

Abstract
Background
Emerging evidence suggests that a subset of FMR1 premutation carriers is at an increased risk for cognitive, emotional, and medical conditions. However, because the premutation is rarely diagnosed at birth, the early developmental trajectories of children with a premutation are not known.
Methods
This exploratory study examined the cognitive, communication, and social-behavioral profiles of 26 infants with a premutation who were identified through participation in a newborn screening for fragile X syndrome pilot study. In this study, families whose newborn screened positive for an FMR1 premutation were invited to participate in a longitudinal study of early development. Twenty-six infants with the premutation and 21 matched, screen-negative comparison babies were assessed using validated standardized measures at 6-month intervals starting as young as 3 months of age. The babies were assessed up to seven times over a 4-year period.
Results
The premutation group was not statistically different from the comparison group on measures of cognitive development, adaptive behavior, temperament, or overall communication. However, the babies with the premutation had a significantly different developmental trajectory on measures of nonverbal communication and hyperresponsivity to sensory experiences. They also were significantly more hyporesponsive at all ages than the comparison group. Cytosine-guanine-guanine repeat length was linearly associated with overall cognitive development.
Conclusions
These results suggest that infants with a premutation may present with subtle developmental differences as young as 12 months of age that may be early markers of later anxiety, social deficits, or other challenges thought to be experienced by a subset of carriers.
Keywords: FMR1 premutation, Early development, Newborn screening
Background
Individuals with premutation range cytosine-guanine-guanine (CGG) repeats (55–200) on the fragile X mental retardation (FMR1) gene were originally considered unaffected carriers whose primary health risk was the chance of offspring with fragile X syndrome (FXS). However, individuals with a premutation (“carriers”) are now known to be vulnerable to two later onset disorders—fragile X-associated tremor/ataxia syndrome (FXTAS) [1, 2] and fragile X-associated primary ovarian insufficiency (FXPOI) [3], and may also be vulnerable to multiple other medical, emotional, and cognitive challenges [4]. Population studies suggest a high prevalence of the premutation, with as many as 1:150 women and 1:450 men estimated to be carriers [5–8]. This increased risk of health concerns and high population prevalence has led to a growing interest in the premutation phenotype [9–12]. However, most research on the premutation has focused on adults. Studies of children or adolescents nearly all involve participants who were identified because of the diagnosis of a close family member with FXS, biasing the sample toward increased likelihood of challenges.
Consistent with referral bias, studies of children with the premutation who were clinically referred (probands) suggest increased rates of developmental problems compared to noncarrier siblings [13–15] and carriers who were nonprobands—identified through cascade testing [16–18]. Further, increased risk for nonspecific developmental delay among children with premutation has been reported [19]. However, other studies report intact cognitive abilities in adults with a premutation without FXTAS [20]. Subtle cognitive difficulties have been reported among adults, most notably in executive functioning [20, 21], working memory [22], visual-spatial perceptual deficits [11], arithmetic [23, 24], speech fluency [25], and pragmatic language [26].
Several studies have documented more attention regulation, anxiety, and autism-like symptoms among carriers compared to controls. Hunter and colleagues [27] found that adult female carriers self-reported significantly more symptoms of inattention, memory, and poor self-concept compared to controls, although they did not score lower on cognitive tests. Anxiety symptoms, including social avoidance, interpersonal sensitivity, shyness, and eye contact avoidance, have been described in carriers, with up to 41 % reporting a lifetime diagnosis of an anxiety disorder [28, 29]. A higher than expected rate of autism spectrum disorders (ASDs) have also been reported for carriers [16, 18, 19, 30], although all these studies had some level of bias. More subtle ASD symptoms, such as social aloofness [28], rigid perfectionism [31], and features of the broad autism phenotype, have been described among carrier women [26]. These findings, along with studies linking the protein encoded by FMR1—the fragile X mental retardation protein (FMRP)—to the regulation of several pathways associated with autism [32–35], suggest a possible link between FMR1 and autism-related phenotypes.
Because the premutation is rarely diagnosed in infancy, only one study has examined early development. This study examined 14 infants with the premutation and found visual processing deficits similar to infants with FXS, although their overall developmental scores were higher than those with FXS [36]. The findings suggested that deficits in spatiotemporal processing and subsequent executive dysfunction in some adults with the premutation may be present very early in life and emphasize the importance of studies examining early development of individuals along the spectrum of FMR1 mutations.
Here, we report findings from a longitudinal study of infants identified with a premutation in a pilot study of newborn screening for FXS. This is a unique sample as the babies were identified and initially assessed within the first few months of life. Further, nearly all families were naïve to FXS or the premutation prior to the birth of their child and therefore had no preconceived information regarding the development of children with a premutation, and none of the babies were clinically referred. We were primarily interested in the very early cognitive, communication, and social-emotional development of infants with the premutation compared with screen-negative comparison infants. Given studies of older children and adults with a premutation, we did not expect to find significant differences in cognitive trajectories; however, we hypothesized that a subsample of infants with the premutation would demonstrate developmental differences compared to infants without a premutation.
Methods
Procedures
With approval from the Institutional Review Board, families were recruited in the postpartum unit within 24 h of the birth of their child. Recruitment took place over 4 years at three university-affiliated hospitals. Details about recruitment procedures and laboratory assessments can be found in previously published reports [8, 37]. To be included, the mother had to be at least 15 years of age, fluent in either Spanish or English, not undergoing stressful medical or personal circumstances, and not have infants in critical care for life-threatening conditions or relinquished for adoption. Newborns with a premutation allele were identified using polymerase chain reaction (PCR) analysis as reported by Tassone et al. [8]. Because the study was intended as a pilot for newborn screening, the molecular data collected reflects only the information necessary to identify an FMR1 mutation; additional molecular measures including activation ratio and methylation status were not assessed.
Out of the 12,709 infants screened across the three sites, one baby screened positive for the presence of a full mutation and 45 screened positive for the presence of a premutation. Families of screen-positive children were called by a medical geneticist or clinician specializing in FXS on the research team and offered a genetic counseling appointment and confirmatory testing. Sixteen families did not have confirmatory testing because they declined further participation (n = 3), did not show up for the diagnostic appointment (n = 2), declined repeat testing (n = 3), or were unable to be reached via phone or mail (n = 8).
All families whose babies had a confirmed expansion following the diagnostic testing were invited to join the longitudinal component of the study. Twenty-eight infants who screened positive for an FMR1 mutation participated in at least one longitudinal assessment. The baby with FXS and one baby who also screened-positive for Klinefelter’s syndrome were not included in the analyses, resulting in a total of 26 infants. A set of comparison tests on demographic variables (maternal age, marital status, race/ethnicity, maternal education) and child CGG repeat length for families who participated in the longitudinal study and those who screened positive but declined participation was conducted, yielding no significant differences between participants and nonparticipants [38] (Table 1).
Table 1.
Demographics of sample
| Premutation sample (n = 26) | Comparison sample (n = 21) | |
|---|---|---|
| Number of assessments | 76 | 48 |
| Gender | 15 females | 15 females |
| 11 males | 6 males | |
| Ethnicity | 14 (54 %) White | 12 (57 %) White |
| 6 (23 %) African American | 5 (24 %) African American | |
| 2 (8 %) Hispanic | 2 (10 %) Hispanic | |
| 4 (15 %) other | 2 (9 %) other | |
| CGG repeat (range) | 55–125 | – |
Following diagnostic confirmation, families who agreed to participate in the follow-up study were scheduled for their first assessment when the child was 6 months of age. Assessments were scheduled at approximately 6-month intervals. All assessments were conducted at the family’s home or at a hospital/clinic-based setting. Assessors were trained by a licensed psychologist on all measures. For most assessments, the assessors were not blind to diagnostic group; however, some measures were double-scored by measure experts who were blind to diagnostic grouping.
Participants
Of the 26 infants with a premutation who participated in at least one assessment, 15 were female. Fifty-four percent of the sample was Caucasian, 23 % was African American, and 8 % was Hispanic. Two sets of twins both with a premutation were included. The range of CGG repeats in infants with the premutation was 55–125, with the majority of the premutation alleles below 70 CGG repeats (77 %).
Because participants entered the study at different times over the course of the 4-year enrollment period, as well as some attrition (n = 6), infants were assessed anywhere between one and seven times (see Table 2). Comparison tests, chi-square frequency tests for categorical variables and t tests for continuous variables, were run to examine potential differences in family demographic and child variables for families who stayed in the study versus those who dropped out after at least one data collection visit. We found no evidence for differences in gender, race, mother’s education, age, or CGG repeat of the infant (all p > .27).
Table 2.
Number of assessments at each age point, by group
| Group | 3–4 months | 5–7 months | 11–12 months | 16–19 months | 22–25 months | 29–31 months | 35–42 months |
|---|---|---|---|---|---|---|---|
| Premutation | 3 | 22 | 18 | 10 | 10 | 5 | 11 |
| Comparison | 0 | 15 | 10 | 8 | 6 | 5 | 4 |
All families who participated in the newborn screening pilot study provided basic demographic information and were told at the time of providing consent that they may be asked to participate in a longitudinal study as a comparison family should their infant screen negative. Comparison infants were recruited from the sample of babies who screened negative for any FMR1 expansion if they were a match for a screen-positive baby based on location (i.e., state of study site), gender, race and ethnicity, maternal education, and family income.
Measures
Eight well-validated, standardized measures were used to assess early cognitive development, language and communication, social-emotional, and sensory behaviors.
Early cognitive development
The Mullen Scales of Early Learning [39] was administered at each assessment. The Mullen is a standardized developmental test for children birth to 68 months, assessing multiple developmental domains including expressive and receptive language, gross and fine motor skills, and visual processing. The Vineland Adaptive Behavior Scales, Second Edition (VABS-II) [40] was also completed in an interview format with the primary caregiver at each assessment point to measure functional behavior. The VABS-II provides parent reported information on functional skills in the areas of daily living, communication, socialization, and motor. Standard scores for all domains, as well as overall composites, were used for analyses for these measures.
Early language and communication
In addition to the broad measures of expressive and receptive language derived from the Mullen Scales of Early Learning (MSEL) and VABS-II, measures of more subtle communication and language development were included in the battery. The parent-reported MacArthur Communicative Development Inventories, Second Edition [41] (CDI) were used to assess more detailed information regarding the infants’ word production and early communicative intent. The CDI consists of two inventories, each with two sections. The CDI/Words and Gestures Inventory is for infants between the ages of 8 and 16 months. The inventory’s word section, which has a 28-item list of phrases and a 396-word checklist, is used to assess the infant’s production and understanding of words and phrases. The gesture section covers 63 gestures for communication, play, imitation of parents and other adults, and activities with objects. The CDI/Words and Sentences Inventory is for toddlers between the ages of 16 and 30 months. The inventory’s word section assesses vocabulary using a 680-word checklist. The second part assesses the toddler’s use of possessives, plurals, and tenses and development of complex sentences. The Words and Gestures Inventory was used at 18, and 24 months, replacing the CDI/Words and Gestures Inventory used at 12 months.
The Communication and Symbolic Behavior Scale (CSBS) [42] was used to assess functional communication. The CSBS is a norm-referenced screening and evaluation tool that helps determine the communicative competence (use of eye gaze, gestures, sounds, words, understanding, and play) of children with a functional communication age between 6 and 24 months (chronological age from about 6 months to 6 years). The scale yields three composite scores—social, speech, and symbolic—and concurrent and predictive validity are well established [42]. The caregiver report form and the directly administered behavioral profile components of the CSBS were administered. All direct administrations were videotaped and double-scored by a speech language pathologist with expertise in CSBS administration and a trained research assistant. Both coders were blind to the diagnostic status of the participants.
Social-emotional and sensory behaviors
Due to reports of an increased incidence of autism, attention, and anxiety symptoms among older children and adults with the premutation, several measures of early social communication and sensory behaviors were included. The Sensory Experiences Questionnaire [43] was completed by the caregivers at each data collection point to assess behavioral responses to common sensory experiences (e.g., child dislikes cuddling). Thirty-three items are used to calculate (as simple means) a total score and subscales for hyperresponsiveness (14 items), hyporesponsiveness (6 items), and sensory seeking (13 items). The Sensory Experiences Questionnaire (SEQ) categorizes scores as “normal,” “at-risk,” or “deficient” based on the number of standard deviations from the mean score for the norming sample [44]. In addition, two measures of autism symptoms were administered. The First Years Inventory (FYI) [45] was completed with participants assessed at 12 months. The FYI was developed to assess the presence of behaviors at 12 months associated with the later development of an autism spectrum disorder. Finally, for those whose last assessment occurred after 24 months, the second edition of the Autism Diagnostic Observation Schedule-2 [46] (ADOS-2) was administered.
Data analysis
Correlations between CGG repeat length and all variables were examined for those in the premutation group, as were comparison (t tests of continuous variables; chi-square for categorical) tests between males and females. For these analyses, we chose the assessment point at the oldest age for each child. For comparison tests between groups, standard scores on all measures were averaged for each participant across all assessments they completed. Despite the small n, longitudinal data analyses were collected at up to seven time points per child for a total of 124 assessments, allowing us to run hierarchical linear models (HLM) on selected variables. Given the large number of potential measures, we selected variables for HLM based on two criteria: (1) variables related to potential differences reported in older individuals with a premutation (social, communication, sensory) and (2) variables demonstrating some divergent patterns upon preliminary descriptive analysis. The primary hypotheses under examination focused on the relationship of child age and diagnostic group to the outcomes of interest. We tested for nonlinear (quadratic) change over time, but there was no evidence that such trends existed, so all models were simplified to include only linear terms. We treated the models for the child outcomes as two-level hierarchical linear models where time is nested within child. Repeated observations of subjects introduce dependence in the data. HLM manages this through the estimation of random effects in addition to traditional regression parameters [47]. The random effects provide estimates for within subject variance to control for this dependence. These models included random effects for the intercept. In all models, age and the moderators were grand mean centered, so any group differences are at the mean of those variables. The data are coded so that the premutation group is the reference. The group parameters in Table 4, then, provide the mean difference between the groups; positive parameters indicate a higher score in the comparison group and negative parameters indicate a higher score in the treatment group. The parameter for age represents the change over time for the comparison group. The interaction indicates how much the premutation group deviated from that rate. The absence of an interaction indicates that the mean difference is constant across all ages and the range of the moderator. Where interactions were suggested, graphs were generated to illustrate the findings.
Table 4.
Parameter estimates and (standard errors) for early development (Mullen, VABS-II), communication (CSBS), and sensory issues (SEQ hyper, hypo, and seeking)
| Effect | Intercept | Group | Age | Group × age |
|---|---|---|---|---|
| Mullen ELC | 98.43 (2.88) | 4.31 (4.27) | .12 (.15) | −.02 (.25) |
| VABS-II ABC | 98.11 (2.05) | 1.88 (2.96) | .11 (.11) | .13 (.19) |
| CSBS-emotion and eye gaze | 10.92 (.77) | 1.12 (1.14) | −.31 (.09)** | .41 (.16)* |
| SEQ-hyporesponsive | 10.96 (.50) | −1.51 (.79)* | .02 (.04) | .00 (.07) |
| SEQ-hyperresponsive | 24.96 (.66) | −.93 (1.06) | −.11 (.05)* | −.21 (.09)* |
| SEQ-seeking | 31.29 (1.32) | 1.64 (2.11) | −.25 (.1) | −.28 (.07) |
*p < .05; **p < .01
Our small sample size provided a substantial barrier to our statistical inferences. Basic statistical theory suggests that parameter estimates in samples of this size are likely to be unreliable (e.g., Gravetter and Wallnau [48, p. 204]). Further, standard error is inversely related to sample size, so power decreases as sample size decreases. Having repeated measurement and testing in HLM framework provides an increase to statistical power [49]. We produced regression diagnostics (e.g., residual and normality plots) for all of the statistical models in the analysis. Examination of these plots indicated that normality and homoscedasticity assumptions were met and provided no suggestion of outliers.
The difficulties arising from the small sample size are compounded by the relatively large number of dependent variables. Typically, the possibility of false discovery as a function of multiple testing leads researchers to make adjustments to the critical values or p values for the results (see Benjamini and Hochberg [50] for a complete discussion). We opted to make no adjustments, however. Adjusting for multiple testing is intended to control for type I error. Given the exploratory nature of this study, we suggest that the uniqueness of this sample and the potential for these results to provide useful effect size estimates for future research makes type II error of greater concern, but readers should keep the possibility of false discovery in mind.
Results
Developmental profiles
Average scores on the Mullen across diagnostic groups were similar over time, with mean scores consistently in the low average to average range for both groups. There were no significant differences between diagnostic groups on any of the MSEL domains (expressive language, receptive language, fine motor, gross motor, visual reception) at any time point. Parent-reported adaptive behaviors measured by the VABS-II composite and domain scores were age appropriate for both groups across all age points. There was no significant age-by-group interaction for any MSEL or VABS-II domain. However, CGG repeat length was negatively correlated with overall (last administered) MSEL scores (−.39; p = .05); children with higher repeats had lower overall development. See Table 3 for mean scores across all assessment points by group on the MSEL and VABS-II.
Table 3.
Means and standard deviations on key measures across all assessment points
| Assessment | Premutation sample: mean (SD) | Comparison sample: mean (SD) | p value for test |
|---|---|---|---|
| Mullen ELCa | 89.1 (14.6) | 92.8 (17.7) | .436 |
| Mullen GMb | 48.7 (11.0) | 49.2 (14.3) | .896 |
| Mullen FM | 46.0 (10.9) | 49.5 (11.2) | .287 |
| Mullen VR | 44.3 (11.4) | 47.9 (12.0) | .301 |
| Mullen RL | 44.9 (11.7) | 45.0 (13.3) | .979 |
| Mullen EL | 41.8 (10.2) | 42.1 (12.3) | .929 |
| VABS-II ABCc | 97.9 (10.7) | 100.3 (9.0) | .408 |
| VABS-II communication | 98.1 (12.3) | 100.4 (8.1) | .446 |
| VABS-II daily living skills | 100.5 (10.9) | 100.5 (13.9) | 1.00 |
| VABS-II motor | 98.0 (14.5) | 100.1 (10.2) | .564 |
| VABS-II socialization | 97.3 (8.7) | 101.1 (7.4) | .113 |
| SEQ hyporesponsivenessd | 11.6 (9.3) | 9.3 (2.0) | .226 |
| SEQ hyperresponsiveness | 24.9 (5.6) | 24.4 (4.3) | .730 |
| SEQ seeking* | 32.8 (9.9) | 22.2 (12.5) | .003 |
| CSBS-parent report totale | 96.7 (14.9) | 98.5 (14.0) | .672 |
| CSBS-parent emotion and eye gazef | 10.9 (3.12) | 11.3 (2.23) | .611 |
| CBSB-parent-communication | 10.9 (4.57) | 11.3 (2.92) | .718 |
| CSBS-parent-gestures* | 11.2 (3.98) | 13.6 (4.12) | .050 |
| CSBS-parent-sounds | 9.6 (3.92) | 11.0 (3.75) | .219 |
| CSBS-parent-words | 11.2 (3.45) | 11.1 (2.93) | .915 |
| CSBS-parent-understanding | 9.0 (3.33) | 9.7 (3.42) | .484 |
| CSBS-parent-object use | 9.7 (3.34) | 9.4 (2.75) | .737 |
| CSBS-parent-social | 10.5 (3.29) | 11.1 (2.77) | .500 |
| CSBS-parent-speech | 9.5 (3.40) | 9.9 (3.42) | .691 |
| CSBS-parent_symbolic | 8.9 (2.71) | 9.1 (2.98) | .813 |
| CSBS-direct total | 88.3 (12.7) | 89.9 (10.0) | .631 |
| CSBS-direct-emotion and eye gaze* | 8.8 (2.59) | 10.7 (3.61) | .049 |
| CBSB-direct-communication | 8.8 (3.27) | 8.7 (2.13) | .900 |
| CSBS-direct-gestures* | 8.3 (2.52) | 10.0 (2.00) | .013 |
| CSBS-direct-sounds | 8.3 (1.89) | 9.1 (2.17) | .190 |
| CSBS-direct-words* | 10.3 (2.50) | 8.9 (2.05) | .041 |
| CSBS-direct-understanding | 9.1 (3.00) | 7.9 (2.22) | .122 |
| CSBS-direct-object use | 7.4 (2.63) | 7.2 (2.96) | .809 |
| CSBS-direct-social | 8.4 (2.52) | 9.6 (1.80) | .064 |
| CSBS-direct-speech | 8.8 (2.60) | 8.9 (2.00) | .882 |
| CSBS-direct-symbolic | 7.4 (2.17) | 7.1 (2.59) | .674 |
aMullen ELC = Early Learning Composite; mean = 100, SD = 15
bMullen GM = gross motor; FM = fine motor; VR = visual reception; RL = receptive language; EL = expressive language; mean = 50, SD = 10
cVABS-II ABC = Vineland Adaptive Behavior Composite; mean for all VABS-II scales = 100, SD = 15
dSEQ = Sensory Experiences Questionnaire; hyporesponsiveness reference mean = 8.7 (1.9); hyperresponsiveness reference mean = 24.1 (4.6); seeking reference mean = 29.7 (8.7) [76]
eCSBS total = Communication Symbolic Behavior Scale; mean = 100, SD = 15
fCSBS subscales (emotion and eye gaze, communication, gestures, sounds, words, understanding, object use, social, speech, symbolic); mean = 10, SD = 3
*significant difference between groups
Language profiles
There were no differences between gender or diagnostic groups at any age point on any of the CDI variables. Scores on the parent report and the direct administered CSBS suggested some differences between the groups however. For the parent report version, across all time points, babies with the premutation were reported to display fewer gestures than the screen-negative babies. On the direct assessment measure, babies with the premutation were rated as having fewer gestures, poorer emotion and eye gaze, and more use of words than the comparison babies (see Table 3). There was a significant interaction between age and group status for the emotion and eye gaze subscale (see Table 4), which measures early social-emotional and nonverbal communication behaviors. While the comparison infants gained skills on this subscale over time, eventually reaching the ceiling by 36 months, babies with the premutation did not show the same increases in skills, resulting in decreasing scaled scores over time (see Fig. 1 and Table 3).
Fig. 1.
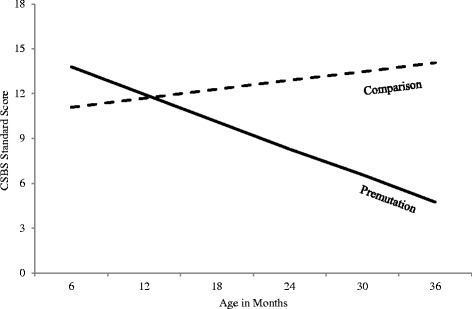
CSBS parent report emotion and eye gaze scaled scores by age and diagnosis
Behavioral profiles
There were no significant differences between males and females with a premutation on any of the behavior domains. However, there were significant differences between diagnostic groups across all assessment points on sensory seeking, with the babies with the premutation displaying more sensory-seeking behaviors. There were also significant differences between groups over time on both hypo- and hyperresponsivity on the SEQ. There were significant main effects for hyporesponsiveness, with the premutation group more hyporesponsive at every age point assessed (see Fig. 2). There was also a significant interaction effect for hyperresponsiveness (see Table 4); while the comparison group became less hyperresponsive as they got older (a normative pattern), the premutation babies became more hyperresponsive over time (see Fig. 3).
Fig. 2.
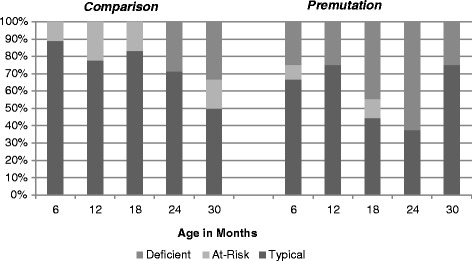
Prevalence of hyporesponsiveness scores across SEQ defined categories
Fig. 3.
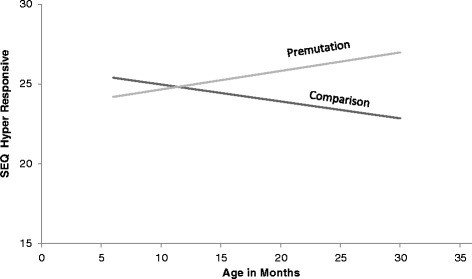
SEQ hyperresponsive scores across time, by group
Autism symptoms
There were no significant differences between groups on any of the scales on the FYI. However, while none of the comparison babies and just 4 % of the norming sample for the FYI [51] scored above 98th percentile on the total risk variable, almost a third of the premutation babies (27 %) scored in this range. Although there were too few participants who completed an ADOS-2 to compare statistically across groups, three out of the eight premutation babies who were tested scored in the elevated range, compared to none of the controls. There were no gender differences on these measures.
Discussion
This exploratory study is the first to examine early developmental profiles of infants with an FMR1 premutation. Amid emerging evidence that premutation CGG expansions on the FMR1 gene are associated with increased risks for social, emotional, and medical difficulties, understanding the early trajectories of these risks is critical. We found that patterns of broad early development did not differ significantly from matched screened-negative babies. This was true for all of the areas of development traditionally assessed by pediatricians during well-baby visits. However, we found significant differences in several important variables related to social and sensory experiences as early as 12 months of age in premutation babies. Infants with the premutation had increased risk for social communication differences as well as both hypo- and hyperresponsiveness to stimuli when compared to both the comparison group, as well as norming samples. These differences suggest a profile of very early regulation challenges which may be related to increased risk for attention, anxiety, or autism symptoms reported for older individuals.
Infants with the premutation were found across all assessment points to be more sensory seeking than their matched screen-negative peers. They were also rated to be more hyporesponsive to sensory stimuli at all ages tested. Hyporesponsivity refers to a lack of or reduced intensity in response to external stimuli. For example, a hyporesponsive infant may not react to loud sounds or may appear to have a very high pain threshold. Patterns of increased hyporesponsivity have been associated with increased risks for autism in both toddlers [52] and older children [53, 54]. Specifically, hyporesponsivity is more strongly associated with the core autism symptoms of social and communication impairments than with repetitive behaviors [55], which are generally more related to hyperresponsivity [56]. Interestingly, the premutation group also became more hyperresponsive over time, whereas the control group became less hyperresponsive as they got older. Hyperresponsivity is used to describe a pattern of exaggerated responses to sensory stimuli—for example avoiding touch or covering ears to block sounds. Individuals with FXS are often described as being hyporesponsive in the first year of life and then become hyperresponsive and hyperaroused in response to external stimuli after the second or third year of life [57]. The hyperresponsive behavior is thought to be related to a lack of GABA inhibition which causes a lack of habituation to sensory stimuli seen in children with FXS [58]. GABA deficits have not only been documented in FXS but also in those with the premutation [59].
This pattern of both hyporesponsiveness and increasing hyperresponsiveness may appear counterintuitive; however, some literature suggests that this co-existing pattern is more common among children with developmental differences, especially autism symptoms [43, 57, 60]. Although it is not uncommon to observe a pattern of co-occurring hyper- and hyporesponsivity in children with developmental differences, other studies have noted that hyperresponsivity may be more strongly related to mental age [53], while hyporesponsivity is more strongly related to core autism features [43], which may explain the co-occurrence in very young cohorts. However, according to the dynamic theory of sensory processing [61], individual responses to stimuli and experiences are partially influenced by two thresholds—one related to orienting to stimuli (orienting threshold) and one associated with tolerance for stimuli (aversion threshold). When in the optimal band of stimulation between orienting and aversion thresholds, an individual is the most capable of attending and engaging. The wider the band between these two thresholds, the greater the person is at adapting to their environment. A hyporesponsive individual will have a higher threshold for orienting (i.e., require more intense input from a stimulus to perceive and attend). Conversely, a hyperresponsive person will have a lower threshold for aversion (i.e., require less intense stimulus to become averse). An individual who is both hypo- and hyperresponsive therefore will have a narrower optimal band of stimulation from which to engage with their world [62]. This pattern may affect the individual’s ability to communicate and interact appropriately with others, leading to increases in anxiety, attention, or autism-like symptoms.
Babies with the premutation also demonstrated elevated social-communication problems as measured by the parent reported CSBS (fewer gestures) and the CSBS direct assessment (fewer gestures, worse emotion and eye gaze). Further, consistent with studies of older individuals with the premutation, just over a third of infants in this study had social-communication difficulties as measured by the parent-reported FYI and the direct assessment on the ADOS-2. The FYI was developed specifically to assess the presence of behaviors at 12 months associated with the later development of an ASD [45]. However, it is also a sensitive tool for identifying early non-ASD developmental differences [63]. Therefore, while elevated scores do not suggest a clear trajectory toward an autism diagnosis, they do suggest increased risk for social communication challenges or other developmental delays.
Gender differences are generally expected given the FMR1 is located on the X chromosome and females, with two X chromosomes, can have a more variable phenotype based on the X-activation ratio (percent of cells with the normal allele on the active X chromosome). We did not find any gender differences on any of our variables of interest. Unfortunately, we did not have additional molecular data (e.g., activation ratio), which could be an important addition to understanding potential gender differences among premutation carriers.
Why some individuals with the premutation develop cognitive, psychiatric, or medical problems and others do not is a key question for FMR1 researchers. In the current sample, higher CGG repeats in the infants were associated with lower developmental scores on the Mullen but not on any of the variables where there was a significant difference from the comparison group. CGG repeat length has been implicated in the onset and severity of several associated features, although the relationship is still unclear. Higher CGG repeats in the premutation range are associated with lower levels of FMRP [64, 65], as well as increases in FMR1 messenger ribonucleic acid (mRNA) levels [66, 67], both of which disrupt normal neuronal functioning. However, while some studies suggest a linear association, particularly for neurological problems (e.g., higher CGG repeats associated with more problems) [22, 66, 68], others have found a nonlinear association (particularly for ovarian or psychiatric problems) with greater severity among those with a CGG repeat number in the mid-premutation range [29, 69–74]. Additional genetic abnormalities (copy number variation) have been found in 20 % of carriers who have ASD or neurological problems [75], and these may have an additive effect, leading to a more severe phenotype. Another possibility is that carriers have a genetic susceptibility to stress [73] which may result in greater impact of every day stressors on the developing brains of these infants. These studies suggest that both molecular and gene-by-environment interaction studies are needed to fully understand who, among premutation carriers, is at greatest risk.
There are several limitations to this study and associated alternative explanations for our findings. Although larger than any other study examining infants with an FMR1 premutation, our findings are still limited by the small sample size. We were underpowered for some of our analyses, especially with the decreasing number of control subjects in older ages. The small sample size and large number of variables does increase the chances for false discoveries, and this is an important limitation to keep in mind. However, in the areas where significant differences were found, the comparison sample tracked along with what would be expected based on norming samples for the instruments, whereas significant differences were observed for the premutation sample. Further, the variation in developmental trajectories found in the premutation infants is not inconsistent with findings for older individuals with a premutation, where developmental differences, especially in social and executive function domains, have been reported in a subsample [16, 18, 19, 30]. Therefore, we argue that our findings from this relatively large, unbiased sample of premutation infants establish an important basis for examining potential molecular and environmental factors which may influence outcomes for individuals with a premutation very early in life.
Although all screen-positive families from the newborn screening study were given the same opportunity to participate, it is possible that more families who had concerns about their child were willing to participate in the study over time, thereby reducing generalizability of the results. However, in our experience, more dysfunctional families tended not to want to participate initially, perhaps biasing the study to higher functioning children. Further, although we were obviously not able to follow the development of those who did not participate, there were no significant differences in family- or child-level variables of those who did not continue in the study after their first assessment and those who were followed for a longer period of time.
In addition, some significant results may be due to parents of the premutation babies being more sensitive to subtle differences as a result of increased anxiety on their part as to the impact of the premutation. However, parents did not report significant differences on any of the broad areas of development or temperament traits. Rather, differences were found in areas that might be expected for children with early regulation difficulties associated with later anxiety, hyperactivity, and other social-emotional challenges. Further, we found significant differences between groups on the direct assessment of the CSBS, which was scored by experts blind to diagnostic group. Finally, these are the same differences commonly reported among larger samples of older individuals with a premutation, suggesting there may be increased risks for some individuals with a premutation which begin very early in life.
Conclusions
This study is the first to report on developmental trajectories of infants and toddlers with an FMR1 premutation and no family history of FXS. Results from this study support the emerging discussion of an endophenotype for individuals on the spectrum of FMR1 involvement—with increased risk for a subset of premutation carriers. Additional genotype-phenotype studies are needed to help identify which FMR1 premutation carriers are at greater risk in order to inform the development of early, supportive interventions to reduce negative outcomes.
Acknowledgments
Funding
This research was supported by NICHD grant numbers HD02274 and HD036071 and Health and Human Services Administration of Developmental Disabilities 90DD0596.
Availability of data and materials
The datasets generated during and/or analyzed during the current study are not publicly available due to the small sample sizes and subsequent risk of breach of confidentiality but are available from the corresponding author on reasonable request.
Authors’ contributions
AW conducted or supervised the longitudinal family assessments, wrote the first draft of the manuscript, integrated all edits by co-authors, and prepared the final manuscript for submission. JS conducted all data analysis and approved the final manuscript as submitted. RH conceptualized and designed the study, oversaw data collection at the UC-Davis site, reviewed and revised the manuscript, and approved the final manuscript as submitted. EB-K conceptualized and designed the study, oversaw the data collection at the Rush site, reviewed and revised the manuscript, and approved the final manuscript as submitted. FT conceptualized and designed the study, carried out the DNA screening test at UC Davis site, reviewed and revised the manuscript, and approved the final manuscript as submitted. DBBJr conceptualized and designed the study, reviewed and revised the manuscript, and approved the final manuscript as submitted. All authors read and approved the final manuscript.
Competing interests
Randi Hagerman and Elizabeth Berry-Kravis have received funding from Novartis, Roche, Alcobra, and Neuren for clinical trials of medications for fragile X syndrome and have consulted with Roche/Genentech and Novartis regarding fragile X treatment and clinical trial design. Elizabeth Berry-Kravis has received funding from Asuragen for creation of DNA standards for FMR1 DNA testing.
Consent for publication
Not applicable.
Ethics approval and consent to participate
This research was approved by the Institutional Review Boards at the University of North Carolina at Chapel Hill, Rush University Medical Center, and The University of California at Davis. All adult participants provided written consent for themselves and their minor children.
Abbreviations
- ADOS
Autism Diagnostic Observation Schedule
- ASD
Autism spectrum disorder
- CDI
MacArthur Communicative Development Inventories
- CGG
Cytosine-guanine-guanine
- CSBS
Communication Symbolic Behavior Scale
- FMR1
Fragile X mental retardation gene
- FMRP
Fragile X mental retardation protein
- FXPOI
Fragile X premature ovarian insufficiency
- FXS
Fragile X syndrome
- FXTAS
Fragile X tremor/ataxia syndrome
- FYI
First Years Inventory
- HLM
Hierarchical linear modeling
- mRNA
Messenger ribonucleic acid
- MSEL
Mullen Scales of Early Learning
- PCR
Polymerase chain reaction
- SEQ
Sensory Experiences Questionnaire
- VABS
Vineland Adaptive Behavior Scales
References
- 1.Berry-Kravis E, Goetz CG, Leehey MA, et al. Neuropathic features in fragile X premutation carriers. Am J Med Genet A. 2007;143A(1):19–26. doi: 10.1002/ajmg.a.31559. [DOI] [PubMed] [Google Scholar]
- 2.Hagerman RJ, Leehey M, Heinrichs W, et al. Intention tremor, parkinsonism, and generalized brain atrophy in male carriers of fragile X. Neurology. 2001;57(1):127–30. doi: 10.1212/WNL.57.1.127. [DOI] [PubMed] [Google Scholar]
- 3.Sullivan SD, Welt C, Sherman S. FMR1 and the continuum of primary ovarian insufficiency. Semin Reprod Med. 2011;29(4):299–307. doi: 10.1055/s-0031-1280915. [DOI] [PubMed] [Google Scholar]
- 4.Wheeler AC, Bailey DB, Jr, Berry-Kravis E, et al. Associated features in females with an FMR1 premutation. J Neurodev Disord. 2014;6(1):30. doi: 10.1186/1866-1955-6-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hantash FM, Goos DM, Crossley B, et al. FMR1 premutation carrier frequency in patients undergoing routine population-based carrier screening: insights into the prevalence of fragile X syndrome, fragile X-associated tremor/ataxia syndrome, and fragile X-associated primary ovarian insufficiency in the United States. Genet Med. 2011;13(1):39–45. doi: 10.1097/GIM.0b013e3181fa9fad. [DOI] [PubMed] [Google Scholar]
- 6.Maenner MJ, Baker MW, Broman KW, et al. FMR1 CGG expansions: prevalence and sex ratios. Am J Med Genet B Neuropsychiatr Genet. 2013;162B(5):466–73. doi: 10.1002/ajmg.b.32176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Seltzer MM, Baker MW, Hong J, Maenner M, Greenberg J, Mandel D. Prevalence of CGG expansions of the FMR1 gene in a US population-based sample. Am J Med Genet B Neuropsychiatr Genet. 2012;159B(5):589–97. doi: 10.1002/ajmg.b.32065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tassone F, Iong KP, Tong TH, et al. FMR1 CGG allele size and prevalence ascertained through newborn screening in the United States. Genome Med. 2012;4(12):100. doi: 10.1186/gm401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Crum-Bailey JM, Dennison DH, Weiner WJ, Hawley JS. The neurology and corresponding genetics of fragile X disorders: insights into the genetics of neurodegeneration. Future Neurol. 2013;8:225–35. doi: 10.2217/fnl.12.92. [DOI] [Google Scholar]
- 10.Hagerman R, Hagerman P. Advances in clinical and molecular understanding of the FMR1 premutation and fragile X-associated tremor/ataxia syndrome. Lancet Neurol. 2013;12(8):786–98. doi: 10.1016/S1474-4422(13)70125-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kraan CM, Hocking DR, Bradshaw JL, et al. Neurobehavioural evidence for the involvement of the FMR1 gene in female carriers of fragile X syndrome. Neurosci Biobehav Rev. 2013;37(3):522–47. doi: 10.1016/j.neubiorev.2013.01.010. [DOI] [PubMed] [Google Scholar]
- 12.Rodriguez-Revenga L, Madrigal I, Pagonabarraga J, et al. Penetrance of FMR1 premutation associated pathologies in fragile X syndrome families. Eur J Hum Genet. 2009;17(10):1359–62. doi: 10.1038/ejhg.2009.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Renda MM, Voigt RG, Babovic-Vuksanovic D, et al. Neurodevelopmental disabilities in children with intermediate and premutation range fragile X cytosine-guanine-guanine expansions. J Child Neurol. 2014;29(3):326–30. doi: 10.1177/0883073812469723. [DOI] [PubMed] [Google Scholar]
- 14.Hagerman R, Lugenbeel K, McLean SD, Taylor A. Learning-disabled males with a fragile X CGG expansion in the upper premutation size range. Pediatrics. 1996;97(1):122–6. [PubMed] [Google Scholar]
- 15.Tassone F, Hagerman RJ, Taylor AK, et al. Clinical involvement and protein expression in individuals with the FMR1 premutation. Am J Med Genet. 2000;91(2):144–52. doi: 10.1002/(SICI)1096-8628(20000313)91:2<144::AID-AJMG14>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 16.Farzin F, Perry H, Hessl D, et al. Autism spectrum disorders and attention-deficit/hyperactivity disorder in boys with the fragile X premutation. J Dev Behav Pediatr. 2006;27(2 Suppl):S137–44. doi: 10.1097/00004703-200604002-00012. [DOI] [PubMed] [Google Scholar]
- 17.Myers GF, Mazzocco MM, Maddalena A, Reiss AL. No widespread psychological effect of the fragile X premutation in childhood: evidence from a preliminary controlled study. J Dev Behav Pediatr. 2001;22(6):353–9. doi: 10.1097/00004703-200112000-00001. [DOI] [PubMed] [Google Scholar]
- 18.Chonchaiya W, Au J, Schneider A, et al. Increased prevalence of seizures in boys who were probands with the FMR1 premutation and co-morbid autism spectrum disorder. Hum Genet. 2012;131(4):581–9. doi: 10.1007/s00439-011-1106-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bailey DB, Jr, Raspa M, Olmsted M, Holiday DB. Co-occurring conditions associated with FMR1 gene variations: findings from a national parent survey. Am J Med Genet A. 2008;146A(16):2060–9. doi: 10.1002/ajmg.a.32439. [DOI] [PubMed] [Google Scholar]
- 20.Loesch DZ, Bui QM, Grigsby J, et al. Effect of the fragile X status categories and the fragile X mental retardation protein levels on executive functioning in males and females with fragile X. Neuropsychology. 2003;17(4):646–57. doi: 10.1037/0894-4105.17.4.646. [DOI] [PubMed] [Google Scholar]
- 21.Grigsby J, Brega AG, Engle K, et al. Cognitive profile of fragile X premutation carriers with and without fragile X-associated tremor/ataxia syndrome. Neuropsychology. 2008;22(1):48–60. doi: 10.1037/0894-4105.22.1.48. [DOI] [PubMed] [Google Scholar]
- 22.Cornish KM, Li L, Kogan CS, et al. Age-dependent cognitive changes in carriers of the fragile X syndrome. Cortex. 2008;44(6):628–36. doi: 10.1016/j.cortex.2006.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lachiewicz AM, Dawson DV, Spiridigliozzi GA, McConkie-Rosell A. Arithmetic difficulties in females with the fragile X premutation. Am J Med Genet A. 2006;140(7):665–72. doi: 10.1002/ajmg.a.31082. [DOI] [PubMed] [Google Scholar]
- 24.Franke P, Leboyer M, Hardt J, et al. Neuropsychological profiles of FMR-1 premutation and full-mutation carrier females. Psychiatry Res. 1999;87(2-3):223–31. doi: 10.1016/S0165-1781(99)00067-0. [DOI] [PubMed] [Google Scholar]
- 25.Sterling AM, Mailick M, Greenberg J, Warren SF, Brady N. Language dysfluencies in females with the FMR1 premutation. Brain Cogn. 2013;82(1):84–9. doi: 10.1016/j.bandc.2013.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Losh M, Klusek J, Martin GE, Sideris J, Parlier M, Piven J. Defining genetically meaningful language and personality traits in relatives of individuals with fragile X syndrome and relatives of individuals with autism. Am J Med Genet B Neuropsychiatr Genet. 2012;159B(6):660–8. doi: 10.1002/ajmg.b.32070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hunter JE, Allen EG, Abramowitz A, et al. No evidence for a difference in neuropsychological profile among carriers and noncarriers of the FMR1 premutation in adults under the age of 50. Am J Hum Genet. 2008;83(6):692–702. doi: 10.1016/j.ajhg.2008.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Franke P, Leboyer M, Gansicke M, et al. Genotype-phenotype relationship in female carriers of the premutation and full mutation of FMR-1. Psychiatry Res. 1998;80(2):113–27. doi: 10.1016/S0165-1781(98)00055-9. [DOI] [PubMed] [Google Scholar]
- 29.Roberts JE, Bailey DB, Jr, Mankowski J, et al. Mood and anxiety disorders in females with the FMR1 premutation. Am J Med Genet B Neuropsychiatr Genet. 2009;150B(1):130–9. doi: 10.1002/ajmg.b.30786. [DOI] [PubMed] [Google Scholar]
- 30.Clifford S, Dissanayake C, Bui QM, Huggins R, Taylor AK, Loesch DZ. Autism spectrum phenotype in males and females with fragile X full mutation and premutation. J Autism Dev Disord. 2007;37(4):738–47. doi: 10.1007/s10803-006-0205-z. [DOI] [PubMed] [Google Scholar]
- 31.Hessl D, Rivera SM, Reiss AL. The neuroanatomy and neuroendocrinology of fragile X syndrome. Ment Retard Dev Disabil Res Rev. 2004;10(1):17–24. doi: 10.1002/mrdd.20004. [DOI] [PubMed] [Google Scholar]
- 32.Huber KM, Gallagher SM, Warren ST, Bear MF. Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc Natl Acad Sci U S A. 2002;99(11):7746–50. doi: 10.1073/pnas.122205699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bear MF, Huber KM, Warren ST. The mGluR theory of fragile X mental retardation. Trends Neurosci. 2004;27(7):370–7. doi: 10.1016/j.tins.2004.04.009. [DOI] [PubMed] [Google Scholar]
- 34.D'Hulst C, Kooy RF. The GABAA receptor: a novel target for treatment of fragile X? Trends Neurosci. 2007;30(8):425–31. doi: 10.1016/j.tins.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 35.Darnell JC, Van Driesche SJ, Zhang C, et al. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell. 2011;146(2):247–61. doi: 10.1016/j.cell.2011.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gallego PK, Burris JL, Rivera SM. Visual motion processing deficits in infants with the fragile X premutation. J Neurodev Disord. 2014;6(1):29. doi: 10.1186/1866-1955-6-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Skinner D, Choudhury S, Sideris J, et al. Parents’ decisions to screen newborns for FMR1 gene expansions in a pilot research project. Pediatrics. 2011;127(6):e1455–63. doi: 10.1542/peds.2010-3078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bailey DB, Wheeler A, Berry-Kravis E, Hagerman R, Tassone F, Powell CM, Sideris J. Maternal Consequences of the Detection of Fragile X Carriers in Newborn Screening. Pediatrics. 2015;136(2):e433–e440. [DOI] [PMC free article] [PubMed]
- 39.Mullen EM. Mullen scales of early learning. Circle Pines: AGS; 1995. [Google Scholar]
- 40.Sparrow SS, Cicchetti DV, Balla DA. Vineland adaptive behavior scales, second edition (Vineland II): survey interview form/caregiver rating form. Livonia: Pearson Assessments; 2005. [Google Scholar]
- 41.Fenson L, Marchman V, Thal D, Dale P, Reznick S, Bates E. MacArthur communicative development inventories: user’s guide and technical manual. 2. Baltimore: Brookes; 2006. [Google Scholar]
- 42.Wetherby AM, Prizant BM. Communication and symbolic behavior scales: developmental profile. Baltimore: Paul H. Brookes Publishing; 2002. [Google Scholar]
- 43.Baranek GT, David FJ, Poe MD, Stone WL, Watson LR. Sensory Experiences Questionnaire: discriminating sensory features in young children with autism, developmental delays, and typical development. J Child Psychol Psychiatry. 2006;47(6):591–601. doi: 10.1111/j.1469-7610.2005.01546.x. [DOI] [PubMed] [Google Scholar]
- 44.Baranek GT, Roberts JE, David FJ, et al. Developmental trajectories and correlates of sensory processing in young boys with fragile X syndrome. Phys Occup Ther Pediatr. 2008;28(1):79–98. doi: 10.1300/J006v28n01_06. [DOI] [PubMed] [Google Scholar]
- 45.Watson LR, Baranek GT, Crais ER, Steven Reznick J, Dykstra J, Perryman T. The first year inventory: retrospective parent responses to a questionnaire designed to identify one-year-olds at risk for autism. J Autism Dev Disord. 2007;37(1):49–61. doi: 10.1007/s10803-006-0334-4. [DOI] [PubMed] [Google Scholar]
- 46.Lord C, Rutter M, DiLavore PC, Risi S, Gotham K, Bishop S. Autism diagnostic observation schedule, second edition (ADOS-2) manual (part 1): modules 1-4. Torrance: Western Psychological Services; 2012. [Google Scholar]
- 47.Raudenbush SW, Bryk AS. Hierarchical linear models: applications and data analysis (advanced quantitative techniques in the social sciences), 2E. New York: Sage Publications; 2001. [Google Scholar]
- 48.Gravetter F, Wallnau L. Statistics for the behavioral sciences. Boston: Cengage Learning; 2016. [Google Scholar]
- 49.Quené H, Van den Bergh H. On multi-level modeling of data from repeated measures designs: a tutorial. Speech Communication. 2004;43(1):103–21. doi: 10.1016/j.specom.2004.02.004. [DOI] [Google Scholar]
- 50.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Roy Stat Soc B. 1995;57:289–300. [Google Scholar]
- 51.Reznick JS, Baranek GT, Reavis S, Watson LR, Crais ER. A parent-report instrument for identifying one-year-olds at risk for an eventual diagnosis of autism: the first year inventory. J Autism Dev Disord. 2007;37(9):1691–710. doi: 10.1007/s10803-006-0303-y. [DOI] [PubMed] [Google Scholar]
- 52.Ben-Sasson A, Cermak SA, Orsmond GI, et al. Extreme sensory modulation behaviors in toddlers with autism spectrum disorders. Am J Occup Ther. 2007;61(5):584–92. doi: 10.5014/ajot.61.5.584. [DOI] [PubMed] [Google Scholar]
- 53.Baranek GT, Boyd BA, Poe MD, David FJ, Watson LR. Hyperresponsive sensory patterns in young children with autism, developmental delay, and typical development. Am J Ment Retard. 2007;112(4):233–45. doi: 10.1352/0895-8017(2007)112[233:HSPIYC]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 54.Liss M, Saulnier C, Fein D, Kinsbourne M. Sensory and attention abnormalities in autistic spectrum disorders. Autism. 2006;10(2):155–72. doi: 10.1177/1362361306062021. [DOI] [PubMed] [Google Scholar]
- 55.Foss-Feig JH, Heacock JL, Cascio CJ. Tactile responsiveness patterns and their association with core features in autism spectrum disorders. Res Autism Spectr Disord. 2012;6(1):337–44. doi: 10.1016/j.rasd.2011.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Boyd BA, Baranek GT, Sideris J, et al. Sensory features and repetitive behaviors in children with autism and developmental delays. Autism Res. 2010;3(2):78–87. doi: 10.1002/aur.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ben-Sasson A, Hen L, Fluss R, Cermak SA, Engel-Yeger B, Gal E. A meta-analysis of sensory modulation symptoms in individuals with autism spectrum disorders. J Autism Dev Disord. 2009;39(1):1–11. doi: 10.1007/s10803-008-0593-3. [DOI] [PubMed] [Google Scholar]
- 58.Miller LJ, McIntosh DN, McGrath J, Shyu V, Lampe M, Taylor AK, et al. Electrodermal responses to sensory stimuli in individuals with fragile X syndrome: a preliminary report. Am J Med Genet. 1999;83:268–79. doi: 10.1002/(SICI)1096-8628(19990402)83:4<268::AID-AJMG7>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 59.Conde V, Palomar FJ, Lama MJ, Martínez R, Carrillo F, Pintado E, Mir P. Abnormal GABA-mediated and cerebellar inhibition in women with the fragile X premutation. J Neurophysiol. 2013;109(5):1315–22. doi: 10.1152/jn.00730.2012. [DOI] [PubMed] [Google Scholar]
- 60.Greenspan SI, Weider S. Developmental patterns and outcomes in infants and children with disorders relating and communicating: a chart review of 200 cases of children with autistic spectrum diagnoses. J Dev Learning Disorders. 1997;1:87–142. [Google Scholar]
- 61.Baranek GT, Reinhartsen DB, Wannamaker SW. Play: engaging children with autism. In: Huebner T, editor. Autism, a sensorimotor approach to management. Philadelphia: F. A. Davis; 2001. [Google Scholar]
- 62.Dunn W. The impact of sensory processing abilities on the daily lives of young children and their families: a conceptual model. Infants Young Child. 1997;9(4):23–35. doi: 10.1097/00001163-199704000-00005. [DOI] [Google Scholar]
- 63.Turner-Brown LM, Baranek GT, Reznick JS, Watson LR, Crais ER. The First Year Inventory: a longitudinal follow-up of 12-month-old to 3-year-old children. Autism. 2013;17(5):527–40. doi: 10.1177/1362361312439633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ludwig AL, Espinal GM, Pretto DI, et al. CNS expression of murine fragile X protein (FMRP) as a function of CGG-repeat size. Hum Mol Genet. 2014;23(12):3228–38. doi: 10.1093/hmg/ddu032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Pretto DI, Mendoza-Morales G, Lo J, et al. CGG allele size somatic mosaicism and methylation in FMR1 premutation alleles. J Med Genet. 2014;51(5):309–18. doi: 10.1136/jmedgenet-2013-102021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Berry-Kravis E, Abrams L, Coffey SM, et al. Fragile X-associated tremor/ataxia syndrome: clinical features, genetics, and testing guidelines. Mov Disord. 2007;22(14):2018–30. doi: 10.1002/mds.21493. [DOI] [PubMed] [Google Scholar]
- 67.Sellier C, Freyermuth F, Tabet R, et al. Sequestration of DROSHA and DGCR8 by expanded CGG RNA repeats alters microRNA processing in fragile X-associated tremor/ataxia syndrome. Cell Rep. 2013;3(3):869–80. doi: 10.1016/j.celrep.2013.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Leehey MA, Berry-Kravis E, Goetz CG, et al. FMR1 CGG repeat length predicts motor dysfunction in premutation carriers. Neurology. 2008;70(16 Pt 2):1397–402. doi: 10.1212/01.wnl.0000281692.98200.f5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sullivan AK, Marcus M, Epstein MP, et al. Association of FMR1 repeat size with ovarian dysfunction. Hum Reprod. 2005;20(2):402–12. doi: 10.1093/humrep/deh635. [DOI] [PubMed] [Google Scholar]
- 70.Ennis S, Ward D, Murray A. Nonlinear association between CGG repeat number and age of menopause in FMR1 premutation carriers. Eur J Hum Genet. 2006;14(2):253–5. doi: 10.1038/sj.ejhg.5201510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Allen EG, Sullivan AK, Marcus M, et al. Examination of reproductive aging milestones among women who carry the FMR1 premutation. Hum Reprod. 2007;22(8):2142–52. doi: 10.1093/humrep/dem148. [DOI] [PubMed] [Google Scholar]
- 72.Tejada MI, Garcia-Alegria E, Bilbao A, et al. Analysis of the molecular parameters that could predict the risk of manifesting premature ovarian failure in female premutation carriers of fragile X syndrome. Menopause. 2008;15(5):945–9. doi: 10.1097/gme.0b013e3181647762. [DOI] [PubMed] [Google Scholar]
- 73.Seltzer MM, Barker ET, Greenberg JS, Hong J, Coe C, Almeida D. Differential sensitivity to life stress in FMR1 premutation carrier mothers of children with fragile X syndrome. Health Psychol. 2012;31(5):612–22. doi: 10.1037/a0026528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Loesch DZ, Bui MQ, Hammersley E, et al. Psychological status in female carriers of premutation FMR1 allele showing a complex relationship with the size of CGG expansion. Clin Genet. 2015;87(2):173–8. doi: 10.1111/cge.12347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lozano R, Hagerman RJ, Duyzend M, Budimirovic DB, Eichler EE, Tassone F. Genomic studies in fragile X premutation carriers. J Neurodev Disord. 2014;6(1):27. doi: 10.1186/1866-1955-6-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Baranek GT. Sensory Experiences Questionnaire (SEQ) University of North Carolina; Chapel Hill: 1999a. Unpublished manuscript.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets generated during and/or analyzed during the current study are not publicly available due to the small sample sizes and subsequent risk of breach of confidentiality but are available from the corresponding author on reasonable request.