

Abstract
Controversy exists regarding the association of bovine leukemia virus (BLV) and breast cancer. PCR-based experimental evidence indicates that BLV DNA is present in breast tissue and that as many as 37% of cancer cases may be attributable to viral exposure. Since this association might have major consequences for human health, we evaluated 51 whole genomes of breast cancer samples for the presence of BLV DNA. Among 32 billion sequencing reads retrieved from the NCBI database of genotype and phenotype, none mapped on different strains of the BLV genome. Controls for sequence divergence and proviral loads further validated the approach. This unbiased analysis thus excludes a clonal insertion of BLV in breast tumor cells and strongly argues against an association between BLV and breast cancer.
Keywords: Breast cancer, Bovine leukemia virus, BLV
Background
BLV naturally infects cattle, water buffalo, yak and zebu [1–4]. Sporadic infections with BLV have occasionally been reported in other species like alpaca [5]. Experimentally, BLV can also be transmitted to a number of species including sheep [6], goats [6], rats [7] and rabbits [8]. BLV infection causes B cell lymphocytosis, leukemia and/or lymphoma in natural and some experimental hosts [1]. There is also controversial evidence suggesting that BLV might infect humans: (1) antibodies against the BLV capsid were detected in 74% of human sera from the Berkeley Community, California [9], (2) BLV DNA was detected in breast tissues using PCR [10–12]. Based on a positive correlation between the rates of BLV infection and tumor frequencies (36–59% compared to 29–45% in normal tissue), as many as 37% of breast cancer cases may be attributable to BLV exposure [12].
Although these observations initiated some skepticism within the scientific community [13], the potential consequences for human health clearly require further investigation.
Results and discussion
To avoid potential experimental artifacts associated with DNA amplification techniques, we directly analyzed whole genomes of breast tumors and adjacent tissues. After retrieval of raw DNA sequences from the NCBI dbGaP [14, 15], paired-reads were probed for alignment on different BLV strains using Bowtie2. As a positive control, a nuclear DNA fragment (chr12: 53,959,600–53,964,000) devoid of repeated sequences that would lead to an overestimation of aligned reads and set to 4.4 kb to fit with the monoploid 8.8 kb BLV genome was selected from the human genome. Alignment of 51 breast tumors genomes on the nuclear control sequence identified between 283 and 1287 paired-reads (illustrated on Fig. 1 and summarized on Table 1). In contrast, no homology was found with 5 different BLV subtypes (highlighted in blue on the phylogenic tree of Fig. 2a). In 19 biopsies adjacent to the breast tumors, 386–1197 paired-reads aligned onto the nuclear DNA sequence whereas none mapped on BLV (Table 1). All DNA samples contained extranuclear DNA as indicated by alignment of a control mitochondrial sequence (NC_012920) (Table 1).
Fig. 1.

Representative alignment of dbGaP sequencing reads to human and BLV DNA. Breast cancer patients were BRC3 from USA (study phs000472), MEX-BR-15 from Mexico and SX1A2 from Vietnam (study phs000369). Aligned reads were visualized using integrative genomics viewer (IGV)
Table 1.
Absence of BLV DNA in 51 whole genomes of breast tumors
| Subject ID | Country | Age | Diagnosis | Sample type | Grade | HER2 status | ER status | PR status | Total no of reads | No. of reads that align on | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Control DNA (nuclear) | Control DNA (mitochondrial) | BLV_AF033818 | BLV_AF257515 | BLV_D00647 | BLV_K02021 | BLV_LC080667 | ||||||||||
| MEX-BR-106 | Mexico | 42 | IDC | Tumor | II | − | + | + | 583,906,975 | 669 | 396,239 | 0 | 0 | 0 | 0 | 0 |
| MEX-BR-116 | Mexico | 92 | IDC | Tumor | III | − | + | − | 577,618,196 | 796 | 1,166,916 | 0 | 0 | 0 | 0 | 0 |
| MEX-BR-15 | Mexico | 45 | IDC | Tumor | II | + | − | + | 571,043,227 | 652 | 1,167,672 | 0 | 0 | 0 | 0 | 0 |
| MEX-BR-154 | Mexico | 52 | IDC | Tumor | III | − | + | + | 700,630,351 | 811 | 400,383 | 0 | 0 | 0 | 0 | 0 |
| MEX-BR-165 | Mexico | 42 | IDC | Tumor | II | − | + | + | 757,323,566 | 737 | 742,646 | 0 | 0 | 0 | 0 | 0 |
| MEX-BR-198 | Mexico | 44 | IDC | Tumor | II | − | + | + | 745,509,529 | 1019 | 1,264,555 | 0 | 0 | 0 | 0 | 0 |
| MEX-BR-50 | Mexico | 47 | IDC | Tumor | II | − | + | + | 605,198,587 | 653 | 958,812 | 0 | 0 | 0 | 0 | 0 |
| MEX-BR-82 | Mexico | 59 | IDC | Tumor | II | − | + | − | 681,881,066 | 687 | 547,863 | 0 | 0 | 0 | 0 | 0 |
| BRC12 | USA | 81 | IDC | Tumor | II | − | U | U | 548,255,169 | 745 | 1,113,306 | 0 | 0 | 0 | 0 | 0 |
| BRC13 | USA | 51 | IDC | Tumor | III | − | 7 | U | 587,461,482 | 686 | 1,106,780 | 0 | 0 | 0 | 0 | 0 |
| BRC14 | USA | 86 | IDC | Tumor | III | − | 7 | U | 755,094,207 | 899 | 1,469,976 | 0 | 0 | 0 | 0 | 0 |
| BRC15 | USA | 83 | IDC | Tumor | II | − | 7 | U | 758,784,262 | 934 | 2,327,824 | 0 | 0 | 0 | 0 | 0 |
| BRC16 | USA | 61 | IDC | Tumor | III | − | 7 | U | 821,134,040 | 1287 | 2,084,782 | 0 | 0 | 0 | 0 | 0 |
| BRC18 | USA | 85 | IDC | Tumor | I | − | 8 | U | 568,355,455 | 677 | 1,395,823 | 0 | 0 | 0 | 0 | 0 |
| BRC19 | USA | 75 | IDC | Tumor | II | − | 8 | U | 596,337,842 | 747 | 1,648,870 | 0 | 0 | 0 | 0 | 0 |
| BRC20 | USA | 61 | IDC | Tumor | III | − | 4 | U | 507,651,900 | 570 | 1,026,830 | 0 | 0 | 0 | 0 | 0 |
| BRC21 | USA | 73 | IDC | Tumor | I | − | 7 | U | 719,742,122 | 817 | 1,710,010 | 0 | 0 | 0 | 0 | 0 |
| BRC22 | USA | 64 | ILC | Tumor | I | − | 6 | U | 608,469,920 | 708 | 953,100 | 0 | 0 | 0 | 0 | 0 |
| BRC23 | USA | 68 | IDC | Tumor | I | − | 7 | U | 613,481,215 | 687 | 1,272,519 | 0 | 0 | 0 | 0 | 0 |
| BRC24 | USA | 51 | IDC | Tumor | II | − | 7 | U | 656,115,800 | 721 | 1,980,030 | 0 | 0 | 0 | 0 | 0 |
| BRC25 | USA | 52 | IDC | Tumor | II | − | 5 | U | 583,560,227 | 712 | 580,203 | 0 | 0 | 0 | 0 | 0 |
| BRC28 | USA | 52 | IDC | Tumor | I | − | 7 | U | 664,667,777 | 781 | 973,990 | 0 | 0 | 0 | 0 | 0 |
| BRC29 | USA | 74 | IDC | Tumor | III | − | 6 | U | 785,019,563 | 596 | 2,085,482 | 0 | 0 | 0 | 0 | 0 |
| BRC3 | USA | 62 | IDC | Tumor | II | − | 8 | U | 695,174,967 | 1026 | 3,134,341 | 0 | 0 | 0 | 0 | 0 |
| BRC30 | USA | 60 | ILC | Tumor | II | − | 5 | U | 663,769,744 | 794 | 1,442,014 | 0 | 0 | 0 | 0 | 0 |
| BRC31 | USA | 66 | IDC | Tumor | II | − | 6 | U | 734,384,352 | 1028 | 1,415,996 | 0 | 0 | 0 | 0 | 0 |
| BRC32 | USA | 54 | IDC | Tumor | I | − | 7 | U | 643,884,178 | 703 | 1,404,436 | 0 | 0 | 0 | 0 | 0 |
| BRC33 | USA | 83 | IDC | Tumor | II | − | 8 | U | 660,668,877 | 819 | 1,284,599 | 0 | 0 | 0 | 0 | 0 |
| BRC34 | USA | 79 | IDC | Tumor | I | − | 7 | U | 572,861,930 | 704 | 1,499,414 | 0 | 0 | 0 | 0 | 0 |
| BRC35 | USA | 76 | IDC | Tumor | II | − | 6 | U | 543,480,474 | 697 | 1,709,943 | 0 | 0 | 0 | 0 | 0 |
| BRC36 | USA | 68 | IDC | Tumor | II | − | 7 | U | 706,448,348 | 804 | 1,501,763 | 0 | 0 | 0 | 0 | 0 |
| BRC40 | USA | 66 | IDC | Tumor | I | − | 8 | U | 600,847,516 | 690 | 1,686,112 | 0 | 0 | 0 | 0 | 0 |
| BRC41 | USA | 55 | IDC | Tumor | II | − | 8 | U | 689,312,217 | 812 | 3,735,591 | 0 | 0 | 0 | 0 | 0 |
| BRC42 | USA | 74 | IDC | Tumor | II | − | U | U | 684,312,302 | 685 | 1,308,948 | 0 | 0 | 0 | 0 | 0 |
| BRC44 | USA | 64 | IDC | Tumor | II | − | 7 | U | 717,390,251 | 891 | 1,430,064 | 0 | 0 | 0 | 0 | 0 |
| BRC47 | USA | 54 | IDC | Tumor | III | − | 5 | U | 580,674,755 | 865 | 960,944 | 0 | 0 | 0 | 0 | 0 |
| BRC48 | USA | 66 | IDC | Tumor | II | − | 6 | U | 782,262,353 | 783 | 1,236,102 | 0 | 0 | 0 | 0 | 0 |
| BRC49 | USA | 56 | IDC | Tumor | II | − | 8 | U | 577,656,003 | 559 | 881,804 | 0 | 0 | 0 | 0 | 0 |
| BRC5 | USA | 72 | IDC | Tumor | II | − | 7 | U | 762,026,860 | 1155 | 2,462,819 | 0 | 0 | 0 | 0 | 0 |
| BRC50 | USA | 78 | ILC | Tumor | I | − | 4 | U | 661,525,693 | 792 | 357,915 | 0 | 0 | 0 | 0 | 0 |
| BRC7 | USA | 78 | IDC | Tumor | II | − | 8 | U | 455,727,994 | 795 | 580,484 | 0 | 0 | 0 | 0 | 0 |
| BRC8 | USA | 87 | IDC | Tumor | I | − | 8 | U | 518,548,285 | 628 | 1,394,439 | 0 | 0 | 0 | 0 | 0 |
| BRC9 | USA | 65 | ILC | Tumor | II | − | 8 | U | 516,702,802 | 697 | 1,759,444 | 0 | 0 | 0 | 0 | 0 |
| 9DDA1 | Vietnam | 60 | IDC | Tumor | III | U | U | U | 706,450,950 | 759 | 1,109,340 | 0 | 0 | 0 | 0 | 0 |
| 9P4X9 | Vietnam | 54 | IDC | Tumor | III | U | U | U | 610,913,537 | 778 | 619,066 | 0 | 0 | 0 | 0 | 0 |
| 9YBUF | Vietnam | 52 | IDC | Tumor | III | U | U | U | 595,959,881 | 616 | 788,058 | 0 | 0 | 0 | 0 | 0 |
| CI5PD | Vietnam | 51 | IDC | Tumor | III | U | U | U | 572,612,309 | 626 | 786,787 | 0 | 0 | 0 | 0 | 0 |
| FYGW6 | Vietnam | 38 | IDC | Tumor | III | U | U | U | 238,201,059 | 282 | 221,942 | 0 | 0 | 0 | 0 | 0 |
| GT33 V | Vietnam | 52 | IDC | Tumor | III | U | U | U | 548,640,325 | 604 | 766,320 | 0 | 0 | 0 | 0 | 0 |
| SX1A2 | Vietnam | 53 | IDC | Tumor | III | U | + | + | 598,405,143 | 693 | 1,002,577 | 0 | 0 | 0 | 0 | 0 |
| UQWDS | Vietnam | 35 | IDC | Tumor | III | U | − | − | 596,126,825 | 665 | 1,285,884 | 0 | 0 | 0 | 0 | 0 |
| 9DDA1 | Vietnam | 60 | IDC | Normal | III | U | U | U | 691,060,649 | 797 | 1,122,133 | 0 | 0 | 0 | 0 | 0 |
| 9P4X9 | Vietnam | 54 | IDC | Normal | III | U | U | U | 601,815,791 | 664 | 694,153 | 0 | 0 | 0 | 0 | 0 |
| 9YBUF | Vietnam | 52 | IDC | Normal | III | U | U | U | 593,968,922 | 646 | 1,202,175 | 0 | 0 | 0 | 0 | 0 |
| CI5PD | Vietnam | 51 | IDC | Normal | III | U | U | U | 566,065,567 | 595 | 911,133 | 0 | 0 | 0 | 0 | 0 |
| FYGW6 | Vietnam | 38 | IDC | Normal | III | U | U | U | 337,274,647 | 386 | 361,063 | 0 | 0 | 0 | 0 | 0 |
| GT33 V | Vietnam | 52 | IDC | Normal | III | U | U | U | 581,403,783 | 652 | 1,189,003 | 0 | 0 | 0 | 0 | 0 |
| SX1A2 | Vietnam | 53 | IDC | Normal | III | U | + | + | 608,739,604 | 700 | 878,362 | 0 | 0 | 0 | 0 | 0 |
| UQWDS | Vietnam | 35 | IDC | Normal | III | U | − | − | 590,387,671 | 685 | 829,847 | 0 | 0 | 0 | 0 | 0 |
| MEX-BR-106 | Mexico | 42 | IDC | Normal | II | − | + | + | 539,137,287 | 526 | 351,034 | 0 | 0 | 0 | 0 | 0 |
| MEX-BR-116 | Mexico | 92 | IDC | Normal | III | − | + | − | 513,833,151 | 520 | 258,287 | 0 | 0 | 0 | 0 | 0 |
| MEX-BR-123 | Mexico | 71 | IDC | Normal | III | − | + | U | 668,026,494 | 761 | 515,501 | 0 | 0 | 0 | 0 | 0 |
| MEX-BR-15 | Mexico | 45 | IDC | Normal | II | + | − | + | 592,958,041 | 670 | 756,778 | 0 | 0 | 0 | 0 | 0 |
| MEX-BR-154 | Mexico | 52 | IDC | Normal | III | − | + | + | 670,289,201 | 929 | 817,446 | 0 | 0 | 0 | 0 | 0 |
| MEX-BR-165 | Mexico | 42 | IDC | Normal | II | − | + | + | 712,308,425 | 706 | 537,516 | 0 | 0 | 0 | 0 | 0 |
| MEX-BR-198 | Mexico | 44 | IDC | Normal | II | − | + | + | 726,225,752 | 831 | 216,109 | 0 | 0 | 0 | 0 | 0 |
| MEX-BR-200 | Mexico | 42 | IDC | Normal | II | − | + | + | 767,097,542 | 1197 | 279,031 | 0 | 0 | 0 | 0 | 0 |
| MEX-BR-28 | Mexico | 79 | MC | Normal | II | − | + | + | 588,561,634 | 607 | 215,022 | 0 | 0 | 0 | 0 | 0 |
| MEX-BR-50 | Mexico | 47 | IDC | Normal | II | − | + | + | 551,537,695 | 618 | 394,842 | 0 | 0 | 0 | 0 | 0 |
| MEX-BR-82 | Mexico | 59 | IDC | Normal | II | − | + | − | 608,849,308 | 719 | 385,789 | 0 | 0 | 0 | 0 | 0 |
Whole genome sequencing data from 51 breast tumors and 19 normal adjacent breast tissues were downloaded from the NCBI dbGaP. Hundreds of millions of paired-reads per sample were probed for alignment on different BLV strains and on nuclear and mitochondrial human control sequences
IDC infiltrating ductal carcinoma, ILC infiltrating lobular carcinoma, MC mixed carcinoma, U unknown
Fig. 2.
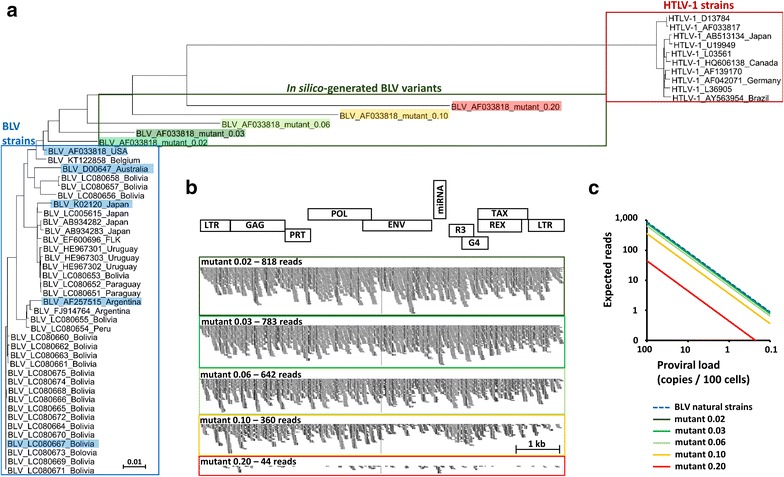
Analysis of sequence variation and proviral load in sequence alignments. a Neighbour-joining phylogenetic tree of BLV and HTLV-1 genomes. b Using the ART simulation tool (NIH), Illumina-like 100 bp paired-reads were generated in silico from the mutants. 880 simulated reads were probed for alignment on BLV AF033818 using Bowtie2 and visualized using IGV. c Correlation between proviral loads and predicted number of reads
Although no paired-read corresponding to five different BLV variants could be identified, the possibility remains that extensive sequence variability impaired detection. On average, the whole genome sequencing procedure generated 660 million reads per sample. Given that the BLV provirus length is 8.8 kb and that a normal human diploid genome is 6.6 billion base pairs, the average number of reads that would be generated by a 8.8 kb-long monoploid sequence is 880 (660,000,000/6600,000,000 × 8800). Providing that the BLV provirus is integrated in a single copy per cell, the whole genome sequencing procedure would thus generate 880 reads on average. If the strain in the sample diverges from the five reference sequences, a fraction of the reads would not be retrieved. Therefore, BLV variants were artificially generated in silico by introducing 2, 3, 6, 10 and 20% nucleotide changes in reference AF033818 (mutants 0.02, 0.03, 0.06, 0.10 and 0.20, respectively). Phylogenetic analysis of Fig. 2a illustrates that in silico generated divergence far exceeds the maximal natural sequence variations observed worldwide [16]. 880 Illumina-like reads were then simulated from these in silico variants using ART simulation tool and mapped on BLV genome AF033818. Most reads (818 of 880) generated from mutant 0.02 aligned on reference sequence AF033818 (Fig. 2b). Even the highly divergent mutant 0.10 still aligned 41% of its 880 reads on the reference. Up to 20% divergence in mutant 0.20 was required to significantly impair detection, although BLV specific reads were still identified (Fig. 2b).
Whole genome analysis thus excludes clonal integration of natural and highly divergent BLV strains in breast tumors. Since only a small proportion of cells may carry the provirus, the sensitivity of the analysis was correlated to the proviral loads. Any natural BLV variant that would infect 10% of the tumor cells is expected to generate about 100 reads (Fig. 2c, dotted blue line). The number of expected reads decreases along with the percentage of infected cells to reach approximately one read with a proviral load of 0.1% (Fig. 2c, dotted blue line). Considering a 59% prevalence of breast tumors positive for BLV [12], 30 samples out of our 51 should be positive. Even with an individual proviral load around 0.1%, this should make about 30 reads (on average one per patient) mapping on BLV, whereas none were found.
Using whole genome analysis, we concluded that there is no evidence for a single BLV-specific or even related sequence. The discrepancies and limitations of this report and others pertain to:
The origin of the samples It is indeed possible that tumor biopsies from previous studies originating from US [11, 12] and Colombia [10] significantly differ from those reported in the dbGaP NCBI database. Even if we restrict our observations on US originating samples (n = 35), the discrepancy remains highly significant. Indeed, Buehring reported 67 breast tumors positive for BLV over 114 cases [12] whereas we found none over 35 cases (the p value for fisher test is 1.12 × 10−6).
The DNA extraction technique In situ PCR suggested that BLV proviral DNA is localized in the cytoplasm [11, 12]. Analysis of mitochondria-specific sequences (Table 1) shows that dbGaP NCBI database includes reads corresponding to 16 kb-long, circular and extranuclear mitochondrial DNA.
The strain divergence Artificial in silico simulation of highly divergent mutants still identified BLV specific reads (Fig. 2b). Since nucleotide substitutions among BLV strains worldwide are limited to 2.3% [16], it remains questionable whether these mutants still belong to the same species. Further analysis show that breast tumor genomes do not map on HTLV-1 sequences (data not shown). Why BLV-conserved sequences were previously identified by PCR remains an enigma.
Viral expression Although BLV is expressed at trace levels in the bovine species, the p24 viral capsid protein was detected in 5% of breast tumors [12]. This observation is inconsistent with RNASeq analysis of 154.7 billion of transcriptome sequencing reads from The Cancer Genome Atlas Research Network [17, 18].
Our present study based on whole genome analysis excludes a clonal insertion of BLV in tumor cells and does not support converging lines of evidence which previously suggested an association between BLV infection and breast cancer.
Methods
Raw DNA sequences from whole genomes of breast tumors and normal breast tissues adjacent the tumor were retrieved from the NCBI database of genotype and phenotype (dbGaP). These sequences were extracted from two studies: (1) estrogen receptor positive breast cancer: aromatase inhibitor response study (accession number phs000472) [14] and (2) sequence analysis of mutations and translocations across breast cancer subtypes (accession number phs000369) [15]. Archive files were downloaded with prefetch v2.5.7 and sequencing reads were extracted with fastdump v2.5.7 using “split-3” option to separate paired reads and single reads (NCBI SRA Toolkit). Paired reads were probed for alignment on different BLV variants (accession numbers: AF033818, AF275515, D00647, K02120, LC080667) and, as positive control, on human genomic sequences using Bowtie2 (version 2.2.5). We used the “very-sensitive” option of Bowtie2 to maximize the likelihood of viral detection. Analyses were performed on computing cluster running on Linux OS. BLV divergent sequences were created in silico by introducing substitutions, deletions or insertions with equal probabilities in 2, 3, 6, 10 and 20% of the reference AF033818 (mutants 0.02, 0.03, 0.06, 0.10 and 0.20, respectively). Neighbor-joining phylogenetic tree was elaborated using Clustal Omega (EMBL-EBI) and visualized by Dendroscope 3. Illumina-like paired-reads were generated from the BLV sequence using the ART simulation tool (version GreatSmokyMountains-04-17-2016, NIH).
Authors’ contributions
NAG and LW designed the experiment, analyzed the data and wrote the paper. Both authors read and approved the final manuscript.
Acknowledgements
We thank David Colignon from CECI (consortium of high-performance computing centres of UCL, ULB, ULg, UMons, and UNamur) and Wouter Coppieters from the GIGA-Genomics platform of the University of Liège for their advice on cluster computing. We are grateful to the NIH dbGaP for providing access to studies phs000369 and phs000472. We thank David Halzen for manuscript editing.
Competing interests
Both authors declare that they have no competing interests.
Availability of data and materials
The datasets analysed during the current study are available from the corresponding author on reasonable request.
Ethics approval and consent to participate
Human DNA sequences were retrieved from the NCBI database of Genotype and Phenotype and processed following the NIH Code of Conduct for Genomic Data Use.
Funding
This work received financial support of the “Fonds National de la Recherche Scientifique” (FNRS), the Télévie, the Interuniversity Attraction Poles (IAP) Program “Virus-host interplay at the early phases of infection” BELVIR initiated by the Belgian Science Policy Office, the Belgian Foundation against Cancer (FBC), the “Centre anticancéreux près ULg” (CAC) and the “Fonds Léon Fredericq” (FLF), the “AgricultureIsLife” project of Gembloux Agrobiotech (GxABT), the “ULg Fonds Spéciaux pour la Recherche”, the COFUND program, the ERA-IB Astinprod and the “Plan Cancer” of the “Service Public Fédéral”. NAG is supported by a grant of the Télévie. LW is a research director of the FNRS.
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Contributor Information
Nicolas A. Gillet, Email: n.gillet@ulg.ac.be
Luc Willems, Email: luc.willems@ulg.ac.be.
References
- 1.Gillet N, Florins A, Boxus M, Burteau C, Nigro A, Vandermeers F, et al. Mechanisms of leukemogenesis induced by bovine leukemia virus: prospects for novel anti-retroviral therapies in human. Retrovirology. 2007;4:18. http://www.ncbi.nlm.nih.gov/pubmed/17362524. [DOI] [PMC free article] [PubMed]
- 2.Meas S, Seto J, Sugimoto C, Bakhsh M, Riaz M, Sato T, et al. Infection of bovine immunodeficiency virus and bovine leukemia virus in water buffalo and cattle populations in Pakistan. J Vet Med Sci. 2000;62:329–31 (cited 20 Jun 2016). http://www.ncbi.nlm.nih.gov/pubmed/10770609. [DOI] [PubMed]
- 3.Jiménez C, Bonilla JA, Dolz G, Rodriguez LR, Herrero L, Bolaños E, et al. Bovine leukaemia-virus infection in Costa Rica. J Vet Med Ser B. 1995;42:385–90 (cited 20 Jun 2016). http://www.ncbi.nlm.nih.gov/pubmed/8594850. [DOI] [PubMed]
- 4.Ma J-G, Zheng W-B, Zhou D-H, Qin S-Y, Yin M-Y, Zhu X-Q, et al. First report of bovine leukemia virus infection in yaks (Bos mutus) in China. Biomed Res Int. 2016;2016:9170167 (cited 2016 Jun 27). http://www.ncbi.nlm.nih.gov/pubmed/27340671. [DOI] [PMC free article] [PubMed]
- 5.Lee LC, Scarratt WK, Buehring GC, Saunders GK. Bovine leukemia virus infection in a juvenile alpaca with multicentric lymphoma. Can Vet J. 2012;53:283–6 (cited 20 Jun 2016). http://www.ncbi.nlm.nih.gov/pubmed/22942445. [PMC free article] [PubMed]
- 6.Kettmann R, Mammerickx M, Portetelle D, Grégoire D, Burny A. Experimental infection of sheep and goat with bovine leukemia virus: localization of proviral information on the target cells. Leuk Res. 1984;8:937–44 (cited 2016 May 25). http://www.ncbi.nlm.nih.gov/pubmed/6096636. [DOI] [PubMed]
- 7.Altanerova V, Portetelle D, Kettmann R, Altaner C. Infection of rats with bovine leukaemia virus: establishment of a virus-producing rat cell line. J Gen Virol. 1989;70(Pt 7):1929–32 (cited 25 May 2016). http://www.ncbi.nlm.nih.gov/pubmed/2544673. [DOI] [PubMed]
- 8.Altanerova V, Ban J, Altaner C. Induction of immune deficiency syndrome in rabbits by bovine leukaemia virus. AIDS. 1989;3:755–8 (cited 25 May 2016). http://www.ncbi.nlm.nih.gov/pubmed/2559751. [DOI] [PubMed]
- 9.Buehring GC, Philpott SM, Choi KY. Humans have antibodies reactive with Bovine leukemia virus. AIDS Res Hum Retrovir. 2003;19:1105–1113. doi: 10.1089/088922203771881202. [DOI] [PubMed] [Google Scholar]
- 10.Giovanna M, Carlos U. Bovine leukemia virus gene segment detected in human breast tissue. Open J Med. 2013;2013:84–90. http://www.scirp.org/journal/PaperInformation.aspx?paperID=28694.
- 11.Buehring GC, Shen HM, Jensen HM, Yeon Choi K, Sun D, Nuovo G. Bovine leukemia virus DNA in human breast tissue. Emerg Infect Dis. 2014;20:772–782. doi: 10.3201/eid2005.131298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Buehring GC, Shen HM, Jensen HM, Jin DL, Hudes M, Block G. Exposure to bovine leukemia virus is associated with breast cancer: a case–control study. PLoS One. 2015;10(9):e0134304–10.1371/journal.pone.0134304. doi: 10.1371/journal.pone.0134304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sinha G. Bovine leukemia virus possibly linked to breast cancer. J Natl Cancer Inst. 2016;108 (cited 22 Apr 2016). http://www.ncbi.nlm.nih.gov/pubmed/26864929. [DOI] [PubMed]
- 14.Ellis MJ, Ding L, Shen D, Luo J, Suman VJ, Wallis JW, et al. Whole-genome analysis informs breast cancer response to aromatase inhibition. Nature. 2012;486:353–360. doi: 10.1038/nature11143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Banerji S, Cibulskis K, Rangel-Escareno C, Brown KK, Carter SL, Frederick AM, et al. Sequence analysis of mutations and translocations across breast cancer subtypes. Nature. 2012;486:405–409. doi: 10.1038/nature11154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Polat M, Takeshima S-N, Hosomichi K, Kim J, Miyasaka T, Yamada K, et al. A new genotype of bovine leukemia virus in South America identified by NGS-based whole genome sequencing and molecular evolutionary genetic analysis. Retrovirology. 2016;13:4 (cited 31 May 2016). http://www.ncbi.nlm.nih.gov/pubmed/26754835. [DOI] [PMC free article] [PubMed]
- 17.Tang KW, Alaei-Mahabadi B, Samuelsson T, Lindh M, Larsson E. The landscape of viral expression and host gene fusion and adaptation in human cancer. Nat Commun. 2013;4:2513. http://www.ncbi.nlm.nih.gov/pubmed/24085110. [DOI] [PMC free article] [PubMed]
- 18.Khoury JD, Tannir NM, Williams MD, Chen Y, Yao H, Zhang J, et al. Landscape of DNA virus associations across human malignant cancers: analysis of 3,775 cases using RNA-Seq. J. Virol. 2013;87:8916–26 (cited 4 May 2016). http://jvi.asm.org/content/early/2013/05/30/JVI.00340-13. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets analysed during the current study are available from the corresponding author on reasonable request.