

Abstract
The increasing number of enzyme applications in chemical synthesis calls for new engineering methods to develop the biocatalysts of the future. An interesting concept in enzyme engineering is the generation of large‐scale mutational data in order to chart protein mutability landscapes. These landscapes allow the important discrimination between beneficial mutations and those that are neutral or detrimental, thus providing detailed insight into sequence–function relationships. As such, mutability landscapes are a powerful tool with which to identify functional hotspots at any place in the amino acid sequence of an enzyme. These hotspots can be used as targets for combinatorial mutagenesis to yield superior enzymes with improved catalytic properties, stability, or even new enzymatic activities. The generation of mutability landscapes for multiple properties of one enzyme provides the exciting opportunity to select mutations that are beneficial either for one or for several of these properties. This review presents an overview of the recent advances in the construction of mutability landscapes and discusses their importance for enzyme engineering.
Keywords: biocatalysis, enzyme engineering, hotspots, mutability landscapes, mutagenesis
1. Advantages of Enzyme Catalysis
Application of enzymes as catalysts in the production of chemicals has the potential to serve as a sustainable and efficient alternative to traditional catalysts used in organic synthesis. Enzymes are nature's catalysts and therefore generally function under mild reaction conditions (i.e., ambient temperatures in aqueous solvent systems). Furthermore, enzymes are biodegradable, nontoxic, and readily available, and their production is not dependent on any rare elements. These features underline the sustainable potential of using enzymes as catalysts. Enzymes are known for their high catalytic rates and excellent regio‐, chemo‐, or stereoselectivity. Enantioselectivity is still a major challenge in traditional catalysis and is highly desirable for the production of pharmaceuticals. Finally, enzymes can be optimized for application in industrial biocatalysis by means of protein engineering. Owing to these advantages, the number of applications for enzyme catalysts in the production of valuable chemicals, especially pharmaceuticals and agrochemicals, is increasing.1, 2, 3
2. Why is Enzyme Engineering Required?
Typical goals of engineering projects in the field of biocatalysis can be divided into three topics.
The first topic has a focus on the catalytic properties of enzymes and includes engineering projects that aim to improve catalytic activity, to alter substrate scope, or to improve (enantio)selectivity. As a result of engineering projects directed towards these goals, there are now many examples of enzymes that carry out industrially relevant transformations, with practical turnover rates.1, 2, 3
The second topic covers enzyme engineering projects that aim to improve enzyme stability. Enzymes can be unstable under process conditions, which might include high temperatures, extreme pH values, high substrate (and product) concentrations, and/or the presence of organic solvents. Major improvements in enzyme stability can be achieved through enzyme engineering.4 Alternatively, solvent engineering or enzyme immobilization can be used to address these stability issues. These methods have recently been reviewed elsewhere.4, 5, 6, 7
The third topic in enzyme engineering is the generation of enzymes that catalyze unnatural chemical transformations. Creating enzymes with new enzymatic activities is currently one of the frontiers in biocatalysis, and there are two main approaches to achieve this. The first of these is the de novo computational design of enzymes, which involves the computational design of an active site and placing it in a suitable protein scaffold.8, 9, 10 Enzyme engineering is required to improve the activity of the initial de novo designed protein to a practical level. The second approach to the creation of enzymes with new activities is to exploit catalytic promiscuity of existing enzymes. Promiscuous activities are enzymatic activities other than the activity for which an enzyme has evolved and that are not part of the organism's physiology.11 It has been long recognized that promiscuous activities can serve as a starting point for natural evolution of new enzymatic functions.12, 13 By using nature's approach, enzyme engineering can be applied to improve promiscuous activities for the generation of new biocatalysts for unnatural chemical transformations.14
3. Hotspot Identification for Enzyme Engineering
Enzyme engineering can be viewed as an iterative procedure that starts with generation of diversity in the wild‐type (WT) enzyme and screening of a collection of mutants for the desired properties. To engineer enzymes efficiently, researchers try to identify hotspot positions in an enzyme where mutations are likely to be beneficial.15 Targeting these sites for combinatorial mutagenesis leads to relatively small libraries with a high percentage of positive hits. The identification of these hotspots requires extensive knowledge of the sequence–function relationships of an enzyme; the main ways to obtain this information are by analyzing the (crystal) structure of the enzyme, multisequence alignments (MSAs) of homologous proteins, or empirical mutational data.
Hotspot identification based on the structure of an enzyme is the most commonly used method in enzyme engineering. Damborski and co‐workers recently published an extensive review on in silico hotspot identification methods that are available as web tools.16 The majority of these tools are structure‐based and therefore require a crystal structure of the enzyme. The computational tools then identify hotspot positions, on the basis of predicted protein–ligand interactions, binding pockets, or residues present in access tunnels of enzymes with buried active sites. Computational tools for the identification of hotspots to improve enzyme stability are mainly based on crystallographic B factors, although computational protein design and consensus methods are gaining momentum in this area.4, 16, 17 Besides these in silico approaches, several experimental, semirational, structure‐based enzyme engineering methods that apply targeted site‐saturation mutagenesis of active‐site residues have been developed. These methods include the highly successful combinatorial active site saturation test (CASTing) method and its derivatives.3, 18, 19
Homology‐based hotspot identification tools require a MSA of homologous proteins to identify the evolutionary conservation of specific amino acid residues in a protein. High conservation scores suggest that a specific residue is important for the structure or function of the protein, whereas low conservation suggests that this residue may be mutated without loss of function. Targeting of positions with mutational robustness therefore increases the chance of obtaining viable mutant enzymes and thereby increases the quality of the library.16
The third basis on which hotspot identification can be conducted is empirical data. These data can be generated by screening libraries created by random mutagenesis methods such as error‐prone PCR. The hotspots identified in these libraries can be targeted by combinatorial site‐saturation mutagenesis.20 The main advantage of this approach is that it does not require extensive prior knowledge of the target enzyme.
Obviously, there are available tools that combine information from all three sources. A successful example of this is the protein sequence–activity relationship (PROSAR) method. Here, a collection of enzyme variants that carry multiple mutations per sequence are generated and empirically tested for the desired activity. The initial pool of enzyme variants covers mutations selected on the basis of a combination of structural information, analysis of MSAs, and random mutagenesis.21 By statistical analysis of the screening results, the PROSAR software tool then evaluates the contribution of each individual mutation in each enzyme variant with multiple mutations. The identified residue positions with beneficial mutations are used for the subsequent rounds of diversification and screening. This cycle is repeated until the engineering goal is met.
4. Protein Mutability Landscapes
An interesting concept in enzyme engineering is the generation and use of mutability landscapes. For this type of analysis, a large number of protein variants are analyzed to determine the effect of each single‐amino‐acid substitution on enzyme activity, selectivity, or stability, thus providing detailed maps of beneficial, neutral, and detrimental amino acids for each residue position and each enzyme property. The generation of mutability landscapes for multiple properties of one enzyme provides different landscapes, with the exciting opportunity to select mutations that are beneficial either for one or for several of these properties and neutral or detrimental for others. Thus, in contrast to other systematic mutagenesis approaches such as gene site‐saturation mutagenesis (GSSM), mutability landscapes provide information not only on beneficial mutations but also on detrimental and neutral mutations. This gives valuable information on sequence–function relationships by revealing regions in the enzyme with mutational robustness as well as functionally important residues and hotspot positions.
The term “mutability landscape” was first used by Rost and co‐workers, who developed the screening for non‐acceptable polymorphisms (SNAP) algorithm to predict the effect of single‐amino‐acid substitutions in disease‐related proteins.22 The predictions of this SNAP algorithm are based on information both from a MSA and from the structural features of the protein of interest.23 Alternatively, the sorting intolerant from tolerant (SIFT) algorithm can be used to make similar predictions based on residue conservation.24 Both methods predict whether an amino acid substitution will be neutral or lead to a functional effect but do not distinguish between detrimental or beneficial effects. This is sufficient when merely looking at pathogenicity because both gain‐of‐function and loss‐of‐function mutations can lead to disease. However, it is of limited use when this mutability landscape is generated for enzyme engineering purposes.
Hecht et al. argue that the lack of comprehensive experimental mutagenesis data seems a crucial problem for the development of better computational tools and that the generation of such experimental data is constrained by the amount of required resources.22 Indeed, available data from experimental protein mutability landscapes are scarce, and the majority of these available studies cover protein–protein interactions or protein–DNA interactions.25, 26, 27 In the last few years, however, there have been several reports on experimentally determined mutability landscapes of enzymes. Here we present an overview of the recent advances in experimentally determined mutability landscapes of enzymes to illustrate how these mutability landscapes were generated and used to gain insight into sequence–function relationships or exploited for enzyme engineering.
5. Generating Mutability Landscapes by Using Defined Collections of Single Mutants
There are two approaches to generating experimental protein mutability landscapes. The first approach involves the characterization of a defined collection of single mutants, and the second is called deep mutational scanning (Figure 1). To construct a defined collection of mutant enzymes that covers (nearly) all possible single‐amino‐acid substitutions of an enzyme requires significant effort and resources, but the characterization of the mutants can be relatively easy because it does not require any oversampling. Therefore, the screening methods are not limited to high‐throughput assays, and this gives more flexibility in the design of the assays and provides access to a broader range of analyses (e.g., HPLC, UV spectroscopy). The following examples of mutability landscapes were generated by this approach.
Figure 1.
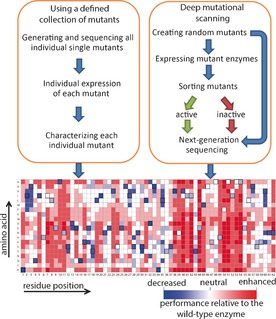
General methods for generating mutability landscapes.
5.1. Protease activity and stability
The usage of “site evaluation libraries”, described in a patent by Estell and Aehle, was basically the first example in which a mutability landscape of an enzyme was generated and applied in enzyme engineering.28 The inventors used a defined collection of single mutants of an alkaline serine protease (ASP) from Cellulomonas strain 69B4, which covered at least 12 variants on each of its 189 residue positions. All members of this collection were screened for protease activity on three substrates (keratin, casein, and succinyl‐alanine‐alanine‐proline‐phenylalanine‐p‐nitroanilide), for thermostability and for stability in the presence of 0.06 % sodium dodecylbenzenesulfonate—a linear alkylbenzenesulfonate (LAS). The performance of each mutant was scored as the apparent change of free energy in the process of interest, relative to WT ASP (ΔΔG app). This value was calculated by using the following formula: ΔΔG app=−RT ln(P var/P wt), where P var is the performance value of the variant and P wt is the performance value of WT ASP. Therefore, negative ΔΔG app values indicate improved performance of the variant, relative to WT ASP. The majority (84–94 %) of the 2851 analyzed single mutants performed worse than WT ASP on the bases of activity or stability. Interestingly, 5–10 % of the positions in ASP contained mutations that were deleterious for all analyzed properties. Because the residues at these positions were also highly conserved in 20 nonredundant homologues of ASP, the authors concluded that these residues are required for the structural fold of the enzyme. Another remarkable finding was that most mutations that led to improved protease activity were at positions located outside the enzyme's active site. For example, the closest residue position at which mutations led to improved protease activity on keratin (Arg14) was 13 Å away from the catalytic Ser137. Therefore, targeted saturation mutagenesis on active site residues would most likely not have led to the identification of improved mutants for this reaction.
One unique advantage of this mutability landscape analysis is that it provides information on mutations that lead to the simultaneous improvement of multiple properties. For example, four positions at which mutations led both to improved protease activity towards keratin and to improved stability in the presence of LAS were identified. These four positions were simultaneously randomized, and the quality of the resulting library was determined on the basis of the performance of 64 randomly picked mutants in both the activity and the LAS stability assay. The average observed performance of these mutants exceeded the expected average performance of the library members, calculated on the basis of the assumption of additive effects of single mutations at the four sites. This indicated that information from the mutability landscape of an enzyme can provide valuable guidance for enzyme engineering.
5.2. Mutability landscapes for improved detergent stability
The large α/β‐hydrolase fold superfamily includes a broad range of synthetically useful enzymes.29 Fulton et al. generated complete mutability landscapes of Bacillus subtilus lipase A (BSLA), an α/β‐hydrolase fold superfamily member, for stability in the presence of different detergents.30 To this end, the authors constructed a defined collection of single mutants, covering each amino acid substitution at each residue position of BSLA. This collection was constructed by performing site‐saturation mutagenesis at each of the 181 residue positions in BSLA. The resulting 181 libraries were subsequently used to transform Escherichia coli cells. From each library, plasmid DNA was isolated from 102 randomly picked colonies and sequenced to determine whether all 19 possible single mutants per residue position were present. Missing single mutants were separately constructed to ensure that the collection of mutants covered all 3439 possible single mutants of BSLA. Subsequently, the residual activity of each mutant was assessed after incubation with varying concentrations of four detergents with different physicochemical properties (i.e., cationic, anionic, zwitterionic, and non‐ionic). The enzymatic activities of the BSLA mutants was measured with the aid of the screening substrate p‐nitrophenyl butyrate (1), which after enzymatic hydrolysis yields p‐nitrophenol (2), which can be detected by UV spectroscopy (Scheme 1 A). By plotting the differences in the residual activity of each mutant relative to that of WT BSLA, the authors were able to identify residue positions at which mutations led to increased tolerance or increased sensitivity towards detergents. By comparing this data with the crystallographic B factors of BSLA, the authors observed that only two of the five regions in BSLA with high B factors contained SDS‐tolerant variants, thus suggesting that B factors are not a good predictor for hotspot positions that can be targeted to enhance detergent stability. Additionally, the authors observed that 84 % of the hotspots for detergent tolerance were located on surface‐exposed sites and that mainly substitutions to aromatic or charged residues, along with cysteine, improved detergent tolerance. This prompted the authors to suggest an optimized mutagenesis strategy based on the use of degenerate codons to introduce only those amino acids at solventexposed sides, for efficiently improving the stability of other (BSLA) α/β‐hydrolase fold enzymes.
Scheme 1.
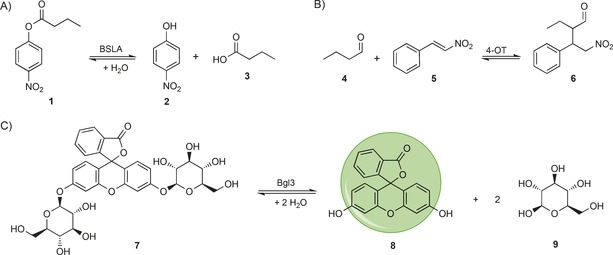
Screening reactions used to generate mutability landscapes for enzymatic activity. A) The BSLA‐catalyzed hydrolysis of p‐nitrophenyl butyrate (1), yielding p‐nitrophenol (2) and butyric acid (3). B) The 4‐OT‐catalyzed Michael‐type addition of butanal (4) to trans‐β‐nitrostyrene (5), yielding chiral γ‐nitroaldehyde 6. C) The Bgl3‐catalyzed glycoside‐bond cleavage of fluorescein di‐(β‐d‐glucopyranoside) (7), yielding fluorescein (8) and β‐d‐glucopyranose (9).
5.3. New catalytic functions and enantioselectivity
Poelarends and co‐workers recently reported the use of mutability landscapes of the promiscuous enzyme 4‐oxalocrotonate tautomerase (4‐OT) to guide the engineering of new biocatalysts for Michael‐type additions.31 The enzyme 4‐OT is extremely promiscuous, and its small monomer size of only 62 residues makes it an ideal template for mutability‐landscape‐guided enzyme engineering.32 One of 4‐OT's promiscuous activities is the Michael‐type addition of unmodified aldehydes to nitroalkenes to yield chiral γ‐nitroaldehydes, which are valuable precursors for γ‐aminobutyric‐acid‐based (GABA‐based) pharmaceuticals.33, 34, 35 To generate the mutability landscapes, a defined collection of 4‐OT genes was constructed; these encoded at least 15 of the 19 possible variants at each residue position. Each member of this collection was individually characterized for the level of soluble protein expression, tautomerase, and “Michaelase” activities, and enantioselectivity.
The level of soluble protein expression was determined for each mutant by quantitative densitometry on SDS gels. After the 4‐OT concentrations in the cell‐free extracts had been quantified, the cell‐free extracts were used in the activity and enantioselectivity assessments. All of the activities were related to the amount of soluble 4‐OT enzyme, thus yielding the specific activities of each mutant. An overview of the effect of each single mutant on both the tautomerase and the “Michaelase” activities (Figure 2 A) provides insight into the numbers of neutral amino acid substitutions, essential residues for one or both activities, and beneficial mutations. The positions at which mutations led to improved “Michaelase” activity (His6, Ala33, Met45, and Phe50) were simultaneously varied in a focused library, which covered only those amino acid substitutions at each position that improved activity. This led to the identification of a triple mutant (H6M/A33E/F50V) that showed an ≈15‐fold improvement in “Michaelase” activity.
Figure 2.
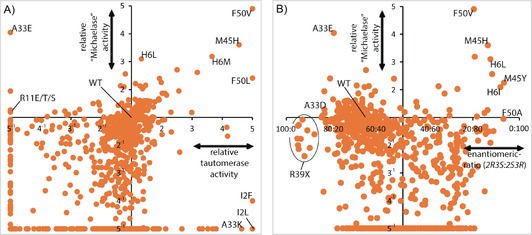
Mutability landscape data derived from van der Meer et al.31 A) Mutational effects on 4‐OT's tautomerase activity, plotted versus the mutational effects on 4‐OT's promiscuous Michael‐type addition activity. B) Mutational effects on 4‐OT's enantioselectivity in the Michael‐type addition reaction, plotted versus the mutational effects on 4‐OT's activity in the Michael‐type addition reaction.
To screen for enantioselectivity, the authors assayed the enzymatic Michael‐type addition of butanal (4) to trans‐β‐nitrostyrene (5, Scheme 1 B). After the progress of the reaction had been followed by UV spectroscopy, the reaction mixtures were cleared by ultrafiltration and directly injected into a RP‐HPLC system with a chiral stationary phase. Each single mutant was individually analyzed in this way, and this allowed for the determination both of the “Michaelase” activity and of the enantiomeric ratio of the enzymatically produced 2‐ethyl‐4‐nitro‐3‐phenylbutanal (6, Scheme 1 B). When the activity data are plotted versus the enantioselectivity data (Figure 2 B) it becomes apparent that single‐amino‐acid substitutions can have significant effects on improving, inverting, or losing the enantioselectivity. In the case of 4‐OT, an inversion in enantioselectivity was required to produce precursors for the biologically more active enantiomers of the GABA analogues. Therefore, the authors made combinations of the single mutants that had displayed the most pronounced inversions in enantioselectivity (H6I, M45Y, and F50A) leading to the identification of 4‐OT M45Y/F50A which produced the 2S,3R enantiomer of 2‐ethyl‐4‐nitro‐3‐phenylbutanal (6) with an er of 96:4. This double mutant also showed inverted enantioselectivity, relative to WT 4‐OT, in the addition of acetaldehyde to various nitroalkenes, producing the pharmaceutically relevant enantiomers of GABA precursors in enantiomeric ratios of up to 97:3. The “Michaelase” activity of M45Y/F50A was also improved relative to WT 4‐OT; this was not surprising because the mutability landscape already indicated that single mutations at these positions led to improved activity (Figure 2 B). Structural analysis of the M45Y/F50A mutant revealed the opening of a hydrophobic pocket capable of accommodating the phenyl group of trans‐β‐nitrostyrene (5) in the active site of 4‐OT. It seems likely that this new binding pocket is related to the inverted enantioselectivity of M45Y/F50A. The simultaneous improvement in activity and enantioselectivity underlines the usefulness of mutability landscapes in enzyme engineering.
6. Generating Mutability Landscapes by Using Deep Mutational Scanning
As mentioned above, it requires significant effort and resources to generate a defined gene collection encoding all single mutants of an enzyme. This bottleneck can be circumvented by using deep mutational scanning. For this, diversity in the WT enzyme is created, followed by high‐throughput sorting of active mutants from inactive mutants (e.g., by flow cytometry, microfluidics, phage display, or growth selection). This allows for the enrichment of active mutants. Conducting next‐generation sequencing enables the comparison of the DNA read counts in the sorted library relative to the unsorted (or preselected) library (Figure 1).36, 37 By this approach, the enrichment factor (E factor, given by the ratio of the DNA read count of a specific variant in the sorted library to that in the unsorted library) of each mutant can be determined and compared to the E factor of the WT enzyme. A mutability landscape based on these E factors can be generated, thus mapping the beneficial, neutral, and detrimental effects of (nearly) all single‐amino‐acid substitutions of an enzyme. However, to obtain full coverage a high degree of oversampling is required, and this demands high throughputs both for the functional sorting and for the sequencing. Several examples of the use of mutability landscapes based on deep mutational scanning to investigate protein–DNA or protein–protein interactions can be found in the literature.25, 26, 27, 36, 37 Recently, the first studies on the generation of mutability landscapes of enzymes by use of deep mutational scanning have been published: these are discussed below.
6.1. Mutability landscape generation by using microfluidics
β‐Glucosidases are enzymes that cleave β‐d‐glucosidic bonds by hydrolysis, which can be an important step in the conversion of biomass into fermentable sugars.38 Romero et al. have generated mutability landscapes of a β‐glucosidase from Streptomyces sp. (Bgl3) by means of a deep mutational scanning approach in combination with a microfluidics‐based sorting system.39 For this, they generated a random mutant library of Bgl3 by error‐prone PCR, with an average of 3.8 mutations per Bgl3 gene. This library was first analyzed by high‐throughput sequencing to establish the DNA read counts in the unsorted library. After this library had been expressed in E. coli BL21(DE3), single E. coli cells were encapsulated in microdroplets containing lysing agents and fluorescein di‐(β‐d‐glucopyranoside) (7), which is a fluorogenic substrate for Bgl3 (Scheme 1 C). Any microdroplet containing an active Bgl3 variant was sorted on the basis of fluorescence, with use of a microfluidics device. In this way the authors achieved a throughput of 100 s−1.
DNA was retrieved from the sorted microdroplets and sequenced by Illumina sequencing. After analysis of 107 variants, the effects of the mutations were determined from the changes in the frequency of occurrence of each mutation before and after the functional sorting. Because of the disadvantage of working with an error‐prone library, mainly those amino acid substitutions that require one nucleotide mutation per codon were accessed in this study. Therefore, only 31 % of all possible single‐amino‐acid substitutions were analyzed. Nevertheless, the generated mutability landscape gave important insights into sequence–function relationships of the enzyme. For example, two essential residues (Lys461 and Asn307) located outside the enzyme's active site were identified in this study. Crystal structure analysis of Bgl3 revealed that Lys461 is part of a network of salt bridges, which suggests that this residue plays a role in the structural stability of the enzyme. Asn307 is within hydrogen‐bonding distance of Glu178, which is the catalytic acid/base in Bgl3 (Scheme 2). It was therefore suggested that Asn307 induced a crucial shift in the pK a of this catalytic residue.
Scheme 2.
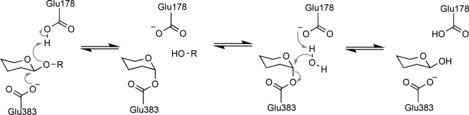
General mechanism of glucosidic‐bond cleavage by Bgl3 (derived from Zechel et al.40).
Single mutations that improve the thermostability of Bgl3 have been identified in a slightly modified microfluidics screening protocol including a heat challenge (65 °C for 10 min). Again, 107 enzyme variants were analyzed, revealing several single mutants with improved thermostability including mutant S325C. Further characterization of this mutant revealed a 5.3 °C increase in T 50 relative to WT Bgl3.
6.2. Mutability landscape generation by using growth selection
Aminoglycoside‐3′‐phosphotransferase II [APH(3′)II] is a kinase involved in antibiotic resistance that catalyzes the phosphorylation of aminoglycoside antibiotics leading to their inactivation. Melnikov et al. performed a single‐substitution mutational scan on APH(3′)II by analyzing the effects of these mutations on the enzyme's activity and substrate specificity, by using kanamycin and five other aminoglycoside antibiotics.41 For this, the genes coding for each single mutant were individually prepared by a microarray‐based DNA synthesis (mutagenesis by integrated tiles “MITE”) approach. All synthesized genes were pooled in equimolar amounts and used to transform E. coli cells. These cells were cultured in liquid medium in the presence of aminoglycoside antibiotics, thereby selecting for cells that express an active APH(3′)II mutant. After this selection, DNA was isolated from the surviving cells and sequenced by an Illumina sequencing approach to determine the frequency of occurrence of each mutant. By determining the difference in abundance of each mutant before and after selection, the authors were able to map the effects of all single‐amino‐acid substitutions on activity on six aminoglycoside antibiotics. From these maps, amino acid substitutions that led to shifts in substrate specificity either towards kanamycin or towards one of the other five tested aminoglycoside antibiotics were identified. By making combinations of these specific amino acid substitutions, the authors engineered five pairs of APH(3′)IIs that either favor or disfavor any of the tested antibiotics over kanamycin. For example, Paro+ and Paro− are a pair of APH(3′)IIs engineered either to favor or to disfavor paromomycin over kanamycin. Paro+ showed unaltered activity for paromomycin (MIC=2000–4000 μg mL−1) relative to WT APH(3′)II but a decreased activity for kanamycin (MIC 31.3 μg mL−1). Paro− had a decreased activity for paromomycin (MIC=62.5 μg mL−1) relative to WT APH(3′)II but unaltered activity towards kanamycin (MIC=2000 μg mL−1) relative to WT APH(3′)II. This remarkable shift in substrate specificity underlines the applicability of mutability landscapes to identification of hotspots for enzyme engineering.
6.3. Mutability landscape generation by use of phage display
E3‐ubiquitin ligases are enzymes that catalyze ubiquitin transfer from E2‐ubiquitin‐conjugating enzymes to lysine residues of substrate proteins. This ubiquitination promotes degradation of the substrate protein, which is a crucial process for homeostasis. Ube4b, for example, functions as an E3‐ubiquitin ligase, which has been linked to cancer pathogenesis because it ubiquitylates the p53 tumor suppressor in vivo.42 A mutability landscape for the activity of the Ube4b enzyme has been generated and analyzed in order to identify the molecular determinants that modulate the ligase activity of these E3 ligases.43 A deep‐mutational scanning approach was conducted on the U‐box domain of Ube4b. This is the active domain of the enzyme, which can perform an auto‐ubiquitination. Libraries with on average two random nucleotide mutations per gene were generated, sequenced, and subsequently displayed on bacteriophages. Bacteriophages that displayed active (auto‐ubiquitinated) U‐box domains were then enriched with the aid of antibodies against (FLAG)‐ubiquitin. Because these antibodies were immobilized on agarose beads, unbound bacteriophages could be washed away (Figure 3). DNA was isolated from enriched bacteriophages and subsequently sequenced by use of Illumina technology. By comparing the DNA read counts of each mutation before and after the enrichment, an E factor was calculated. In this way, 98 289 unique mutant enzymes were characterized, 932 of them single mutants. Mapping of the E factors of these single mutants revealed that some regions (e.g., loops 1 and 2, as well as helix 1) were less tolerant to mutations than other portions of the U‐box domain. Interestingly, several single mutants with improved activity relative to WT could be identified from this mutability landscape. Combining these beneficial single mutations had a synergistic effect and resulted in two double mutants (M1124V/N1142T and D1139N/N1142T) each with a 22‐fold‐enhanced ubiquitin ligase activity relative to the WT U‐box domain. Mechanistic studies on these improved single and double mutants revealed that all beneficial mutations either enhanced the ligase activity by improving binding of the U‐box domain to the E2‐ubiquitin complex or by improving allosteric activation of the E2‐ubiquitin complex. This illustrates the fact that beneficial mutations can be useful both for the generation of superior enzymes and to provide useful insight into enzyme mechanisms.
Figure 3.
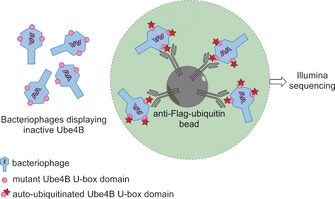
Enrichment procedure for bacteriophages displaying active (auto‐ubiquitylated) U‐box domains of Ube4b. Bacteriophages displaying inactive Ube4B U‐box domains do not bind to anti‐Flag‐ubiquitin beads and are washed away. Only the bacteriophages that display active auto‐ubiquitylated Ube4B U‐box domains bind to the anti‐Flag‐ubiquitin beads and are sequenced.43
7. Summary and Outlook
Currently, most studies on enzyme mutability landscapes have focused on small enzymes (Table 1); this reflects the required costs and effort to generate a mutability landscape. When using a defined collection of single mutants, the bottleneck lies in the generation of this defined mutant gene collection. Currently, PCR‐based site‐directed mutagenesis techniques are mostly used for the generation of the mutants. Other more recently developed mutagenesis techniques include chemoenzymatic methods (e.g., sequence saturation mutagenesis “SeSaM”),44 microarray‐based DNA synthesis (e.g., MITE)41 or nonsense‐suppressor tRNA methods.25 The development of these methods might reduce the required amount of effort and costs to generate a defined collection of single mutants. Moreover, because of the ever decreasing costs of commercially available synthetic DNA, the most economical way to obtain a defined collection of single mutants of an enzyme might be DNA synthesis.45 In the case of deep mutational scanning the bottleneck for generating mutability landscapes lies in the high‐throughput sequencing and high‐throughput screening. Both of these techniques are rapidly evolving,36, 37, 46 which might facilitate the generation of mutability landscapes by use of deep mutational scanning.
Table 1.
Available studies on experimental mutability landscape analyses of enzymes.
| Type of | Defined | Deep | Investigated | Used for | Size of | Ref. |
|---|---|---|---|---|---|---|
| enzyme | mutant | mutational | enzymatic | hotspot | enzyme | |
| collection | scanning | property[a] | identification[b] | |||
| protease | X | A, S, SS | X | 189 | 28 | |
| lipase | X | A, S | 181 | 30 | ||
| tautomerase/ | X | A, E, ES, SS | X | 62 | 31 | |
| “Michaelase” | ||||||
| glucosidase | X | A, S | 500 | 39 | ||
| kinase | X | A, SS | X | 263 | 41 | |
| ligase | X | A | X | 102[c] | 43 |
[a] S: stability. A: activity. E: expression. ES: enantioselectivity. SS: substrate specificity. [b] The box is checked when combinatorial mutagenesis was conducted on hotspots that were identified in the mutability landscape. [c] Only the U‐box domain of Ube4b was analyzed.
In conclusion, mutability landscapes are a powerful tool with which to identify “hotspots” at any place in the amino acid sequence of an enzyme. These “hotspots” can be used as targets for combinatorial mutagenesis to yield superior enzymes with improved catalytic properties, stability, or even new enzymatic activities. The generation of mutability landscapes for several properties of one enzyme (e.g., stability and activity or activity and enantioselectivity) provides a unique opportunity to select mutations that are beneficial for either one or both of these properties. Furthermore, mutability landscapes can be used to advance our understanding of sequence–function relationships in enzymes because they provide systematic information on neutral, beneficial, and detrimental amino acid substitutions. Both detrimental and beneficial mutations can be extremely helpful for elucidation of enzyme mechanisms. Neutral mutations are thought to have an important role in natural enzyme evolution, because they may result in “neutral drift”.47, 48 Owing to these advantages, combined with the technical advances in high‐throughput screening and DNA sequencing, we expect that mutability landscape analysis will become accessible for larger enzymes, and more commonly used for enzyme engineering in the coming years.
Acknowledgements
The authors acknowledge funding from the Division of Earth and Life Sciences of the Netherlands Organisation of Scientific Research (ALW grant 820.02.021), the European Research Council under the European Community's Seventh Framework Programme (FP7/2007‐2013)/ERC Grant agreement no. 242293, and the European Union's Horizon 2020 research and innovation programme under grant agreement no. 635595.
J.-Y. van der Meer, L. Biewenga, G. J. Poelarends, ChemBioChem 2016, 17, 1792.
References
- 1. Bornscheuer U. T., Huisman G. W., Kazlauskas R. J., Lutz S., Moore J. C., Robins K., Nature 2012, 485, 185–194. [DOI] [PubMed] [Google Scholar]
- 2. Nestl B. M., Hammer S. C., Nebel B. A., Hauer B., Angew. Chem. Int. Ed. 2014, 53, 3070–3095; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 3132–3158. [Google Scholar]
- 3. Reetz M. T., Angew. Chem. Int. Ed. 2011, 50, 138–174; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 144–182. [Google Scholar]
- 4. Bommarius A. S., Paye M. F., Chem. Soc. Rev. 2013, 42, 6534–6565. [DOI] [PubMed] [Google Scholar]
- 5. Homaei A. A., Sariri R., Vianello F., Stevanato R., J. Chem. Biol. 2013, 6, 185–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Illanes A., Cauerhff A., Wilson L., Castro G. R., Bioresour. Technol. 2012, 115, 48–57. [DOI] [PubMed] [Google Scholar]
- 7. Sheldon R. A., van Pelt S., Chem. Soc. Rev. 2013, 42, 6223–6235. [DOI] [PubMed] [Google Scholar]
- 8. Kiss G., Çelebi-Ölçüm N., Moretti R., Baker D., Houk K. N., Angew. Chem. Int. Ed. 2013, 52, 5700–5725; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 5810–5836. [Google Scholar]
- 9. Kries H., Blomberg R., Hilvert D., Curr. Opin. Chem. Biol. 2013, 17, 221–228. [DOI] [PubMed] [Google Scholar]
- 10. Zanghellini A., Curr. Opin. Biotechnol. 2014, 29, 132–138. [DOI] [PubMed] [Google Scholar]
- 11. Khersonsky O., Tawfik D. S., Annu. Rev. Biochem. 2010, 79, 471–505. [DOI] [PubMed] [Google Scholar]
- 12. Jensen R. A., Annu. Rev. Microbiol. 1976, 30, 409–425. [DOI] [PubMed] [Google Scholar]
- 13. O'Brien P. J., Herschlag D., Chem. Biol. 1999, 6, R91–R105. [DOI] [PubMed] [Google Scholar]
- 14. Renata H., Wang Z. J., Arnold F. H., Angew. Chem. Int. Ed. 2015, 54, 3351–3367; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 3408–3426. [Google Scholar]
- 15. Turner N. J., Nat.Chem.Biol. 2009, 5, 567–573. [DOI] [PubMed] [Google Scholar]
- 16. Sebestova E., Bendl J., Brezovsky J., Damborsky J. in Methods in Molecular Biology, Vol. 1179: Directed Evolution Library Creation: Methods and Protocols (Eds.: E. M. J. Gillam, J. N. Copp, D. Ackerley), Humana, Totowa, 2014, pp. 291–314. [DOI] [PubMed] [Google Scholar]
- 17. Wijma H. J., Floor R. J., Janssen D. B., Curr. Opin. Struct. Biol. 2013, 23, 588–594. [DOI] [PubMed] [Google Scholar]
- 18. Reetz M. T., Wang L., Bocola M., Angew. Chem. Int. Ed. 2006, 45, 1236–1241; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 1258–1263. [Google Scholar]
- 19. Reetz M. T., Kahakeaw D., Sanchis J., Molecular BioSystems 2009, 5, 115–122. [DOI] [PubMed] [Google Scholar]
- 20. Chica R. A., Doucet N., Pelletier J. N., Curr. Opin. Biotechnol. 2005, 16, 378–384. [DOI] [PubMed] [Google Scholar]
- 21. Fox R. J., Davis S. C., Mundorff E. C., Newman L. M., Gavrilovic V., Ma S. K., Chung L. M., Ching C., Tam S., Muley S., Grate J., Gruber J., Whitman J. C., Sheldon R. A., Huisman G. W., Nat. Biotechnol. 2007, 25, 338–344. [DOI] [PubMed] [Google Scholar]
- 22. Hecht M., Bromberg Y., Rost B., J. Mol. Biol. 2013, 425, 3937–3948. [DOI] [PubMed] [Google Scholar]
- 23. Hecht M., Bromberg Y., Rost B., BMC Genomics 2015, 16 Suppl. 8, S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sim N., Kumar P., Hu J., Henikoff S., Schneider G., Ng P. C., Nucleic Acids Res. 2012, 40, W452–W457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Markiewicz P., Kleina L. G., Cruz C., Ehret S., Miller J. H., J. Mol. Biol. 1994, 240, 421–433. [DOI] [PubMed] [Google Scholar]
- 26. Whitehead T. A., Chevalier A., Song Y., Dreyfus C., Fleishman S. J., De Mattos C., Myers C. A., Kamisetty H., Blair P., Wilson I. A., Baker D., Nat. Biotechnol. 2012, 30, 543–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. R. N. McLaughlin, Jr. , Poelwijk F. J., Raman A., Gosal W. S., Ranganathan R., Nature 2012, 491, 138–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Estell D. A., Aehle W., U.S. patent 12/778 915, 2011.
- 29. Jochens H., Hesseler M., Stiba K., Padhi S. K., Kazlauskas R. J., Bornscheuer U. T., ChemBioChem 2011, 12, 1508–1517. [DOI] [PubMed] [Google Scholar]
- 30. Fulton A., Frauenkron-Machedjou V. J., Skoczinski P., Wilhelm S., Zhu L., Schwaneberg U., Jaeger K. E., ChemBioChem 2015, 16, 930–936. [DOI] [PubMed] [Google Scholar]
- 31. van der Meer J. Y., Poddar H., Baas B. J., Miao Y., Rahimi M., Kunzendorf A., van Merkerk R., Tepper P. G., Geertsema E. M., Thunnissen A. M., Quax W. J., Poelarends G. J., Nat. Commun. 2016, 7, 10911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Baas B. J., Zandvoort E., Geertsema E. M., Poelarends G. J., ChemBioChem 2013, 14, 917–926. [DOI] [PubMed] [Google Scholar]
- 33. Geertsema E. M., Miao Y., Tepper P. G., de Haan P., Zandvoort E., Poelarends G. J., Chem. Eur. J. 2013, 19, 14407–14410. [DOI] [PubMed] [Google Scholar]
- 34. Miao Y., Geertsema E. M., Tepper P. G., Zandvoort E., Poelarends G. J., ChemBioChem 2013, 14, 191–194. [DOI] [PubMed] [Google Scholar]
- 35. Zandvoort E., Geertsema E. M., Baas J. B., Quax W. J., Poelarends G. J., Angew. Chem. Int. Ed. 2012, 51, 1240–1243; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 1266–1269. [Google Scholar]
- 36. Fowler D. M., Fields S., Nat. Methods 2014, 11, 801–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shin H., Cho B., Int. J. Mol. Sci. 2015, 16, 23094–23110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Perez-Pons J. A., Cayetano A., Rebordosa X., Lloberas J., Guasch A., Querol E., FEBS J. 1994, 223, 557–565. [DOI] [PubMed] [Google Scholar]
- 39. Romero P. A., Tran T. M., Abate A. R., Proc. Natl. Acad. Sci. USA 2015, 112, 7159–7164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zechel D. L., Withers S. G., Acc. Chem. Res. 2000, 33, 11–18. [DOI] [PubMed] [Google Scholar]
- 41. Melnikov A., Rogov P., Wang L., Gnirke A., Mikkelsen T. S., Nucleic Acids Res. 2014, 42, e112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wu H., Pomeroy S. L., Ferreira M., Teider N., Mariani J., Nakayama K. I., Hatakeyama S., Tron V. A., Saltibus L. F., Spyracopoulos L., Leng R. P., Nat. Med. 2011, 17, 347–355. [DOI] [PubMed] [Google Scholar]
- 43. Starita L. M., Pruneda J. N., Lo R. S., Fowler D. M., Kim H. J., Hiatt J. B., Shendure J., Brzovic P. S., Fields S., Klevit R. E., Proc. Natl. Acad. Sci. USA 2013, 110, E1263–E1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ruff A. J., Kardashliev T., Dennig A., Schwaneberg U. in Methods in Molecular Biology, Vol. 1179: Directed Evolution Library Creation (Eds.: E. M. J. Gillam, J. N. Copp, D. Ackerley), Humana, Totowa, 2014, pp. 45–68. [DOI] [PubMed] [Google Scholar]
- 45. Carlson R., Nat. Biotechnol. 2009, 27, 1091–1094. [DOI] [PubMed] [Google Scholar]
- 46. Wójcik M., Telzerow A., Quax W. J., Boersma Y. L., Int. J. Mol. Sci. 2015, 16, 24918–24945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bloom J. D., Romero P. A., Lu Z., Arnold F. H., Biol. Direct 2007, 2, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Soskine M., Tawfik D. S., Nat. Rev. Genet. 2010, 11, 572–582. [DOI] [PubMed] [Google Scholar]