

Abstract
New Findings
-
What is the central question of this study?
What is the functional relevance of OPN isoform expression in muscle pathology?
-
What is the main finding and its importance?
The full‐length human OPN‐a isoform is the most pro‐inflammatory isoform in the muscle microenvironment, acting on macrophages and myoblasts in an RGD‐integrin‐dependent manner. OPN‐a upregulates expression of tenascin‐C (TNC), a known Toll‐like receptor 4 (TLR4) agonist. Blocking TLR4 signalling inhibits the pro‐inflammatory effects of OPN‐a, suggesting that a potential mechanism of OPN action is by promoting TNC–TLR4 signalling.
Although osteopontin (OPN) is an important mediator of muscle remodelling in health and disease, functional differences in human spliced OPN variants in the muscle microenvironment have not been characterized. We thus sought to define the pro‐inflammatory activities of human OPN isoforms (OPN‐a, OPN‐b and OPN‐c) on cells present in regenerating muscle. OPN transcripts were quantified in normal and dystrophic human and dog muscle. Human macrophages and myoblasts were stimulated with recombinant human OPN protein isoforms, and cytokine mRNA and protein induction was assayed. OPN isoforms were greatly increased in dystrophic human (OPN‐a > OPN‐b > OPN‐c) and dog muscle (OPN‐a = OPN‐c). In healthy human muscle, mechanical loading also upregulated OPN‐a expression (eightfold; P < 0.01), but did not significantly upregulate OPN‐c expression (twofold; P > 0.05). In vitro, OPN‐a displayed the most pronounced pro‐inflammatory activity among isoforms, acting on both macrophages and myoblasts. In vitro and in vivo data revealed that OPN‐a upregulated tenascin‐C (TNC), a known Toll‐like receptor 4 (TLR4) agonist. Inhibition of TLR4 signalling attenuated OPN‐mediated macrophage cytokine production. In summary, OPN‐a is the most abundant and functionally active human spliced isoform in the skeletal muscle microenvironment. Here, OPN‐a promotes pro‐inflammatory signalling in both macrophages and myoblasts, possibly through induction of TNC–TLR4 signalling. Together, our findings suggest that specific targeting of OPN‐a and/or TNC signalling in the damaged muscle microenvironment may be of therapeutic relevance.
Keywords: Duchenne muscular dystrophy, muscle inflammation, OPN‐a, osteopontin, skeletal muscle, TNC
Introduction
Osteopontin (OPN), or secreted phosphoprotein 1 (SPP1), is a member of the small integrin‐binding ligand N‐linked glycoprotein (SIBLING) protein family. SIBLING family proteins are structurally characterized by their small flexible structure and the presence of an Arg‐Gly‐Asp (RGD) tripeptide, an authentic integrin‐binding sequence (Fisher et al. 2001). OPN is a secreted protein with diverse physiological functions influenced by multiple pre‐ and post‐translational modifications. Increasing importance has been attributed to OPN as an immunoregulatory protein in inflammation and tissue remodelling (acute and chronic) and cancer (Shin, 2012; Bandopadhyay et al. 2014). However, the amino acid sequence is relatively poorly conserved between mice and humans (∼63% homology), with the greatest degree of conservation occurring around receptor binding sites (e.g. RGD domain) and proteolytic cleavage sites. The human but not mouse gene shows three alternatively spliced isoforms (Gimba & Tilli, 2013). Full‐length OPN protein is designated OPN‐a, with a molecular weight of ∼54 kDa prior to extensive post‐translational modifications. OPN‐b lacks exon 5 (∼50 kDa), and OPN‐c lacks exon 4 (∼47 kDa) of the seven exons.
All human OPN isoforms contain the RGD and SVVYGLR integrin‐binding peptide sequences (Yokosaki et al. 1999), a CD44 binding domain, and thrombin and matrix metalloproteinase cleavage sites. Unlike the RGD integrin‐binding peptide sequence, the SVVYGLR integrin‐binding site is exposed only upon thrombin cleavage of OPN (Yokosaki et al. 1999). The three spliced isoforms differ in protein cross‐linking sites (Gln residues) that undergo covalent polymerization by transglutaminase 2, a difference that significantly alters OPN function (Higashikawa et al. 2007; Nishimichi et al. 2011). Transglutaminase 2 catalyses OPN polymerization at multiple Gln residues coded by exons 2–5 (Christensen et al. 2014). The deleted exons 5 (OPN‐b) and 4 (OPN‐c) contain one and three Gln residues, respectively, making the degree of isoform polymerization proportional to the number of transglutamination sites, with OPN‐a > OPN‐b > OPN‐c (Nishimichi et al. 2011). Transglutamination of OPN is thought to modify protein function by altering the conformational state of OPN, thereby increasing its cell‐binding and chemotactic abilities (Kaartinen et al. 1999; Nishimichi et al. 2011).
Despite the widely characterized role of OPN as a pro‐inflammatory protein, little research has investigated the function or expression patterns of OPN spliced isoforms outside of the field of cancer biology. In tumour cells, differential OPN isoform expression patterns are related to cancer cell behaviours and pathological outcomes such as proliferation, migration and metastasis (Gimba & Tilli, 2013). In breast cancer cells, OPN‐a‐ but not OPN‐c‐transfected cells display signal transducer and activator of transcription 1 & 3 (STAT1/3) DNA binding (Shi et al. 2014). STAT1/3 activation occurs in response to cytokines, such as interleukin (IL)‐6, suggesting that OPN isoforms may differentially modulate inflammatory cytokine induction. However, the effects of OPN isoforms on inflammatory signalling in other cellular microenvironments, such as skeletal muscle, remain unclear.
The role of OPN in muscle remodelling has received increased interest as a result of three recent observations. First, ablation of OPN expression in the mouse model of Duchenne muscular dystrophy (dystrophin‐deficient mdx mice) improves disease pathology (Vetrone et al. 2009). Second, a genetic polymorphism in the human OPN gene altering gene transcription is associated with disease severity in Duchenne muscular dystrophy (DMD; Pegoraro et al. 2011; Bello et al. 2015). Finally, the same genetic polymorphism is associated with increased muscle size (Hoffman et al. 2013) and increased damage in response to eccentric contraction in young adults (Barfield et al. 2014). In normal skeletal muscle in homeostatic conditions, OPN expression is very low or undetectable. Expression of OPN is rapidly induced in acute and chronic muscle injury, with expression primarily localized to infiltrating immune cells and proliferating myogenic cells (Uaesoontrachoon et al. 2008; Zanotti et al. 2011; Paliwal et al. 2012; Pagel et al. 2014). In myogenic cells, OPN expression is upregulated with inflammatory stimuli and degeneration/remodelling (Uaesoontrachoon et al. 2008; Paliwal et al. 2012). However, functional differences between spliced isoforms in the regenerating muscle microenvironment have not been characterized.
In healthy and diseased muscle (e.g. DMD), macrophages are the predominant cellular infiltrate and express high levels of OPN (Hirata et al. 2003). Despite increasing importance being attributed to OPN in muscle remodelling, the relative contribution of spliced isoforms to muscle remodelling has not been studied in either health or disease. We thus sought to provide better definition of the role of OPN spliced isoforms in the skeletal muscle microenvironment by studying macrophage and myoblast/muscle inflammatory signalling. We observed that OPN‐a is the most abundantly expressed isoform in human muscle. In both macrophages and myoblasts, OPN‐a was the most pro‐inflammatory spliced isoform.
Methods
Ethical approval
All tissue samples were obtained by protocols approved by the Institutional Animal Care and Use Committee or Institutional Review Board, for respective animal and human studies at respective institutions. For human studies, all participants consented to study participation at the enrolling institution, and all study procedures were conducted according to standards set forth by the Declaration of Helsinki.
Human and canine skeletal muscle OPN expression
Human
All human muscle biopsies were obtained by fine‐needle biopsy of the vastus lateralis muscle and flash‐frozen for processing. Briefly, samples were collected using local anaesthetic (1% lidocaine). Duchenne muscular dystrophy, Becker's muscular dystrophy (BMD) and control human muscle biopsies were archival specimens from a tissue bank at the Children's National Medical Center. These samples were obtained from consenting subjects and/or their legal guardian, if under the age of 18 years. All subjects were male owing to the X‐linked nature of DMD. For OPN isoform detection, the human forward primer (ATTGCAGTGATTTGCTTTTGC) was designed against human exon 2 and the reverse primer (GCAACCGAAGTTTTCACTCC) against exon 6.
For loading stress studies, biopsies were obtained from healthy male (n = 11) and female (n = 11) human subjects (age 57 ± 3 years) immediately before and 24 h after loading‐induced stress. Here, subjects performed unaccustomed resistance exercise, by performing 9–10 repetitions at ∼65% one‐repetition maximum. All subjects were > 18 years old and consented to study participation at the University of Alabama, Birmingham (Stec et al. 2015). qPCR was performed by Taqman gene array on a StepOnePlus Real‐Time PCR System (Applied Biosystems, Foster City, CA, USA). Here, glyceraldehyde‐6‐phosphate dehydrogenase (GAPDH) was used as an endogenous control.
Canine
Muscle samples were taken at either biopsy or post mortem from the cranial sartorius (CS), vastus lateralis (VL) and long digital extensor (LDE) from the opposite limbs of golden retriever muscular dystrophy (GRMD) dystrophin‐deficient dogs (n = 8) and wild‐type littermates (n = 4) at 4–9 weeks (early stage disease) and 6 months (symptomatic disease), as previously described (Nghiem et al. 2013). All dogs were used and cared for according to principles outlined in the National Research Council Guide for the Care and Use of Laboratory Animals. Dogs were housed at the Kornegay laboratory at either the University of Missouri or the University of North Carolina at Chapel Hill. All animals were given ad libitum access to food and water. The GRMD dogs were identified at 1 day of age based on a dramatic elevation of serum creatine kinase. Genotype was confirmed by PCR when creatine kinase results were ambiguous. Characteristic clinical signs subsequently developed. For muscle biopsy, dogs were given a general preanaesthetic regimen comprising acepromazine maleate, butorphanol and atropine sulfate and masked and maintained under general anesthesia with sevoflurane. On average, 400 mg of tissue was sampled per dog.
For dystrophic human and dog samples, hypoxanthine phosphoribosyltransferase 1 (HPRT1) was used as an endogenous control for expression of OPN isoforms. RNA was isolated by TRIzol extraction, and cDNA synthesis was performed using an Applied Biosystems’ High Capacity cDNA Reverse Transcription Kit (Life Technologies) according to methods described previously (Nghiem et al. 2013). Briefly, for PCR analyses, cDNA was incubated at 90°C for 1 min; a multiplex reaction was then carried out with OPN and HPRT1 probes according to the manufacturer's instructions using AccuPrime SuperMix II (Invitrogen). Reactions were performed in triplicate for samples from both normal (n = 4; all male) and GRMD (n = 8; n = 4 male and n = 4 female) cDNA (CS, LDE and VL) at 4–9 weeks and 6 months. The PCR conditions for human samples were 30 cycles at 94°C for 5 min, 94°C for 30 s, 60°C for 1 min, 72°C for 30 s, and 72°C for 7 min. OPN mRNA isoform bands were detected with GeneSnap software and quantified and normalized to HPRT1 in GeneTools software (Syngene, Frederick, MD).
Human primary monocyte‐derived macrophages
Consenting healthy adult men not taking anti‐inflammatory or immunoregulating medications underwent venipuncture for the isolation of primary human monocytes by negative selection. All subjects consented to participate in this study conducted at the Children's National Medical Center. To obtain monocytes, venous blood was collected in sodium heparin vacutainers and immediately processed for isolation of peripheral blood‐derived mononuclear cells via density centrifugation using Ficoll Paque Plus (GE Lifesciences, Marlborough, MA, USA). Monocytes were then isolated from the peripheral blood‐derived mononuclear cell layer via negative magnetic selection for CD14+ cells, without CD16+ cell depletion (StemCell Technologies, Vancouver, BC, Canada). Monocyte cell identity was confirmed via staining for human CD14 and CD16 (BD Biosciences). Negative selection for monocytes resulted in a cell population of ∼94% purity as defined as follows: CD14hi CD16lo, CD14lo CD16hi and/or CD14hi CD16hi as determined by fluorescence‐activated cell sorting (FACS). Monocytes were plated at a density of ∼100,000 cells per well in 96‐well plates for culture in DMEM–F12 media containing 1% penicillin–streptomycin and 10% fetal bovine serum, and cultured with 50 ng ml−1 human recombinant macrophage colony‐stimulating factor (M‐CSF) (R&D Systems, Minneapolis, MN, USA). Cells were cultured for 5 days at 37 °C in air supplemented with 5% CO2, in order to differentiate cells into primary human macrophages. Human monocyte‐derived macrophages were phenotypically characterized by an increase in size and the formation of cytoplasmic projections.
Recombinant protein derivation
Human OPN isoforms (OPN‐a, ‐b and ‐c) were expressed as glutathione S‐transferase (GST) fusion proteins in Escherichia coli with pGEX6P plasmid, affinity purified and cleaved from GST according to previously described methods (Nishimichi et al. 2011). Recombinant human OPN‐a proteins containing: (i) RGD (arginine‐glycine‐aspartic) acid residues; and (ii) OPN‐a with the RGD domain mutated to KAE [lysine‐alanine‐glutamic; ΔRGD→KAE (OPN‐a‐KAE)] were a kind gift from Dr Larry Fisher at the National Institutes of Health. Briefly, recombinant proteins were generated by subcloning OPN‐a‐RGD and OPN‐a‐KAE cDNA into pAd/CMV/V5‐DEST adenovirus vectors in HEK293 cells. Viral transfection of human stromal fibroblasts was then used to generate recombinant proteins, which were purified via column chromatography (Fedarko et al. 2000). Briefly, the proteins were purified using Sartobind Q strong basic anion exchanger (Sartorius, Göttingen, Germany). Here, the Sartobind Q 15 membrane was equilibrated with 50 ml PBS prior to sample filtration. The membrane was then washed with 8 m urea, 0.2% β‐mercaptoethanol–PBS and 100 ml PBS. A linear salt gradient to 2.0 m NaCl–PBS was used to purify proteins to 95% purity as measured by Stains‐All (Sigma, St Louis, MO, USA).
Human macrophage stimulation
Human recombinant OPN proteins showed evidence of endotoxin contamination (Pierce LAL assay; Thermo Scientific, Waltham, MA, USA). To inhibit endotoxin effects on assays, we pretreated all samples with polymixin‐B (PMB; InvivoGen, San Diego, CA, USA). This was shown to inhibit the effects of E. coli‐derived lipopolysaccharide (LPS; tested range 10–100 ng ml−1) on human macrophages. Macrophages were pretreated with 100 μg ml filtered PMB sulfate in PBS 30 min prior to stimulation with OPN (InvivoGen). Polymixin‐B is a positively charged cyclic polypeptide that binds to and inhibits the actions of the negatively charged and bioactive lipid A tail of endotoxin (Cavaillon & Haeffner‐Cavaillon, 1986). The pretreatment of cells with PMB was optimized through testing of IL‐6 and tumour necrosis factor‐α secretion from human macrophages assayed by enzyme‐linked immunosorbent assay ELISA and FACS bead array. Endotoxin‐depleted recombinant human and murine OPN‐a was also obtained for testing and optimization of inflammatory responses (R&D Systems).
For mRNA assays, macrophages were treated with OPN proteins for 4 h. For protein assays, macrophages were treated with OPN proteins for 24 h. For protein assay by ELISA, R&D Duo Set kits were used (R&D Systems).
Nanostring expression profiling
After culturing for 5 days with 50 ng ml−1 human M‐CSF, fresh human monocyte medium was added, and primary human monocyte‐derived macrophages were stimulated for 4 h with human recombinant OPN proteins for expression profiling by Nanostring according to methods described previously (Dillingham et al. 2015). Briefly, cells were harvested in cell stripper (Cellgro, Manassas, VA, USA), washed and resuspended in RLT Buffer (Qiagen, Hilden, Germany). Messenger RNA molecule counting was performed on 10,000 macrophages per treatment condition using a custom Nanostring probe set and a NanoString nCounter analytics system (Nanostring Technologies, Seattle, WA, USA). Eight negative control probes (targeting RNA sequences not expressed in humans) were used to adjust for background noise. Six positive probes (targeting housekeeping gene transcripts) were included in each probe set for initial content normalization, using geometric means of control transcript counts as a normalization factor. Additional normalization was performed against reference genes that were abundantly expressed (>2 SD over negative control probes) and did not display variability between treatment conditions and donors (P > 0.05). Based on these normalization procedures, GIGYF2, RMBX2, STK35 and TOX4 were used as normalization controls for macrophage studies, and HDAC3, RBMX2, STK35, TOX4 and USP4 for muscle studies.
Human myogenic cell culture
Immortalized human myoblasts were a gift from Dr Vincent Mouly at the Centre for Research in Myology in Paris, France. These primary human myoblasts were derived from a healthy 25‐year‐old female subject and immortalized according to previously described methods (Di Donna et al. 2003). Muscle was obtained via fine‐needle biopsy of the vastus lateralis for cell isolation. Once immortalized, cells were grown in skeletal muscle cell media (Promocell, Heidelberg, Germany) supplemented with 20% fetal bovine serum at 37°C in air supplemented with 5% CO2. Cells were plated overnight prior to stimulation with OPN.
Phagocytosis assay
Freshly isolated primary human monocytes were cultured in human macrophage media without added M‐CSF and left to adhere overnight at 37°C in air supplemented with 5% CO2. Cells were then washed, and fresh medium was added 1 h before 30 min pretreatment with PMB. Cells were then stimulated for 2 h with recombinant OPN proteins. Following stimulation with OPN, fluorescently conjugated E. coli particles (K‐12 strain) were suspended in Hanks’ balanced salt solution and added to the solution, according to the manufacturer's instructions (Vybrant Phagocytosis Assay Kit; Molecular Probes, Eugene, OR, USA). After an additional 2 h incubation period, cells were washed to remove non‐phagocytosed E. coli particles, and cellular fluorescence of non‐viable cells was quenched with Trypan Blue for 1 min. The Trypan Blue solution was then aspirated. To determine cellular uptake of fluorescent E. coli particles, fluorescence was measured first on a fluorescence plate reader using wavelengths of 485 nm for excitation and 525 nm for emission, and a repeat experiment was performed by FACS using the fluorescein isothiocyanate (FITC) channel (FL1). All experiments were run in technical replicates of n = 5, according to the manufacturer's instructions. For FACS reading, cells were harvested with cell stripper, centrifuged, and resuspended in FACS buffer. All fluorescence measures were normalized for background fluorescence (E. coli particles only) prior to normalization to vehicle control cells (PMB treated).
Intramuscular OPN transfection and expression profiling
Use of mice for research was approved and conducted according to the Children's National Medical Center Institutional Animal Care and Use Committee guidelines. Mice were given ad libitum access to food and water. Animals were euthanized by CO2 overdose. For transfection experiments, murine OPN‐a plasmid (GeneCopoeia; 50 μg total in 100 μl PBS) was injected into the right tibialis anterior (TA) muscle of 3‐month‐old male BLA/J mice (n = 3). The TA muscle was chosen because it is accessible by injection without surgery and is relatively homogeneous in fibre‐type composition (predominantly fast twitch). Given that surgery is well known to induce inflammation, we wished to avoid this added confounding variable, because inflammation was a key end point. The contralateral left TA muscle was injected with 50 μg of scrambled (empty) plasmid (GeneCopoeia, Rockville, MD, USA) and used as a vehicle and injection control. Tibialis anterior muscles were harvested 7 days postinjection for microarray expression profiling. The bellies of the TA muscles were sectioned, and total RNA was extracted from ∼50 muscle sections using TRI reagent (Sigma‐Aldrich, St Louis, MO, USA). Illumina beadchip microarray was performed on the extracted mRNA according to previously described methods (Uaesoontrachoon et al. 2013). Data were analysed through use of Ingenuity Pathway Analysis software. A P value of ≤0.05 and fold change of ≥1.5 between vector‐ and OPN‐a‐injected groups was used as a cut point for pathway analysis.
Statistical analyses
Statistical significance between treatment conditions was assessed via Student's t tests, unless otherwise indicated. Statistical significance was set at P < 0.05. For Nanostring expression profiling, ANOVA was performed, and differences between groups were assessed using Bonferonni correction for multiple testing (cytokines and doses) through use of Partek Genomics Suite software, version 6.6 (St. Louis, MO, USA.). Additionally, Student's paired t tests were used to examine single‐dose responsiveness between donors, where paired comparisons between treatment per donor was performed. For transient transfection experiments, statistical powering for microarray was based on initial measures of inflammation (inflammatory foci per tissue cross‐section). For these analyses, a sample size of n = 3 was considered sufficient for statistical powering (Cohen's d = 2.9).
Results
Prominent expression of OPN isoforms in dystrophic muscle
Spliced OPN isoform (Fig. 1 A) expression patterns in dystrophic muscle were determined by RT‐PCR in dystrophin‐deficient human and dog muscle. Consistent with previous reports, OPN was scarcely expressed in dystrophin‐sufficient muscle but highly expressed in dystrophic muscle, in both humans and dogs (Porter et al. 2004; Zanotti et al. 2011; Kornegay et al. 2014; Fig. 1 B and C). In patients with DMD, all three OPN RNA isoforms were observed; OPN‐a (full length), OPN‐b (minus exon 5) and OPN‐c (minus exon 4), with relative levels of OPN‐a > OPN‐b > OPN‐c (Fig. 1 B). In GRMD dog muscle, OPN‐a and OPN‐c were expressed at similar levels, and OPN‐b was not recognized (Fig. 1 C).
Figure 1. Alternatively spliced osteopontin isoforms are expressed in dystrophic muscle .
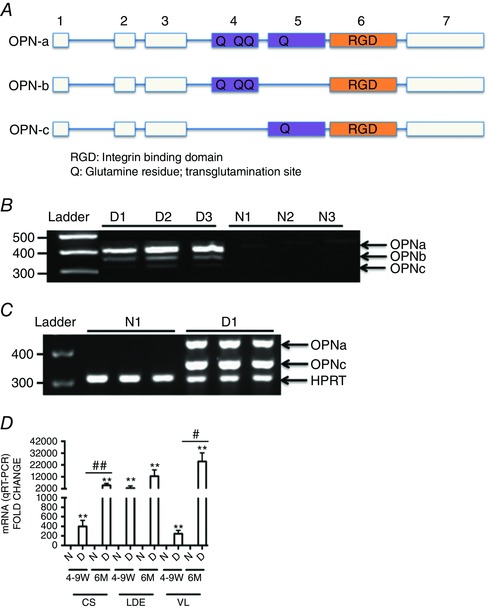
A, OPN spliced isoform transcript structure. The blocks in this figure represent exons and the lines represent introns. Here, OPN‐a is full length (top), OPN‐b is missing exon 5 (middle), and OPN‐c is missing exon 4 (bottom). B, osteopontin isoform transcript expression in muscle biopsies of three dystrophin‐deficient Duchenne muscular dystrophy (DMD) patients [D1–D3], and three dystrophin‐sufficient control subjects [N1–N3] as determined by RT‐PCR. Minimal expression is detected in dystrophin‐sufficient muscle, whereas all three isoforms are expressed in DMD muscle (full‐length OPN‐a, OPN‐b lacking exon 5, and OPN‐c lacking exon 4). C, dog dystrophin‐deficient muscle likewise shows no detectable expression in dystrophin‐sufficient littermates (N1), but high expression of OPN‐a and OPN‐c in dystrophin‐deficient golden retriever muscular dystrophy (GRMD) vastus lateralis muscle (D1). D, RT‐PCR analysis of muscle biopsies from three muscles and two age points (4‐9 W, 4–9 weeks old; and 6 M, 6 months old) in dystrophin‐sufficient (N) and ‐deficient (D) dogs shows elevated OPN mRNA in all dystrophic muscles at all age points. An increase in OPN‐a mRNA levels with age is seen in the three muscle groups tested (CS, cranial sartorius; LDE, long digital extensor; and VL, vastus lateralis). Here, OPN‐a mRNA levels were correlated with both age and severity of muscle involvement (CS, mildly affected; and LDE and VL, severely affected). Triplicates are shown per sample. Significant difference between dystrophic and non‐dystrophic littermates: ** P < 0.01. Significant differences with age per muscle group: # P ≤ 0.05 and ## P ≤ 0.01 (GRMD, n = 8; and dystrophin‐sufficient littermates, n = 4).
To determine whether the relative levels of these isoforms changed as a function of age or muscle histological severity, we studied the GRMD dog model of DMD, a spontaneously occurring large‐animal model that shares many clinical and histological features of the human disease (Kornegay et al. 2012). Expression of OPN in the severely affected VL and LDE and the mildly affected CS of GRMD dogs (Nghiem et al. 2013) was assessed by qRT‐PCR. This differential involvement of muscles in GRMD is shared with human DMD patients (Li et al. 2015). OPN transcripts increased as a function of both age and muscle histological severity (Fig. 1 D). Here, OPN‐c expression closely paralleled OPN‐a expression (not shown).
Human OPN‐a and OPN‐c isoform ratios change with DMD severity and mechanical loading
To confirm the relevance of our PCR findings, we performed Nanostring expression profiling on VL biopsies from a larger cohort of human DMD (dystrophin deficient) and BMD patients (partly dystrophin deficient). Total OPN (all isoforms) and OPN‐c levels were quantified because the sequences of OPN‐a and OPN‐b displayed too high an overlap for accurate sequence coverage by Nanostring expressing profiling. Histology (Haematoxylin and Eosin) was performed on muscle biopsy sections prior to analyses. Histology was used to group samples according to histological severity as determined by the number of degenerating and regenerating fibres and inflammatory foci per image field. No significant differences in total OPN (sequence specific to OPN exon 7) or OPN‐c (sequence specific to exons 3–5, with exon 4 deleted) were observed between patients based on diagnosis (DMD versus BMD) or histological severity (mild versus severe; not shown). However, when comparing the ratio of OPN‐c expression relative to all forms of OPN, DMD patients displayed a higher ratio of total OPN relative to OPN‐c expression. Additionally, when grouping DMD and BMD patients based on histological severity, increased severity was associated with a higher ratio of total OPN relative to OPN‐c expression (Fig. 2 A). This suggests that the expression of OPN‐a and OPN‐b isoforms, not OPN‐c, is more closely associated with dystrophic pathological severity.
Figure 2. Human OPN‐a and OPN‐c isoform ratios change with dystrophic severity and mechanical loading .
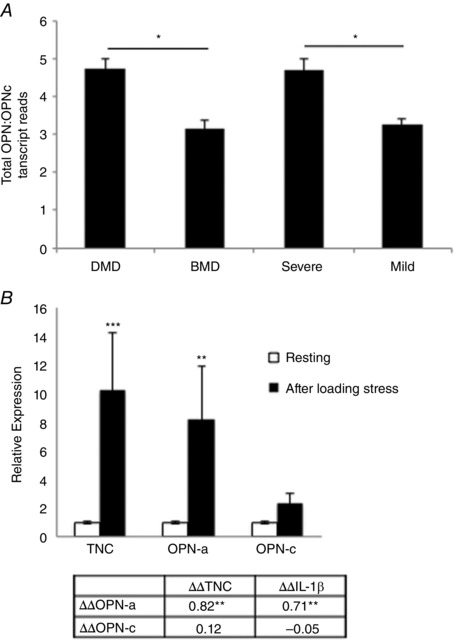
A, the expression ratio of total OPN (using primers specific to exon 7) to OPN‐c (specific to mRNA sequence spanning from exons 3–5, with exon 4 deleted) in Duchenne muscular dystrophy (DMD) and Becker's muscular dystrophy (BMD) vastus lateralis human muscle biopsies (n = 18; all male). Patients were grouped by histological severity prior to Nanostring mRNA molecule counting, as indicated by total fibre degeneration and regeneration and inflammatory foci, as assessed via Haematoxylin and Eosin staining. B, transcript fold changes (ΔΔC t) were quantified before and 24 h after mechanical loading (n = 22, matched for sex) by qRT‐PCR. IL‐1β and OPN‐c were undetectable in some subjects (n = 3 and n = 4, respectively) at either baseline or at 24 h and were excluded from analysis. The table below displays Pearson product–moment correlation coefficients between fold changes in transcripts (ΔΔC t values) in all subjects. * P < 0.05 and ** P < 0.01 between groups.
Given that OPN is expressed during muscle degeneration and remodelling, which is a prominent feature of DMD, we sought to characterize OPN isoform expression in healthy human muscle in response to a mild inflammatory stimulus. Subjects performed one bout of unaccustomed mechanical loading (knee extension), and muscle biopsies of the vastus lateralis muscle were taken before and 24 h after loading. This stimulus was previously observed to induce mild muscle damage and inflammation (Merritt et al. 2013). Here, we observed, similar to DMD patient muscle biopsies, that OPN‐a, not OPN‐c, was the most prominent isoform upregulated in response to mechanical loading. Expression of OPN‐a increased approximately eightfold (P < 0.05) with mechanical loading. Expression of OPN‐c increased approximately two‐fold, but did not reach statistical significance (P > 0.05). Expression levels of OPN‐a were also correlated with induction of the pro‐inflammatory proteins tenascin‐C (TNC), a Toll‐like receptor 4 (TLR4) agonist (Midwood et al. 2009), and IL‐1β. Levels of OPN‐c were not correlated with expression of either of these pro‐inflammatory proteins (Fig. 2 B).
Differential cytokine induction by OPN‐a relative to OPN‐b and OPN‐c
To understand the role of elevated OPN isoform expression in dystrophic muscle, we next observed the effects of each human OPN isoform on macrophage activation, because macrophages extensively infiltrate dystrophic muscle and play crucial roles in muscle remodelling. Primary human macrophages derived from circulating monocytes from normal volunteers (n = 4 healthy male donors) were incubated with recombinant human OPN protein isoforms (Yokosaki et al. 1999). Cells were pretreated with PMB to inhibit the effects of any endotoxin contamination within the recombinant proteins. Cells were then stimulated with recombinant OPN isoforms (1.0 μg ml−1) or vehicle (PMB) for 4 h. Messenger RNA molecule counting (Nanostring) was performed to quantify the expression of the following pro‐inflammatory and pro‐fibrotic transcripts: CCL5, IL‐1β, IL‐10, IL‐15, IFITM1, TNC and transforming growth factor β (TGFβ) isoforms 1–3. The transcripts included in the custom Nanostring panel were chosen based on previous microarray data demonstrating the acute upregulation of these transcripts in response to pro‐inflammatory and pro‐fibrotic stimuli and have been published elsewhere (Dillingham et al. 2015). OPN‐a induced the greatest amount of pro‐inflammatory transcript induction (Fig. 3 A). Significant main and between‐treatment effects were observed when comparing differences in cytokine production induced by OPN‐a, OPN‐b and OPN‐c as determined by ANOVA testing, with Bonferroni correction (main effects: CCL5, P < 0.001; IL‐1β, P < 0.001; IL‐10, P = 0.009; TNC, P = 0.015; and IFITM1, P = 0.010; and between‐group effects for all proteins: OPN‐a versus OPN‐b, P < 0.01; and OPN‐a versus OPN‐c P < 0.001). Expression of TGFβ‐1 did not change in response to OPN treatment and TGFβ‐2/3 isoform expression was not detectable (not shown). Together these data suggest that OPN‐a promotes strong cytokine induction from human macrophages, whereas OPN‐b and OPN‐c induce less inflammatory activity.
Figure 3. Human primary macrophages show a pro‐inflammatory response to recombinant OPN‐a in an RGD‐dependent manner .
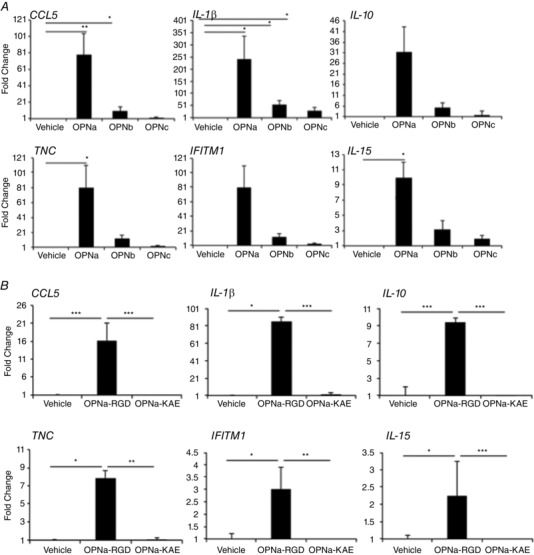
Shown are fold changes of immunoregulatory transcripts from primary human macrophages treated with 1 μg ml−1 OPN isoforms (A) or RGD‐sufficient (OPN‐a‐RGD) and RGD‐deficient [ΔRGD→KAE (OPN‐a‐KAE)] OPN (B). A, OPN‐a increased pro‐inflammatory transcripts, whereas OPN‐b and OPN‐c show less activity. Statistical significance is presented relative to vehicle control. B, macrophages stimulated with 2 μg ml−1 OPN‐a‐RGD or OPN‐a‐KAE show that the pro‐inflammatory activity of OPN‐a requires RGD‐mediated binding to integrins on macrophages. Statistical significance is compared between treatment conditions. Expression values after treatment with OPN isoforms are presented as the average fold change relative to vehicle [polymixin‐B (PMB)] treatment for n = 4 healthy male donors. * P < 0.05, ** P < 0.01 and *** P < 0.001 as assessed via Student's paired t tests (average donor response).
OPN contains an RGD integrin‐binding domain on exon 6, which is conserved in all three spliced isoforms. To determine whether the pro‐inflammatory activity of OPN was mediated by integrins that bind to the RGD site, we used mutant OPN, in which RGD was changed to KAE (OPN‐a‐KAE; Young et al. 1990). RGD‐sufficient OPN‐a significantly increased the expression of inflammatory cytokines, whereas OPN‐a‐KAE displayed attenuated effects (Fig. 3 B).
So far, we have shown that OPN is upregulated in dystrophic muscle and exerted pro‐inflammatory effects on macrophages. To gain a greater understanding of the roles of OPN in muscle remodelling, we examined the pro‐inflammatory effects of OPN on myoblasts. After incubating myoblasts in the presence of OPN isoforms, secreted cytokine protein was assessed by IL‐6 ELISA. Here, OPN‐a was the most potent inducer of IL‐6 production (Fig. 4 A). This also appeared to be mediated by RGD‐integrin binding because, as on macrophages, the OPN‐a RGD→KAE mutant induced less IL‐6 production (Fig. 4 B).
Figure 4. OPN isoforms differentially induce pro‐inflammatory cytokine production from human myoblasts in an RGD‐dependent manner .
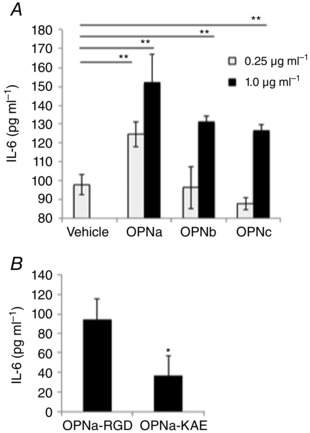
Immortalized human myoblasts were cultured for 48 h with recombinant human OPN proteins treated with 0.25 or 1.0 μg ml−1 human OPN‐a, OPN‐b or OPN‐c (A) or 1.0 μg ml−1 recombinant human OPNa‐RGD or OPNa‐KAE (B). Interleukin‐6 (IL‐6) concentrations in the secreted media were quantified after 48 h by enzyme‐linked immunosorbent assay. Samples were run in replicates of n = 5.
Human OPN‐a cytokine induction is attenuated in macrophages upon inhibition of TLR4 signalling
Given that we observed OPN‐induced TNC expression from macrophages and TNC is a known TLR4 agonist (Midwood et al. 2009), we sought to determine whether OPN promoted macrophage cytokine production in a TLR4‐dependent manner. To determine whether OPN‐RGD pro‐inflammatory activity occurs via TLR4, a small molecule intracellular inhibitor of TLR4 signalling, TAK‐242, was used. TAK‐242 decreased ∼98% of IL‐6 expression induced by 100 (not shown) and 10 ng ml−1 LPS (Fig. 5) in primary human macrophages. TAK‐242 did not affect cell viability (not shown). Induction of IL‐6 by human OPN‐a‐RGD was likewise blocked by TAK‐242 (∼93% decrease in IL‐6 production), whereas OPN‐a‐KAE did not significantly increase IL‐6 production from macrophages. With OPN‐a‐KAE treatment, the IL‐6 concentrations were 5.3 pg ml−1 (vehicle) and 4.0 pg ml−1 (TAK‐242; Fig. 5).
Figure 5. Pretreatment of human macrophages with TAK‐242, an inhibitor of Toll‐like receptor 4 (TLR4) signalling, inhibits human OPN‐a‐mediated IL‐6 production .
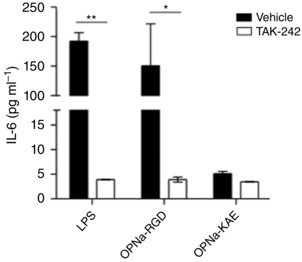
Primary human macrophages were treated with TAK‐242 (100 nm) or vehicle (DMSO) for 1 h before stimulation with 1 μg ml−1 OPN‐a‐RGD and OPN‐a‐KAE or 10 ng ml−1 lipopolysaccharide (LPS; positive control). Interleukin‐6 concentrations in the secreted media were measured at 24 h by enzyme‐linked immunosorbent assay. The pro‐inflammatory activity of OPN‐a was largely blocked by TAK‐242. TAK‐242 had little effect on OPN‐a‐KAE activity. Samples were run in replicates of n = 4.
Enhancement of monocyte phagocytosis equally by OPN‐a, OPN‐b or OPN‐c
Extensive macrophage infiltration is a feature of dystrophic muscle, where macrophages phagocytose cellular debris from necrotic myofibres. We tested the effects of OPN isoforms on macrophage phagocytosis (Fig. 6). All OPN spliced isoforms increased phagocytosis of fluorescently conjugated bacterial particles by ∼6‐8‐fold (Fig. 6 A). OPN‐a‐RGD and OPN‐a‐KAE induced phagocytosis equally, with 8‐9‐fold increase in macrophage phagocytosis relative to vehicle (PMB; Fig. 6 B). These results suggest that, in contrast to cytokine induction, stimulation of phagocytosis is not mediated by RGD‐integrins, and another OPN domain is required for the phagocytosis that is shared and equally accessible between the three isoforms.
Figure 6. Human OPN‐mediated monocyte phagocytosis is isoform and RGD independent .
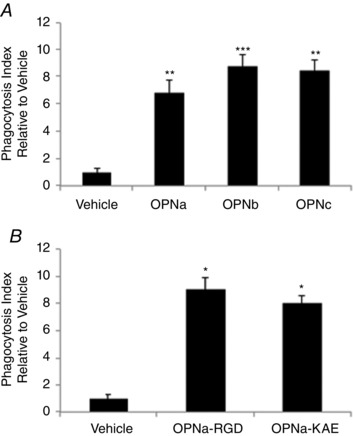
After 30 min pretreatment with polymixin‐B (PMB), primary human monocytes were treated for 2 h with recombinant OPN proteins or vehicle (PMB) in the presence of fluorescently conjugated Escherichia coli particles. Cells were washed, and cellular fluorescence of non‐viable cells was quenched with Trypan Blue. Phagocytosis is presented as the fold increase in fluorescence from viable cells relative to vehicle control cells (PMB). All conditions were normalized for background fluorescence (E. coli particles only) before calculation of the fold change relative to vehicle control. A, phagocytosis induced by 0.5 μg ml−1 OPN spliced isoforms relative to vehicle (PMB). B, phagocytosis induced by 1.0 μg ml−1 OPN‐a‐RGD and OPN‐a‐KAE. All samples were run in replicates of n = 5, according to the manufacturer's instructions (Vybrant Phagocytosis Assay Kit; Life Technologies). Significance is presented relative to the vehicle control.
Intramuscle OPN‐a expression upregulates pathways indicative of macrophage recruitment and TNC–TLR4 signalling
In response to acute muscle injury by cardiotoxin, pro‐inflammatory macrophages express OPN (Paliwal et al. 2012). To provide an insight into the role of OPN‐a expression on skeletal muscle transcriptomics, we performed transcriptional profiling on muscle tissue sections following intramuscular OPN‐a overexpression in 3‐month‐old BLA/J mice. We observed that OPN‐a overexpression upregulated transcripts indicative of pro‐inflammatory macrophage recruitment and activation. When comparing the expression of genes in TA muscles of mice injected with either OPN‐a or empty plasmid, 432 genes were differentially expressed at the P < 0.01 level at 7 days postinjection. Overexpression of OPN‐a led to a significant increase in its own transcript (SPP1; 3.8‐fold; P = 0.008; Fig. 7). OPN‐a induction also increased expression of its RGD binding receptors: integrin αV (1.6‐fold; P = 0.005) and integrin α5 (1.5‐fold; P = 0.003; not shown). At 7 days postinjection, the most highly upregulated pro‐inflammatory transcripts by OPN‐a included TNC (3.9‐fold, P = 0.004), IL‐18 (1.8‐fold; P = 0.010), IL‐17 receptor A (1.5‐fold; P = 0.007) and CD40 (1.9‐fold; P = 0.008; Fig. 7). CX3CR1 expression was also upregulated 1.9‐fold (P = 0.005), suggesting increased macrophage infiltration after acute injury in response to OPN‐a relative to the empty vector control. The marker of alternatively activated (less pro‐inflammatory) macrophages, CD163, was also downregulated 2.3‐fold (P = 0.008). Through network analysis, GM‐CSF appeared to be a major upstream regulator of OPN‐a‐inducible transcripts within the skeletal muscle (z‐score 4.95; P value of overlap 1.04 × 10−9). Together, data from these two experiments suggest that: (i) OPN‐a‐inducible proteins are upregulated in acute muscle injury; and (ii) intramuscular overexpression of OPN‐a promotes macrophage chemotaxis and pro‐inflammatory activation within the skeletal muscle.
Figure 7. OPN‐a promotes pro‐inflammatory macrophage infiltration in vivo .
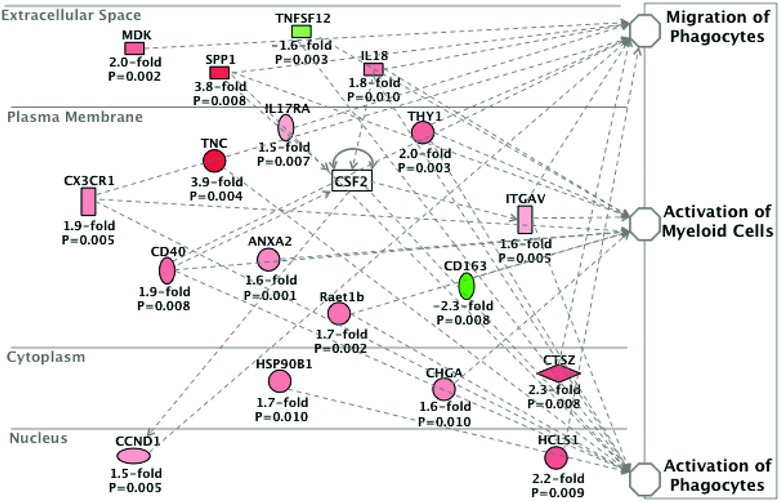
BLA/J mice (n = 3) were injected with OPN‐a and an empty vector in contralateral tibialis anterior (TA) muscles. The TA muscles were harvested 7 days postinjection, and Illumina mRNA expression profiling was performed. Data are represented as the average transcript fold change by OPN‐a relative to empty vector control. Changes in all transcripts shown were statistically significant (P < 0.05). CSF2, granulocyte‐macrophage colony‐stimulating factor (GM‐CSF); SPP1, osteopontin; TNC, tenascin‐C.
Discussion
OPN is increasingly recognized as an important mediator of muscle inflammation after damage. A genetic study showed that gene polymorphisms in OPN modulate the response of adult volunteer muscle to inflammation detected by magnetic resonance imaging after damaging contractions (Barfield et al. 2014), followed by another study showing that these same polymorphisms alter the clinical severity of DMD (Bello et al. 2015). In the present study, we found prominent upregulation of OPN isoforms in dystrophic muscles and with mechanical loading, a mild inflammatory stimulus. This led us to define the pro‐inflammatory effects of OPN spliced isoforms on the skeletal muscle microenvironment. Macrophages, an extensively infiltrating immune cell type in dystrophic muscle, were found to be highly responsive to OPN‐a as indicated by increased pro‐inflammatory cytokine production. OPN‐a also exerted some pro‐inflammatory effects on myoblasts. Among the three human OPN isoforms, OPN‐a, OPN‐b and OPN‐c, cytokine induction was most prominent in response to OPN‐a in macrophages and myoblasts. In contrast, each of the three isoforms stimulated monocyte phagocytosis equally well. Through expression profiling, we observed that intramuscular overexpression of OPN‐a induced expression of TNC, an endogenous activator of TLR4 signalling. Furthermore, OPN‐a upregulated TNC expression in human macrophages in vitro. Upon examining the effects of TLR4 signalling ablation on OPN‐mediated cytokine production, we observed that blockade of TLR4 signalling attenuated OPN‐a‐mediated cytokine production in human macrophages. Together, our findings suggest that OPN‐a acts as the most pro‐inflammatory isoform in the muscle microenvironment, possibly through induction of TNC–TLR4 signalling.
In the present study, we compared OPN splice variants with each other because of distinct properties of the variants in tumour cells and increased isoform expression in dystrophic human muscle. Mutating the RGD sequence of OPN‐a revealed that the pro‐inflammatory effect of OPN‐a, on both macrophages and myoblasts, is mediated by the RGD sequence that binds to integrins including αvβ3, αvβ5 and α5β1. Structural differences between these isoforms include the presence or absence of exon 4 or exon 5, while the RGD and encompassing sequence, TYDGRGDSVVYGLR, in exon 6 is consistently present in the isoforms (Fig. 1 A). The distinct pro‐inflammatory effects of the OPN isoforms observed in this study would probably not occur unless exon 4 or exon 5 influenced the interaction between OPN and integrins. The ability of exon 4 or exon 5 to affect OPN–integrin interactions is supported by several observations. First, OPN displays enhanced integrin‐mediated function upon polymerization by a cross‐linking enzyme, transglutaminase 2, that covalently links Gln and Lys residues. OPN‐b and OPN‐c are less polymerized than OPN‐a (Nishimichi et al. 2011) owing to loss of Gln residues at exons 4 and 5, which serve as cross‐linking sites. Given that transglutaminase 2 is abundantly expressed by macrophages (Zakrzewicz et al. 2015) and skeletal muscle (Park et al. 1994), it is possible that the OPN isoforms were polymerized during incubation with macrophages and myoblasts. OPN‐a might polymerize sufficiently for cytokine induction owing to its enhanced ability to undergo polymerization. Further, polymeric OPN forms a new binding site for another integrin, α9β1, whose signals might also be less intensive in OPN‐b and OPN‐c than OPN‐a. Second, OPN is thought to bind with many proteins through its naturally disordered domain, including the regions encompassed at exons 4 and 5. If some proteins in the culture supernatant from cells or fetal bovine serum bind differentially to the isoforms, the interactions between OPN and integrins would not be the same between isoforms. In contrast, observations of RGD‐independent and equally enhanced monocyte phagocytosis among isoforms strongly suggest that another OPN receptor, whose binding site is equally accessible among isoforms, promotes monocyte phagocytosis. The αxβ2 integrin might serve such a function because it is abundantly expressed on macrophages and documented to promote murine macrophage phagocytosis (Schack et al. 2009).
From expression profiling, our data suggest that OPN‐a promotes TNC‐ and TLR4‐responsive cytokines (IL‐1β and IL‐18) in humans and mice. This was evident in that OPN‐a induced TNC and IL‐1β expression from human macrophages. Furthermore, OPN‐a expression was correlated with TNC and IL‐1β expression levels in human muscle following mechanical loading. In mice, overexpression of OPN‐a also increased expression of TNC and IL‐18, another TLR4‐responsive cytokine (Rhee et al. 2013). This led us to examine whether OPN‐a induced cytokine production via TLR4 signalling. We observed that the pro‐inflammatory activity of OPN‐a on human macrophages was attenuated by pretreatment with a small molecule inhibitor of TLR4 signalling, TAK‐242. In murine RAW264.7 macrophages, TAK‐242 inhibits TLR4 signalling by binding to the TIR domain (Cys747) of TLR4 to inhibit downstream signalling activation (Matsunaga et al. 2011). Our in vitro and in vivo profiling data suggest that OPN upregulates TNC expression. TNC is an extracellular matrix protein upregulated in response to acute injury that promotes TLR4 signalling (Midwood et al. 2009). Thus, a possible mechanism of OPN‐mediated cytokine production may be induction of TNC–TLR4 signalling. Previous reports have also observed that OPN ablation attenuates TNC and IL‐1β expression levels in a model of lung injury (Sabo‐Attwood et al. 2011), which further supports this hypothesis. It is additionally possible that OPN could be acting as a TLR4 agonist itself, because numerous endogenous activators of TLR4 signalling have been reported.
In contrast to our findings, OPN isoforms derived from tumour (MCF‐7) cells do not display pro‐inflammatory activity (Sun et al. 2013). Here, Sun et al. observed no effect of OPN isoforms on LPS‐mediated IL‐8, IL‐6 or IL‐12 production from primary human monocytes, whereas all isoforms decreased tumour necrosis factor‐α and increased IL‐10 production. Only OPN‐c was observed to increase the expression of CD163 on monocytes, a marker of alternative macrophage activation (Kowal et al. 2011), suggesting potential anti‐inflammatory activity of OPN‐c. We did not observe an anti‐inflammatory effect of OPN‐c per se. However, we observed that OPN‐c displayed the least pro‐inflammatory activity of all of the OPN‐a isoforms. The differential activities of OPN isoforms may vary widely among tissues and cell types as a result of multiple post‐translational modifications of OPN and differential OPN receptor expression patterns (i.e. CD44 versus integrins). Such differences may explain varying outcomes between studies and highlight a need to investigate isoform‐specific post‐translational effects of OPN in different tissues.
In dystrophic human muscle, we observed that higher OPN‐c expression levels, relative to total OPN, were associated with reduced histological severity. This suggests that within the skeletal muscle, OPN‐a is likely to be the primary contributor to the pathological effects of OPN. Targeting of OPN‐a may thus be a useful approach to reducing OPN‐mediated muscle inflammation. Previously, inhibition of OPN has been shown to reduce skeletal muscle inflammation in dystrophic mice (Vetrone et al. 2009) and promote regeneration following acute injury in aged mice (Paliwal et al. 2012). Targeting OPN‐a, but not OPN‐c, signalling in human skeletal muscle might be a useful therapeutic strategy, because OPN does exert some positive effects on skeletal muscle regeneration. Substratum OPN promotes myoblast attachment in murine cells (Uaesoontrachoon et al. 2008). Conversely, soluble OPN‐a inhibits myotube fusion. These differential effects of OPN on myoblast proliferation and myotube fusion are congruent with the pro‐inflammatory effects of OPN, because inflammatory stimuli typically promote myoblast proliferation and inhibit myotube fusion (Arnold et al. 2007). As the beneficial effects of OPN ablation in dystrophic and ageing muscle are presumably attributable to inhibition of its inflammatory activities (OPN‐a is the only isoform expressed in mice), inhibiting the inflammatory action of OPN‐a while maintaining some activity of OPN‐c might serve as a useful therapy in humans. Future studies examining the effects of OPN isoforms on myogenesis (proliferation and fusion) may further identify the therapeutic utility of OPN isoform targeting in muscle disease.
In mdx mice, ablation of OPN attenuates muscle fibrosis and TGFβ expression (Vetrone et al. 2009). However, we did not observe a direct effect of OPN on TGFβ production in macrophages or myoblasts (not shown). Based on our findings, the ability of OPN to promote fibrosis is likely to be attributable to its pro‐inflammatory, chemotactic and phagocytic activities, rather than directly inducing TGFβ production from myoblasts or macrophages. This is supported, because genetic ablation of TLR4 reduces both inflammation and fibrosis in mdx mice, suggesting that inflammation, particularly innate immunity, contributes to muscle fibrosis in the context of dystrophin deficiency (Giordano et al. 2015). However, it is possible that OPN expression in the muscle microenvironment promotes TGFβ secretion by another cell type.
Limitations
In dystrophic human muscle, this study was limited by a conservative sample size. Future profiling of OPN‐a and OPN‐c expression levels and their correlation with muscle pathology might elucidate the utility of OPN isoform ratios as a biomarker of disease severity. We did not specifically examine the effects of inhibiting TNC on OPN‐mediated inflammatory cytokine production. Further studies investigating OPN–TNC signalling are thus warranted. Additionally, it is unknown whether OPN isoform expression and function are fibre‐type specific. Future studies examining the effects of OPN overexpression in murine muscle groups with varying fibre‐type composition may thus be of interest.
Together, our data suggest that OPN is an important mediator of muscle inflammation, whereby its expression in response to mechanical loading and in dystrophin deficiency contributes to both macrophage and muscle cytokine production. Furthermore, our data support a body of literature indicating that localized myofibre OPN‐a expression increases inflammation by promoting pro‐inflammatory monocyte recruitment and macrophage activation to damaged myofibres. The pro‐inflammatory activity of OPN‐a may be mediated, in part, by induction of TNC–TLR4 signalling. Our data further suggest differential activities of human spliced OPN isoforms and suggest that targeting RGD‐mediated integrin signalling and/or transglutamination might attenuate the pro‐inflammatory activity of OPN‐a within the skeletal muscle microenvironment.
Additional information
Competing interests
None declared.
Author contributions
G.M.M., K.U., P.P.N., L.B., S.D. and Y.Y. contributed to data acquisition, analysis and/or interpretation. G.M.M., Y.Y., J.M.D., H.B.C., J.N.K., M.M.B., D.M.M., K.N. and E.P.H. contributed to study design and data analysis and interpretation. All authors approved the final version of the manuscript and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
E.P.H.’s work was supported by NIH grants 5P50AR060836 and 5R01NS029525 and by the Clark Charitable Foundation, Inc. G.M.M. was supported by T32HD071866 and the Clark Charitable Foundation, Inc. K.N.’s contribution to this project was provided by the following NIH grants: 5K26OD011171 and 1P50AR060836.
Acknowledgements
We would like to thank Dr Larry Fisher's laboratory for facilitating production of recombinant OPN‐a‐RGD and OPN‐a‐KAE proteins. We also thank Dr Vincent Mouly for supplying the immortalized human myoblasts and the platform for immortalization of human cells at the Myology Institute in Paris. We kindly thank Dr Isabelle Richard for providing the BLA/J mice. We thank Mamta Giri and Jaya Punetha for their technical assistance with Nanostring profiling.
Edited by: Dawn A. Lowe
References
- Arnold L, Henry A, Poron F, Baba‐Amer Y, van Rooijen N, Plonquet A, Gherardi RK & Chazaud B (2007). Inflammatory monocytes recruited after skeletal muscle injury switch into antiinflammatory macrophages to support myogenesis. J Exp Med 204, 1057–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandopadhyay M, Bulbule A, Butti R, Chakraborty G, Ghorpade P, Ghosh P, Gorain M, Kale S, Kumar D, Kumar S, Totakura KV, Roy G, Sharma P, Shetti D, Soundararajan G, Thorat D, Tomar D, Nalukurthi R, Raja R, Mishra R, Yadav AS & Kundu GC (2014). Osteopontin as a therapeutic target for cancer. Expert Opin Ther Targets 18, 883–895. [DOI] [PubMed] [Google Scholar]
- Barfield WL, Uaesoontrachoon K, Wu CS, Lin S, Chen Y, Wang PC, Kanaan Y, Bond V & Hoffman EP (2014). Eccentric muscle challenge shows osteopontin polymorphism modulation of muscle damage. Hum Mol Genet 23, 4043–4050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bello L, Kesari A, Gordish‐Dressman H, Cnaan A, Morgenroth LP, Punetha J, Duong T, Henricson EK, Pegoraro E, McDonald CM, Hoffman EP & Cooperative International Neuromuscular Research Group Investigators (2015). Genetic modifiers of ambulation in the Cooperative International Neuromuscular Research Group Duchenne Natural History Study. Ann Neurol 77, 684–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavaillon JM & Haeffner‐Cavaillon N (1986). Polymyxin‐B inhibition of LPS‐induced interleukin‐1 secretion by human monocytes is dependent upon the LPS origin. Mol Immunol 23, 965–969. [DOI] [PubMed] [Google Scholar]
- Christensen B, Zachariae ED, Scavenius C, Thybo M, Callesen MM, Kløverpris S, Oxvig C, Enghild JJ & Sørensen ES (2014). Identification of transglutaminase reactive residues in human osteopontin and their role in polymerization. PLoS One 9, e113650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Donna S, Mamchaoui K, Cooper RN, Seigneurin‐Venin S, Tremblay J, Butler‐Browne GS & Mouly V (2003). Telomerase can extend the proliferative capacity of human myoblasts, but does not lead to their immortalization. Mol Cancer Res 1, 643–653. [PubMed] [Google Scholar]
- Dillingham BC, Knoblach SM, Many GM, Harmon BT, Mullen AM, Heier CR, Bello L, McCall JM, Hoffman EP, Connor EM, Nagaraju K, Reeves EK & Damsker JM (2015). VBP15, a novel anti‐inflammatory, is effective at reducing the severity of murine experimental autoimmune encephalomyelitis. Cell Mol Neurobiol 35, 377–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedarko NS, Fohr B, Robey PG, Young MF & Fisher LW (2000). Factor H binding to bone sialoprotein and osteopontin enables tumor cell evasion of complement‐mediated attack. J Biol Chem 275, 16666–16672. [DOI] [PubMed] [Google Scholar]
- Fisher LW, Torchia DA, Fohr B, Young MF & Fedarko NS (2001). Flexible structures of SIBLING proteins, bone sialoprotein, and osteopontin. Biochem Biophys Res Commun 280, 460–465. [DOI] [PubMed] [Google Scholar]
- Gimba ER & Tilli TM (2013). Human osteopontin splicing isoforms: known roles, potential clinical applications and activated signaling pathways. Cancer Lett 331, 11–17. [DOI] [PubMed] [Google Scholar]
- Giordano C, Mojumdar K, Liang F, Lemaire C, Li T, Richardson J, Divangahi M, Qureshi S & Petrof BJ (2015). Toll‐like receptor 4 ablation in mdx mice reveals innate immunity as a therapeutic target in Duchenne muscular dystrophy. Hum Mol Genet 24, 2147–2162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higashikawa F, Eboshida A & Yokosaki Y (2007). Enhanced biological activity of polymeric osteopontin. FEBS Lett 581, 2697–2701. [DOI] [PubMed] [Google Scholar]
- Hirata A, Masuda S, Tamura T, Kai K, Ojima K, Fukase A, Motoyoshi K, Kamakura K, Miyagoe‐Suzuki Y & Takeda S (2003). Expression profiling of cytokines and related genes in regenerating skeletal muscle after cardiotoxin injection: a role for osteopontin. Am J Pathol 163, 203–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman EP, Gordish‐Dressman H, McLane VD, Devaney JM, Thompson PD, Visich P, Gordon PM, Pescatello LS, Zoeller RF, Moyna NM, Angelopoulos TJ, Pegoraro E, Cox GA & Clarkson PM (2013). Alterations in osteopontin modify muscle size in females in both humans and mice. Med Sci Sports Exerc 45, 1060–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaartinen MT, Pirhonen A, Linnala‐Kankkunen A & Mäenpää PH (1999). Cross‐linking of osteopontin by tissue transglutaminase increases its collagen binding properties. J Biol Chem 274, 1729–1735. [DOI] [PubMed] [Google Scholar]
- Kornegay JN, Bogan JR, Bogan DJ, Childers MK, Li J, Nghiem P, Detwiler DA, Larsen CA, Grange RW, Bhavaraju‐Sanka RK, Tou S, Keene BP, Howard JF Jr, Wang J, Fan Z, Schatzberg SJ, Styner MA, Flanigan KM, Xiao X & Hoffman EP (2012). Canine models of Duchenne muscular dystrophy and their use in therapeutic strategies. Mamm Genome 23, 85–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornegay JN, Spurney CF, Nghiem PP, Brinkmeyer‐Langford CL, Hoffman EP & Nagaraju K (2014). Pharmacologic management of Duchenne muscular dystrophy: target identification and preclinical trials. ILAR J 55, 119–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowal K, Silver R, Slawinska E, Bielecki M, Chyczewski L & Kowal‐Bielecka O (2011). CD163 and its role in inflammation. Folia Histochem Cytobiol 49, 365–374. [DOI] [PubMed] [Google Scholar]
- Li W, Zheng Y, Zhang W, Wang Z, Xiao J & Yuan Y (2015). Progression and variation of fatty infiltration of the thigh muscles in Duchenne muscular dystrophy, a muscle magnetic resonance imaging study. Neuromuscul Disord 25, 375–380. [DOI] [PubMed] [Google Scholar]
- Matsunaga N, Tsuchimori N, Matsumoto T & Ii M (2011). TAK‐242 (resatorvid), a small‐molecule inhibitor of Toll‐like receptor (TLR) 4 signaling, binds selectively to TLR4 and interferes with interactions between TLR4 and its adaptor molecules. Mol Pharmacol 79, 34–41. [DOI] [PubMed] [Google Scholar]
- Merritt EK, Stec MJ, Thalacker‐Mercer A, Windham ST, Cross JM, Shelley DP, Craig Tuggle S, Kosek DJ, Kim JS & Bamman MM (2013). Heightened muscle inflammation susceptibility may impair regenerative capacity in aging humans. J Appl Physiol 115, 937–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Midwood K, Sacre S, Piccinini AM, Inglis J, Trebaul A, Chan E, Drexler S, Sofat N, Kashiwagi M, Orend G, Brennan F & Foxwell B (2009). Tenascin‐C is an endogenous activator of Toll‐like receptor 4 that is essential for maintaining inflammation in arthritic joint disease. Nat Med 15, 774–780. [DOI] [PubMed] [Google Scholar]
- Nghiem PP, Hoffman EP, Mittal P, Brown KJ, Schatzberg SJ, Ghimbovschi S, Wang Z & Kornegay JN (2013). Sparing of the dystrophin‐deficient cranial sartorius muscle is associated with classical and novel hypertrophy pathways in GRMD dogs. Am J Pathol 183, 1411–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimichi N, Hayashita‐Kinoh H, Chen C, Matsuda H, Sheppard D & Yokosaki Y (2011). Osteopontin undergoes polymerization in vivo and gains chemotactic activity for neutrophils mediated by integrin α9β1. J Biol Chem 286, 11170–11178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagel CN, Wasgewatte Wijesinghe DK, Taghavi Esfandouni N & Mackie EJ (2014). Osteopontin, inflammation and myogenesis: influencing regeneration, fibrosis and size of skeletal muscle. J Cell Commun Signal 8, 95–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paliwal P, Pishesha N, Wijaya D & Conboy IM (2012). Age dependent increase in the levels of osteopontin inhibits skeletal muscle regeneration. Aging (Albany NY) 4, 553–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SC, Kim WH, Lee MC, Seong SC, Song KY & Choe MA (1994). Modulation of transglutaminase expression in rat skeletal muscle by induction of atrophy and endurance training. J Korean Med Sci 9, 490–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pegoraro E, Hoffman EP, Piva L, Gavassini BF, Cagnin S, Ermani M, Bello L, Soraru G, Pacchioni B, Bonifati MD, Lanfranchi G, Angelini C, Kesari A, Lee I, Gordish‐Dressman H, Devaney JM, McDonald CM & Cooperative International Neuromuscular Research Group (2011). SPP1 genotype is a determinant of disease severity in Duchenne muscular dystrophy. Neurology 76, 219–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter JD, Merriam AP, Leahy P, Gong B, Feuerman J, Cheng G & Khanna S (2004). Temporal gene expression profiling of dystrophin‐deficient (mdx) mouse diaphragm identifies conserved and muscle group‐specific mechanisms in the pathogenesis of muscular dystrophy. Hum Mol Genet 13, 257–269. [DOI] [PubMed] [Google Scholar]
- Rhee AC, Cain AL, Hile KL, Zhang H, Matsui F & Meldrum KK (2013). IL‐18 activation is dependent on Toll‐like receptor 4 during renal obstruction. J Surg Res 183, 278–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabo‐Attwood T, Ramos‐Nino ME, Eugenia‐Ariza M, Macpherson MB, Butnor KJ, Vacek PC, McGee SP, Clark JC, Steele C & Mossman BT (2011). Osteopontin modulates inflammation, mucin production, and gene expression signatures after inhalation of asbestos in a murine model of fibrosis. Am J Pathol 178, 1975–1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schack L, Stapulionis R, Christensen B, Kofod‐Olsen E, Skov Sørensen UB, Vorup‐Jensen T, Sørensen ES & Höllsberg P (2009). Osteopontin enhances phagocytosis through a novel osteopontin receptor, the αXβ2 integrin. J Immunol 182, 6943–6950. [DOI] [PubMed] [Google Scholar]
- Shi Z, Mirza M, Wang B, Kennedy MA & Weber GF (2014). Osteopontin‐a alters glucose homeostasis in anchorage‐independent breast cancer cells. Cancer Lett 344, 47–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin T (2012). Osteopontin as a two‐sided mediator in acute neuroinflammation in rat models. Acta Histochem 114, 749–754. [DOI] [PubMed] [Google Scholar]
- Stec MJ, Mayhew DL & Bamman MM (2015). The effects of age and resistance loading on skeletal muscle ribosome biogenesis. J Appl Physiol 119, 851–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Feng A, Chen S, Zhang Y, Xie Q, Yang M, Shao Q, Liu J, Yang Q, Kong B & Qu X (2013). Osteopontin splice variants expressed by breast tumors regulate monocyte activation via MCP‐1 and TGF‐β1. Cell Mol Immunol 10, 176–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uaesoontrachoon K, Wasgewatte Wijesinghe DK, Mackie EJ & Pagel CN (2013). Osteopontin deficiency delays inflammatory infiltration and the onset of muscle regeneration in a mouse model of muscle injury. Dis Model Mech 6, 197–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uaesoontrachoon K, Yoo HJ, Tudor EM, Pike RN, Mackie EJ & Pagel CN (2008). Osteopontin and skeletal muscle myoblasts: association with muscle regeneration and regulation of myoblast function in vitro . Int J Biochem Cell Biol 40, 2303–2314. [DOI] [PubMed] [Google Scholar]
- Vetrone SA, Montecino‐Rodriguez E, Kudryashova E, Kramerova I, Hoffman EP, Liu SD, Miceli MC & Spencer MJ (2009). Osteopontin promotes fibrosis in dystrophic mouse muscle by modulating immune cell subsets and intramuscular TGF‐β. J Clin Invest 119, 1583–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokosaki Y, Matsuura N, Sasaki T, Murakami I, Schneider H, Higashiyama S, Saitoh Y, Yamakido M, Taooka Y & Sheppard D (1999). The integrin α9β1 binds to a novel recognition sequence (SVVYGLR) in the thrombin‐cleaved amino‐terminal fragment of osteopontin. J Biol Chem 274, 36328–36334. [DOI] [PubMed] [Google Scholar]
- Young MF, Kerr JM, Termine JD, Wewer UM, Wang MG, McBride OW & Fisher LW (1990). cDNA cloning, mRNA distribution and heterogeneity, chromosomal location, and RFLP analysis of human osteopontin (OPN). Genomics 7, 491–502. [DOI] [PubMed] [Google Scholar]
- Zakrzewicz A, Atanasova S, Padberg W & Grau V (2015). Monocytic tissue transglutaminase in a rat model for reversible acute rejection and chronic renal allograft injury. Mediators Inflamm 2015, 429653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanotti S, Gibertini S, Di Blasi C, Cappelletti C, Bernasconi P, Mantegazza R, Morandi L & Mora M (2011). Osteopontin is highly expressed in severely dystrophic muscle and seems to play a role in muscle regeneration and fibrosis. Histopathology 59, 1215–1228. [DOI] [PubMed] [Google Scholar]