

Abstract
There have been remarkable developments in the field of autoinflammatory diseases over the last 20 years. Research has led to definitions of new conditions, increased understanding of disease mechanisms and specific treatment. The polygenic autoinflammatory condition of periodic fever, aphthous stomatitis, pharyngitis and cervical adenitis (PFAPA) is the most common autoinflammatory disorder among children in many parts of the world. The clinical features often include clockwork regularity of episodes, prompt responses to corticosteroids and therapeutic effects of tonsillectomy, but the disease mechanisms are largely unknown.
Conclusion
This review discusses the emerging understanding of autoinflammatory diseases, with special emphasis on PFAPA.
Keywords: Aphthous stomatitis, Autoinflammatory diseases, Disease mechanisms, Periodic fever, Pharyngitis and cervical adenitis
Abbreviations
- CAPS
Cryopyrin‐associated periodic syndrome
- CINCA
Chronic infantile neurological, cutaneous and articular syndromes
- DIRA
Deficiency of IL‐1 receptor antagonist
- FCAS
Familial cold autoinflammatory syndrome
- FMF
Familial Mediterranean fever
- HIDS
Hyperimmunoglobulinaemia D with periodic fever syndrome
- MKD
Mevalonate kinase deficiency
- MWS
Muckle–Wells syndrome
- PFAPA
Periodic fever, aphthous stomatitis, pharyngitis and cervical adenitis
- TRAPS
Tumour necrosis factor receptor‐associated periodic syndrome
Key notes.
There have been remarkable developments in the field of autoinflammatory diseases over the last 20 years.
This review discusses the emerging understanding of autoinflammatory diseases, with special emphasis on the polygenic autoinflammatory condition of periodic fever, aphthous stomatitis, pharyngitis and cervical adenitis (PFAPA) syndrome.
The clinical features of PFAPA often include clockwork regularity of episodes, prompt responses to corticosteroids and therapeutic effects of tonsillectomy, but the disease mechanisms are largely unknown.
Introduction
When Michael McDermott and Daniel Kastner coined the concept of autoinflammatory diseases in 1999, it marked a paradigm shift as it depicted an entirely new group of immunological diseases in conceptual, clinical and mechanistic terms. Autoinflammatory diseases were defined as ‘conditions characterised by seemingly unprovoked episodes of inflammation, without high‐titre autoantibodies or antigen‐specific T‐cells' 1, 2. The concept was suggested in the same paper that described the genetic background of the TNF receptor‐associated periodic syndrome (TRAPS), but was also linked to the identification, two years earlier, of mutations that cause familial Mediterranean fever (FMF) (Table 1) 1, 3. This review discusses the emerging field of autoinflammatory conditions, with a special emphasis on periodic fever, aphthous stomatitis, pharyngitis and cervical adenitis (PFAPA) syndrome. PFAPA is a relative common and important differential diagnosis for many infections occurring in preschool children, and every paediatrician will encounter PFAPA several times during her or his career.
Table 1.
Clinical characteristics of (some) monogenic autoinflammatory diseases and PFAPA
| Clinical classification | Fever (length of episodes) | Main organs involved/symptoms | Skin rash | Gene (inheritance) | Protein | Suggested treatment |
|---|---|---|---|---|---|---|
| Periodic fever syndrome | ||||||
| TRAPS | Yes (1–6 weeks) |
Eyes: conjunctivitis, periorbital oedema, pleuritis, pericarditis CNS: headache Abdominal pain Splenomegaly Arthritis/arthralgia, myalgia |
Migrating erysipelas‐like, painful | TNFRSF1A (AD) | TNF receptor 1 (TNFR1) |
IL‐1 blockade Corticosteroids Etanercept NSAID Colchicine |
| FMF | Yes (½–3 days) | Large joints: serositis, peritonitis, pleuritis and/or arthritis | Sometimes erysipelas‐like erythema, most often in the ankle region | MEFV (AR) | Pyrin | Colchicine (IL‐1 blockade in refractory cases or colchicine side‐effects) |
| FCAS (CAPS) |
Yes (12–24 hours) Triggered by cold exposure |
Eyes: conjunctivitis Joints: arthralgia |
Urticaria‐like rash | NLRP3 (AD) | NLRP3 | IL‐1 blockade |
| MWS (CAPS) | Yes (24–48 hours) |
CNS: hearing loss, headache Eyes: conjunctivitis Joints: arthralgia/arthritis |
Urticaria‐like rash | NLRP3 (AD) | NLRP3 | IL‐1 blockade |
| CINCA/NOMID (CAPS) | Continuous with fever during flares |
CNS: meningitis, hearing loss, headache Eyes: conjunctivitis, uveitis and papilloedema Bone: bony overgrowth Joints: arthralgia/arthritis |
Urticaria‐like rash increase with flares | NLRP3 (AD) | NLRP3 | IL‐1 blockade |
| HIDS/MKD |
Yes (3–7 days) Flares triggered by vaccinations |
Abdominal pain, diarrhoea, lymphadenopathy, splenomegaly Headache Oral and/or genital ulcers Arthritis/arthralgia |
Exanthema (Maculopapular or purpuric) | MVK (AR) | Mevalonate kinase (MK) |
IL‐1 blockade NSAID Corticosteroids TNF blockade |
| FCAS2 | Yes, flares triggered by cold |
Arthralgia, headaches, aphthous stomatitis, abdominal pain Hearing loss |
Urticarial rash | NLRP12 (AD) | NLRP12 |
NSAID Corticosteroids |
| PFAPA | Yes (3‐7 days, most commonly 4‐5 days) | Recurrent attacks of fever often regularly associated with sign and symptoms according to the acronym PFAPA in the absence of upper respiratory tract infection | No, in our hands the diagnosis is questioned in patients with skin rash | N/A | N/A |
NSAID Tonsillectomy Colchicine Corticosteroids |
| Diseases with pyogenic lesions | ||||||
| DIRA | No | Multifocal osteomyelitis periarticular soft‐tissue swelling | Pustular | IL‐1RN (AR) | IL1 receptor antagonist | IL‐1 blockade |
| Diseases with granulomatous lesions | ||||||
| BS/PGA | Rarely, not a dominant feature |
Arthritis Uveitis |
Tan coloured rash. Ichthyosis‐like exanthema | NOD2 (AD) | CARD15 |
TNF blockade Corticosteroids Methotrexate |
| Diseases with panniculitis‐induced lipodystrophy | ||||||
| CANDLE/PRAAS | Yes (often continuous) |
Myositis, arthritis/arthralgia Joint contractures, muscle atrophy Hepatomegaly, splenomegaly Basal ganglia calcifications, Anaemia |
Panniculitis, lipodystrophy Nodular exanthema |
PSMB8 (AR), PSMA3, PSMB4, PSMB9, POMP | PSB8, PSMA3, PSMA4, PSMB9 and POMP |
Corticosteroids? JAK inhibition? |
| Diseases with psoriatic lesions | ||||||
| DITRA | Flares with fever | Arthritis, geographic tongue, cholangitis nail dystrophy | Generalised pustular psoriasis (often without psoriasis vulgaris) | IL36RN (AR) | IL‐36Ra |
IL‐1 blockade? TNF blockade? Corticosteroids Methotrexate |
| CAMPS | Not a distinct feature | Psoriatic arthritis |
Generalised pustular psoriasis (often with psoriasis vulgaris) Some cases of familial pityriasis rubra pilaris och psoriasis vulgaris Erythrodermic psoriasis |
CARD14 (AD) | CARD14 |
IL‐12/23 blockade? or IL‐17 blockade? |
| Diseases with neurologic and dermatologic manifestations | ||||||
| AGS | Rarely | Progressive brain disease | Chilblains | Seven different genes (AD or AR) | Several different proteins | No established treatment |
| Diseases with IBD | ||||||
| EO‐IBD (IL‐10 receptor deficiency, IL‐10 deficiency) | Recurrent fever rare | Early‐onset enterocolitis with haematochezia, colonic abscesses, perianal fistulas, oral ulcers and failure to thrive | Recurrent folliculitis | IL‐10, IL‐10RA and IL‐10RB | IL‐10, IL‐10RA and IL‐10RB | Beyond the scope of this review |
TRAPS = Tumour necrosis factor receptor‐associated periodic syndrome; FMF = Familial Mediterranean fever; FCAS = Familial cold autoinflammatory syndrome; MWS = Muckle–Wells syndrome; CINCA = Chronic infantile neurological, cutaneous and articular syndromes; CAPS = Cryopyrin‐associated periodic syndrome; HIDS = Hyperimmunoglobulinaemia D with periodic fever syndrome; MKD = Mevalonate kinase deficiency; FCAS2 = Familial cold autoinflammatory syndrome 2; PFAPA = Periodic fever, aphthous stomatitis, pharyngitis and cervical adenitis; DIRA = Deficiency of IL‐I receptor antagonist; BS = Blau syndrome; PGA = Paediatric granulomatous arthritis; CANDLE = Chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature; PRAAS = Proteasome‐associated autoinflammatory syndrome; DITRA = Deficiency of IL‐36 receptor antagonist; CAMPS = CARD14‐mediated psoriasis; AGS = Aicardi–Goutières syndrome; EO‐IBD = Early‐onset inflammatory bowel disease; AD = Autosomal dominant; AR = autosomal recessive.
Clinical features of autoinflammatory diseases
Autoinflammatory diseases or periodic fever syndromes are clinically characterised by recurrent episodes of fever, systemic inflammation and symptoms such as skin rashes, abdominal pain, chest pain, lymphadenopathy or arthritis. Autoinflammatory diseases that have been more recently defined often have symptoms similar to those of the classical autoinflammatory diseases, but in a continuous manner rather than in episodes and, or, with milder systemic inflammation 4. Phenotypes of the different diseases are used to classify autoinflammatory diseases (Table 1), with clinical signs and symptoms forming the basis of the differential diagnosis and guiding the genetic analysis 5.
Autoinflammatory conditions are caused by dysregulation of innate immunity
It has become clear that autoinflammation is caused by dysregulation of innate immunity, as described by Kastner et al. 6, who proposed that autoinflammatory diseases are ‘clinical disorders marked by abnormally increased inflammation, mediated predominantly by cells and molecules of the innate immune system, with a significant host predisposition’. This definition was closely related to the identification and pathophysiological understanding of mutations in the NOD‐like receptor 3 gene (NLRP3) that causes cryopyrin‐associated periodic syndromes (CAPS) 4, 7. Under normal conditions, NLRP3 recognises unique microbial and danger components and forms a NLRP3 inflammasome that is essential for the defence against infections and the innate immune response. The formation of the NLRP3 inflammasome leads to processing of procaspase‐1 into caspase‐1 that, in turn, cleaves to pro‐ IL‐1β to IL‐1β (Fig. 1). Mutations in NLRP3 can result in three autoinflammatory phenotypes, which often are overlapping. In order of increasing severity, these are familial cold autoinflammatory syndrome (FCAS), Muckle–Wells syndrome (MWS) and chronic infantile neurological, cutaneous and arthritis (CINCA), also known as neonatal‐onset multisystem inflammatory disease (NOMID) (Table 1) 3, 4. Intriguingly, the same CAPS‐associated mutation can give rise to more than one disease phenotype – FCAS, MWS or CINCA – and mutation negative patients often have a somatic mutation in some of the cells tested, which is known as somatic mosaicism 8.
Figure 1.
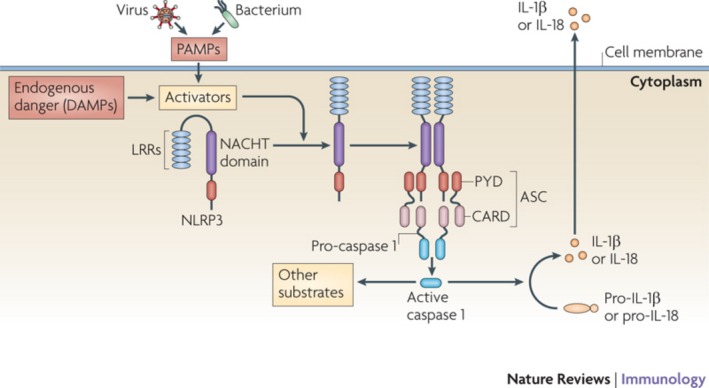
NLRP3 inflammasome activation. Under healthy conditions, NOD‐like receptor family, pyrin domain containing 3 (NLRP3) is auto‐repressed through interaction between the NACHT domain and the leucine‐rich repeat domain (LRR). Cellular activation by PAMPs and DAMPs removes this auto‐repression. As a result, NLRP3 unfolds itself and the NACHT domain is exposed. This leads to oligomerisation of NLRP3 that recruits apoptosis‐associated speck‐like protein containing a CARD (ASC) and pro‐caspase 1, inducing activation of caspase 1, which in turn activates IL‐1 and IL‐18. Tschopp & Schroder K. Nat Rev Immunol 2010; 10:210–5. With permission (3802450071000). PAMPs, pathogen‐associated molecular patterns; DAMPs, damage‐associated molecular patterns; LRRs, leucine‐rich repeats; NACHT, NAIP (neuronal apoptosis inhibitory protein), CIITA (MHC class II transcription activator), HET‐E (incompatibility locus protein from Podosporina anserina) and TP1 (telomerase‐associated protein); ASC, apoptosis‐associated speck‐like protein containing a CARD; PYD, pyrin domain; CARD, caspase‐recruitment domain.
The importance of the balance between IL‐1 and its receptor antagonist IL‐1Ra
IL‐1β is a potent proinflammatory cytokine that initiates an inflammatory response, including secretion of IL‐6 from the liver. Although IL‐1β is necessary for the disease pathology in many autoinflammatory conditions, increased serum concentrations of IL‐1β are rarely measurable. The common way to demonstrate the pathogenic role of IL‐1β in a specific autoinflammatory disease in vivo is by a IL‐1 blockade, exemplified by the treatment of CAPS. The effect of IL‐1β is balanced by the presence of the IL‐1 receptor antagonist (IL‐1Ra). This is exemplified by the autosomal recessive disease deficiency of the IL‐1 receptor antagonist (DIRA), where the lack of IL‐1Ra leads to unopposed normal levels of IL‐1α and IL‐1β, in contrast to the hypersecretion of IL‐1β in CAPS 9. This difference in disease mechanism may explain the different clinical features in DIRA and CAPS, with a pustular rash and sterile osteomyelitis in DIRA and urticaria‐like rash and bony overgrowth in CAPS (Table 1). Just like CAPS, DIRA can be efficiently treated by an IL‐1 blockade.
Current classification according to mechanisms in monogenic autoinflammatory conditions
Although much of the focus in the autoinflammatory field has been on the importance of IL‐1β, a number of conditions with other defining cytokines, or pathways, have been described, including conditions mediated by NF‐κB. The conditions include familial cold autoinflammatory syndrome 2 (FCAS2) 10, Blau syndrome or paediatric granulomatous arthritis 11, 12 and CARD14‐mediated psoriasis (CAMPS); diseases mediated by type I interferon, such as chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature (CANDLE) or proteasome‐associated autoinflammatory syndrome (PRAAS) 13, Aicardi‐Goutières syndrome 14; disorders mediated by IL‐36, for example deficiency of the IL‐36 receptor antagonist (DITRA) 15 and conditions due to decreased IL‐10 activity such as early‐onset enterocolitis 16, allowing for classification according to a defining cytokine as depicted in Table 2 4. See Table 1 for the clinical classifications.
Table 2.
The main cytokines involved in major monogenic autoinflammatory diseases
| Defining cytokine |
|---|
| IL‐1β |
| CAPS (FCAS, MWS, CINCA) |
| DIRA |
| HIDS/MKD |
| IL‐1β and others |
| TRAPS |
| FMF |
| Multiple cytokines via NF‐κB activation |
| FCAS 2 |
| BS/PGA |
| CAMPS |
| INF type I |
| PRAAS/CANDLE |
| AGS 1/2/3/4/5/6/7 |
| Deficiency of IL‐10 signalling |
| EO‐IBD (IL‐10 receptor deficiency, IL‐10 deficiency) |
| IL‐36 |
| DITRA |
CAPS = Cryopyrin‐associated periodic syndrome; FCAS = Familial cold autoinflammatory syndrome; MWS = Muckle–Wells syndrome; CINCA = Chronic infantile neurological, cutaneous and articular syndromes; DIRA = Deficiency of IL‐I receptor antagonist; HIDS = Hyperimmunoglobulinaemia D with periodic fever syndrome; MKD = Mevalonate kinase deficiency; TRAPS = Tumour necrosis factor receptor‐associated periodic syndrome; FMF = Familial Mediterranean fever; FCAS2 = Familial cold autoinflammatory syndrome 2; BS = Blau syndrome; PGA = Paediatric granulomatous arthritis; CAMPS = CARD14‐mediated psoriasis; CANDLE = Chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature; PRAAS = Proteasome‐associated autoinflammatory syndrome; AGS = Aicardi‐Goutieres syndrome; EO‐IBD = Early‐onset inflammatory bowel disease; DITRA = Deficiency of IL‐36 receptor antagonist.
Monogenic autoinflammatory diseases have also been classified according to pathogenic mechanisms, in other words, in terms of how molecules of innate immunity are dysregulated. Such mechanisms include intracellular sensor function defects, accumulation of intracellular triggers, loss of a negative regulator of inflammation and effects on signalling molecules that upregulate innate immune cell function. A classification of autoinflammatory diseases according to their pathogenic mechanisms is shown in Table 3. Some conditions could be assigned to more than one category.
Table 3.
The mechanisms in monogenic autoinflammatory diseases
| Innate mechanism | Component | Mechanism |
|---|---|---|
| Intracellular sensor function defects | ||
| CAPS (FCAS, MWS, NOMID/CINCA) | NLRP3 (cryopyrin) | Activation of NLRP3 inflammasome (gain‐of‐function) leading to IL‐1β production |
| FMF | Pyrin | Inflammasome activation, increased IL‐1β production |
| BS/PGA | NOD2 | NF‐κB and RIP2K activation |
| CAMPS | Adaptor molecule C CARD14 | Increased NF‐κB |
| Accumulation of intracellular triggers | ||
| TRAPS | Folding defect and accumulation of TNFR1 | MAPK activation, Increased production of mROS, ER stress |
| CANDLE/PRAAS | Proteasome dysfunction | IFN response gene induction |
| HIDS/MKD | Mevalonate kinase | Lack of prenylation leads to cytoskeletal changes and inflammasome activation |
| Loss of a negative regulator of inflammation | ||
| DIRA | Loss of IL‐1 antagonism | Uncontrolled IL‐1 signalling |
| DITRA | Loss of IL‐36 antagonism | Uncontrolled IL‐36 signalling |
| EO‐IBD | Loss of IL‐10 or IL‐10 receptor antagonist | Decreased IL‐10 signalling |
| Effects on signalling molecules that upregulate innate immune cell function | ||
| AGS | Type I interferonopathy‐related proteins | Increased INF type I production |
CAPS = Cryopyrin‐associated periodic syndrome; FCAS = Familial cold autoinflammatory syndrome; MWS = Muckle–Wells syndrome; CINCA = Chronic infantile neurological, cutaneous and articular syndromes; FMF = Familial Mediterranean fever; BS = Blau syndrome; PGA = Paediatric granulomatous arthritis; AGS = Aicardi–Goutières syndrome; CAMPS = CARD14‐mediated psoriasis; TRAPS = Tumour necrosis factor receptor‐associated periodic syndrome; CANDLE = Chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature; PRAAS = Proteasome‐associated autoinflammatory syndrome; HIDS = Hyperimmunoglobulinaemia D with periodic fever syndrome; MKD = Mevalonate kinase deficiency; DIRA = Deficiency of IL‐I receptor antagonist; DITRA = Deficiency of IL‐36 receptor antagonist; EO‐IBD = Early‐onset inflammatory bowel disease.
The continuum model of immunological diseases
In the first definition of autoinflammatory diseases, McDermott made a clear distinction between autoinflammation and autoimmunity. Today, there is an emerging understanding that innate and adaptive immunity are closely linked and that autoinflammatory conditions may have an adaptive or autoimmune component 4. In 2006, McDermott and McGonagle proposed that immunological diseases should be conceived as a continuum with ‘pure monogenic autoinflammatory diseases’ at one end and ‘pure monogenic autoimmune diseases’ at the other, as illustrated in Figure 2 17.
Figure 2.
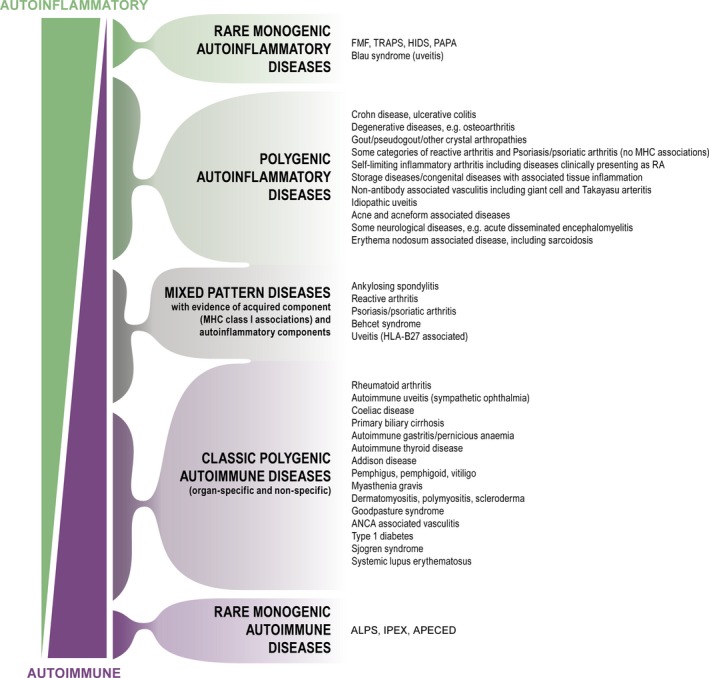
The immunological disease continuum. McGonagle and McDermott's continuum model moved the understanding of immunological diseases forward by integrating the concept of autoinflammation with that of autoimmunity and also applied the concept of autoinflammation to polygenic diseases and to diseases that may have both an autoinflammatory and an autoimmune component. McGonagle & McDermott 17. With permission. TRAPS, tumour necrosis factor receptor‐associated periodic syndrome; FMF, familial Mediterranean fever; HIDS, hyperimmunoglobulinaemia D with periodic fever syndrome; PAPA, pyogenic arthritis, pyoderma gangrenosum (PG) and acne syndrome; ANCA, antineutrophil cytoplasmic antibodies; APLS, autoimmune lymphoproliferative syndrome; IPEX, immunodysregulation polyendocrinopathy enteropathy x‐linked syndrome; APECED, autoimmune polyendocrinopathy‐candidiasis‐ectodermal dystrophy.
Since the continuum model was formulated, it has become clear that even ‘pure monogenic autoinflammatory diseases’ 17 activates the adaptive immune system. One such example is the Th17 differentiation of CD4+ cells in CAPS, a response that diminishes in response to the IL‐1 blockade 18, 19. There are rare monogenic autoinflammatory conditions, which have clinical phenotypes that combine autoinflammation with immunodeficiency or autoimmunity, but these are beyond the scope of this review 4, 6.
The complex pathophysiology of polygenic or multifactorial autoinflammatory diseases
Examples of polygenic or multifactorial autoinflammatory diseases in addition to PFAPA are chronic nonbacterial osteomyelitis, systemic‐onset juvenile idiopathic arthritis and adult onset Still's disease. As illustrated above, the search for disease mechanisms in monogenic autoinflammatory diseases has been a success story, where the definition of disease‐causing mutations has led to the identification of disease mechanisms and sometimes to specific treatment. In other cases, effective responses to specific treatment have led to the identification of candidate genes and disease‐causing mutations 9. In polygenic diseases, genetic characterisation is complex because of the many genes involved. The defining cytokines in polygenic diseases are seldom known, if any, and specific treatment is rarely available. Therefore, the approaches used to define and understand monogenic conditions have been of limited value when applied to the study of polygenic autoinflammatory diseases. Researchers trying to define disease mechanisms in polygenic conditions often depend on sets of clues from cellular and cytokine studies.
Periodic fever, aphthous stomatitis, pharyngitis and cervical adenitis syndrome
The most common autoinflammatory condition among children, outside regions with a high prevalence of FMF, is PFAPA, which is polygenic or multifactorial. The clinical phenotype of PFAPA has all the features of a classical periodic fever syndrome, with unprovoked febrile episodes and systemic as well as localised inflammation 20. Even though studies have shed new light on the pathophysiology of PFAPA, the disease mechanisms are largely unknown. Families, paediatricians and researchers are still intrigued by the often clockwork regularity with which typical episodes appear, the prompt response to corticosteroids and the therapeutic effect of tonsillectomy.
Epidemiology
In early studies, it was estimated that no more than one case of PFAPA would be diagnosed during an entire paediatric career. Today, PFAPA is diagnosed at a much higher frequency in children below the age of five years. However, the syndrome is also recognised in older children and even in adults 21, 22, 23. PFAPA has been described in many parts of the world, but the only estimate of annual incidence is from Norway, which was 2.3 per 10 000 children up to five years old 24. In western Sweden, approximately 300 children were given a definite or probable diagnosis of PFAPA during a 10‐year period (Berg S and Wekell P,unpublished data). This corresponded to an annual incidence of at least three per 10 000 children below the age of five years, which is similar to the data from Norway. A predominance of boys has been described in several PFAPA cohorts 24, 25, 26.
Clinical manifestations
Patients who suffer from PFAPA have recurrent attacks of fever and symptoms associated with the core features of the disease – aphthous stomatitis, pharyngitis and adenitis – in the absence of upper respiratory tract infection. Between the episodes, children with PFAPA are healthy, with normal growth and development, as delineated in the classical diagnostic criteria (Table 4) 27.
Table 4.
Classical diagnostic criteria for periodic fever, aphthous stomatitis, pharyngitis and cervical adenitis (PFAPA) as defined by Thomas et al. 27
| Diagnostic criteria for PFAPA |
| Regularly recurring fevers with an early age of onset (<5 years of age) |
| Symptoms in the absence of upper respiratory tract infection with at least one of the following clinical signs: |
| Aphthous stomatitis |
| Cervical lymphadenitis |
| Pharyngitis |
| Exclusion of cyclic neutropenia |
| Completely asymptomatic interval between episodes |
| Normal growth and development |
The duration of the fever attacks is usually three to seven days most commonly four to five days, with an interval of two to eight weeks, most commonly three to six weeks 24, 27, 28, 29, 30. At some stage of the disease, the episodes characteristically occur with a regular interval, ‘sometimes to a point that the parents can predict the time for an episode and inform their employer’ according to Federici and Galtorno 31. The regularity may disappear over time, and the child then experiences longer intervals and shorter or milder episodes (or both) 22. In our clinical experience, children with PFAPA appear to have fewer upper respiratory infections than other children as long as their PFAPA is active, but the upper respiratory infections seem to re‐emerge as the child grows out of the episodes (Wekell P, Berg S and Fasth A, unpublished observation).
In addition to the signs and symptoms included in the classical criteria, children with PFAPA often have mild abdominal pain, leg pain and nausea and vomiting during episodes 27, 30. The spectrum of additional symptoms was even more striking in a study by Hofer et al. 26 that rightly stressed the importance of establishing new disease criteria with better specificity. Even though children with PFAPA are, by definition, free of symptoms between episodes, aphthous stomatitis is not confined to the episodes and a few children with very frequent attacks do not recover completely between attacks 32.
Diagnosis
The diagnosis of PFAPA is still largely based on recognition of the clinical features delineated in the classical PFAPA criteria (Table 4) 27. These criteria are not totally exclusive to other conditions, including hereditary periodic fever syndromes and need to be refined to improve specificity, in particular, but also sensitivity 20, 28. Until a gold standard has been defined, for example in terms of genetic predisposition or disease mechanism, it is hard to see how the validity of different sets of clinical criteria can be accurately evaluated.
Recurrent infections also need to be considered as a differential diagnosis of PFAPA, namely repeated streptococcal infections, urinary tract infections and viral infections involving the throat and causing raised inflammatory markers, for example adenovirus. Over time, several characteristics of PFAPA will make an infectious aetiology less likely, as the often clockwork episodes, the presence of aphthous stomatitis, the lack of response to antibiotics, the distinct response to corticosteroids, the lack of infections among family members and negative cultures all point towards another diagnosis.
The exclusion of the very rare disease cyclic neutropenia, in which the blood neutrophils typically oscillate with a 21‐day interval, is included in the classical criteria. On occasions, cyclic neutropenia cannot be excluded on clinical grounds as the nadir of the neutrophil count may occur before the onset of fever. In such cases, analysing the neutrophil elastase gene (ELANE) or repeated neutrophil counts three times a week for six weeks can confirm the diagnosis and thus differentiate it from PFAPA 33.
Gattorno et al. 21 proposed a diagnostic score with the aim of predicting the likelihood that a child with PFAPA‐like symptoms could actually have a hereditary periodic fever syndrome instead. It is important to remember that the accuracy of such a prediction will always depend on the prevalence of relevant conditions in the population, for example the prevalence of PFAPA and FMF, respectively, in a specific context. In addition, it is not clear how somatic mosaicism of monogenic autoinflammatory disorders would influence the validity of the diagnostic score developed by Gattorno.
PFAPA also needs to be distinguished from monogenic periodic fever syndromes, such as FMF, MKD and TRAPS, and the diagnosis of PFAPA should be challenged in children who fulfil the criteria but show additional signs and symptoms suggestive of hereditary periodic fever syndromes. These include skin rashes, arthritis, severe abdominal pain, diarrhoea, chest pain and splenomegaly, fever episodes longer than seven days, a history of hearing loss or symptoms secondary to cold exposure 21, 34, 35. The above‐mentioned diagnostic score could be useful for this purpose 21.
The diagnostic workup for PFAPA can be particularly challenging in children who originate from a population with a high prevalence of FMF. Another hereditary periodic fever (HPF) to bear in mind is MKD, also called hyperimmunoglobulinaemia D with periodic fever syndrome (HIDS), that commonly has the same length of episodes, but is often associated with diarrhoea and exanthema as well as flares triggered by vaccinations. In the differential, it is important to note that hyper‐IgD is not a specific indicator for HIDS, as increased IgD concentrations are also seen in other inflammatory conditions, including autoinflammatory diseases such as FMF, TRAPS and PFAPA 29, 36. Increased excretion of mevalonate acid during an inflammatory episode is a valid screening test for MKD, provided that the laboratory can detect very low concentrations of the substance in urine. If indicated, MKD is diagnosed by the identification of mutations in the MVK gene.
Innate immune dysregulation leads to activation of adaptive immunity in PFAPA
Dysregulation of innate immunity in PFAPA is indicated by the clinical phenotype, increase of proinflammatory cytokines, neutrophilia, prompt response to corticosteroids, lack of response to antibiotics and a possible response to the IL‐1 blockade. The role of the fever‐mediating cytokine IL‐1β needs to be further examined 37. A study of whole blood gene expression on the messenger ribonucleic acid level showed increased expression of the IL‐1‐related genes IL1B, IL1RN, CASP1 and I18RAP during PFAPA episodes 38. Studies of ex vivo LPS‐stimulated pheripheral blood mononuclear cells (PBMCs) and purified monocytes from febrile patients with PFAPA showed that the cells had an increased IL‐1β secretion compared to cells from afebrile patients 39. One small case series suggested that PFAPA flares were responsive to IL1‐blockade 38. The role of IL‐1β is still not settled in PFAPA and a well‐designed study of IL‐1 blockade would be an important step forward to demonstrate whether IL‐1β has any role in PFAPA.
A number of other proinflammatory cytokines in addition to IL‐1 have been evaluated in PFAPA with the expectation that inflammatory mediators are increased during the febrile episode. IL‐6 is indeed increased in serum during disease flares 24, 37, 38, 39, 40. One study also showed increased IL‐18 levels during febrile episodes 38. In addition, IL‐1β and the INF‐γ‐related cytokine IP‐10/CXCL10 were increased in PFAPA attacks compared to the afebrile phase 38, 39, 40, 41, as were MIG/CXCL9 38 and MIP‐1& #xF062;/CCL4 38, 41. In summary, this cytokine pattern supports an increased INF‐γ secretion somewhere during the PFAPA cycle, with an adjoining Th1 differentiation of CD4+ T cells. The Th1 differentiation in PFAPA may be due to IL‐18, which, in the presence of IL‐12, induces IFN‐γ and promotes a Th1 response 37. These findings indicate that there is an activation of both the innate and the adaptive immune system in PFAPA with a Th1 differentiation of CD4+ cells during febrile episodes.
Febrile attacks were associated with lymphopenia, and it is not clear whether this was a result of increased myelopoiesis paralleled by a decrease in lymphopoiesis as in acute inflammation or was a consequence of homing of lymphocytes to lymphatic tissue including cervical lymph nodes and tonsils 42. It has also been shown that neutrophil function was altered in PFAPA, including apoptosis, priming and generation of an intracellular oxidative burst during febrile attacks 43.
The role of the tonsils in PFAPA is unknown, but has been highlighted by the often positive response to tonsillectomy. One can speculate that the tonsils are either the primary centres for immune dysregulation or that they harbour a trigger of the immune dysregulation that is a trigger in individuals with a predisposition for PFAPA. One important limitation in studying the role of tonsils in PFAPA is that studies are limited to analyses of tonsils in the afebrile phase. None of the available studies have been able to delineate a plausible mechanism or probable trigger 44, 45, 46, 47. One interesting study showed that the microbiota of the tonsils differed in patients with PFAPA and postulated that the microbiota may have played a role in triggering the inflammatory processes 48.
Biomarkers in PFAPA
Inflammatory variables are significantly increased during PFAPA episodes, including the acute phase proteins CRP and serum amyloid A (SAA). SAA is a useful inflammatory indicator in the management of children with autoinflammatory conditions for three reasons: firstly to measure inflammation during attacks and low‐grade inflammation between attacks, secondly to monitor effectiveness of anti‐inflammatory treatment and thirdly to estimate the risk of developing amyloidosis 49. In PFAPA, both CRP and SAA should normalise between episodes, while in FMF SAA often remains elevated between fever episodes 49.
Several small studies have suggested that procalcitonin, a marker associated with infection, is not markedly elevated during febrile episodes of PFAPA 40, 50. These results parallel the findings in FMF, in which a slight but significant increase in procalcitonin levels was observed during inflammatory episodes 51. Hence, procalcitonin is of limited use when it comes to distinguishing PFAPA from FMF. There is an increase of S100A8/A9 and S100A12 in febrile PFAPA that seems to normalise in the afebrile phase 39, 43. High levels of S100 proteins are characteristic of active systemic juvenile idiopathic arthritis and FMF 52.
There is no indication that the immunomodulator galectin‐3 is increased during febrile attacks of PFAPA or during the afebrile phase 43. This is in contrast to the increased serum levels of galectin‐3 during attack‐free periods in FMF 53. Furthermore, a study of plasma galectin‐3 showed increased concentrations in septicaemia but not in viral infections or in attacks of nondetermined autoinflammatory syndromes, as compared to healthy controls 54. The increase of galectin‐3 in several other inflammatory conditions has been reported, including systemic lupus erythematosus (SLE) 55, Behçet's disease 56 and juvenile idiopathic arthritis 57, as well as in Crohn's disease and ulcerative colitis 58. If the findings of increased plasma levels of galectin‐3 in other inflammatory diseases and low levels in PFAPA are verified, galectin‐3 might be used as tool to distinguish these conditions from PFAPA, particularly with regard to young children who originate in regions with a high prevalence of FMF.
Management
Non‐steroidal anti‐inflammatory drugs (nsaids) are more efficient than paracetamol in reducing fever during PFAPA episodes 32. Corticosteroids abort a PFAPA flare with such extreme efficiency, usually resolving fever within hours, that clinicians can question the PFAPA diagnosis if the effect fails to materialise 25, 27, 30, 32, 59. For unknown reasons, a significant proportion of children with PFAPA experience shortening of the intervals after treatment with corticosteroids 23, 25, 30.
In western Sweden, steroid treatment is primarily used to postpone a febrile episode that occurs at an unsuitable time for the child or the family, whereas in some parts of the world the chosen approach is to treat each episode with corticosteroids 60. Tonsillectomy has turned out to be the most attractive treatment alternative, resolving 80–90% of episodes in cases in an initial case series 61. The effectiveness of tonsillectomy in PFAPA has been supported in two small randomised control trials 62 and in meta‐analyses 63. Treatment by tonsillectomy is based on clinical experience, and so far, no pathophysiological ground has been found. That is why it is important to carefully evaluate, in discussion with the parents, the balance of risks and benefits for each child, bearing in mind the age of the child as well as the length, intensity and frequency of the episodes and the likely time to resolution without treatment 63.
Cimetidine, a histamine type 2 receptor antagonist, has previously been reported to induce remission of PFAPA 27, but a recent report found it to be ineffective in the majority of patients 60. Colchicine, which is the first choice of treatment for FMF, has been evaluated in a few patients with PFAPA, resulting in an increased interval between the episodes, but not in resolution of the disease. The role of colchicine needs to be further investigated 64. We tend to use colchicine in children with atypical PFAPA, namely children who have additional symptoms but are not suffering from a hereditary periodic fever syndrome, or in children who did not improve following tonsillectomy. Another role for colchicine might be treating children with a predominance of aphthous stomatitis, although this still has to be evaluated. A small case series indicated that PFAPA flares were responsive to an IL‐1 blockade, as discussed above 38.
Prognosis
Children with PFAPA are usually healthy between flares, and the symptoms disappear within three to five years after disease onset or in adolescence (6, 27, 28, 29, 32, 59). It is important to note that relapses are well described in PFAPA 30, and we have seen late relapses in our clinic that occurred after several years (Wekell P and Berg S, unpublished observation). Despite the good prognosis, our clinical experience and preliminary data from parental interviews indicate that the disease considerably influences the quality of life of the child and the family as a whole, as long as the episodes persist (Sparud Lundin C and Wekell P, manuscript in preparation).
Discussion
For a long time, innate immunity was just seen as a basic, noncomplex way for the body to defend itself against microbes, by eliciting an inflammatory reaction. The more complex immune mechanisms involved in long‐term immunity, immune deficiency and autoimmunity were all attributed entirely to the adaptive immune system. Charles Janeway first foresaw the existence and function of innate immunity in a classic paper in 1989 called Approaching the asymptote? Evolution and revolution in immunology 65. In this study, he proposed the idea of pattern recognition, a general principle of innate immune recognition, and his work provided a conceptual framework for the integration of innate and acquired immunity 66.
The innate immune response is a first line of defence, which is rapid and short‐lived and recognises exogenous and endogenous danger, by a limited number of germline‐encoded evolutionarily conserved receptors. As for all biological systems, dysfunction or dysregulation may lead to disease. In innate immunity, the field of autoinflammatory diseases has been defined as conditions associated with increased inflammation due to a significant host predisposition.
Over the last 20 years, an increasing number of monogenic autoinflammatory conditions have been characterised: their genetic background has been defined in 36 clinical conditions to date and a large proportion of these are represented in Table 1 4. Although there are still substantial clinical and mechanistic knowledge gaps, these conditions are more accurately diagnosed, treated and their disease mechanism is better understood today. Even though, there are some clues as to which genes and molecular mechanisms are involved in polygenic or multifactorial diseases, over time it has been shown that it is more difficult to delineate the mechanisms of innate dysregulation in polygenic or multifactorial diseases. This is exemplified by PFAPA, the most common autoinflammatory condition among children outside regions with very high prevalence of FMF.
During two decades of remarkable developments in the field, research on autoinflammatory diseases has not only led to an increased understanding of disease mechanisms per se, but it has also been indispensable for the understanding of innate immunity as a whole, as well as shedding light on the importance of these mechanisms in other disease conditions. Today, researchers at one end of the autoinflammatory disease spectrum discuss rare monogenic conditions, while discussions at the other end of the spectrum concern the role of inflammatory pathways in common diseases such as mellitus type 2, Alzheimer's disease and atherosclerosis 3, 4. This is a development that no one could have foreseen 17 years ago, when the concept of autoinflammation was coined.
Funding
This review was funded by the Department of Research and Development of the NU Hospital Organization, Region Västra Götaland, the Fyrbodal Research and Development Council, and the Health and Medical Care Committee of the Regional Executive Board, the Swedish Medical Research Council, the IngaBritt and Arne Lundberg Research Foundation, the King Gustaf V 80‐Year Memorial Foundation, the Swedish Rheumatism Association.
References
- 1. McDermott MF, Aksentijevich I, Galon J, McDermott EM, Ogunkolade BW, Centola M, et al. Germline mutations in the extracellular domains of the 55 kDa TNF receptor, TNFR1, define a family of dominantly inherited autoinflammatory syndromes. Cell 1999; 97: 133–44. [DOI] [PubMed] [Google Scholar]
- 2. Galon J, Aksentijevich I, McDermott MF, O'Shea JJ, Kastner DL. TNFRSF1A mutations and autoinflammatory syndromes. Curr Opin Immunol 2000; 12: 479–86. [DOI] [PubMed] [Google Scholar]
- 3. Masters SL, Simon A, Aksentijevich I, Kastner DL. Horror autoinflammaticus: the molecular pathophysiology of autoinflammatory disease. Annu Rev Immunol 2009; 27: 621–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. de Jesus AA, Canna SW, Liu Y, Goldbach‐Mansky R. Molecular mechanisms in genetically defined autoinflammatory diseases: disorders of amplified danger signaling. Annu Rev Immunol 2015; 33: 823–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shinar Y, Obici L, Aksentijevich I, Bennetts B, Austrup F, Ceccherini I, et al. Guidelines for the genetic diagnosis of hereditary recurrent fevers. Ann Rheum Dis 2012; 71: 1599–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kastner DL, Aksentijevich I, Goldbach‐Mansky R. Autoinflammatory disease reloaded: a clinical perspective. Cell 2010; 140: 784–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hoffman HM, Mueller JL, Broide DH, Wanderer AA, Kolodner RD. Mutation of a new gene encoding a putative pyrin‐like protein causes familial cold autoinflammatory syndrome and Muckle‐Wells syndrome. Nat Genet 2001; 29: 301–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Saito M, Nishikomori R, Kambe N, Fujisawa A, Tanizaki H, Takeichi K, et al. Disease‐associated CIAS1 mutations induce monocyte death, revealing low‐level mosaicism in mutation‐negative cryopyrin‐associated periodic syndrome patients. Blood 2008; 111: 2132–41. [DOI] [PubMed] [Google Scholar]
- 9. Aksentijevich I, Masters SL, Ferguson PJ, Dancey P, Frenkel J, van Royen‐Kerkhoff A, et al. An autoinflammatory disease with deficiency of the interleukin‐1‐receptor antagonist. N Engl J Med 2009; 360: 2426–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jeru I, Duquesnoy P, Fernandes‐Alnemri T, Cochet E, Yu JW, Lackmy‐Port‐Lis M, et al. Mutations in NALP12 cause hereditary periodic fever syndromes. Proc Natl Acad Sci USA 2008; 105: 1614–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Miceli‐Richard C, Lesage S, Rybojad M, Prieur AM, Manouvrier‐Hanu S, Hafner R, et al. CARD15 mutations in Blau syndrome. Nat Genet 2001; 29: 19–20. [DOI] [PubMed] [Google Scholar]
- 12. Kanazawa N, Okafuji I, Kambe N, Nishikomori R, Nakata‐Hizume M, Nagai S, et al. Early‐onset sarcoidosis and CARD15 mutations with constitutive nuclear factor‐kappaB activation: common genetic etiology with Blau syndrome. Blood 2005; 105: 1195–7. [DOI] [PubMed] [Google Scholar]
- 13. Agarwal AK, Xing C, DeMartino GN, Mizrachi D, Hernandez MD, Sousa AB, et al. PSMB8 encoding the beta5i proteasome subunit is mutated in joint contractures, muscle atrophy, microcytic anemia, and panniculitis‐induced lipodystrophy syndrome. Am J Hum Genet 2010; 87: 866–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Crow YJ. Type I interferonopathies: a novel set of inborn errors of immunity. Ann N Y Acad Sci 2011; 1238: 91–8. [DOI] [PubMed] [Google Scholar]
- 15. Onoufriadis A, Simpson MA, Pink AE, Di Meglio P, Smith CH, Pullabhatla V, et al. Mutations in IL36RN/IL1F5 are associated with the severe episodic inflammatory skin disease known as generalized pustular psoriasis. Am J Hum Genet 2011; 89: 432–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Glocker EO, Kotlarz D, Boztug K, Gertz EM, Schaffer AA, Noyan F, et al. Inflammatory bowel disease and mutations affecting the interleukin‐10 receptor. N Engl J Med 2009; 361: 2033–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. McGonagle D, McDermott MF. A proposed classification of the immunological diseases. PLoS Med 2006; 3: e297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Meng G, Zhang F, Fuss I, Kitani A, Strober W. A mutation in the Nlrp3 gene causing inflammasome hyperactivation potentiates Th17 cell‐dominant immune responses. Immunity 2009; 30: 860–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lasiglie D, Traggiai E, Federici S, Alessio M, Buoncompagni A, Accogli A, et al. Role of IL‐1 beta in the development of human T(H)17 cells: lesson from NLPR3 mutated patients. PLoS ONE 2011; 6: e20014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Marshall GS. Prolonged and recurrent fevers in children. J Infect 2014; 68(Suppl. 1): S83–93. [DOI] [PubMed] [Google Scholar]
- 21. Gattorno M, Caorsi R, Meini A, Cattalini M, Federici S, Zulian F, et al. Differentiating PFAPA syndrome from monogenic periodic fevers. Pediatrics 2009; 124: e721–8. [DOI] [PubMed] [Google Scholar]
- 22. Ozen S, Bilginer Y. A clinical guide to autoinflammatory diseases: familial Mediterranean fever and next‐of‐kin. Nat Rev Rheumatol 2014; 10: 135–47. [DOI] [PubMed] [Google Scholar]
- 23. Padeh S, Stoffman N, Berkun Y. Periodic fever accompanied by aphthous stomatitis, pharyngitis and cervical adenitis syndrome (PFAPA syndrome) in adults. Isr Med Assoc J 2008; 10: 358–60. [PubMed] [Google Scholar]
- 24. Forsvoll J, Kristoffersen EK, Oymar K. Incidence, clinical characteristics and outcome in Norwegian children with periodic fever, aphthous stomatitis, pharyngitis and cervical adenitis syndrome; a population‐based study. Acta Paediatr 2013; 102: 187–92. [DOI] [PubMed] [Google Scholar]
- 25. Feder HM, Salazar JC. A clinical review of 105 patients with PFAPA (a periodic fever syndrome). Acta Paediatr 2010; 99: 178–84. [DOI] [PubMed] [Google Scholar]
- 26. Hofer M, Pillet P, Cochard MM, Berg S, Krol P, Kone‐Paut I, et al. International periodic fever, aphthous stomatitis, pharyngitis, cervical adenitis syndrome cohort: description of distinct phenotypes in 301 patients. Rheumatology 2014; 53: 1125–9. [DOI] [PubMed] [Google Scholar]
- 27. Thomas KT, Feder HM Jr, Lawton AR, Edwards KM. Periodic fever syndrome in children. J Pediatr 1999; 135: 15–21. [DOI] [PubMed] [Google Scholar]
- 28. Marshall GS, Edwards KM, Lawton AR. PFAPA syndrome. Pediatr Infect Dis J 1989; 8: 658–9. [DOI] [PubMed] [Google Scholar]
- 29. Padeh S, Brezniak N, Zemer D, Pras E, Livneh A, Langevitz P, et al. Periodic fever, aphthous stomatitis, pharyngitis, and adenopathy syndrome: clinical characteristics and outcome. J Pediatr 1999; 135: 98–101. [DOI] [PubMed] [Google Scholar]
- 30. Tasher D, Somekh E, Dalal I. PFAPA syndrome: new clinical aspects disclosed. Arch Dis Child 2006; 91: 981–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Federici S, Gattorno M. A practical approach to the diagnosis of autoinflammatory diseases in childhood. Best Pract Res Clin Rheumatol 2014; 28: 263–76. [DOI] [PubMed] [Google Scholar]
- 32. Ter Haar N, Lachmann H, Ozen S, Woo P, Uziel Y, Modesto C, et al. Treatment of autoinflammatory diseases: results from the Eurofever Registry and a literature review. Ann Rheum Dis 2013; 72: 678–85. [DOI] [PubMed] [Google Scholar]
- 33. Dale DC, Person RE, Bolyard AA, Aprikyan AG, Bos C, Bonilla MA, et al. Mutations in the gene encoding neutrophil elastase in congenital and cyclic neutropenia. Blood 2000; 96: 2317–22. [PubMed] [Google Scholar]
- 34. Samuels J, Ozen S. Familial Mediterranean fever and the other autoinflammatory syndromes: evaluation of the patient with recurrent fever. Curr Opin Rheumatol 2006; 18: 108–17. [DOI] [PubMed] [Google Scholar]
- 35. Wekell P, Fasth A, Berg S. Autoinflammatory disorders In Aghamohammadi A, Rezaei N, editors. Clinical cases in primary immunodeficiency diseases a problem‐solving approach. Berlin Heidelberg: Springer, 2012: 309–24. [Google Scholar]
- 36. Gattorno M, Federici S, Pelagatti MA, Caorsi R, Brisca G, Malattia C, et al. Diagnosis and management of autoinflammatory diseases in childhood. J Clin Immunol 2008; 28(Suppl. 1): S73–83. [DOI] [PubMed] [Google Scholar]
- 37. Stojanov S, Hoffmann F, Kery A, Renner ED, Hartl D, Lohse P, et al. Cytokine profile in PFAPA syndrome suggests continuous inflammation and reduced anti‐inflammatory response. Eur Cytokine Netw 2006; 17: 90–7. [PubMed] [Google Scholar]
- 38. Stojanov S, Lapidus S, Chitkara P, Feder H, Salazar JC, Fleisher TA, et al. Periodic fever, aphthous stomatitis, pharyngitis, and adenitis (PFAPA) is a disorder of innate immunity and Th1 activation responsive to IL‐1 blockade. Proc Natl Acad Sci USA 2011; 108: 7148–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kolly L, Busso N, von Scheven‐Gete A, Bagnoud N, Moix I, Holzinger D, et al. Periodic fever, aphthous stomatitis, pharyngitis, cervical adenitis syndrome is linked to dysregulated monocyte IL‐1beta production. J Allergy Clin Immunol 2013; 131: 1635–43. [DOI] [PubMed] [Google Scholar]
- 40. Brown KL, Wekell P, Osla V, Sundqvist M, Savman K, Fasth A, et al. Profile of blood cells and inflammatory mediators in periodic fever, aphthous stomatitis, pharyngitis and adenitis (PFAPA) syndrome. BMC Pediatr 2010; 10: 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Forsvoll J, Kristoffersen EK, Oymar K. Elevated levels of CXCL10 in the periodic fever, aphthous stomatitis, pharyngitis and cervical adenitis syndrome (PFAPA) during and between febrile episodes; an indication of a persistent activation of the innate immune system. Pediatr Rheumatol Online J 2013; 11: 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Manz MG, Boettcher S. Emergency granulopoiesis. Nat Rev Immunol 2014; 14: 302–14. [DOI] [PubMed] [Google Scholar]
- 43. Sundqvist M, Wekell P, Osla V, Bylund J, Christenson K, Savman K, et al. Increased intracellular oxygen radical production in neutrophils during febrile episodes of periodic fever, aphthous stomatitis, pharyngitis, and cervical adenitis syndrome. Arthritis Rheum 2013; 65: 2971–83. [DOI] [PubMed] [Google Scholar]
- 44. Peridis S, Koudoumnakis E, Theodoridis A, Stefanaki K, Helmis G, Houlakis M. Surgical outcomes and histology findings after tonsillectomy in children with periodic fever, aphthous stomatitis, pharyngitis, and cervical adenitis syndrome. Am J Otolaryngol 2010; 31: 472–5. [DOI] [PubMed] [Google Scholar]
- 45. Dytrych P, Krol P, Kotrova M, Kuzilkova D, Hubacek P, Krol L, et al. Polyclonal, newly derived T cells with low expression of inhibitory molecule PD‐1 in tonsils define the phenotype of lymphocytes in children with periodic fever, aphtous stomatitis, pharyngitis and adenitis (PFAPA) syndrome. Mol Immunol 2015; 65: 139–47. [DOI] [PubMed] [Google Scholar]
- 46. Valenzuela PM, Araya A, Perez CI, Maul X, Serrano C, Beltran C, et al. Profile of inflammatory mediators in tonsils of patients with periodic fever, aphthous stomatitis, pharyngitis, and cervical adenitis (PFAPA) syndrome. Clin Rheumatol 2013; 32: 1743–9. [DOI] [PubMed] [Google Scholar]
- 47. Forsvoll J, Janssen EA, Moller I, Wathne N, Skaland I, Klos J, et al. Reduced number of CD8 + cells in tonsillar germinal centres in children with the periodic fever, aphthous stomatitis, pharyngitis and cervical adenitis syndrome. Scand J Immunol 2015; 82: 76–83. [DOI] [PubMed] [Google Scholar]
- 48. Tejesvi MV, Uhari M, Tapiainen T, Pirttila AM, Suokas M, Lantto U, et al. Tonsillar microbiota in children with PFAPA (periodic fever, aphthous stomatitis, pharyngitis, and adenitis) syndrome. Eur J Clin Microbiol Infect Dis 2016; 35: 963–70. [DOI] [PubMed] [Google Scholar]
- 49. Lachmann HJ, Sengul B, Yavuzsen TU, Booth DR, Booth SE, Bybee A, et al. Clinical and subclinical inflammation in patients with familial Mediterranean fever and in heterozygous carriers of MEFV mutations. Rheumatology 2006; 45: 746–50. [DOI] [PubMed] [Google Scholar]
- 50. Yazgan H, Keles E, Yazgan Z, Gebesce A, Demirdoven M. C‐reactive protein and procalcitonin during febril attacks in PFAPA syndrome. Int J Pediatr Otorhinolaryngol 2012; 76: 1145–7. [DOI] [PubMed] [Google Scholar]
- 51. Yuksel S, Ekim M, Ozcakar ZB, Yalcinkaya F, Acar B, Oztuna D, et al. The value of procalcitonin measurements in children with familial Mediterranean fever. Rheumatol Int 2012; 32: 3443–7. [DOI] [PubMed] [Google Scholar]
- 52. Holzinger D, Kessel C, Omenetti A, Gattorno M. From bench to bedside and back again: translational research in autoinflammation. Nat Rev Rheumatol 2015; 11: 573–85. [DOI] [PubMed] [Google Scholar]
- 53. Yilmaz H, Inan O, Darcin T, Bilgic MA, Akcay A. Serum galectin‐3 levels were associated with proteinuria in patients with Familial Mediterranean Fever. Clin Exp Nephrol 2015; 19: 436–42. [DOI] [PubMed] [Google Scholar]
- 54. ten Oever J, Giamarellos‐Bourboulis EJ, van de Veerdonk FL, Stelma FF, Simon A, Janssen M, et al. Circulating galectin‐3 in infections and non‐infectious inflammatory diseases. Eur J Clin Microbiol Infect Dis 2013; 32: 1605–10. [DOI] [PubMed] [Google Scholar]
- 55. Kang EH, Moon KC, Lee EY, Lee YJ, Lee EB, Ahn C, et al. Renal expression of galectin‐3 in systemic lupus erythematosus patients with nephritis. Lupus 2009; 18: 22–8. [DOI] [PubMed] [Google Scholar]
- 56. Lee YJ, Kang SW, Song JK, Park JJ, Bae YD, Lee EY, et al. Serum galectin‐3 and galectin‐3 binding protein levels in Behcet's disease and their association with disease activity. Clin Exp Rheumatol 2007; 25(Suppl. 45): S41–5. [PubMed] [Google Scholar]
- 57. Ezzat MH, El‐Gammasy TM, Shaheen KY, Osman AO. Elevated production of galectin‐3 is correlated with juvenile idiopathic arthritis disease activity, severity, and progression. Int J Rheum Dis 2011; 14: 345–52. [DOI] [PubMed] [Google Scholar]
- 58. Frol'ova L, Smetana K Jr, Borovska D, Kitanovicova A, Klimesova K, Janatkova I, et al. Detection of galectin‐3 in patients with inflammatory bowel diseases: new serum marker of active forms of IBD? Inflamm Res 2009; 58: 503–12. [DOI] [PubMed] [Google Scholar]
- 59. Wurster VM, Carlucci JG, Feder HM Jr, Edwards KM. Long‐term follow‐up of children with periodic fever, aphthous stomatitis, pharyngitis, and cervical adenitis syndrome. J Pediatr 2011; 159: 958–64. [DOI] [PubMed] [Google Scholar]
- 60. Ter Haar NM, Oswald M, Jeyaratnam J, Anton J, Barron KS, Brogan PA, et al. Recommendations for the management of autoinflammatory diseases. Ann Rheum Dis 2015; 74: 1636–44. [DOI] [PubMed] [Google Scholar]
- 61. Galanakis E, Papadakis CE, Giannoussi E, Karatzanis AD, Bitsori M, Helidonis ES. PFAPA syndrome in children evaluated for tonsillectomy. Arch Dis Child 2002; 86: 434–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Garavello W, Romagnoli M, Gaini RM. Effectiveness of adenotonsillectomy in PFAPA syndrome: a randomized study. J Pediatr 2009; 155: 250–3. [DOI] [PubMed] [Google Scholar]
- 63. Burton MJ, Pollard AJ, Ramsden JD, Chong LY, Venekamp RP. Tonsillectomy for periodic fever, aphthous stomatitis, pharyngitis and cervical adenitis syndrome (PFAPA). Cochrane Database Syst Rev 2014; 9: CD008669. [DOI] [PubMed] [Google Scholar]
- 64. Tasher D, Stein M, Dalal I, Somekh E. Colchicine prophylaxis for frequent periodic fever, aphthous stomatitis, pharyngitis and adenitis episodes. Acta Paediatr 2008; 97: 1090–2. [DOI] [PubMed] [Google Scholar]
- 65. Janeway CA Jr. Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb Symp Quant Biol 1989; 54(Pt 1): 1–13. [DOI] [PubMed] [Google Scholar]
- 66. Medzhitov R. Approaching the asymptote: 20 years later. Immunity 2009; 30: 766–75. [DOI] [PubMed] [Google Scholar]