

Abstract
Background
Kidney injury molecule‐1 (Kim‐1) has been validated as a urinary biomarker for acute and chronic renal damage. The expression of Kim‐1 mRNA is also activated by acute kidney injury induced by cisplatin in rodents and humans. To date, the measurement of Kim‐1 expression has not fully allowed the detection of in vitro cisplatin nephrotoxicity in immortalized culture cells, such as human kidney‐2 cells and immortalized proximal tubular epithelial cells.
Methods
We measured the augmentation of Kim‐1 mRNA expression after the addition of cisplatin using immortalized S3 cells established from the kidneys of transgenic mice harboring temperature‐sensitive large T antigen from Simian virus 40.
Results
A mouse Kim‐1 gene luciferase reporter in conjunction with an Hprt gene reporter detected cisplatin‐induced nephrotoxicity in S3 cells. These two reporter genes were contained in a mouse artificial chromosome, and two luciferases that emitted different wavelengths were used to monitor the respective gene expression. However, the Kim‐1 reporter gene failed to respond to cisplatin in A9 fibroblast cells that contained the same reporter mouse artificial chromosome, suggesting cell type‐specificity for activation of the reporter.
Conclusions
We report the feasibility of measuring in vitro cisplatin nephrotoxicity using a Kim‐1 reporter gene in S3 cells.
Keywords: cisplatin, Kim‐1, mouse artificial chromosome, nephrotoxicity, reporter gene, S3 cells
1. INTRODUCTION
The kidney is a major organ for maintaining homeostasis in the body and is susceptible to certain drugs, environmental toxicants and protein overload.1, 2, 3 In vivo kidney damage is identified by monitoring serum creatinine or blood urea nitrogen levels, although the correlation of these markers with injury is sometimes poor.4 Thus, there has been an intensive effort to identify in vivo biomarkers that can be selectively used to monitor kidney damage at an earlier stage.5 Among these markers, kidney injury molecule‐1 [Kim‐1, also known as T cell immunoglobulin and mucin‐1 (TIM‐1) and hepatitis A virus cellular receptor 1 (Havcr1)] is a type I cell membrane glycoprotein.6, 7 Kim‐1 mRNA levels are elevated more than any known gene in rodents and humans after the initiation of kidney injury.8, 9, 10, 11 The ectodomain of Kim‐1 protein is shed from proximal tubular kidney epithelial cells into the urine after injury. Urinary Kim‐1 has been shown to be a sensitive and early diagnostic indicator of renal injury in a variety of acute and chronic rodent kidney injury models.12
Currently, in vitro screening for nephrotoxicity is based on measuring cell death, which is indicative of nonspecific cytotoxicity rather than injury or stress specific to kidney cells.13, 14, 15 Recent studies investigated the use of measuring the mRNA expression or protein levels of Kim‐1 and other biomarkers to detect nephrotoxicity in vitro. 16, 17, 18 However, Kim‐1 expression was not markedly induced after cisplatin treatment in immortalized human cell lines such as human kidney‐2 cells (HK‐2) or immortalized renal proximal tubular epithelial cells (IM‐PTECs).19, 20 To date, immortalized kidney cell lines, in which Kim‐1 expression is reproducibly activated in response to cisplatin, have not been reported under monolayer culture conditions.
Hosoyamada et al.21 and Takeda et al.22 reported immortalized mouse S1, S2 and S3 cell lines from transgenic mice harboring temperature sensitive Simian virus 40 (SV40) T antigen. In their studies, the proximal tubule region (S1, S2 and S3) of the kidney was microdissected and cell lines from each segment were established.21, 22 These cells were shown to partially retain some kidney characteristics23, 24 and were used in nephrotoxicity studies.25, 26, 27 S3 cells were shown to be more sensitive to cisplatin compared to S1 or S2 cells, which is consistent with the S3 segment of rat kidney being more susceptible to cisplatin compared to the S1 and S2 segments.21, 23, 28, 29
The mouse artificial chromosome (MAC) is derived from mouse chromosome 11 and was established by deleting known or predicted genes via telomere truncation methods.30 MAC is maintained in cells as an independent chromosome. Therefore, it has unique characteristics and comprises a large fragment. In addition, introduced fragments containing adequate cis‐elements are assumed to be intrinsically expressed. Because genes in the MAC are not integrated into random loci in the host genome, they are less affected by surrounding chromatin states than genes in other vectors, such as conventional plasmids and retrovirus.31, 32 Moreover, reporter expression from MAC is similar among independent cell clones that contain the same reporter genes,31 and this feature is beneficial to establishing reporter cells. We employed a MAC vector containing Kim‐1 and Hprt reporter genes in the present study.
2. MATERIALS AND METHODS
2.1. Cell culture, compound treatment and cell viability assay
Immortalized mouse S1, S2 and S3 cell lines were cultured in Dulbecco's modified Eagle's medium (DMEM)/F‐12 (Gibco, Gaithersburg, MD, USA) supplemented with 1 μg/ml insulin (Sigma, St Louis, MO, USA), 10 μg/ml transferrin (Gibco), 10 ng/ml recombinant human epidermal growth factor (Peprotech, Rocky Hill, NJ, USA), 5% fetal bovine serum (FBS), 100 U/mL penicillin and 100 μg/mL streptomycin.21 Cells were maintained in 5% CO2 at 33 °C. A9 cells with multiple integration site‐containing mouse artificial chromosome (MI‐MAC) were cultured in DMEM supplemented with 10% FBS and penicillin/streptomycin at 37 °C.
Cisplatin (Wako Pure Chemicals, Osaka, Japan) was dissolved in saline (5 mM) at the time of use. Gentamicin, acetaminophen (AAP) and mannitol were purchased from Wako Pure Chemicals or Sigma, and were dissolved in culture medium (50 mg/ml), DMSO (1 M) and water (1 M), respectively. Cells were seeded at density of 2–10 × 104/well in a 96‐well cell culture plate (#165306; Thermo Scientific, Rochester, NY, USA), 24 h prior to assay initiation. Compounds diluted in culture medium were incubated with cells for 72 h. The cells were washed with phosphate‐buffered saline (PBS) and incubated with fresh medium containing 10% Alamar Blue (AbD Serotec, Oxford, UK) and then cultured for 1 h, followed by measuring fluorescence (excitation =560 nm, emission =590 nm) to monitor the number of living cells. In some experiments, medium containing chemicals was removed and cells were washed with PBS at 6 or 16 h after the addition of compound. Cells were then cultured in 100 μl of fresh medium for a further 42–56 h for a reverse transcription‐polymerase chain reaction (RT‐PCR) or a luciferase assay.
2.2. Real‐time RT‐PCR
Total RNA was prepared using RNeasy PLUS (Qiagen, Hilden, Germany) and 1 μg of RNA was used to synthesize cDNA using a Superscript VILO kit (Invitrogen, Carlsbad, CA, USA) in accordance with manufacturer's instructions. Real‐time PCR reactions were carried out with Thunderbird SYBR qPCR Mix (Toyobo, Osaka, Japan) and a StepOne Plus Real Time PCR System (Applied Biosystems, Carlsbad, CA, USA). We generated standard curves using a dilution series of cDNA for quantification. PCR primers are listed in the Supporting information (Table S1).
2.3. Reporter construction and microcell‐mediated chromosome transfer (MMCT).
Detailed procedures for the construction of a MAC vector containing mouse Kim‐1 reporter gene are described in the Supporting information (Doc. S1). Details on the mouse Hprt reporter gene have been described previously.33 Briefly, SLG and SLR3 luciferase reporter units34 (Toyobo), which emit green and red light, respectively, were inserted into a modified (or retrieved) P1 artificial chromosome (PAC) containing mouse Kim‐1 gene promoter (11 kb) or Hprt gene promoter (20 kb), respectively, together with the insulator sequence of a DNase I hypersensitive site 4 (HS4) from the chicken β‐globin gene,35, 36 an antibiotic expression cassette and an integrase recognition site. The coding sequence of SLR3 was modified to reduce the binding sites of transcription factors in its cDNA sequence, without changing the amino acid sequence of PhRED [I212L/N351 K].34 Using integrases (phiC31 for Kim‐1‐SLG and R4 for Hprt‐SLR3), these reporters were sequentially inserted into MI‐MAC in A9 cells.36, 37 The resulting reporter MAC was verified by genomic PCR and transferred into immortalized S3 cells via the MMCT method.36, 37 G418 (400 μg/ml) resistant clones were isolated, and genomic PCR and fluorescence in situ hybridization (FISH) were carried out to confirm the retention of the reporter MAC.36 PCR primers are listed in the Supporting information (Table S2).
2.4. Luciferase assay
We used a cell culture‐treated black well plate (#655090; Greiner, Frickenhausen, Germany) for the luciferase assays. Cells were treated with chemicals and washed with PBS, and then culture medium (50 μl) and an equal volume of Tripluc Luciferase Assay Reagent (Toyobo) were added to the cells. Different wavelengths of luciferase activities from SLG and SLR3 were measured using a Phelios luminometer (ATTO, Tokyo, Japan) equipped with a spectral filter.38 Transmission coefficients of SLG and SLR3 for the F2 (R60) filter were 0.0868 and 0.5817, respectively. At least three independent wells were used to determine the luciferase activity.
3. RESULTS
3.1. Cisplatin represses the cell growth of immortalized S3 cells
We first investigated the cellular toxicity of cisplatin, AAP, gentamicin and mannitol in immortalized S1, S2 and S3 cells. We also used A9 (fibroblast) cells, which are not derived from kidney and which are tumorigenic in nude mice.39 We incubated the cells with compounds for 72 h and performed Alamar Blue assays. Similar to the MTT [3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide] assay, the Alamar Blue reagent (resazurin) detects reduced substrates in response to metabolism in living cells.40 Cisplatin is a widely used chemotherapeutic drug for various cancers that is often limited by its side effect of nephrotoxicity, especially at concentrations greater than 20 μM in the plasma.41, 42 AAP is not only a hepatotoxicant, but also is nephrotoxic.43 Repeated gentamicin treatment for more than 1 week induced kidney injury in rats.11, 12 We used mannitol as a negative control because it does not induce nephrotoxicity in general. As shown in Figure 1, S3 cells were more sensitive to cisplatin compared to S1 and S2 cells and showed a similar susceptibility to A9 cells. At 10 μM of cisplatin, the growth of S1 and S2 cells was marginally repressed and the growth of S3 and A9 cells was severely compromised (Figure 1, left). High concentrations of AAP slightly decreased the cell viability of S1 and S2 cells and moderately decreased the cell viability of S3 and A9 cells. This suggested a cell type‐specific cellular toxicity of AAP in S3 and A9 cells (Figure 1, left). High concentrations of gentamicin (10 mg/ml) moderately inhibited cell growth in all the tested cells (Figure 1, right). Mannitol did not affect cell growth or viability as determined by the Alamar Blue assay (Figure 1, right). Based on these results, we chose S3 cells for further studies of in vitro nephrotoxicity using cisplatin.
Figure 1.
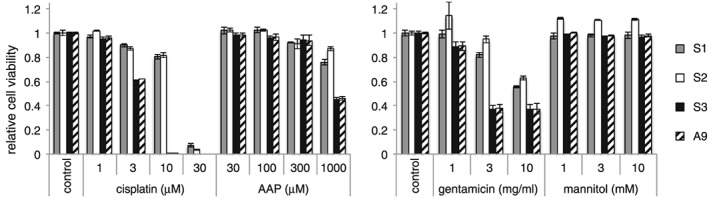
Immortalized S3 cells are more susceptible to cisplatin than S1 and S2 cells. S1, S2, S3 and A9 cells were incubated with the indicated compounds at the indicated concentrations for 72 h, and a proliferation/viability assay using Alamar Blue reagent was performed. Data are the mean ± SD (n = 3)
3.2. Cisplatin enhances Kim‐1 gene expression in S3 cells
The expression of Kim‐1 mRNA was previously reported to be markedly induced in rats in response to renal injury caused by cisplatin.10, 11 We incubated S3 cells with cisplatin for 16 h and then cultured them with fresh medium that did not contain cisplatin for a further 56 h. Using real‐time RT‐PCR, we evaluated the gene expression of Kim‐1 in S3 cells 72 h after the addition of cisplatin, as well as the expression of Gapdh, Hprt and Aqp1 genes. Gapdh and Hprt are housekeeping genes that are commonly used as references in gene expression studies. Aqp1 is a proximal tubule marker of kidney cells.44, 45 As shown in Figure 2, Kim‐1 gene expression was enhanced after cisplatin treatment, although the degree of enhancement might not be as high as in an animal model.10, 11 The addition of 10 μM cisplatin resulted in 20‐ and 13‐fold activation of Kim‐1 when normalized by Hprt and Aqp1, respectively (Figure 2A and 2B). At 30 μM, Kim‐1 mRNA was induced by seven‐ and 15‐fold with reference to Hprt and Aqp1, respectively (Figure 2A and 2B). We did not observe cell death or Kim‐1 gene activation in response to gentamicin in these conditions (data not shown and Figure 2A and 2B).
Figure 2.
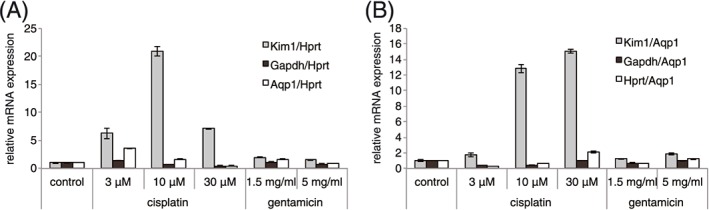
Cisplatin increases Kim‐1 mRNA expression in S3 cells. Cells were incubated with cisplatin or gentamicin for 16 h, and then washed and cultured for a further 56 h in fresh medium without cisplatin or gentamicin. Expression of Kim‐1, Gapdh, Hprt and Aqp1 mRNA was determined by real‐time RT‐PCR. Expression of mRNA was normalized by Hprt (A) or Aqp1 (B). Error bars indicate the SD
We also examined the time course expression of Kim‐1 mRNA after cisplatin treatment. We seeded S3 cells and, after 24 h, we added cisplatin and incubated the cells for an additional 12, 24, 48 and 72 h. Cells were collected at each time point and then real‐time RT‐PCR analysis was performed. Kim‐1 mRNA expression was already enriched in the control sample (0 μM of cisplatin) at 12 and 24 h (see Supporting infromation, Figure S1). This result was consistent with studies reporting that a high level of Kim‐1 mRNA expression was also observed in proliferating or dedifferentiated proximal tubular cells.7, 46, 47 We also found that 12–24 h of treatment with cisplatin did not increase Kim‐1 mRNA expression but rather reduced Kim‐1 expression. Kim‐1 expression in control samples (0 μM of cisplatin) at 48 or 72 h was decreased compared to control samples of 12 or 24 h, and cisplatin enhanced Kim‐1 expression after 48–72 h (see Supporting information, Figure S1). These results are in agreement with previous studies reporting that robust Kim‐1 mRNA and protein induction was observed after 48–72 h of cisplatin treatment in the rat kidney.10, 11, 48 We also examined Kim‐1 protein expression in S3 cell lysate using western blotting (see Supporting information, Figure S2). The expression of Kim‐1 full‐length protein in cell lysate showed a similar time course trend as Kim‐1 mRNA (see Supporting information, Figure S1) when we used Kim‐1 antibody raised against the middle part of Kim‐1 protein (see Supporting information, Figure S2).
3.3. Construction of Kim‐1 and Hprt reporter genes
Having confirmed that cisplatin upregulates Kim‐1 gene expression in S3 cells, we constructed a reporter gene using SLG (green luciferase) under the control of mouse Kim‐1 promoter to detect nephrotoxicity more easily. Because little information regarding Kim‐1 gene regulation has been reported,49, 50 we used a relatively long mouse genomic sequence comprising the putative Kim‐1 promoter region that should be conserved in mammals (see Supporting information, Figure S3). We also used a mouse Hprt promoter‐fused SLR3 (red luciferase) reporter for normalization. These two reporters were inserted into the MAC in A9 cells using a multi‐integrase system (Figure 3; see also Supporting information, Figure S1–S5).30, 33, 37 We transferred the reporter‐containing MAC into S3 cells and selected G418 resistant clones. We confirmed these clones by genomic PCR and FISH to demonstrate that transferred MAC retained Kim‐1 and Hprt reporter genes (Figure 3) and used a representative clone for subsequent assays.
Figure 3.
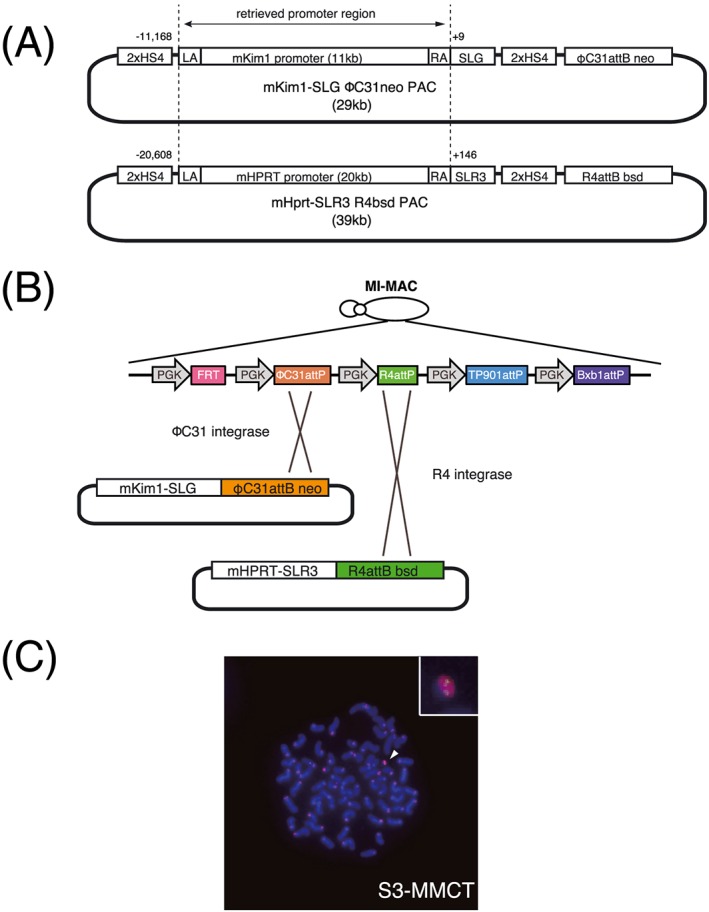
Construction of Kim‐1 and Hprt reporter genes. Schematic map of PAC vectors (A). Vector contains 2 × HS4 insulator, left arm for retrieving, retrieved promoter region from a BAC clone (dotted line), right arm, luciferase reporter, 2 × HS4, and integrase attB‐drug resistant gene. Numbers beside dotted line indicate nucleotide position of each promoter (transcription start site is defined as +1). Detailed map of multi‐integrase platform on MI‐MAC and integrated PAC vector array (B). MI platform has five recognition sites for recombinase/integrase with PGK promoter. The first vector (Hprt‐ SLR3) was integrated at the R4 site and the second (Kim‐1‐SLG) was integrated at the ΦC31 site. FISH analysis of MI‐MAC transferred S3 cells (C). Probes of biotin‐labeled Kim‐1‐SLG and Hprt‐SLR3 containing PAC vector region (green), and digoxigenin‐labeled mouse minor satellite DNA (red) were used to detect reporter MAC. S3 cells with reporter MAC were used. The arrowhead indicates the detected MAC reporter. The insert shows the enlarged MAC reporter vector
3.4. Kim‐1 reporter gene is activated in cisplatin‐treated S3 cells
We treated reporter S3 cells with cisplatin, AAP, gentamicin or mannitol for 72 h and performed luciferase assays to measure both SLG and SLR3 activity (Figure 4A and 4B). A continuous 72 h of cisplatin treatment resulted in decreased SLR3 activity (Hprt gene reporter) in a dose‐dependent manner (Figure 4A), reflecting the number of living S3 cells (Figure 1). However, 5 μM of cisplatin treatment increased the raw SLG count (Kim‐1 gene reporter activity) by approximately four‐fold (Figure 4A), as well as the relative activity of the Kim‐1 reporter gene by 50‐fold when normalized by Hprt reporter activity (SLR3) (Figure 4B). High concentrations (100–500 μM) of AAP also activated the Kim‐1 reporter gene to some extent (Figure 4B). Gentamicin treatment decreased Hprt reporter activity (SLR3), as well as Kim‐1 activity (SLG), and the relative SLG activity as normalized by SLR3 did not change (Figure 4B, lower left). Mannitol did not alter SLR3 or SLG activity, which is consistent with its lack of toxicity in S3 cells (Figure 4A and 4B).
Figure 4.
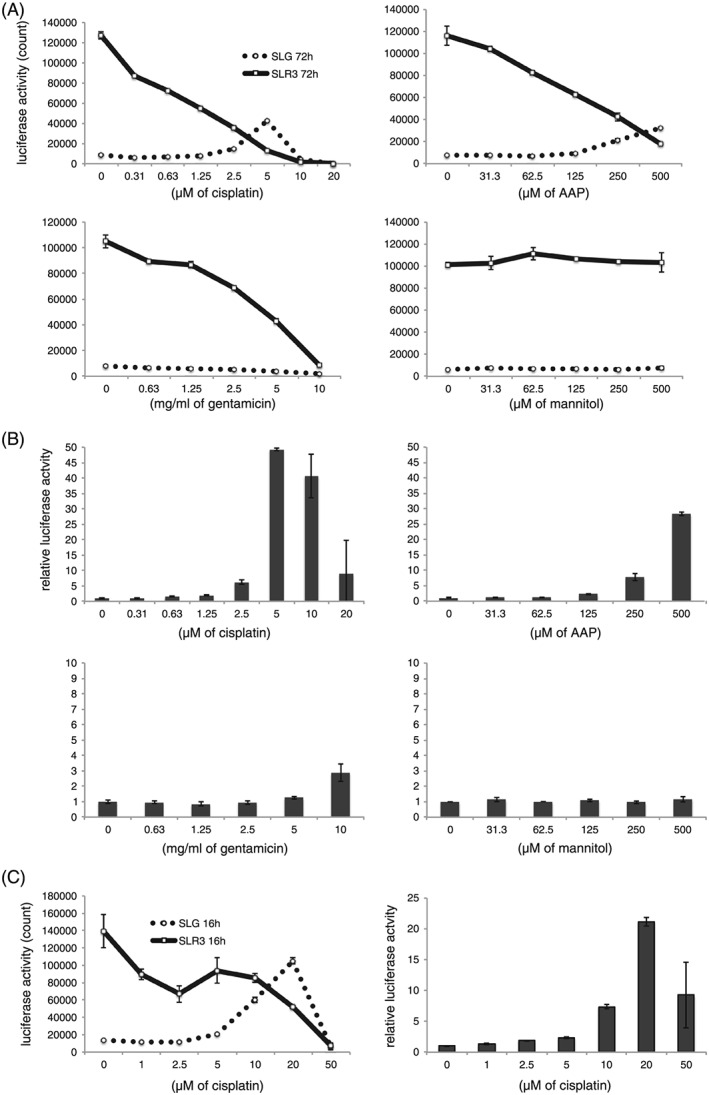
Cisplatin augments the expression of Kim‐1 reporter gene in S3 cells. S3 cells that harbored stable Kim1‐SLG and Hprt‐SLR3 reporters were incubated with chemicals at the indicated concentrations for 72 h, and luciferase assays were performed (A and B). Luciferase activities measured by luminometer are indicated (A). SLG counts were divided by SLR3 counts and the relative activity was defined as 1 at 0 μM (B). Reporter S3 cells were incubated with cisplatin or gentamicin for 16 h and luciferase activity was determined at 72 h after the addition of chemicals (C). Luciferase counts upon cisplatin treatment are shown (left). Relative luciferase activity normalized by Hprt‐SLR3 is shown (right). Error bars indicate the SD
We found that continuous 48‐h cisplatin treatment (10–25 μM) only moderately (by two‐ to eight‐fold) activated the Kim‐1 reporter gene (48 h in the Supporting information, Figure S6 A and S6B). In parallel, we washed chemically‐treated cells with PBS 6 h after the addition of compound, and incubated cells with fresh medium for a further 42 h (6 h in the Supporting information, Figure S4 A and S4B). Kim‐1 reporter (SLG) was activated by approximately three‐fold at 25 μM of cisplatin and by more than 15‐fold at 50 μM in the 6‐h treated group (see Supporting information, Figure S6B, white column). We noted that treatment with 10–50 μM cisplatin for 6 h increased the gross SLG activity (raw counts) compared to treatment for 48 h (see Supporting information, Figure S6 A, thin dotted and thick dotted lines). Therefore, we investigated whether washing conditions could improve Kim‐1 reporter gene activation. The results showed that 16 h of incubation with 10–20 μM cisplatin dramatically enhanced raw SLG counts (by up to seven‐fold) as indicated by the luciferase assay at 72 h after the addition of cisplatin (Figure 4C, left). In addition, we detected a relative luciferase augmentation by up to 22‐fold (Figure 4C, right). The degree of relative activation of Kim‐1 reporter by cisplatin was comparable to the mRNA induction measured by real‐time RT‐PCR (Figure 2A). Gentamicin did not induce SLG or SLR3 activation as a result of 16 h of treatment (data not shown) and this was consistent with the result of the RT‐PCR (Figure 2). These results suggest that the Kim‐1 reporter gene activity reflects the endogenous Kim‐1 mRNA expression induced by cisplatin in S3 cells.
3.5. Kim‐1 gene reporter is not activated in cisplatin‐treated A9 cells
Next, we examined whether the same reporter set responded to cisplatin or other chemicals in A9 cells, which are not derived from the kidney. We incubated reporter‐containing A9 cells with compounds for 72 h and found no obvious induction of the Kim‐1 reporter gene by cisplatin treatment (Figure 5), in contrast to S3 cells (Figure 4). Gentamicin and mannitol did not alter SLG or SLR3 activities in A9 cells (Figure 5) and these results were similar to those obtained using S3 cells (Figure 4).
Figure 5.
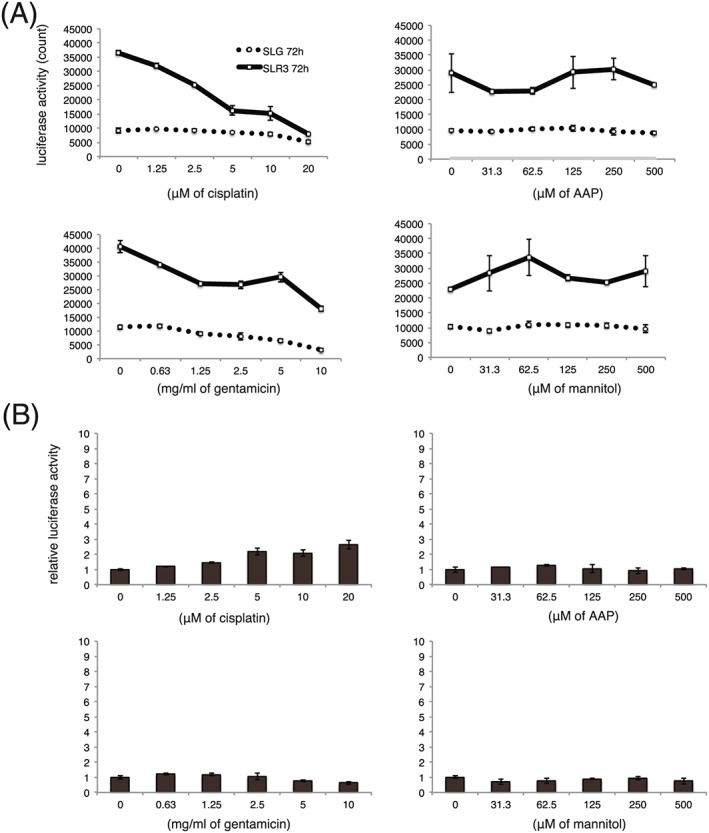
Kim‐1 gene reporter does not respond to cisplatin in A9 cells. A9 cells that contained Kim‐1 and Hprt reporter genes were incubated with chemicals for 72 h and subjected to a luciferase assay as shown in Figure 4A and 4B
4. DISCUSSION
In the present study, we demonstrated that Kim‐1, which has been identified as an in vivo biomarker for nephrotoxicity, could be used as an in vitro biomarker for the assessment of renal toxicity induced by cisplatin in S3 cells. To date, cisplatin has failed to induce Kim‐1 mRNA expression in most immortalized kidney cell lines, such as HK‐2 and IM‐PTECs, although cisplatin causes cytotoxicity as measured by MTT (or related) assays in these cell lines.19, 20
We also demonstrated that Kim‐1 protein expression was upregulated after cisplatin treatment for 72 h in S3 cells (see Supporting information, Figure S2). Because Kim‐1 protein expression in S3 cell lysates, as well as Kim‐1 mRNA (Figures 2; see also Supporting information, Figure S1), was at high levels in the control sample (0 μM of cisplatin) at 12–24 h, Kim‐1 expression was considered to be also associated with dedifferentiation of kidney cell caused by proliferation.
We report that a combination of Kim‐1 and Hprt reporter genes reproducibly detected cisplatin‐induced nephrotoxicity in S3 cells (Figure 4). Assays that measure cellular toxicity via MTT (or related) or apoptosis have been widely used for in vitro toxicity assays.13, 14, 15 However, these assays mainly focus on assessing metabolic activity and cell death rather than renal cell injury or stress. Because Kim‐1 expression is associated with nephrotoxicity in vivo, 3 upregulation of the Kim‐1 gene is considered an indication of kidney cell damage in S3 cells. We confirmed that Hprt reporter activity correlated with cell number (cell viability) in A9 cells33 and in S3 cells (data not shown). Therefore, although the use of either Kim‐1 or Hprt reporter activity was suggestive of toxicity, Kim‐1 reporter activity normalized by Hprt was more indicative. Because cisplatin causes both nephrotoxicity and cytotoxicity, a reduction in cell number caused by cytotoxicity appears to be unavoidable (Figures 1 and 4). Within this context, in A9 cells, cisplatin treatment resulted in a decrease of Hprt‐reporter activity (i.e. cytotoxicity) but did not increase normalized Kim‐1 reporter activity, indicating the cell type specificity of Kim‐1 reporter genes in response to nephrotoxicity in S3 cells (Figures 4 and 5).
Gentamicin did not activate Kim‐1 mRNA expression or the Kim‐1 reporter gene in S3 cells (Figures 2 and 4). This is not unexpected because the repeated administration of gentamicin (for more than 1 week) was reported to be required for Kim‐1 induction in rats.11, 12, 51 In the case of NKi‐2 cells, gentamicin treatment for 3–5 days induced cell damage when the cells formed a kidney‐like structure (3D culture) in vitro. 52 These reports imply that a longer incubation time and complex cell–cell interactions may be required for gentamicin to induce nephrotoxicity. Therefore, we consider that a reduction of Hprt‐SLR3 activity by high concentrations of gentamicin treatment should be caused by relatively nonspecific cytotoxicity in S3 cells (Figure 4A). This speculation is consistent with the lack of Kim‐1 reporter gene activation after gentamicin treatment for 72 h (Figure 4B), which was similar to that using A9 cells (Figure 5).
In the present study, we identified the promoter region of mouse Kim‐1, at least partially, and developed a Kim‐1 gene luciferase reporter in conjunction with an Hprt gene reporter to detect cisplatin‐induced nephrotoxicity in immortalized mouse kidney S3 cells in vitro. We demonstrated that Kim‐1, which was identified as an in vivo biomarker for nephrotoxicity, was useful for the in vitro assessment of cisplatin‐induced renal toxicity of S3 cells. These results should aid the mechanistic analysis of cisplatin‐induced nephrotoxicity and might also help with the development of a method for assessing the nephrotoxicity of novel drugs in vitro.
Supporting information
Supporting info item
ACKNOWLEDGEMENTS
We are grateful to Dr Toshiaki Inoue, Dr Seiichiro Himeno and Dr Hitomi Fujishiro for their valuable discussions. We thank Dr Natsumi Noda and Dr Yoshihiro Nakajima for reporter construction. We also thank Dr Promsuk Jutabha for maintenance of S1, S2 and S3 cells, as well as Ms Miyuki Nomura for technical assistance. This research was supported by a grant from Ministry of Economy, Trade and Industry (METI), KAKENHI Grants‐in‐Aid for Scientific Research, and Regional Innovation Strategy Support Program from the Ministry of Education, Culture, Sports, Science and Technology of Japan. The authors declare that there is no conflict of interest.
Kokura, K. , Kuromi, Y. , Endo, T. , Anzai, N. , Kazuki, Y. , Oshimura, M. , and Ohbayashi, T. (2016) A kidney injury molecule‐1 (Kim‐1) gene reporter in a mouse artificial chromosome: the responsiveness to cisplatin toxicity in immortalized mouse kidney S3 cells. J Gene Med, 18: 273–281. doi: 10.1002/jgm.2925.
REFERENCES
- 1. Pfaller W, Gstraunthaler G. Nephrotoxicity testing in vitro – what we know and what we need to know. Environ Health Perspect. 1998;106(Suppl 2):559–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. van Timmeren MM, Bakker SJL, Vaidya VS, et al. Tubular kidney injury molecule‐1 in protein‐overload nephropathy. Am J Physiol Renal Physiol. 2006;291:F456–F464. [DOI] [PubMed] [Google Scholar]
- 3. Wasung ME, Chawla LS, Madero M. Biomarkers of renal function, which and when? Clin Chim Acta. 2015;438:350–357. [DOI] [PubMed] [Google Scholar]
- 4. Bonventre JV, Vaidya VS, Schmouder R, Feig P, Dieterle F. Next‐generation biomarkers for detecting kidney toxicity. Nat Biotechnol. 2010;28:436–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. van Meer L, Moerland M, Cohen AF, Burggraaf J. Urinary kidney biomarkers for early detection of nephrotoxicity in clinical drug development. Br J Clin Pharmacol. 2014;77:947–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yang L, Brooks CR, Xiao S, Sabbisetti V, et al. KIM‐1‐mediated phagocytosis reduces acute injury to the kidney. J Clin Invest. 2015;125:1620–1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lim AI, Tang SCW, Lai KN, Leung JCK. Kidney injury molecule‐1: more than just an injury marker of tubular epithelial cells? J Cell Physiol. 2013;228:917–924. [DOI] [PubMed] [Google Scholar]
- 8. Amin RP, Vickers AE, Sistare F, et al. Identification of putative gene based markers of renal toxicity. Environ Health Perspect. 2004;112:465–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chiusolo A, Defazio R, Zanetti E, et al. Kidney injury molecule‐1 expression in rat proximal tubule after treatment with segment‐specific nephrotoxicants: a tool for early screening of potential kidney toxicity. Toxicol Pathol. 2010;38:338–345. [DOI] [PubMed] [Google Scholar]
- 10. Ichimura T, Bonventre JV, Bailly V, et al. Kidney injury molecule‐1 (KIM‐1), a putative epithelial cell adhesion molecule containing a novel immunoglobulin domain, is up‐regulated in renal cells after injury. J Biol Chem. 1998;273:4135–4142. [DOI] [PubMed] [Google Scholar]
- 11. Vaidya VS, Ozer JS, Dieterle F, et al. Kidney injury molecule‐1 outperforms traditional biomarkers of kidney injury in preclinical biomarker qualification studies. Nat Biotechnol. 2010;28:478–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sasaki D, Yamada A, Umeno H, et al. Comparison of the course of biomarker changes and kidney injury in a rat model of drug‐induced acute kidney injury. Biomarkers. 2011;16:553–566. [DOI] [PubMed] [Google Scholar]
- 13. Kohda Y, Kawai Y, Iwamoto N, et al. Serum thymic factor, FTS, attenuates cisplatin nephrotoxicity by suppressing cisplatin‐induced ERK activation. Biochem Pharmacol. 2005;70:1408–1416. [DOI] [PubMed] [Google Scholar]
- 14. Park MS, De Leon M, Devarajan P. Cisplatin induces apoptosis in LLC‐PK1 cells via activation of mitochondrial pathways. J Am Soc Nephrol. 2002;13:858–865. [DOI] [PubMed] [Google Scholar]
- 15. Sancho‐Martínez SM, Piedrafita FJ, Cannata‐Andía JB, López‐Novoa JM, López‐Hernández FJ. Necrotic concentrations of cisplatin activate the apoptotic machinery but inhibit effector caspases and interfere with the execution of apoptosis. Toxicol Sci. 2011;122:73–85. [DOI] [PubMed] [Google Scholar]
- 16. Kim SY, Sohn SJ, Won AJ, Kim HS, Moon A. Identification of noninvasive biomarkers for nephrotoxicity using HK‐2 human kidney epithelial cells. Toxicol Sci. 2014;140:247–258. [DOI] [PubMed] [Google Scholar]
- 17. Rached E, Hoffmann D, Blumbach K, Weber K, Dekant W, Mally A. Evaluation of putative biomarkers of nephrotoxicity after exposure to ochratoxin a in vivo and in vitro. Toxicol Sci. 2008;103:371–381. [DOI] [PubMed] [Google Scholar]
- 18. Sohn S‐J, Kim SY, Kim HS, et al. In vitro evaluation of biomarkers for cisplatin‐induced nephrotoxicity using HK‐2 human kidney epithelial cells. Toxicol Lett. 2013;217:235–242. [DOI] [PubMed] [Google Scholar]
- 19. Benedetti G, Fredriksson L, Herpers B, Meerman J, van de Water B, de Graauw M. TNF‐α‐mediated NF‐κB survival signaling impairment by cisplatin enhances JNK activation allowing synergistic apoptosis of renal proximal tubular cells. Biochem Pharmacol. 2013;85:274–286. [DOI] [PubMed] [Google Scholar]
- 20. Huang JX, Kaeslin G, Ranall MV, et al. Evaluation of biomarkers for in vitro prediction of drug‐induced nephrotoxicity: comparison of HK‐2, immortalized human proximal tubule epithelial, and primary cultures of human proximal tubular cells. Pharmacol Res Perspect. 2015;3: e00148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hosoyamada M, Obinata M, Suzuki M, Endou H. Cisplatin‐induced toxicity in immortalized renal cell lines established from transgenic mice harboring temperature sensitive SV40 large T‐antigen gene. Arch Toxicol. 1996;70:284–292. [DOI] [PubMed] [Google Scholar]
- 22. Takeda M, Kobayashi M, Shirato I, Osaki T, Endou H. Cisplatin‐induced apoptosis of immortalized mouse proximal tubule cells is mediated by interleukin‐1 beta converting enzyme (ICE) family of proteases but inhibited by overexpression of Bcl‐2. Arch Toxicol. 1997;71:612–621. [DOI] [PubMed] [Google Scholar]
- 23. Lee WK, Jang S‐B, Cha SH, et al. Different sensitivity to nephrotoxic agents and osmotic stress in proximal tubular and collecting duct cell lines derived from transgenic mice. Toxicol in Vitro. 2002;16:55–62. [DOI] [PubMed] [Google Scholar]
- 24. Takeda M, Tojo A, Sekine T, Hosoyamada M, Kanai Y, Endou H. Role of organic anion transporter 1 (OAT1) in cephaloridine (CER)‐induced nephrotoxicity. Kidney Int. 1999;56:2128–2136. [DOI] [PubMed] [Google Scholar]
- 25. Khamdang S, Takeda M, Babu E, et al. Interaction of human and rat organic anion transporter 2 with various cephalosporin antibiotics. Eur J Pharmacol. 2003;465:1–7. [DOI] [PubMed] [Google Scholar]
- 26. Shiraya K, Hirata T, Hatano R, et al. A novel transporter of SLC22 family specifically transports prostaglandins and co‐localizes with 15‐hydroxyprostaglandin dehydrogenase in renal proximal tubules. J Biol Chem. 2010;285:22141–22151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Takeda M, Khamdang S, Narikawa S, et al. Characterization of methotrexate transport and its drug interactions with human organic anion transporters. J Pharmacol Exp Ther. 2002;302:666–671. [DOI] [PubMed] [Google Scholar]
- 28. Mashhadi MA, Arab MR, Azizi F, Shahraki MR. Histological study of toxic effects of cisplatin single dose injection on rat kidney. Gene Cell Tissue. 2014;1:1–4. [Google Scholar]
- 29. Osman AM, El‐Sayed EM, El‐Demerdash E, et al. Prevention of cisplatin‐induced nephrotoxicity by methimazole. Pharmacol Res. 2000;41:115–121. [DOI] [PubMed] [Google Scholar]
- 30. Takiguchi M, Kazuki Y, Hiramatsu K, et al. A novel and stable mouse artificial chromosome vector. ACS Synth Biol. 2014;3:903–914. [DOI] [PubMed] [Google Scholar]
- 31. Narai T, Katoh M, Inoue T, et al. Construction of a luciferase reporter system to monitor osteogenic differentiation of mesenchymal stem cells by using a mammalian artificial chromosome vector. Yonago Acta Med. 2015;58:23–29. [PMC free article] [PubMed] [Google Scholar]
- 32. Oshimura M, Uno N, Kazuki Y, Katoh M, Inoue T. A pathway from chromosome transfer to engineering resulting in human and mouse artificial chromosomes for a variety of applications to bio‐medical challenges. Chromosom Res. 2015;23:111–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Endo T, Noda N, Kuromi Y, et al. Evaluation of an Hprt‐luciferase reporter gene on a mammalian artificial chromosome in response to cytotoxicity. Yonago Acta Med. 2016;59:174–182. [PMC free article] [PubMed] [Google Scholar]
- 34. Li X, Nakajima Y, Niwa K, Viviani VR, Ohmiya Y. Enhanced red‐emitting railroad worm luciferase for bioassays and bioimaging. Protein Sci. 2010;19:26–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hiratsuka M, Uno N, Ueda K, et al. Integration‐free iPS cells engineered using human artificial chromosome vectors. PLoS One. 2011;6: e25961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yoshimura Y, Nakamura K, Endo T, et al. Mouse embryonic stem cells with a multi‐integrase mouse artificial chromosome for transchromosomic mouse generation. Transgenic Res. 2015;24:717–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yamaguchi S, Kazuki Y, Nakayama Y, Nanba E, Oshimura M, Ohbayashi T. A method for producing transgenic cells using a multi‐integrase system on a human artificial chromosome vector. PLoS One. 2011;6: e17267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Nakajima Y, Kimura T, Sugata K, et al. Multicolor luciferase assay system: one‐step monitoring of multiple gene expressions with a single substrate. BioTechniques. 2005;38:891–894. [DOI] [PubMed] [Google Scholar]
- 39. Kucherlapati R, Shin SI. Genetic control of tumorigenicity in interspecific mammalian cell hybrids. Cell. 1979;16:639–648. [DOI] [PubMed] [Google Scholar]
- 40. Hamid R, Rotshteyn Y, Rabadi L, Parikh R, Bullock P. Comparison of alamar blue and MTT assays for high through‐put screening. Toxicol in Vitro. 2004;18:703–710. [DOI] [PubMed] [Google Scholar]
- 41. Karasawa T, Steyger PS. An integrated view of cisplatin‐induced nephrotoxicity and ototoxicity. Toxicol Lett. 2015;237:219–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kelsen DP, Alcock N, Young CW. Cisplatin nephrotoxicity. Correlation with plasma platinum concentrations. Am J Clin Oncol. 1985;8:77–80. [PubMed] [Google Scholar]
- 43. Ahmad ST, Arjumand W, Nafees S, et al. Hesperidin alleviates acetaminophen induced toxicity in Wistar rats by abrogation of oxidative stress, apoptosis and inflammation. Toxicol Lett. 2012;208:149–161. [DOI] [PubMed] [Google Scholar]
- 44. Kitamura S, Sakurai H, Makino H. Single adult kidney stem/progenitor cells reconstitute three‐dimensional nephron structures in vitro. Stem Cells. 2015;33:774–784. [DOI] [PubMed] [Google Scholar]
- 45. Kitamura S, Yamasaki Y, Kinomura M, et al. Establishment and characterization of renal progenitor like cells from S3 segment of nephron in rat adult kidney. FASEB J. 2005;19:1789–1797. [DOI] [PubMed] [Google Scholar]
- 46. Kandasamy K, Chuah JKC, Su R, et al. Prediction of drug‐induced nephrotoxicity and injury mechanisms with human induced pluripotent stem cell‐derived cells and machine learning methods. Sci Rep. 2015;5:12337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ichimura T, Asseldonk EJPV, Humphreys BD, et al. Kidney injury molecule‐1 is a phosphatidylserine receptor that confers a phagocytic phenotype on epithelial cells. J Clin Invest. 2008;118:1657–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ichimura T, Hung CC, Yang SA, Stevens JL, Bonventre JV. Kidney injury molecule‐1: a tissue and urinary biomarker for nephrotoxicant‐induced renal injury. Am J Physiol Renal Physiol. 2004;286:F552–F563. [DOI] [PubMed] [Google Scholar]
- 49. Ajay AK, Kim T‐M, Ramirez‐Gonzalez V, Park PJ, Frank DA, Vaidya VS. A bioinformatics approach identifies signal transducer and activator of transcription‐3 and checkpoint kinase 1 as upstream regulators of kidney injury molecule‐1 after kidney injury. J Am Soc Nephrol. 2014;25:105–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chen F, Smith R, Gu Y‐Z, Collins ND, Nioi P. Toxicoepigenetic alteration of the kidney injury molecule 1 gene in gentamicin‐exposed rat kidney. Toxicol Sci. 2010;117:375–380. [DOI] [PubMed] [Google Scholar]
- 51. Wang E‐J, Snyder RD, Fielden MR, Smith RJ, Gu Y‐Z. Validation of putative genomic biomarkers of nephrotoxicity in rats. Toxicology. 2008;246:91–100. [DOI] [PubMed] [Google Scholar]
- 52. DesRochers TM, Suter L, Roth A, Kaplan DL. Bioengineered 3D human kidney tissue, a platform for the determination of nephrotoxicity. PLoS One. 2013;8:e59219. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting info item