate Alzheimer's disease. N. Engl. J. Med. 370, 322–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saul B., Kuner T., Sobetzko D., Brune W., Hanefeld F., Meinck H. M. and Becker C. M. (1999) Novel GLRA1 missense mutation (P250T) in dominant hyperekplexia defines an intracellular determinant of glycine receptor channel gating. J. Neurosci. 19, 869–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sbai O., Ferhat L., Bernard A. et al (2008) Vesicular trafficking and secretion of matrix metalloproteinases‐2, ‐9 and tissue inhibitor of metalloproteinases‐1 in neuronal cells. Mol. Cell Neurosci. 39, 549–568. [DOI] [PubMed] [Google Scholar]
- Schaefer N., Kluck C. J., Price K. L. et al (2015) Disturbed neuronal ER‐golgi sorting of unassembled glycine receptors suggests altered subcellular processing is a cause of human hyperekplexia. J. Neurosci. 35, 422–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schell M. J., Molliver M. E. and Snyder S. H. (1995) D‐serine, an endogenous synaptic modulator: localization to astrocytes and glutamate‐stimulated release. Proc. Natl Acad. Sci. USA 92, 3948–3952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmeisser M. J., Ey E., Wegener S. et al (2012) Autistic‐like behaviours and hyperactivity in mice lacking ProSAP1/Shank2. Nature 486, 256–260. [DOI] [PubMed] [Google Scholar]
- Selkoe D. J. (2002) Alzheimer's disease is a synaptic failure. Science 298, 789–791. [DOI] [PubMed] [Google Scholar]
- Selkoe D. J. (2011) Resolving controversies on the path to Alzheimer's therapeutics. Nat. Med. 17, 1060–1065. [DOI] [PubMed] [Google Scholar]
- Selkoe D., Mandelkow E. and Holtzman D. (2012) Deciphering Alzheimer disease. Cold Spring Harb. Perspect Med. 2, a011460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaked I., Meerson A., Wolf Y., Avni R., Greenberg D., Gilboa‐Geffen A. and Soreq H. (2009) MicroRNA‐132 potentiates cholinergic anti‐inflammatory signaling by targeting acetylcholinesterase. Immunity 31, 965–973. [DOI] [PubMed] [Google Scholar]
- Shankar G. M., Li S., Mehta T. H. et al (2008) Amyloid‐beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat. Med. 14, 837–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen X., Chen J., Li J., Kofler J. and Herrup K. (2016) Neurons in vulnerable regions of the Alzheimer's disease brain display reduced ATM signaling. eNeuro 3, pii: ENEURO.0124‐15.2016. doi: 10.1523/ENEURO.0124‐15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng M. and Kim E. (2011) The postsynaptic organization of synapses. Cold Spring Harb. Perspect. Biol. 3, pii: a005678. doi: 10.1101/cshperspect.a005678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherrington R., Rogaev E. I., Liang Y. et al (1995) Cloning of a gene bearing missense mutations in early‐onset familial Alzheimer's disease. Nature 375, 754–760. [DOI] [PubMed] [Google Scholar]
- Shiang R., Ryan S. G., Zhu Y. Z., Hahn A. F., O'Connell P. and Wasmuth J. J. (1993) Mutations in the alpha 1 subunit of the inhibitory glycine receptor cause the dominant neurologic disorder, hyperekplexia. Nat. Genet. 5, 351–358. [DOI] [PubMed] [Google Scholar]
- Shin N., Jeong H., Kwon J. et al (2008) LRRK2 regulates synaptic vesicle endocytosis. Exp. Cell Res. 314, 2055–2065. [DOI] [PubMed] [Google Scholar]
- Shipman S. L., Schnell E., Hirai T., Chen B. S., Roche K. W. and Nicoll R. A. (2011) Functional dependence of neuroligin on a new non‐PDZ intracellular domain. Nat. Neurosci. 14, 718–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siarey R. J., Carlson E. J., Epstein C. J., Balbo A., Rapoport S. I. and Galdzicki Z. (1999) Increased synaptic depression in the Ts65Dn mouse, a model for mental retardation in Down syndrome. Neuropharmacology 38, 1917–1920. [DOI] [PubMed] [Google Scholar]
- Siarey R. J., Kline‐Burgess A., Cho M., Balbo A., Best T. K., Harashima C., Klann E. and Galdzicki Z. (2006) Altered signaling pathways underlying abnormal hippocampal synaptic plasticity in the Ts65Dn mouse model of Down syndrome. J. Neurochem. 98, 1266–1277. [DOI] [PubMed] [Google Scholar]
- Siddiqui A., Lacroix T., Stasko M. R., Scott‐McKean J. J., Costa A. C. and Gardiner K. J. (2008) Molecular responses of the Ts65Dn and Ts1Cje mouse models of Down syndrome to MK‐801. Genes Brain Behav. 7, 810–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh S. P., He X., McNamara J. O. and Danzer S. C. (2013) Morphological changes among hippocampal dentate granule cells exposed to early kindling‐epileptogenesis. Hippocampus 23, 1309–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith P. Y., Hernandez‐Rapp J., Jolivette F. et al (2015) miR‐132/212 deficiency impairs tau metabolism and promotes pathological aggregation in vivo. Hum. Mol. Genet. 24, 6721–6735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soltesz I., Alger B. E., Kano M., Lee S. H., Lovinger D. M., Ohno‐Shosaku T. and Watanabe M. (2015) Weeding out bad waves: towards selective cannabinoid circuit control in epilepsy. Nat. Rev. Neurosci. 16, 264–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sontag J. M. and Sontag E. (2014) Protein phosphatase 2A dysfunction in Alzheimer's disease. Fron. Mol. Neurosci. 7, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotthibundhu A., Sykes A. M., Fox B., Underwood C. K., Thangnipon W. and Coulson E. J. (2008) Beta‐amyloid(1‐42) induces neuronal death through the p75 neurotrophin receptor. J. Neurosci. 28, 3941–3946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soukupova M., Binaschi A., Falcicchia C., Zucchini S., Roncon P., Palma E., Magri E., Grandi E. and Simonato M. (2014) Impairment of GABA release in the hippocampus at the time of the first spontaneous seizure in the pilocarpine model of temporal lobe epilepsy. Exp. Neurol. 257, 39–49. [DOI] [PubMed] [Google Scholar]
- Soukupova M., Binaschi A., Falcicchia C., Palma E., Roncon P., Zucchini S. and Simonato M. (2015) Increased extracellular levels of glutamate in the hippocampus of chronically epileptic rats. Neuroscience 301, 246–253. [DOI] [PubMed] [Google Scholar]
- Spillantini M. G., Schmidt M. L., Lee V. M., Trojanowski J. Q., Jakes R. and Goedert M. (1997) Alpha‐synuclein in Lewy bodies. Nature 388, 839–840. [DOI] [PubMed] [Google Scholar]
- Spires‐Jones T. L. and Hyman B. T. (2014) The intersection of amyloid beta and tau at synapses in Alzheimer's disease. Neuron 82, 756–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staley K. (2015) Molecular mechanisms of epilepsy. Nat. Neurosci. 18, 367–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strack S. and Colbran R. J. (1998) Autophosphorylation‐dependent targeting of calcium/calmodulin‐dependent protein kinase II by the NR2B subunit of the N‐methyl‐ D‐aspartate receptor. J. Biol. Chem. 273, 20689–20692. [DOI] [PubMed] [Google Scholar]
- Suhren O., Bruyn G. and Tuynman A. (1966) Hyperexplexia, a heriditary startle syndrome. J. Neurol. Sci. 3, 577–605. [Google Scholar]
- Sweet E. S., Saunier‐Rebori B., Yue Z. and Blitzer R. D. (2015) The Parkinson's disease‐associated mutation LRRK2‐G2019S impairs synaptic plasticity in mouse hippocampus. J. Neurosci. 35, 11190–11195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sykova E. and Nicholson C. (2008) Diffusion in brain extracellular space. Physiol. Rev. 88, 1277–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takashima S., Becker L. E., Armstrong D. L. and Chan F. (1981) Abnormal neuronal development in the visual cortex of the human fetus and infant with down's syndrome. A quantitative and qualitative Golgi study. Brain Res. 225, 1–21. [DOI] [PubMed] [Google Scholar]
- Talantova M., Sanz‐Blasco S., Zhang X. et al (2013) Aβ induces astrocytic glutamate release, extrasynaptic NMDA receptor activation, and synaptic loss. Proc. Natl Acad. Sci. USA 110, E2518–E2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatavarty V., Sun Q. and Turrigiano G. G. (2013) How to scale down postsynaptic strength. J. Neurosci. 33, 13179–13189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terry R. D., Masliah E., Salmon D. P., Butters N., DeTeresa R., Hill R., Hansen L. A. and Katzman R. (1991) Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann. Neurol. 30, 572–580. [DOI] [PubMed] [Google Scholar]
- Thomas R. H., Chung S. K., Wood S. E., Cushion T. D., Drew C. J., Hammond C. L., Vanbellinghen J. F., Mullins J. G. and Rees M. I. (2013) Genotype‐phenotype correlations in hyperekplexia: apnoeas, learning difficulties and speech delay. Brain 136, 3085–3095. [DOI] [PubMed] [Google Scholar]
- Tijssen M. A., Vergouwe M. N., van Dijk J. G., Rees M., Frants R. R. and Brown P. (2002) Major and minor form of hereditary hyperekplexia. Mov. Disord. 17, 826–830. [DOI] [PubMed] [Google Scholar]
- Tokuda T., Qureshi M. M., Ardah M. T. et al (2010) Detection of elevated levels of alpha ‐synuclein oligomers in CSF from patients with Parkinson disease. Neurology 75, 1766–1772. [DOI] [PubMed] [Google Scholar]
- Traynelis S. F., Wollmuth L. P., McBain C. J. et al (2010) Glutamate receptor ion channels: structure, regulation, and function. Pharmacol. Rev. 62, 405–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai C. H., Chang F. C., Su Y. C., Tsai F. J., Lu M. K., Lee C. C., Kuo C. C., Yang Y. W. and Lu C. S. (2004) Two novel mutations of the glycine receptor gene in a Taiwanese hyperekplexia family. Neurology 63, 893–896. [DOI] [PubMed] [Google Scholar]
- Tu S., Okamoto S., Lipton S. A. and Xu H. (2014) Oligomeric Abeta‐induced synaptic dysfunction in Alzheimer's disease. Mol. Neurodegener. 9, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano G. (2012) Homeostatic synaptic plasticity: local and global mechanisms for stabilizing neuronal function. Cold Spring Harb. Perspect. Biol. 4, a005736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Um J. W., Kaufman A. C., Kostylev M. et al (2013) Metabotropic glutamate receptor 5 is a coreceptor for Alzheimer abeta oligomer bound to cellular prion protein. Neuron 79, 887–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbano M., Okwara L., Manser P., Hartmann K., Herndon A. and Deutsch S. I. (2014) A trial of D‐cycloserine to treat stereotypies in older adolescents and young adults with autism spectrum disorder. Clin. Neuropharmacol. 37, 69–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vafadari B., Salamian A. and Kaczmarek L. (2015) MMP‐9 in Translation: From Molecule to Brain Physiology, Pathology and Therapy. J. Neurochem. doi: 10.1111/jnc.13415. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Valenzuela J. C., Heise C., Franken G., Singh J., Schweitzer B., Seidenbecher C. I. and Frischknecht R. (2014) Hyaluronan‐based extracellular matrix under conditions of homeostatic plasticity. Philos. Trans. R. Soc. Lond. B Biol. Sci. 369, 20130606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varoqueaux F., Aramuni G., Rawson R. L., Mohrmann R., Missler M., Gottmann K., Zhang W., Sudhof T. C. and Brose N. (2006) Neuroligins determine synapse maturation and function. Neuron 51, 741–754. [DOI] [PubMed] [Google Scholar]
- Varvel N. H., Bhaskar K., Patil A. R., Pimplikar S. W., Herrup K. and Lamb B. T. (2008) Abeta oligomers induce neuronal cell cycle events in Alzheimer's disease. J. Neurosci. 28, 10786–10793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villmann C., Oertel J., Ma‐Hogemeier Z. L., Hollmann M., Sprengel R., Becker K., Breitinger H. G. and Becker C. M. (2009) Functional complementation of Glra1(spd‐ot), a glycine receptor subunit mutant, by independently expressed C‐terminal domains. J. Neurosci. 29, 2440–2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vivo L., Landi S., Panniello M. et al (2013) Extracellular matrix inhibits structural and functional plasticity of dendritic spines in the adult visual cortex. Nat. Commun. 4, 1484. [DOI] [PubMed] [Google Scholar]
- Walsh D. M., Klyubin I., Fadeeva J. V., Cullen W. K., Anwyl R., Wolfe M. S., Rowan M. J. and Selkoe D. J. (2002) Naturally secreted oligomers of the amyloid beta protein potently inhibit long‐term potentiation in vivo. Nature 416, 535–539. [DOI] [PubMed] [Google Scholar]
- Wang H. Y., Lee D. H., D'Andrea M. R., Peterson P. A., Shank R. P. and Reitz A. B. (2000) beta‐Amyloid(1‐42) binds to alpha7 nicotinic acetylcholine receptor with high affinity. Implications for Alzheimer's disease pathology. J. Biol. Chem. 275, 5626–5632. [DOI] [PubMed] [Google Scholar]
- Werner C., Pauli M., Doose S. et al (2016) Human autoantibodies to amphiphysin induce defective presynaptic vesicle dynamics and composition. Brain 139, 365–379. [DOI] [PubMed] [Google Scholar]
- Wetherington J., Serrano G. and Dingledine R. (2008) Astrocytes in the epileptic brain. Neuron 58, 168–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winner B., Jappelli R., Maji S. K. et al (2011) In vivo demonstration that [alpha]‐synuclein oligomers are toxic. Proc. Natl Acad. Sci. USA, 108, 4194–4199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittner L., Eross L., Szabo Z., Toth S., Czirjak S., Halasz P., Freund T. F. and Magloczky Z. S. (2002) Synaptic reorganization of calbindin‐positive neurons in the human hippocampal CA1 region in temporal lobe epilepsy. Neuroscience 115, 961–978. [DOI] [PubMed] [Google Scholar]
- Won H., Lee H. R., Gee H. Y. et al (2012) Autistic‐like social behaviour in Shank2‐mutant mice improved by restoring NMDA receptor function. Nature 486, 261–265. [DOI] [PubMed] [Google Scholar]
- Wu G. M. and Hou X. Y. (2010) Oligomerized Abeta25‐35 induces increased tyrosine phosphorylation of NMDA receptor subunit 2A in rat hippocampal CA1 subfield. Brain Res. 1343, 186–193. [DOI] [PubMed] [Google Scholar]
- Xia W., Yang T., Shankar G. M., Smith I. M., Shen Y., Walsh D. and Selkoe D. J. (2009) A specific enzyme‐linked immunosorbent assay for measuring β‐amyloid protein oligomers in human plasma and brain tissue of patients with Alzheimer disease. Arch. Neurol. 66, 190–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong Y., Coombes C. E., Kilaru A., Li X., Gitler A. D., Bowers W. J., Dawson V. L., Dawson T. M. and Moore D. J. (2010) GTPase activity plays a key role in the pathobiology of LRRK2. PLoS Genet. 6, e1000902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaar M., Zhai S., Pilch P. F., Doyle S. M., Eisenhauer P. B., Fine R. E. and Gilchrest B. A. (1997) Binding of beta‐amyloid to the p75 neurotrophin receptor induces apoptosis. A possible mechanism for Alzheimer's disease. J. Clin. Investig. 100, 2333–2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamakawa K., Huot Y. K., Haendelt M. A., Hubert R., Chen X. N., Lyons G. E. and Korenberg J. R. (1998) DSCAM: a novel member of the immunoglobulin superfamily maps in a Down syndrome region and is involved in the development of the nervous system. Hum. Mol. Genet. 7, 227–237. [DOI] [PubMed] [Google Scholar]
- Zelano J., Lundberg R. G., Baars L., Hedegard E. and Kumlien E. (2015) Clinical course of poststroke epilepsy: a retrospective nested case‐control study. Brain Behav 5, e00366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H., Sun S., Herreman A., De Strooper B. and Bezprozvanny I. (2010) Role of presenilins in neuronal calcium homeostasis. J. Neurosci. 30, 8566–8580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou C., Sun H., Klein P. M. and Jensen F. E. (2015) Neonatal seizures alter NMDA glutamate receptor GluN2A and 3A subunit expression and function in hippocampal CA1 neurons. Front. Cell Neurosci. 9, 362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y., Nwabuisi‐Heath E., Dumanis S. B., Tai L. M., Yu C., Rebeck G. W. and LaDu M. J. (2012) APOE genotype alters glial activation and loss of synaptic markers in mice. Glia 60, 559–569. [DOI] [PMC free article] [PubMed] [Google Scholar]