

Abstract
The amyloid beta peptide 42 (Aβ42) is an aggregation‐prone peptide that plays a pivotal role in Alzheimer′s disease. We report that a subtle perturbation to the peptide through a single chirality change at glutamate 22 leads to a pronounced delay in the β‐sheet adoption of the peptide. This was accompanied by an attenuated propensity of the peptide to form fibrils, which was correlated with changes at the level of the fibrillary architecture. Strikingly, the incorporation of d‐glutamate was found to stabilize a soluble, ordered macromolecular assembly with enhanced cytotoxicity to PC12 cells, highlighting the importance of advanced prefibrillary Aβ aggregates in neurotoxicity.
Keywords: aggregation, Alzheimer′s disease, amyloid beta peptide, chirality, neurotoxicity
Alzheimer′s disease (AD) is a major neurodegenerative disorder that affects over 35 million people worldwide.1 Reflecting the increase in life expectancy, these numbers continue to rise, while no cure exists.2 Amyloid β (Aβ) is an aggregation‐prone peptide of 36–43 amino acids in length and has been strongly implicated in the mechanism of AD.3 The Aβ42 peptide is widely regarded as the most toxic Aβ entity in AD, which has been attributed to its high aggregation propensity.4, 5 The aggregation profile is complex, with diverse oligomeric, pre‐fibrillary, and fibrillary states being formed. Over the past decade, diffusible oligomers have been recognized as particularly neurotoxic species.4, 5
Familial AD can arise from diverse mutations within the Aβ42 sequence.6 Over ten Aβ42 mutations have been identified, most of which are disease‐causing single amino acid alterations. Strikingly, from those mutations, four AD‐accelerating variants are positioned on one specific amino acid—glutamate 22 (E22)—which identifies the residue as particularly important in the context of Aβ42 neurotoxicity. The four E22‐borne familial mutations have in common that they alter the charge at that residue, either through amino‐acid substitution (E22G, Arctic, G=glycine; E22K, Italian, K=lysine; E22Q, Dutch, Q=glutamine), or amino‐acid deletion (E22Δ, Osaka).6 Biophysical experiments demonstrated that those substitutions enhance the Aβ propensity towards oligomer,7 or fibril formation.8 To further examine the role of residue 22 of Aβ42 on structure and function of the peptide, we have created the E22e chiral mutant 2 (e=d‐glutamate). This subtle molecular edit enables for an alteration of the sidechain disposition of the peptide without affecting its physical properties, such as size, charge distribution, and polarizability (Figure 1).
Figure 1.

The Aβ sequence with glutamate 22 highlighted in blue. The differences between Aβ40 and Aβ42 are highlighted in red. Replacement of l‐glutamate with d‐glutamate at position 22 enables for a subtle alteration of the sidechain disposition.
We studied the effect of the introduction of d‐glutamate at position 22 on the aggregation propensity of Aβ42 by conducting thioflavin T binding experiments (Figure 2 A). Remarkably, the chiral E22e mutant 2 exhibited a fivefold reduction in the fibril‐formation rate compared to the wildtype (WT) peptide 1 (t 1/2[E22e]=65.6 min; t 1/2[WT]=13.4 min). The rate of the fibril formation of 1 was comparable to those previously reported in the literature.9, 10, 11 The aggregation ability of the Aβ42 peptide is believed to stem from its propensity to undertake a secondary structural transition from a random‐coil‐like structure to a β‐sheet configuration.3 We therefore examined the time‐resolved circular dichroism spectra of the peptides 1 and 2 over a period of 24 h. In agreement with the thioflavin T binding results, a delay in the random coil to β‐sheet configuration of the Aβ E22e peptide 2 was observed (see Supporting Information). These results demonstrate a reduced propensity of the peptide 2 for aggregation at the fibrillary endpoint, as well as at prefibrillary stages.
Figure 2.
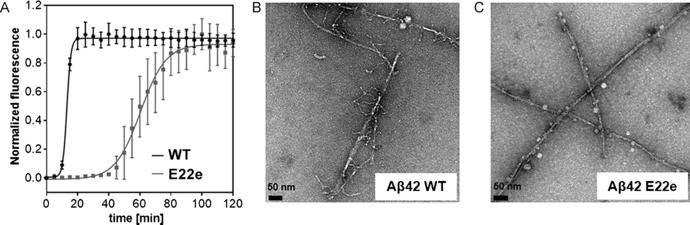
A) Aggregation kinetics of the Aβ42 wildtype peptide 1 (black) and Aβ42 E22e peptide 2 (grey) at 20 μm, monitored by the Thioflavin T (ThT) fluorescence (λ em=444 nm, λ ex=485 nm) at 37 °C. B, C) Representative transmission electron microscopy (TEM) images of the fibrillary architectures of the Aβ42 wildtype peptide 1 (B) and the Aβ42 E22e peptide 2 (C). The samples were incubated in phosphate buffer (20 mm, pH=7.4) at 222 μm before being diluted to 200 nm for imaging.
The delayed aggregation kinetics of the E22e peptide 2 led us to investigate whether the fibrillary assemblies of 2 were altered compared to 1. To do this, both Aβ42 WT 1 and Aβ42 E22e 2 fibrils were grown for 7 days at 37 °C following protocols by Tycko et al (see Supporting Information for details).9 Transmission electron microscopy (TEM) images of the wildtype Aβ42 fibrils (Figure 2 B) showed a distinct fibrillary architecture, characterized by the presence of numerous branches extending from the main fibril. This is of particular interest, given recent reports suggesting that the Aβ42‐fibril formation is a secondary nucleation‐dependent process.10 In contrast, peptide 2 displayed more elongated, organized fibrillary structures devoid of branches (Figure 2 C). Analogous TEM experiments were conducted, following an incubation of the peptides 1 and 2 for 2 h. The results were consistent in terms of branching, which was observable for Aβ42 WT, but not for the E22e chiral variant (see Supporting Information for images and further details).
The difference in the fibrillary morphologies between the peptides 1 and 2 led us to further investigate whether alterations in the prefibrillary structural assemblies could account for the striking differences. Photochemically induced crosslinking of unmodified proteins (PICUP) experiments were carried out to gain insight into the distribution of the oligomeric states.12 Comparative analyses of the wildtype 1 and the E22e Aβ42 peptide 2 were conducted at two time points, either immediately upon reconstitution or following an incubation for 24 h. The oligomerization profiles of the two scaffolds 1 and 2 showed no statistically significant difference in the population states of the oligomers (dimer–heptamer), indicating that any differences in the fibrillary assembly of the two peptides occurred at more advanced stages of the aggregation process (Figure 3 A, B, see Supporting Information for details). To investigate these late‐stage prefibrillary structures we employed small‐angle X‐ray scattering (SAXS) analysis. SAXS has been shown to be a powerful technique for monitoring amyloid‐related structural features.13 We examined the SAXS curves of both wildtype 1 and E22e Aβ42 peptide 2 after initial reconstitution and following 24 h incubation at 37 °C (Figure 3 C, D). For both time points, SAXS analysis of the peptide 2 demonstrated a Bragg reflection corresponding to a species with a periodicity of 3.7 nm. This value is consistent with the dimensions of a single unit of the wildtype Aβ42 sequence found within a fibril using NMR and in silico structural models.14 No Bragg reflection and an increase in the heterogeneity of the sample was observed for peptide 1, reflected by the large variance at high Q values.
Figure 3.
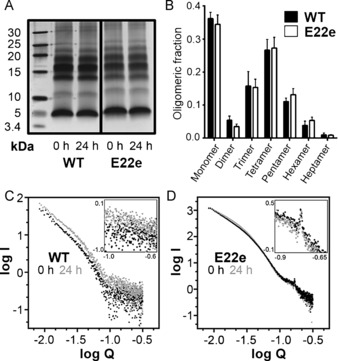
All experiments were carried out in phosphate buffer (20 mm, pH=7.4) A) Representative PICUP (photochemically induced crosslinking of unmodified proteins) gels at both t=0 h and t=24 h. All PICUP experiments were carried out in phosphate buffer at 50 μm, either directly after reconstitution, or following incubation for 24 h. Corresponding experiments were also performed at 20 μm (see the Supporting Information). B) Densitometric analysis of oligomeric band intensity at t=0 h, (see Supporting Information for t=24 h). C) Small‐angle X‐ray scattering (SAXS) measurements of the Aβ42 wildtype peptide 1 at t=0 h (black) and t=24 h (grey). D) SAXS measurements of the Aβ42 E22e peptide 2 at t=0 h (black) and t=24 h (grey).
The mechanism underlying the toxicity of Aβ42 remains a subject of active research. Diverse modes of cytotoxicity have been proposed, including membrane disruption, induction of tau hyperphosphorylation, oxidative stress mediated through copper complexation, brain insulin resistance/signaling, and mitochondrial toxicity.15 The original (fibril‐centric) amyloid‐cascade hypothesis was reformulated when diffusible Aβ42 oligomers emerged as the more toxic species.4c To test whether the prefibrillary stabilized structure of peptide 2 exhibited an increase in cytotoxicity, we monitored the effects of varying concentrations of the wildtype peptide 1 and the E22e peptide 2 on rat pheochromocytoma PC12 cells (Figure 4). Addition of either peptide resulted in a reduction in the cellular viability, determined by the cell proliferation reagent WST‐1. At 20 μm, a 30 % reduction in the cellular viability was observed when dosing with the wildtype peptide 1 (Figure 4, WT). However, addition of the same concentration of the E22e peptide 2 resulted in an 80 % reduction in the viability (Figure 4, E22e). The cellular viability of the PC12 cells was also found to be lower when peptide 2 was dosed at 10 μm, with a reduction in the cellular viability close to 65 %, compared with a 20 % reduction when dosing with the same concentration of peptide 1 (for detailed graphical analysis see Supporting Information). Preincubation (2 h or 4 h) of the peptides prior to administration did not affect their cytotoxicity (see Supporting Information). Our E22e variant offers a unique way of trapping an advanced aggregation intermediate of Aβ42 with enhanced toxicity, and highlights how a subtle structural change—a single chiral substitution—can have profound effects on aggregation and neurotoxicity.
Figure 4.
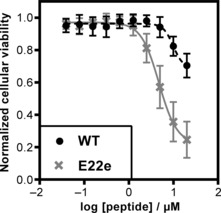
Dose‐response curves of both wildtype Aβ42 1 (black) and Aβ42 E22e 2 (grey) peptides against the rat pheochromocytoma PC12 adhesive cell line. Cells were plated at 5000 cells per well and allowed to adhere for 24 h prior to peptide addition, followed by incubation for additional 72 h. The cellular viability was determined at the endpoint of the assay using the cell proliferation reagent WST‐1.
In conclusion, incorporation of d‐glutamate at position 22 of Aβ42 resulted in a peptide with attenuated propensity for misfolding and aggregation. Transmission electron microscopy showed a striking difference in the fibril morphology. The E22e peptide 2 exhibited elongated, ordered amyloid‐beta fibrils. This is in stark contrast to the Aβ42 WT peptide 1, which displayed a fibrillary architecture, characterized by the presence of a large number of sidechains protruding from the main fibril. No difference in the population density of the oligomers (dimer–heptamer) between the two peptides was observed. However, SAXS analysis of the E22e peptide 2 showed the presence of a unique Bragg reflection corresponding to a soluble species with a periodicity of 3.7 nm. Cell culture studies established a three‐ to fourfold increase in the cytotoxicity in response to the E22e substitution in Aβ42. This subtle molecular edit therewith offers a tool to improve our understanding of the Aβ42 neurotoxicity.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We are grateful to Prof. Maya Koronyo‐Hamaoui, Prof. Glenn Millhauser and Prof. James Nowick for helpful discussions, to Prof. David Kliger for his assistance in interpretation of CD spectra, as well as to Rafael Palomino and Kate Markham from the Millhauser laboratory for their help with initial Aβ42 syntheses. Victoria Klein is acknowledged for her assistance with initial gel electrophoresis experiments. We acknowledge the NIH S10OD016246‐01A1 award for purchase of the JASCO J1500 CD.
C. J. A. Warner, S. Dutta, A. R. Foley, J. A. Raskatov, Chem. Eur. J. 2016, 22, 11967.
References
- 1. Querfurth H. W., LaFerla F. M., N. Engl. J. Med. 2010, 362, 329–344. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Hardy J., Selkoe D. J., Science 2002, 297, 353–356; [DOI] [PubMed] [Google Scholar]
- 2b. Selkoe D. J., Nat. Med. 2011, 17, 1060–1065. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Chiti F., Dobson C. M., Annu. Rev. Biochem. 2006, 75, 333–366; [DOI] [PubMed] [Google Scholar]
- 3b. Masters C. L., Weinman N. A., Multhaup G., McDonald B. L., Beyreuther K., Proc. Natl. Acad. Sci. USA 1985, 82, 4245–4249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.
- 4a. Gong Y. S., Chang L., Viola K. L., Lacor P. N., Lambert M. P., Finch C. E., Krafft G. A., Klein W. L., Proc. Natl. Acad. Sci. USA 2003, 100, 10417–10422; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4b. Lacor P. N., Buniel M. C., Chang L., Fernandez S. J., Gong Y. S., Viola K. L., Lambert M. P., Velasco P. T., Bigio E. H., Finch C. E., Krafft G. A., Klein W. L., J. Neurosci. 2004, 24, 10191–10200; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4c. Haass C., Selkoe D. J., Nat. Rev. Mol. Cell. Bio. 2007, 8, 101–112; [DOI] [PubMed] [Google Scholar]
- 4d. Selkoe D. J., Behav. Brain Res. 2008, 192, 106–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.
- 5a. Lesné S., Koh M. T., Kotilinek L., Kayed R., Glabe C. G., Yang A., Gallagher M., Ashe K. H., Nature 2006, 440, 352–357; [DOI] [PubMed] [Google Scholar]
- 5b. Haupt C., Leppert J., Röenicke R., Meinhardt J., Yadav J. K., Ramachandran R., Ohlenscläger O., Reymann K. G., Görlach M., Fändrich M., Angew. Chem. Int. Ed. 2012, 51, 1576–1579; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 1608–1611. [Google Scholar]
- 6. Benilova I., Karran E., De Strooper B., Nat. Neurosci. 2012, 15, 349–357. [DOI] [PubMed] [Google Scholar]
- 7. Nilsberth C., Westlind-Danielsson A., Eckman C. B., Condron M. M., Axelman K., Forsell C., Stenh C., Luthman J., Teplow D. B., Younkin S. G., Näslund J., Lannfelt L., Nat. Neurosci. 2001, 4, 887–893. [DOI] [PubMed] [Google Scholar]
- 8. Baumketner A., Krone M. G., Shea J. E., Proc. Natl. Acad. Sci. USA 2008, 105, 6027–6032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Petkova A. T., Leapman R. D., Guo Z., Yau W.-M., Mattson M. P., Tycko R., Science 2005, 307, 262–265. [DOI] [PubMed] [Google Scholar]
- 10. Cohen S. I. A., Linse S., Luheshi L. M., Hellstrand E., White D. A., Rajah L., Otzen D. E., Vendruscolo M., Dobson C. M., Knowles T. P. J., Proc. Natl. Acad. Sci. USA 2013, 110, 9758–9763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Finder V. H., Vodopivec I., Nitsch R. M., Glockshuber R., J. Mol. Biol. 2010, 396, 9–18. [DOI] [PubMed] [Google Scholar]
- 12.
- 12a. Bitan G., Lomakin A., Teplow D. B., J. Biol. Chem. 2001, 276, 35176–35184; [DOI] [PubMed] [Google Scholar]
- 12b. Bitan G., Kirkitadze M. D., Lomakin A., Vollers S. S., Benedek G. B., Teplow D. B., Proc. Natl. Acad. Sci. USA 2003, 100, 330–335; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12c. Bitan G., Teplow D. B., Acc. Chem. Res. 2004, 37, 357–364. [DOI] [PubMed] [Google Scholar]
- 13. Langklide A. E., Vestergaard B., FEBS Lett. 2009, 583, 2600–2609. [DOI] [PubMed] [Google Scholar]
- 14.
- 14a. Coles M., Bicknell W., Watson A. A., Fairlie D. P., Craik D. J., Biochemistry 1998, 37, 11064–11077; [DOI] [PubMed] [Google Scholar]
- 14b. Lührs T., Ritter C., Adrian M., Riek-Loher D., Bohrmann B., Döbeli H., Schubert D., Riek R., Proc. Natl. Acad. Sci. USA 2005, 102, 17342–17347; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14c. Qiang W., Yau W. M., Luo Y., Mattson M. P., Tycko R., Proc. Natl. Acad. Sci. USA 2012, 109, 4443–4448; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14d. Xiao Y., Ma B., Parthasarathy S., Long F., Hoshi M., Nussinov R., Ishii Y., Nat. Struct. Mol. Biol. 2015, 22, 499–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.
- 15a. Ittner L. M., Götz J., Nat. Rev. Neurosci. 2011, 12, 67–72; [DOI] [PubMed] [Google Scholar]
- 15b. Hung Y. H., Bush A. I., Cherny R. A., J. Biol. Inorg. Chem. 2010, 15, 61–76. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary