

Abstract
Molecular solar‐thermal energy storage systems are based on molecular switches that reversibly convert solar energy into chemical energy. Herein, we report the synthesis, characterization, and computational evaluation of a series of low molecular weight (193–260 g mol−1) norbornadiene–quadricyclane systems. The molecules feature cyano acceptor and ethynyl‐substituted aromatic donor groups, leading to a good match with solar irradiation, quantitative photo‐thermal conversion between the norbornadiene and quadricyclane, as well as high energy storage densities (396–629 kJ kg−1). The spectroscopic properties and energy storage capability have been further evaluated through density functional theory calculations, which indicate that the ethynyl moiety plays a critical role in obtaining the high oscillator strengths seen for these molecules.
Keywords: donor–acceptor systems, energy conversion, molecular switches, norbornadiene, quadricyclane
Introduction
Reversible photoinduced isomerization of organic or organometallic compounds into metastable isomers has been recognized as a means of storing solar energy,1 a strategy frequently referred to as molecular solar‐thermal (MOST) energy storage.2 A number of different systems have been proposed, including anthracene,3 stilbene,4 azobenzene,5 dihydroazulene,6 and tetracarbonyl‐fulvalene‐diruthenium7 derivatives. The system that has received most attention, as a large amount of energy can be stored in a small ring, is probably norbornadiene (1), which undergoes an endothermic photoinduced [2+2] cycloaddition to its valence isomer quadricyclane (2).8 The reaction is reversible, and by thermal or catalytic induction the reverse reaction regenerates norbornadiene with release of heat (Scheme 1).1
Scheme 1.
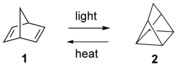
Photoinduced isomerization of norbornadiene 1 to quadricyclane 2 and the back conversion.
To design a practically useful MOST system, several requirements have to be fulfilled: 1) solar spectrum match for the absorption of the parent compound (in this case norbornadiene), 2) high photoisomerization quantum yield, 3) no spectral overlap between the parent compound and the photoisomer (the photoisomer should not compete for photons) and, 4) a highly endergonic reaction profile with a high activation energy for the reverse reaction.1c Another important factor affecting the performance is the molecular weight, which together with the storage energy gives the storage density. Moreover, for practical flow device applications, a liquid is required, which means that the solubilities of both the parent compound and the photoisomer become crucial.9
The absorption onset of unsubstituted norbornadiene 1 is 267 nm, but since the intensity of solar radiation below around 300 nm is very low at sea level, norbornadiene is essentially inert to sunlight. To prepare quadricyclane, high‐power ultraviolet lamps are employed, typically in the presence of a photosensitizer. Approaches to red‐shift the absorption of 1 have previously been studied, and one method is to introduce electron‐donating and electron‐accepting substituents to create a push–pull conjugated system.8d However, the introduction of large substituents is at the expense of a high molecular weight, which in turn lowers the energy storage density. Computational work has shown that the molar storage energy is largely independent of substitution pattern, implying that the molecular weight is the most important optimization parameter in this regard.10 In previous studies by our group, a series of electron‐donating and electron‐accepting diaryl‐substituted norbornadienes with an improved solar spectrum match compared to 1 was synthesized (Table 1).11 The most red‐shifted compound (3 e) has an absorption maximum at 365 nm and an absorption onset of 462 nm. However, this significant red‐shift comes at the expense of a rather high molecular weight, 355 g mol−1 for 3 e, compared to 92 g mol−1 for 1 (Table 1).1
Table 1.
Diaryl‐substituted norbornadienes (3 a–e) previously designed in our group, the absorption maxima, absorption onsets, half‐lives of the corresponding quadricyclanes, and molecular weights.11
| ||||||
|---|---|---|---|---|---|---|
| Entry | R1 | R2 | A max [nm] | A onset [a] [nm] | t 1/2 [h] | M W [g mol−1] |
| 3 a | H | H | 308 | 389 | 1030 | 244 |
| 3 b | H | OMe | 309 | 402 | 751 | 274 |
| 3 c | CF3 | OMe | 318 | 414 | 70 | 342 |
| 3 d | CN | OMe | 350 | 431 | 209 | 299 |
| 3 e | CF3 | NMe2 | 365 | 462 | 1.9 | 355 |
[a] Absorption onset defined as log ϵ=2.
Although compound 3 e allowed the evaluation of a solar‐collecting device, there is plenty of room for improvement in terms of molecular weight, half‐life of the photoisomer, solubility, as well as solar spectrum match. As mentioned above, a recent computational study suggested that the molecular weight is a key optimization parameter when aiming for high energy density in these systems.10 Inspired by this hypothesis, our approach here was to find a robust low molecular weight electron‐accepting substituent to replace the p‐substituted phenyl groups in compounds 3 a–e. Herein, we report a series of new cyano‐substituted norbornadiene derivatives with improved photochemical properties yet significantly reduced molecular weight.
Results and Discussion
Since the cyano group is one of the smallest electron‐accepting groups, with a molecular weight of only 27 g mol−1 compared to 103 g mol−1 for the p‐C6H4CN group, our first choice for modification of the previously reported norbornadiene system (Table 1, 3 a–e) was to attach a cyano group directly at a vinyl carbon atom (C2) thereof. 2,3‐Dicyanonorbornadienes with aliphatic substituents in different positions have been reported to have high solubility in organic solvents.12 2,3‐Dicyanonorbornadiene has been investigated as an MOST candidate, both in the free state13 and as a ligand in ruthenium complexes,14 but to the best of our knowledge, 2‐cyanonorbornadienes bearing electron‐donating aryl substituents at the 3‐position have not hitherto been described. Therefore, a new synthetic route had to be developed.
Conformational analysis of 3 a–e revealed that steric hindrance forces the aromatic substituents out of plane, reducing the overlap between the π‐systems of these substituents and the norbornadiene carbon‐carbon double bond.15 We hypothesized that reduced steric hindrance would allow for greater orbital overlap and hence a more red‐shifted absorption spectrum. Based on this analysis, an ethynyl linker was introduced between the aromatic donor group and the norbornadiene C3 atom. This has the additional effect of extending the conjugated system and thus further red‐shifting the absorption. Only a few examples of ethynyl‐substituted norbornadienes have been reported, which have included conjugated polymers,16 photochromic bridges to link dinuclear ruthenium complexes,17 and starting materials for the synthesis of ethynyl cyclopentadiene derivatives by retro‐Diels–Alder reactions.18 To the best of our knowledge, no application of these compounds for MOST energy storage has been proposed. Thus, this new series of norbornadiene derivatives is characterized by a cyano group in the 2‐position and a variety of aromatic donor groups attached via a −C≡C− linker to the C3 carbon atom. The selected target compounds 4 a–d are depicted in Figure 1. In addition, compound 5 was prepared to allow direct comparison between the systems with and without ethynyl substitution.1
Figure 1.
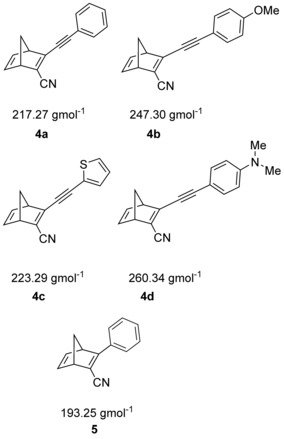
Second‐generation norbornadienes 4 a–d and 5 with their molecular weights.
Synthesis
Norbornadienes 4 a–d could be synthesized through a Sonogashira19 cross‐coupling reaction between 2‐cyano‐3‐chloronorbornadiene (6) and the corresponding donor‐substituted acetylene (Scheme 2 a), whereas 5 could be obtained by a Suzuki20 cross‐coupling reaction between 6 and phenylboronic acid (Scheme 2 b).2
Scheme 2.
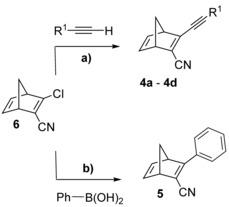
a) Sonogashira cross‐couplings to obtain 4 a–d. For 4 a: R1=phenyl, 4 b: R1=p‐methoxyphenyl, 4 c: R1=2‐thiophenyl, 4 d: R1=p‐dimethylaminophenyl. b) Suzuki cross‐coupling to obtain 5.
Consequently, an efficient synthesis of 6 was essential. Only one synthetic procedure for 6 has been described in the literature.21 In this procedure, a four‐step synthetic route affords 1,1‐dichloro‐2,2‐dicyanoethylene (12),21a,21b which is allowed to react with cyclopentadiene through a Diels–Alder reaction to provide the norbornene derivative 14. Finally, treating 14 with potassium hydroxide gives 6.21c The overall procedure is thus a six‐step route resulting in a moderate overall yield (22 %) of 6, and includes steps that appeared less attractive to us, such as bubbling highly toxic chlorine gas through a solution of 10 for 8 h. It has recently been reported that 2,3‐dibromonorbornadiene reacts with copper(I) cyanide to give 2‐bromo‐3‐cyanonorbornadiene in 35 % yield,22 and since we have previously reported23 a one‐pot route to 2‐bromo‐3‐chloro‐norbornadiene, 15, it appeared logical to use 15 as a precursor for 6. Indeed, 15 was found to react with copper(I) cyanide in N‐methylpyrrolidone to give the desired product 6 (Scheme 3). In order to suppress the formation of 1,3‐dicyanonorbornadiene, an excess of 15 was used, which could be easily recovered and recycled. The reaction mixture proved to be easy to separate. It was applied to a silica pad and consecutively eluted, first with pentane to recover unreacted 15, which could be reused without further purification, and then with dichloromethane to afford 6. The yield, based on consumed 15, was 88 %.3
Scheme 3.
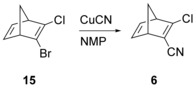
New route to 2‐chloro‐3‐cyanonorbornadiene.
With 6 in hand, the next step towards 4 a–d and 5 was to perform Sonogashira19 or Suzuki20 cross‐couplings (Scheme 2). The Sonogashira reaction to obtain symmetrical norbornadiene‐2,3‐diynes from 2,3‐dichloronorbornadienes was first explored in 1997 by Durr et al.24 as an alternative to previous methods in which 2,3‐diiodonium norbornadiene salts were treated with lithium alkynyl‐cuprates.25 An alternative route, in which triflate groups are utilized as coupling partner instead of a halogen, has also been explored.26 Syntheses of symmetrical and unsymmetrical norbornadiene‐2,3‐diynes through palladium‐catalyzed Sonagashira reactions using dichlorobis(triphenylphosphine)palladium(II), copper(I) iodide, and trimethylamine have been reported by Tranmer et al.27 For the double‐Sonogashira coupling reaction leading to symmetrical norbornadiene‐2,3‐diynes, the reactions were reported to be more selective using toluene as solvent compared to THF, whereas for the monocoupled products the opposite effect was observed and THF provided purer products. Moreover, higher yields were obtained by rigorously excluding dioxygen and water. Therefore, dry degassed THF became our solvent of choice. The Sonogashira reactions proceeded smoothly in THF for all of the ethynyl derivatives using dichlorobis(triphenylphosphine)palladium(II), copper(I) iodide, and trimethylamine or diisopropylamine as the base. At room temperature, the reactions were completed within 2–4 h and the products were easily purified by flash column chromatography to obtain 4 a–d in satisfactory yields (56–77 %). The Suzuki coupling between 6 and phenylboronic acid was performed in THF at reflux temperature with cesium fluoride, tri‐tert‐butylphosphine, and tris(dibenzylideneacetone)dipalladium(0). In order to obtain a satisfactory yield, plenty of the catalyst was added (26 mol %) and after purification 5 was obtained in 57 % yield.
Photoisomerization
The UV/Vis absorption spectra of 4 a–d and 5 in toluene are shown in Figure 2 a, and the absorption onsets, absorption maxima, and molar extinction coefficients are listed in Table 2. The absorption maximum for 5 is 309 nm and the onset is 358 nm, whereas the maxima for 4 a–d are all in the range 331–398 nm with onsets in the range 374–456 nm. The most red‐shifted absorption was observed for 4 d, with a maximum at 398 nm and an onset at 456 nm. Thus, among the synthesized compounds, 4 d is the norbornadiene that best meets the requirements of solar spectrum match. For comparison, 2,3‐dicyanonorbornadiene has an onset of around 360 nm and a maximum close to 300 nm.13, 2, 2
Figure 2.
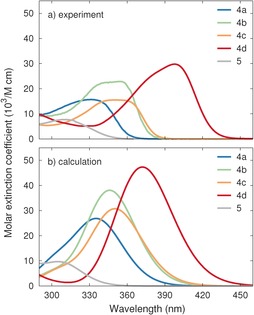
A) UV/Vis absorption spectra for 4 a–4 d, and 5 recorded in toluene. B) Absorption spectra of 4 a–4 d and 5 calculated at the B3LYP level (top) in comparison with experiments (bottom). A scissors shift of 0.3 eV as well as broadening of 0.20 eV were applied.
Table 2.
Absorption onsets, absorption maxima, molar extinction coefficients, and quantum yields of conversion to the corresponding quadricyclanes for 4 a–d and 5, determined in toluene.[a]
| Entry | ϵ max [m −1 cm−1] | A max [nm] | A onset [b] [nm] | Φ [%] |
|---|---|---|---|---|
| 4 a | 15.7×103 | 331 | 374 | 39 |
| 4 b | 22.9×103 | 355 | 391 | 38 |
| 4 c | 15.5×103 | 340 | 395 | 47 |
| 4 d | 29.8×103 | 398 | 456 | 28 |
| 5 | 7.72×103 | 309 | 358 | 58 |
[a] Quantum yields (Φ) for the photoisomerization of norbornadienes 4 a–d and 5 to quadricyclanes 16 a–d and 17 reported as averages from two to three individual measurements. Samples were irradiated at 365 nm (4 b, 4 c) or 310 nm (4 a, 5), each dissolved in toluene. [b] Absorption onset defined as log ϵ=2.
By comparing the absorption spectra of 4 a and 5, it becomes clear that the ethynyl unit not only exerts a significant effect on the absorption onset, but also doubles the extinction coefficient. The top candidate in our previous studies regarding solar spectrum match (3 e, Figure 1) showed an absorption maximum of 365 nm and an onset of 462 nm. Thus, compared to 3 e, compound 4 d has a sharper absorption peak with a significant red‐shift of the absorption maximum (398 nm; +33 nm). The difference in onset of absorbance is smaller (456 nm; −6 nm), which reflects the comparable spectrally wider absorption band of 3 e compared to that of 4 d. This could be a consequence of the steric hindrance between the phenyl groups as well as thermally activated rotation of the side groups in 3 e, which results in a distribution of phenyl rotation angles, and thus electron communication between the donor and acceptor groups leading to spectral broadening. The overall solar spectrum match is roughly the same for the two compounds; however, the molar extinction coefficient is about three times higher for 4 d than for 3 e, which should be taken into account in future device implementation.
To study the photoisomerization of 4 a–d and 5 to the corresponding quadricyclanes (16 a–d and 17, Scheme 4), the norbornadienes were irradiated with a metal‐halide lamp. The formation of the quadricyclanes was monitored by both NMR and UV/Vis spectroscopies. In the UV/Vis absorption spectra (Figure 3), no spectral overlap between the norbornadienes and the quadricyclanes was observed in the visible region, allowing for quantitative conversion between the two isomers and thus meeting one of the important requirements for an efficient MOST system.4, 3
Scheme 4.
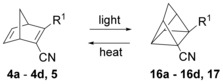
Photoisomerization of 4 a–d and 5 to obtain 16 a–d and 17. For 4 a, 16 a: R1=phenylethynyl; 4 b, 16 b: R1=(p‐methoxyphenyl)ethynyl; 4 c, 16 c: R1=(2‐thiophenyl)ethynyl; 4 d, 16 d: R1=(p‐dimethylaminophenyl)ethynyl; 5, 17: R1=phenyl.
Figure 3.
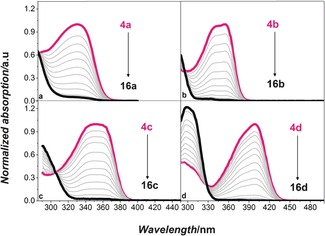
UV/Vis spectra of the formation of quadricyclanes (thick black lines) by irradiation of norbornadienes (red lines): a) formation of 16 a from 4 a, b) formation of 16 b from 4 b, c) formation of 16 c from 4 c, d) formation of 16 d from 4 d.
Furthermore, isosbestic points were detected in the UV/Vis spectra of 4 a–d, indicating that only two species were present in the solution (Figure 3). For 5, with the least red‐shifted spectrum, the expected isosbestic point could not be observed in toluene due to overlap with the absorption onset of the solvent. However, NMR observation indicated that photoisomerization and back‐conversion proceeded with little or no degradation for 5 as well. To demonstrate that the compounds could be fully converted by unfiltered solar light, a solution of 4 d in a quartz cuvette was exposed to 1.5 AM standard solar spectrum in a solar simulator. After about 10 s, it was possible to observe full conversion of 4 d to 16 d. This experiment showed how 4 d has the potential to be employed as an MOST system in testing devices with sunlight‐induced conversion (see Figure SI6 in the Supporting Information).
Photoisomerization quantum yields were determined in toluene using potassium ferrioxalate as a chemical actinometer.28 The measurements were carried out using a 365 nm or 310 nm light‐emitting diode and the quantum yields are reported as an average of two or three determinations (Table 2). The highest quantum yields were observed for 5 and 4 c at 59 % and 47 %, respectively, whereas those for 4 a and 4 b were lower at 39 % and 38 %, respectively. The top candidate with regard to solar spectrum match (4 d) has a quantum yield of around 28 %.
Back conversion
To evaluate the thermal stabilities of the photoisomers (16 a–d and 17), a kinetic study was performed to determine the enthalpies and entropies of activation for the thermally induced reverse reactions. The experiment was carried out by determining the rate constants, k, at six different temperatures (Supporting Information, Figure S3). The norbornadienes were converted to the quadricyclanes by irradiation, and the back‐conversion was studied by measuring the increase in absorbance by UV/Vis spectrophotometry. From the rate constants at these different temperatures, the enthalpies and entropies of activation were estimated using the Eyring equation (Table 3).3
Table 3.
Thermodynamic data for the thermal reactions from quadricyclanes 16 a–d and 17 to norbornadienes 4 a–d and 5, determined in toluene.
| Entry | ΔH ≠ [kJ mol−1] | ΔS ≠ [J K−1 mol−1] | t 1/2 [a] [h] | |
|---|---|---|---|---|
|
16 a
|
104 | 5.57 | 22.0 | |
|
16 b
|
102 | 3.86 | 15.8 | |
|
16 c
|
101 | 5.65 | 7.43 | |
|
16 d
|
92.5 | −19.1 | 5.05 | |
|
17
|
112 | −1.31 | 1320 |
[a] Half‐lives of the photoisomers 16 a–d and 17 determined from Eyring parameters at 25 °C.
The experiments showed 17, without an ethynyl unit, to be much more stable than 16 a–d, with a half‐life of around 55 days at room temperature. Among the norbornadienes with the ethynyl linker, 16 a and 16 b have the longest half‐lives of approximately 22 and 16 h, respectively, whereas 16 c and 16 d are less stable, with half‐lives of approximately 7 and 5 h, at room temperature. Thus, back‐conversions of 16 c to 4 c and of 16 d to 4 d proceed much more rapidly than those of 16 a and 16 b. Nevertheless, 16 d has a 2.6 times longer half‐life than the previously studied photoisomer of 3 e, which has a storage half‐life of 1.9 h.
Energy storage
All compounds were photoisomerized to the corresponding quadricyclanes, and the enthalpies of the back‐conversion to the norbornadienes were determined by differential scanning calorimetry (DSC). In addition, NMR spectroscopy and thermal gravimetric analyses were performed to verify that photoisomerization and heat release occurred with little or no degradation.
All of the compounds show exothermic peaks upon isomerization to the norbornadienes. Isomerizations of 16 d to 4 d and of 17 to 5 each show one exothermic peak (Figure 4), and the measured values of the heat release are 103 and 122 kJ mol−1, respectively, corresponding to energy storage densities of 396–629 kJ kg−1 (Table 4).4, 4
Figure 4.
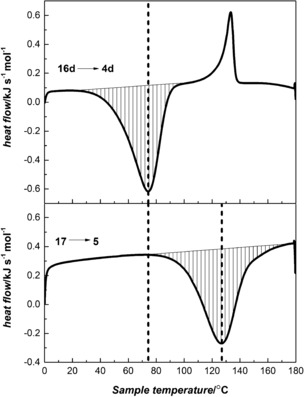
DSC thermograms showing the heat release peaks for the thermal back‐conversion of quadricyclanes 16 d and 17 to the corresponding norbornadienes. Melting of 16 d was observed at around 120 °C.
Table 4.
Energy storage densities for 16 d and 17, determined by DSC.
| Entry | ΔH [kJ mol−1] | ΔH [kJ kg−1] | ΔH [kcal mol−1] | |
|---|---|---|---|---|
|
16 d
|
103 | 396 | 25 | |
|
17
|
122 | 629 | 29 |
However, for compounds 16 a, 16 b, and 16 c, two exothermic peaks were observed for the thermal back‐conversion (Supporting Information, Figure S4), which we speculate may be due to complex phase behavior in the mixture of norbornadiene and quadricyclane. Since distinguishing the heat release peak from other eventual phase transitions was troublesome for these compounds, it was not possible to precisely determine the values, and hence they are not reported here.
Insight from electronic structure calculations
To elucidate the features of the measured absorption spectrum and to rationalize the experimental findings, we performed regular as well as time‐dependent density functional theory (TD‐DFT) calculations at the B3LYP/6‐311+G* level29 on each of the compounds. Based on a systematic comparison of different exchange‐correlation functionals with MP2 as well as complete active space calculations,10 we have previously established that the B3LYP functional yields absorption spectra and geometries in good agreement with both experiment and higher‐level calculations.
The calculated absorption spectra are generally in good agreement with experiment (Figure 2 b). The most notable difference with respect to experimental spectra is the absence of a shoulder on the high‐energy side of the first excitation peak for compounds 4 a–d. We suggest that this feature can be attributed to the presence of the ethynyl bridge in these compounds. The ground‐state geometry maximizes the conjugation of the π‐system that extends from the double bond in the norbornadiene part of the molecule via the ethynyl bridge to the functionalized phenyl group. Rotations about the triple bond in the bridge are very soft and therefore readily sampled under ambient conditions. At the same time, these rotations reduce the conjugation, causing a blue‐shift of the first excitation maximum, which is dominated by the HOMO–LUMO transition and is therefore most sensitive to changes in the π‐system. If these effects are superimposed, they give rise to a pronounced shoulder on the high‐energy side of the first absorption maximum. This interpretation is supported by the observation that the shoulder feature is specific to the compounds that contain an ethynyl bridge (4 a–d) and absent in compounds that have aryl substituents attached via single bonds (compound 5 in the present work as well as the compounds in our previous report11).
Most crucially, the calculations correctly capture all of the relevant trends, namely the increasing red‐shift on going from 4 a to 4 d and the large difference in the magnitude of the extinction coefficient between equivalent compounds with (4 a) and without (5) an ethynyl bridge.
The shift of the absorption spectrum with the strength of the donor species is in line with our earlier calculations.10, 15 More interesting in the present context is the strong enhancement of the dipole oscillator strength (Table 5) that results from insertion of an ethynyl bridge on going from 4 a to 5, and gives rise to a large increase in the extinction coefficient. The lowest excitation in both cases is dominated by HOMO–LUMO transitions. The dipole oscillator strength depends on the transition dipole moment, , which illustrates that the transition strength is sensitive to the spatial overlap between the orbitals involved, in particular near the nodes. It is therefore instructive to compare the relative alignment of the HOMO and LUMO states in 4 a and 5 (Figure 5). This reveals a near‐perfect alignment of the orbitals across the triple bond in 4 a, with the node in the LUMO being located precisely at the midpoint between the two C atoms. In contrast, in the case of 5, the bond angle across the C=C double bond to the phenyl ring distorts both the LUMO and HOMO, and shifts the LUMO node away from the maximum of the HOMO state at the center of the C−C bond. The ethynyl group in 4 a thus minimizes the distortion of the conjugated π‐system between the parent compound and the donor group, which enhances both the red‐shift and the dipole strength.5, 5
Table 5.
Calculated storage energies (ΔH) and dipole oscillator strengths for the first excitation (f) for compounds 4 a–d and 5.
| Entry | ΔH [kJ mol−1] | f [a.u.] |
|---|---|---|
| 4 a | 118.46 | 0.44 |
| 4 b [a] | 121.00 | 0.66/0.64 |
| 4 c [a] | 119.03 | 0.53/0.52 |
| 4 d | 124.46 | 0.83 |
| 5 | 114.27 | 0.16 |
[a] Data for both rotamers are provided.
Figure 5.
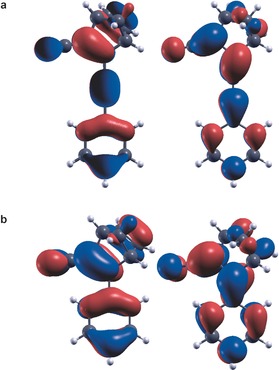
HOMO (left) and LUMO (right) isosurfaces for: a) 4 a, and b) 5. The stronger distortion of the orbitals across the bridging bond between parent compound and donor group in the case of 5 relative to 4 a reduces the dipole strength of the HOMO–LUMO transition in the former case and leads to a lower overall attenuation coefficient.
After geometry relaxation, vibrational contributions to storage enthalpies were calculated for each rotamer and the resulting ΔH values are reported in Table 5.
For systems with multiple rotamers, the enthalpies of the norbornadiene and quadricyclane systems were Boltzmann‐weighted before calculating the final storage enthalpy. For the molecules 4 d and 5, for which energy differences between the quadricyclane and norbornadiene form are available, the calculated energies show good agreement with experimental observation.
Conclusions
A series of substituted donor–acceptor norbornadienes with cyano acceptors and ethynyl‐aryl donor units has been synthesized and evaluated in the context of molecular solar‐thermal energy storage. A key step in the synthesis is a procedure for the formation of 2‐cyano‐3‐chloronorbornadiene from readily available starting materials. Carbon‐carbon bond‐forming reactions based on Suzuki or Sonogashira cross‐couplings have been established in order to prepare the target compounds in 57–77 % yields from 2‐cyano‐3‐chloronorbornadiene. The photophysical properties of the compounds have been investigated, which confirmed that the applied substitution pattern is effective in red‐shifting the absorption of norbornadiene (1) by up to 189 nm (absorption onset 456 nm) for compound 4 d, this being obtained in a system with a molecular weight of only 260 g mol−1. In comparison, previously reported 3 e shows a similar onset of absorption (462 nm), but at the expense of a much higher molecular weight of 355 g mol−1. Nevertheless, the absorption onset of 4 d is still 200 nm away from the “optimal” onset of absorption of 656 nm, which could be a parameter for future optimization.9 The storage density recorded upon heat‐releasing conversion of quadricyclane to norbornadiene showed energy densities of up to 629 kJ kg−1, exceeding the energy densities typically found in phase‐change materials.30 The half‐lives of the photoisomers 16 a–d are in the range 5–22 h, which means that for future applications whereby long‐term storage as well as an optimal solar spectrum match are needed, the molecular system still needs some optimization and compounds akin to 17 may be more suitable.
The compounds 4 a–d developed in the present study show significantly higher extinction coefficients than the systems 3 a–e that we investigated previously.11 Through first principles calculations, we have shown that the ethynyl group is responsible for this enhancement as it reduces the distortion of HOMO and LUMO states across the bond between the parent compound and the donor group. This interpretation has been confirmed by the experimental realization of compound 5, the ethynyl‐free analogue of 4 a. The maximum extinction coefficient of 5 is indeed both blue‐shifted and halved compared to that for 4. This finding suggests that the insertion of triple bonds could be employed for similar purposes in other chromophore systems.
Experimental Section
General
All commercial chemicals were used as received. 2‐Bromo‐3‐chloronorbornadiene was prepared according to published procedures. Tetrahydrofuran (THF) was dried using an MBraun MB SPS‐800 solvent purification system. All glassware was dried overnight at 150 °C. Column chromatography was performed on a Biotage Isolera One instrument using pre‐packed silica columns (25 g or 50 g Biotage® SNAP Cartridge). All spectrophotometric analyses were performed using a Cary 50 Bio or a Cary 100 UV/Vis spectrophotometer. Quantum yields were determined using potassium ferrioxalate as a chemical actinometer and a fiber‐coupled LED (M365 F1 (365 nm) or M310 L3 (310 nm)) for irradiation. 1H and 13C NMR spectra were obtained at 400 and 100 MHz, respectively, on a Varian 400/54 spectrometer. 13C NMR spectra of the quadricyclanes 16 a–d were obtained on a Varian 500 MHz spectrometer. Chemical shifts are reported in ppm with residual protonated solvent as an internal standard (CHCl3 δ H=7.26 ppm, CHCl3 δ C=77.16 ppm). Elemental analyses were performed at the Mikroanalytisches Laboratorium Kolbe, Mülheim, Germany. IR analyses were carried out on a Perkin‐Elmer Frontier FTIR spectrometer. Differential scanning calorimetry (DSC) experiments were performed on a Mettler Toledo DSC 2 apparatus.
Synthesis
2‐Cyano‐3‐chloronorbornadiene (6): 2‐Bromo‐3‐chloronorbornadiene (15; 10.2 g, 0.0500 mol) was dissolved in N‐methylpyrrolidone (20 mL) and the solution was degassed. Copper(I) cyanide (3.58 g, 0.0400 mol) was added, and the mixture was stirred at 100 °C under nitrogen for 7 h. The reaction mixture was cooled and mixed with 1 m aqueous sodium cyanide solution (100 mL) and diethyl ether (100 mL). Vigorous shaking may result in an emulsion. The phases were separated, the aqueous phase was extracted with diethyl ether (5×50 mL), and the volume of the combined organic phases was reduced to about 30 mL in vacuo. The solution was washed with water (3×10 mL) and brine (10 mL), and then concentrated. The product was diluted with pentane to about twice the volume and applied to silica gel moistened with pentane (ca. 25 mm silica layer in a Ø 65 mm sintered glass funnel). The product was eluted from the silica with pentane (150 mL) followed by dichloromethane (200 mL). The pentane fraction was concentrated to give 15 (2.7 g) as a colorless liquid. The dichloromethane fraction was dried over Na2SO4 and concentrated to give 6 as a slightly yellow liquid (4.92 g; 88 % yield based on consumed 15). Analytical data were consistent with previous reports. 1H NMR (CDCl3): δ=6.92 (ddd, J=5.1, 2.9, 0.7 Hz, 1 H), 6.86 (ddd, J=5.1, 3.0, 0.9 Hz, 1 H), 3.86 (dddd, J=4.4, 2.7, 1.7, 0.9 Hz, 1 H), 3.62 (ddtd, J=3.1, 2.4, 1.6, 0.7 Hz, 1 H), 2.41 (dt, J=6.9, 1.6 Hz, 1 H), 2.25 ppm (dt, J=6.9, 1.7 Hz, 1 H); 13C NMR (CDCl3): δ=165.58, 142.87, 140.43, 120.37, 114.51, 72.85, 58.22, 53.84 ppm.
Sonogashira cross‐coupling (general procedure): 2‐Cyano‐3‐chloronorbornadiene (1 equiv.), copper(I) iodide (10 mol %), and dichlorobis(triphenylphosphine)palladium(II) (5 mol %) were taken up in THF (5 mL mmol−1) in a dry flask under nitrogen atmosphere. Dry triethylamine or diisopropylamine (0.33 mL mmol−1) was added dropwise over 15 min to the reaction mixture. A solution of the acetylene derivative (1.1 equiv.) in THF (1 mL) was slowly added, and the resulting mixture was stirred at room temperature for 2–4 h. It was then diluted with dichloromethane (30 mL) and filtered through a short silica plug. The solvents were evaporated, the residue was dissolved in diethyl ether (30 mL), and the solution was washed with water (10×3 mL) and brine (10 mL). The crude product was purified by automated column chromatography.
2‐Cyano‐3‐(phenylethynyl)norbornadiene (4 a): 2‐Cyano‐3‐chloronorbornadiene (400 mg, 2.6 mmol, 1 equiv.), copper(I) iodide (50 mg, 0.26 mmol, 10 mol %), bis(triphenylphosphine)palladium(II) dichloride (92 mg, 0.13 mmol, 5 mol %), diisopropylamine (0.82 mL), and phenylacetylene (290 mg, 2.8 mmol, 1.1 equiv.) were reacted according to the general procedure. The crude product was purified by automated column chromatography (CH2Cl2/heptane, 3:7) to provide 2‐cyano‐3‐(phenylethynyl)norbornadiene 4 a (405 mg, 1.9 mmol, 73 %) as an off‐white solid. 1H NMR (CDCl3): δ=7.54–7.48 (m, 2 H), 7.42–7.32 (m, 3 H), 6.87 (qdd, J=5.1, 2.9, 0.9 Hz, 2 H), 3.90 (ddt, J=2.3, 1.7, 1.0 Hz, 1 H), 3.85 (dtd, J=2.9, 1.5, 0.7 Hz, 1 H), 2.33 (dt, J=7.0, 1.5 Hz, 1 H), 2.23 ppm (dt, J=7.0, 1.6 Hz, 1 H); 13C NMR (CDCl3): δ=154.20, 142.27, 141.39, 132.02, 129.69, 128.62, 128.45, 122.12, 116.47, 107.90, 83.26, 72.91, 57.34, 54.19 ppm; IR: =3005.8, 2978.1, 2907.1, 2859.8, 2812.4, 2107.0, 2153.6, 1607.3, 1567.8, 1516.5, 1437.6, 1358.7, 1287.7, 1226.6, 1058.9, 1171.3, 1102.3, 1017.5 (m), 936.57, 818.2, 806.37, 739.3, 693.93, 668.29, 561.76, 530.2, 472.99, 453.26 cm−1; elemental analysis calcd (%) for C16H11N: C 88.45, H 5.10, N 6.45; found: C 87.69, H 5.26, N 6.49.
2‐Cyano‐3‐((4‐methoxyphenyl)ethynyl)norbornadiene (4 b): 2‐Cyano‐3‐chloronorbornadiene (110 mg, 0.73 mmol, 1 equiv.), copper(I) iodide (12.5 mg, 0.07 mmol, 10 mol %), bis(triphenylphosphine)palladium(II) dichloride (23 mg, 0.03 mmol, 4 mol %), triethylamine (0.27 mL), and 1‐ethynyl‐4‐methoxybenzene (110 mg, 0.83 mmol, 1.1 equiv.) were reacted according to the general procedure. The crude product was purified by automated column chromatography (CH2Cl2/hexane, 1:1) to provide 2‐cyano‐3‐((4‐methoxyphenyl)ethynyl)norbornadiene 4 b (139 mg, 0.56 mmol, 77 %) as an off‐white/yellow oil. 1H NMR (CDCl3): δ=7.48–7.43 (m, 2 H), 6.90–6.83 (m, 4 H), 3.89–3.87 (m, 1 H), 3.83 (s, 3 H), 3.83–3.81 (m, 1 H), 2.31 (dt, J=7.0, 1.7 Hz, 1 H), 2.21 ppm (dt, J=7.0, 1.6 Hz, 1 H); 13C NMR (CDCl3): δ=160.82, 154.51, 142.32, 141.30, 133.75, 126.97, 116.75, 114.34, 114.21, 108.54, 82.64, 72.70, 57.35, 55.54, 54.07 ppm; IR: =3002.5, 2940.4, 2874.4, 2835.6, 2206.8, 2160.3, 1604.7, 1569.7, 1499.9, 1487.8, 1463.0, 1296.1, 1247.6, 1172.0, 1104.0, 1057.4, 1022.5, 927.4, 869.18, 834.25, 810.96, 791.55, 733.33, 706.16, 673.17, 640.18, 620.78, 580.02, 558.68, 531.5, 506.28, 453.88 cm−1; elemental analysis calcd (%) for C17H13NO: C 82.57, H 5.30, N 5.66; found: C 82.37, H 5.37, N 5.74.
2‐Cyano‐3‐(thiophen‐2‐ylethynyl)norbornadiene (4 c): 2‐Cyano‐3‐chloronorbornadiene (120 mg, 0.79 mmol. 1 equiv.), copper(I) iodide (12.5 mg, 0.07 mmol, 9 mol %), and bis(triphenylphosphine)palladium(II) dichloride (23 mg, 0.03 mmol, 4 mol %), triethylamine (0.27 mL), and 2‐ethynylthiophene (78 mg, 0.72 mmol, 0.9 equiv.) were reacted according to the general procedure. The crude product was purified by automated column chromatography (CH2Cl2/hexane, 3:7) to provide 2‐cyano‐3‐(thiophen‐2‐ylethynyl)norbornadiene 4 c (90 mg, 0.40 mmol, 56 %) as a yellow powder. 1H NMR (CDCl3): δ=7.40 (dt, J=5.1, 1.0 Hz, 1 H), 7.34 (dt, J=3.7, 1.0 Hz, 1 H), 7.04 (ddd, J=5.1, 3.7, 0.9 Hz, 1 H), 6.90–6.84 (m, 2 H), 3.90 (dtd, J=3.0, 1.5, 0.8 Hz, 1 H), 3.86 (dtq, J=3.0, 1.6, 0.7 Hz, 1 H), 2.33–2.30 (m, 1 H), 2.22 ppm (dt, J=7.0, 1.6 Hz, 1 H); 13C NMR (CDCl3): δ=153.54, 142.31, 141.32, 133.92, 129.79, 127.79, 127.69, 121.96, 116.49, 101.17, 87.29, 72.75, 57.33, 54.27 ppm; IR: =3095.7, 2979.2, 2940.4, 2913.2, 2866.7, 2203.0, 2160.3, 1571.7, 1554.2, 1503.8, 1416.4, 1305.8, 1284.5, 1230.1, 1203.0, 1171.0, 1113.7, 1098.2, 1036.0, 1014.7, 855.6, 834.25, 820.66, 797.37, 743.04, 711.99, 688.7, 624.66, 605.25, 566.44, 506.28, 483.0, 463.58, 451.94 cm−1; elemental analysis calcd (%) for C14H9NS: C 75.31, H 4.06, N 6.27; found: C 75.39, H 3.93, N 6.07.
2‐Cyano‐3‐((4(dimethylamino)phenyl)ethynyl)norbornadiene (4 d): 2‐Cyano‐3‐chloronorbornadiene (100 mg, 0.66 mmol, 1 equiv.), copper(I) iodide (17 mg, 0.09 mmol, 14 mol %), bis(triphenylphosphine)palladium(II) dichloride (25 mg, 0.04 mmol, 6 mol %), triethylamine (0.22 mL), and 4‐ethynyl‐N,N‐dimethylaniline (105 mg, 0.73 mmol, 1.1 equiv.) were reacted according to the general procedure. The crude product was purified by automated column chromatography (CH2Cl2/hexane, 1:1) to provide cyano‐3‐((4‐(dimethylamino)phenyl)ethynyl)norbornadiene 4 d (120 mg, 0.46 mmol, 70 %) as a yellow powder. 1H NMR (CDCl3): δ=7.41–7.36 (m, 2 H), 6.89–6.81 (m, 2 H), 6.63 (dd, J=8.9, 1.8 Hz, 2 H), 3.89–3.84 (m, 1 H), 3.83–3.80 (m, 1 H), 3.01 (s, 6 H), 2.29 (dt, J=6.9, 1.6 Hz, 1 H), 2.18 ppm (dt, J=6.9, 1.7 Hz, 1 H); 13C NMR (CDCl3): δ=154.89, 151.02, 142.35, 141.18, 133.58, 124.61, 117.22, 111.75, 110.80, 108.50, 82.79, 72.34, 57.48, 53.91, 40.22 ppm; IR: =3072.4, 3002.5, 2936.5, 2979.2, 2866.7, 2203.0, 2187.4, 1598.9, 1575.6, 1556.2, 1482.4, 1435.8, 1329.1, 1286.4, 1226.3, 1203.0, 1160.3, 1117.6, 1065.2, 1010.8, 925.46, 906.05, 871.12, 830.37, 760.50, 721.69, 688.7, 628.5, 597.5, 570.32, 523.74, 471.35, 455.82 cm−1; elemental analysis calcd (%) for C18H16N2: C 83.04, H 6.19, N 10.76; found: C 83.35, H 6.32, N 10.76.
2‐Cyano‐3‐phenylnorbornadiene (5): Cesium fluoride (0.5 g, 3.3 mmol), phenylboronic acid (146 mg, 1.2 mmol), and tris(dibenzylideneacetone)dipalladium(0) (150 mg, 0.26 mmol, 26 mol %) were dissolved in dry THF (2 mL) under nitrogen. 1‐Chloro‐2‐cyanonorbornadiene (150 mg, 1 mmol) was added, followed by tri‐tert‐butylphosphine (1 m in toluene, 0.20 mL, 0.2 mmol). The mixture was heated under reflux for 24 h then allowed to cool. The product was filtered through a silica pad, which was eluted with dichloromethane until the eluate was colorless; concentration of the combined filtrate and eluate gave a red oil. The crude product was purified by automated column chromatography (CH2Cl2/hexane, 3:7) to provide 2‐cyano‐3‐phenylnorbornadiene 5 (110 mg, 0.57 mmol, 57 %) as a clear oil. 1H NMR (CDCl3): δ=7.75–7.70 (m, 2 H), 7.48–7.36 (m, 3 H), 6.96–6.92 (m, 1 H), 6.87 (ddd, J=5.1, 3.1, 0.8 Hz, 1 H), 4.13 (ddtd, J=3.3, 2.4, 1.6, 0.8 Hz, 1 H), 3.94 (ddtt, J=3.0, 2.2, 1.5, 0.7 Hz, 1 H), 2.29 (dt, J=6.9, 1.7 Hz, 1 H), 2.20 ppm (dt, J=6.9, 1.6 Hz, 1 H); 13C NMR (CDCl3): δ=170.95, 143.24, 140.45, 133.23, 130.25, 129.03, 126.58, 118.55, 117.07, 71.36, 55.13, 54.33 ppm; IR: =3056.5, 2997.7, 2939.0, 2876.1, 2192.6, 1586.8, 1559.5, 1485.1, 1440.0, 1320.5, 1226.1, 1203.0, 1161.1, 1077.3, 1031.1, 1001.8, 880.16, 819.35, 762.74, 721.1, 687.26, 662.10, 582.42, 481.77, 456.61 cm−1; elemental analysis calcd (%) for C14H11N: C 87.01, H 5.74, N 7.25; found: C 87.02, H 5.75, N 7.21.
Preparative photoisomerization (general procedure): The norbornadiene was dissolved in degassed chloroform or toluene and irradiated with a 150 W HQI lamp (Osram) for 30–50 min. The photoisomerization was confirmed by 1H and 13C NMR spectroscopies; the product was not isolated.
2‐Cyano‐3‐(phenylethynyl)quadricyclane (16 a): 2‐Cyano‐3‐(phenylethynyl)quadricyclane was obtained according to the general procedure. 1H NMR (CDCl3): δ=7.42 (ddt, J=5.3, 3.1, 1.3 Hz, 2 H), 7.31–7.27 (m, 3 H), 2.57 (ddd, J=5.0, 2.5, 1.0 Hz, 1 H), 2.45 (dd, J=12.1, 1.4 Hz, 1 H), 2.37 (ddd, J=4.9, 2.6, 1.0 Hz, 1 H), 2.32 (dt, J=4.9, 1.4 Hz, 1 H), 2.21 (dd, J=12.1, 1.4 Hz, 1 H), 2.09 ppm (dt, J=4.8, 1.4 Hz, 1 H); 13C NMR (500 MHz, CDCl3): δ=131.81, 129.69, 128.38, 128.28, 123.04, 122.14, 118.22, 85.79, 83.99, 83.27, 33.54, 32.37, 25.65, 25.06, 18.43, 14.80 ppm.
2‐Cyano‐3‐((4‐methoxyphenyl)ethynyl)quadricyclane (16 b): 2‐Cyano‐3‐((4‐methoxyphenyl)ethynyl)quadricyclane was obtained according to the general procedure. 1H NMR (CDCl3): δ=7.38–7.34 (m, 2 H), 6.84–6.80 (m, 2 H), 3.80 (s, 3 H), 2.56 (dd, J=5.0, 2.6 Hz, 1 H), 2.45 (dt, J=12.0, 1.4 Hz, 1 H), 2.34 (dd, J=4.9, 2.6 Hz, 1 H), 2.31 (dq, J=5.0, 1.4 Hz, 1 H), 2.19 (dt, J=12.1, 1.5 Hz, 1 H), 2.06 ppm (dq, J=4.9, 1.4 Hz, 1 H). 13C NMR (500 MHz, CDCl3): δ=159.69, 133.36, 118.34, 115.15, 114.04, 85.61, 82.40, 55.42, 33.59, 32.36, 32.22, 25.64, 24.86, 18.52, 14.85 ppm.
2‐Cyano‐3‐(thiophen‐2‐ylethynyl)quadricyclane (16 c): 2‐Cyano‐3‐(thiophen‐2‐ylethynyl)quadricyclane was obtained according to the general procedure. 1H NMR (CDCl3): δ=7.23 (ddd, J=5.2, 1.2, 0.3 Hz, 1 H), 7.20 (ddd, J=3.6, 1.2, 0.3 Hz, 1 H), 6.95 (ddd, J=5.2, 3.7, 0.3 Hz, 1 H), 2.58 (dd, J=5.0, 2.5 Hz, 1 H), 2.46 (dt, J=12.1, 1.4 Hz, 1 H), 2.40 (dd, J=4.9, 2.6 Hz, 1 H), 2.33 (dq, J=5.0, 1.4 Hz, 1 H), 2.21 (dt, J=12.1, 1.5 Hz, 1 H), 2.11 ppm (dq, J=4.9, 1.4 Hz, 1 H); 13C NMR (500 MHz, CDCl3): δ=123.59, 127.31, 127.05, 123.14, 118.12, 110.16, 88.01, 78.58, 33.53, 32.78, 32.36, 25.56, 18.50, 14.83 ppm.
2‐Cyano‐3‐((4‐(dimethylamino)phenyl)ethynyl)quadricyclane (16 d): 2‐Cyano‐3‐((4‐(dimethylamino)phenyl)ethynyl)quadricyclane was obtained according to the general procedure. 1H NMR (CDCl3): δ=7.32–7.29 (m, 2 H), 6.63–6.58 (m, 2 H), 2.96 (s, 3 H), 2.54 (dd, J=5.0, 2.5 Hz, 1 H), 2.44 (dt, J=12.0, 1.4 Hz, 1 H), 2.33–2.28 (m, 2 H), 2.18 (dt, J=12.0, 1.5 Hz, 1 H), 2.04 ppm (dq, J=5.0, 1.5 Hz, 1 H); 13C NMR (500 MHz, CDCl3): δ=133.66, 118.02, 112.57, 87.80, 82.13, 40.00, 33.31, 32.38, 32.20, 25.16, 24.88, 19.47, 15.78 ppm.
2‐Cyano‐3‐phenylquadricyclane (17): 2‐Cyano‐3‐phenylquadricyclane was obtained according to the general procedure. 1H NMR (CDCl3): δ=7.40–7.32 (m, 2 H), 7.30–7.22 (m, 3 H), 2.68 (dt, J=4.9, 2.4 Hz, 1 H), 2.48 (dt, J=11.9, 1.3 Hz, 1 H), 2.42 (dq, J=5.0, 1.3 Hz, 1 H), 2.32–2.24 (m, 2 H), 1.94 ppm (dq, J=5.1, 1.4 Hz, 1 H); 13C NMR (CDCl3): δ=136.02, 128.65, 126.61, 126.09, 119.62, 35.39, 32.40, 31.85, 31.25, 26.77, 22.21, 14.33 ppm.
Calculations
Regular and time‐dependent density functional theory (TD‐DFT) calculations were carried out at the B3LYP/6–311+G* level29 for all compounds. Based on a systematic comparison of different exchange‐correlation functionals with MP2 as well as complete active space calculations,10 we have previously established that the B3LYP functional yields absorption spectra and geometries in good agreement with both experiment and higher‐level calculations (Figure 2).
For compound 4 b, we consider the two isomers (rotamers) that result from 180° rotations of the aryl group with respect to the bridging oxygen atom. The spectra were subsequently obtained by averaging over the isomers weighted by their respective Boltzmann factors.15 All calculations were carried out using the NWChem 6.5 software package.31
The molar attenuation coefficient ( ) was obtained from the calculated oscillator strengths ( ) and energies ( ) of vertical excitations at minimum geometry according to:
where ) is a normalized Gaussian function. An artificial broadening of 0.2 eV was employed to simulate the effect of vibrations.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
The authors would like to thank the Swedish Research Council, the Knut and Alice Wallenberg Foundation, and the Swedish Foundation for Strategic Research for financial support.
M. Quant, A. Lennartson, A. Dreos, M. Kuisma, P. Erhart, K. Börjesson, K. Moth-Poulsen, Chem. Eur. J. 2016, 22, 13265.
References
- 1.
- 1a. Weigert F., Jahrbuch fur Photographie, Kinematographie und Reproduktionsverfahren 1909, 109; [Google Scholar]
- 1b. Scharf H. D., Fleischhauer J., Leismann H., Ressler I., Schleker W., Weitz R., Angew. Chem. Int. Ed. Engl. 1979, 18, 652–662; [Google Scholar]; Angew. Chem. 1979, 91, 696–707; [Google Scholar]
- 1c. Yoshida Z. I., J. Photochem. 1985, 29, 27–40; [Google Scholar]
- 1d. Kucharski T. J., Tian Y., Akbulatov S., Boulatov R., Energy Environ. Sci. 2011, 4, 4449–4472; [Google Scholar]
- 1e. Moth-Poulsen K. in Organic Synthesis and Molecular Engineering, Wiley, Hoboken: 2014, pp. 179–196; [Google Scholar]
- 1f. Lennartson A., Roffey A., Moth-Poulsen K., Tetrahedron Lett. 2015, 56, 1457–1465; [Google Scholar]
- 1g. Dinda M., Chakraborty S., Kanti Si M., Samanta S., Ganguly B., Maiti S., Ghosh P. K., RSC Adv. 2014, 4, 54558–54564; [Google Scholar]
- 1h. Zhitomirsky D., Cho E., Grossman J. C., Adv. Energy Mater. 2016, 6, n/a. [Google Scholar]
- 2. Moth-Poulsen K., Coso D., Börjesson K., Vinokurov N., Meier S. K., Majumdar A., Vollhardt K. P. C., Segalman R. A., Energy Environ. Sci. 2012, 5, 8534–8537. [Google Scholar]
- 3.
- 3a. Jones G., Reinhardt T. E., Bergmark W. R., Sol. Energy 1978, 20, 241–248; [Google Scholar]
- 3b. Fritzsche, J. Prakt. Chem. 1867, 101, 333–343; [Google Scholar]
- 3c. Weigert F., Ber. Dtsch. Chem. Ges. 1909, 42, 850–862. [Google Scholar]
- 4.
- 4a. Caia V., Cum G., Gallo R., Mancini V., Pitoni E., Tetrahedron Lett. 1983, 24, 3903–3904; [Google Scholar]
- 4b. Bastianelli C., Caia V., Cum G., Gallo R., Mancini V., J. Chem. Soc. Perkin Trans. 2 1991, 679–683; [Google Scholar]
- 4c. Stoermer R., Ber. Dtsch. Chem. Ges. 1909, 42, 4865–4871. [Google Scholar]
- 5.
- 5a. Bandara H. M. D., Burdette S. C., Chem. Soc. Rev. 2012, 41, 1809–1825; [DOI] [PubMed] [Google Scholar]
- 5b. Slavov C., Yang C., Schweighauser L., Boumrifak C., Dreuw A., Wegner H. A., Wachtveitl J., Phys. Chem. Chem. Phys. 2016, 18, 14795–14804; [DOI] [PubMed] [Google Scholar]
- 5c. Masutani K., Morikawa M.-a., Kimizuka N., Chem. Commun. 2014, 50, 15803–15806; [DOI] [PubMed] [Google Scholar]
- 5d. Hartley G. S., Nature 1937, 140, 281; [Google Scholar]
- 5e. Hartley G. S., J. Chem. Soc. 1938, 633–642; [Google Scholar]
- 5f. Adamson A. W., Vogler A., Kunkely H., Wachter R., J. Am. Chem. Soc. 1978, 100, 1298–1300. [Google Scholar]
- 6.
- 6a. Broman S. L., Petersen M. A., Tortzen C. G., Kadziola A., Kilsaa K., Nielsen M. B., J. Am. Chem. Soc. 2010, 132, 9165–9174; [DOI] [PubMed] [Google Scholar]
- 6b. Broman S. L., Nielsen M. B., Phys. Chem. Chem. Phys. 2014, 16, 21172–21182. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Vollhardt K. P. C., Weidman T. W., J. Am. Chem. Soc. 1983, 105, 1676–1677; [Google Scholar]
- 7b. Boese R., Cammack J. K., Matzger A. J., Pflug K., Tolman W. B., Vollhardt K. P. C., Weidman T. W., J. Am. Chem. Soc. 1997, 119, 6757–6773; [Google Scholar]
- 7c. Harpham M. R., Nguyen S. C., Hou Z., Grossman J. C., Harris C. B., Mara M. W., Stickrath A. B., Kanai Y., Kolpak A. M., Lee D., Liu D.-J., Lomont J. P., Moth-Poulsen K., Vinokurov N., Chen L. X., Vollhardt K. P. C., Angew. Chem. Int. Ed. 2012, 51, 7692–7696; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 7812–7816; [Google Scholar]
- 7d. Lennartson A., Lundin A., Boerjesson K., Gray V., Moth-Poulsen K., Dalton Trans. 2016, 45, 8740–8744. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Hammond G. S., Turro N. J., Fischer A., J. Am. Chem. Soc. 1961, 83, 4674; [Google Scholar]
- 8b. Cristol S. J., Snell R. L., J. Am. Chem. Soc. 1958, 80, 1950–1952; [Google Scholar]
- 8c. Dauben W. G., Cargill R. L., Tetrahedron 1961, 15, 197; [Google Scholar]
- 8d. Bren V. A. D., Minkin A. D., Minkin V. I., Chernoivanov V. A., Russ. Chem. Rev. 1991, 60, 451–469; [Google Scholar]
- 8e. Dubonosov A. D., Bren V. A., Chernoivanov V. A., Russ. Chem. Rev. 2002, 71, 917–927; [Google Scholar]
- 8f. Brummel O., Besold D., Doepper T., Wu Y., Bochmann S., Lazzari F., Waidhas F., Bauer U., Bachmann P., Papp C., Steinrueck H.-P., Goerling A., Libuda J., Bachmann J., ChemSusChem 2016, 9, 1424–1432. [DOI] [PubMed] [Google Scholar]
- 9. Börjesson K., Lennartson A., Moth-Poulsen K., ACS Sustainable Chem. Eng. 2013, 1, 585–590. [Google Scholar]
- 10. Kuisma M. J., Lundin A. M., Moth-Poulsen K., Hyldgaard P., Erhart P., J. Phys. Chem. C 2016, 120, 3635–3645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gray V., Lennartson A., Ratanalert P., Boerjesson K., Moth-Poulsen K., Chem. Commun. 2014, 50, 5330–5332. [DOI] [PubMed] [Google Scholar]
- 12. Zhou Y., Miki S., Yoshida Z., Chem. Express 1989, 4, 53–56. [Google Scholar]
- 13. Harel Y., Adamson A. W., Kutal C., Grutsch P. A., Yasufuku K., J. Phys. Chem. 1987, 91, 901–904. [Google Scholar]
- 14. Lainé P., Marvaud V., Gourdon A., Launay J. P., Argazzi R., Bignozzi C. A., Inorg. Chem. 1996, 35, 711–714. [Google Scholar]
- 15. Kuisma M., Lundin A., Moth-Poulsen K., Hyldgård P., Erhart P., ChemSusChem 2016, 9, 1786–1794. [DOI] [PubMed] [Google Scholar]
- 16. Babudri F., Bilancia G., Cardone A., Coppo P., De Cola L., Farinola G. M., Hofstraat J. W., Naso F., Photochem. Photobiol. Sci. 2007, 6, 361–364. [DOI] [PubMed] [Google Scholar]
- 17. Fraysse S., Coudret C., Launay J.-P., Eur. J. Inorg. Chem. 2000, 1581–1590. [Google Scholar]
- 18. Dalkilic E., Dastan A., Tetrahedron 2015, 71, 1966–1970. [Google Scholar]
- 19. Sonogashira K., Tohda Y., Hagihara N., Tetrahedron Lett. 1975, 16, 4467–4470. [Google Scholar]
- 20. Suzuki A., J. Organomet. Chem. 1999, 576, 147–168. [Google Scholar]
- 21.
- 21a. Josey A. D., C. L. Dickinson, Jr. , Dewhirst K. C., McKusick B. C., J. Org. Chem. 1967, 32, 1941–1944; [Google Scholar]
- 21b. Moore J. A., Mehta P. G., Macromolecules 1993, 26, 916–920; [Google Scholar]
- 21c. Friedrich K., Bechtold K., Willin L., Fritz H., J. Prakt. Chem. 2000, 342, 819–824. [Google Scholar]
- 22. Gunes Y., Arcelik N., Sahin E., Fleming F. F., Altundas R., Eur. J. Org. Chem. 2015, 6679–6686. [Google Scholar]
- 23. Lennartson A., Quant M., Moth-Poulsen K., Synlett 2015, 1501–1504. [Google Scholar]
- 24. Durr R., Cossu S., De Lucchi O., Synth. Commun. 1997, 27, 1369–1372. [Google Scholar]
- 25.
- 25a. Ryan J. H., Stang P. J., J. Org. Chem. 1996, 61, 6162–6165; [DOI] [PubMed] [Google Scholar]
- 25b. Stang P. J., Blume T., Zhdankin V. V., Synthesis 1993, 35–36. [Google Scholar]
- 26. Tsui G. C., Le Marquand P., Allen A., Tam W., Synthesis 2009, 609–619. [Google Scholar]
- 27. Tranmer G. K., Tam W., Synthesis 2002, 1675–1682. [Google Scholar]
- 28.
- 28a. Parker C. A., Proc. R. Soc. London Ser. A 1953, 220, 104–116; [Google Scholar]
- 28b. Hatchard C. G., Parker C. A., Proc. R. Soc. London Ser. A 1956, 235, 518–536. [Google Scholar]
- 29.
- 29a. Becke A. D., J. Chem. Phys. 1993, 98, 5648–5652; [Google Scholar]
- 29b. McLean A. D., Chandler G. S., J. Chem. Phys. 1980, 72, 5639–5648; [Google Scholar]
- 29c. Krishnan R., Binkley J. S., Seeger R., Pople J. A., J. Chem. Phys. 1980, 72, 650–654; [Google Scholar]
- 29d. Lee C., Yang W., Parr R. G., Phys. Rev. B 1988, 37, 785–789. [DOI] [PubMed] [Google Scholar]
- 30. Handbook of Chemistry and Physics, CRC, Boca Raton, 2015, p. 6–145. [Google Scholar]
- 31. Valiev M., Bylaska E. J., Govind N., Kowalski K., Straatsma T. P., Van Dam H. J. J., Wang D., Nieplocha J., Apra E., Windus T. L., de Jong W. A., Comput. Phys. Commun. 2010, 181, 1477–1489. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary