

Abstract
Combining agents has the potential to attenuate resistance in metastatic cancer. However, knowledge of appropriate starting doses for novel drug combinations in clinical trials and practice is lacking. Analysis of 372 published studies was used to ascertain safe starting doses for doublets involving a cytotoxic and targeted agent. Phase I–III adult oncology clinical trial publications (January 1, 2010 to December 31, 2013) were identified (PubMed). The dose of drug used in each combination was compared to the single agent recommended dose [FDA‐approved/recommended phase 2 dose (RP2D)/maximum tolerated dose (MTD)]. Dose percentages were calculated as: (safe dose of drug in combination/dose of drug as single agent at FDA/RP2D/MTD) × 100. Additive dose percentages were the sum of the dose percentage for each drug. A total of 24,326 patients (248 drug combinations) were analyzed. In 38% of studies, both drugs could be administered at 100% of their FDA‐approved/RP2D/MTD dose. The lowest safe additive dose percentage was 41% with poly‐ADP ribose polymerase (PARP) or histone deacetylase inhibitors as the targeted agents; 82%, in the absence of these agents; and 97%, with an antibody in the combination. If one drug was administered at 100% of the single agent dose, the lowest safe dose percentage for the second drug was 17% (cytotoxic at 100%) or 36% (targeted at 100%) of the FDA‐approved/RP2D/MTD dose. The current findings can help inform safe starting doses for novel two‐drug combinations (cytotoxic and targeted agents) in the context of clinical trials and practice.
Keywords: targeted therapy, cytotoxic chemotherapy, maximum tolerated dose, recommended Phase 2 dose, precision medicine
Short abstract
What's new?
Cytotoxic and targeted cancer drugs act through distinct mechanisms, and when used in combination they can potentially augment therapeutic effectiveness while minimally impacting toxicity. However, whereas algorithms for safe starting doses for new single‐agent therapies are well established, there are few guidelines for combination therapies. Here, analyses of data from published Phase I–III clinical trials shows that about 38% of patients tolerated combinations in which both drugs were administered at full starting doses. In the majority of patients, significant dose reductions were required to guard against toxicity. Intrapatient dose escalation is possible, however, potentially allowing for increased efficacy.
Metastatic carcinomas have complicated and heterogenous molecular and biologic landscapes.1 Biologic heterogeneity exists between histologies, within the same diagnostic group, and even within individual patients.2, 3, 4, 5, 6 As an example, The Cancer Genome Atlas Study identified numerous genes that can be altered in carcinomas. Most malignancies have several alterations, which can vary by tumor type.3 Hence, tailored targeted therapies are emerging as an important modality for treating metastatic malignancies. However, experience suggests that targeted monotherapy is unlikely to eradicate metastatic carcinomas or even result in prolonged remissions.
Cytotoxic chemotherapy has been the mainstay of established therapeutic regimens for many tumors, with improvement in outcomes having been demonstrated in randomized clinical trials. Furthermore, combination treatment approaches have dramatically changed outcomes for numerous malignancies, with high cure rates seen in Hodgkin's lymphoma, acute lymphocytic leukemia and testicular cancer.7 Combinations of cytotoxic chemotherapy and targeted agents are therefore being increasingly pursued.
Determining safe starting doses for therapeutic combinations of anticancer drugs in clinical trials and practice can be challenging. While drugs are combined routinely and safely outside of the cancer field based on algorithms for patients with multiple comorbidities and the average cancer patient is on a median of eight drugs for other health problems prior to starting treatment for malignancy,8 oncology drugs may require a more cautious approach due to the narrower therapeutic windows for many cytotoxic chemotherapeutic agents.9 Given that over 300 anticancer drugs are approved or in advanced clinical trials, there are over 45,000 possible two‐drug combinations. It is not feasible to conduct a clinical trial for every possible drug combination, dose and cancer type. The current analysis therefore evaluated previously published oncology clinical trials of two drug combinations in which a targeted agent and a cytotoxic were included to determine the lowest safe starting doses required for de novo combinations in clinical trials and practice.
Material and Methods
Identification of publications
Phase I–III oncology clinical trials of two‐drug combination therapy where one agent was targeted and one was a cytotoxic agent were identified by PubMed search for “cancer, phase, combination or combined” among “clinical trials.” Studies were limited to those published between January 1, 2010, and December 31, 2013. Targeted agents are often cytostatic and designed to impact signals in the cancer cell that are differentially expressed or abnormal as compared to those in normal tissue. They include antibodies targeting a specific protein or small molecule inhibitors with a low nM IC50 for a specific protein (concentration required to produce 50% inhibition of enzyme function). Cytotoxic agents are defined as chemical substances that kill rapidly dividing cells, but have the potential to harm rapidly dividing healthy tissue including bone marrow, the gastrointestinal tract and hair follicles. Studies involving hormonal therapy, immunotherapy, radiation, special populations such as pediatric, elderly or organ dysfunction patients, and those administering the dose of drug in the combination >100% of the standard dose of the agent were excluded from the analysis (Fig. 1).
Figure 1.
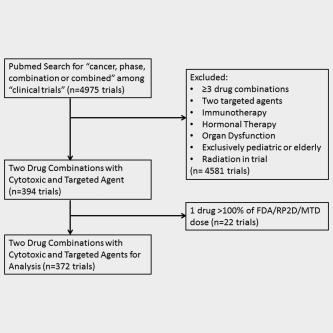
Consort diagram. Articles were identified by PubMed search and screened to identify two drug combinations with a cytotoxic and targeted agent. Studies of immunotherapy, radiation, organ dysfunction, pediatric patients, elderly patients, or those where one drug was given at greater than the FDA‐approved/RP2D/MTD dose were excluded from the analysis.
Data analysis
The published manuscripts were reviewed to determine drugs used in the combination, disease evaluated, number of participants, dose of each drug in the combination, recommended phase 2 dose (RP2D) or maximum tolerated dose (MTD) achieved in the study for Phase I trials, (or Food and Drug Administration recommended dose for approved drugs), dose limiting toxicities and Grade 3 or 4 toxicities. For trials that tested more than one drug combination, each combination was considered a separate study in the analysis. A “dose percentage” was calculated for each drug in the combination. The dose of drug was compared to the single agent recommended dose which was defined as either the FDA‐approved dose or for non‐FDA‐approved drugs, either the RP2D or the MTD. RP2D was given preference over MTD, and FDA‐approved dose was given highest priority.
Dose percentage was calculated as follows: (safe dose of drug in combination/dose of drug as a single agent at FDA‐approved dose or RP2D or MTD) × 100. Additive dose percentages for two drugs were calculated by adding the dose percentage for each drug; thus the maximum additive dose percentage was 200% (100% of each drug).
Results
During the 4‐year period of publications (January 1, 2010, through December 31, 2013), the total number of studies of a targeted drug combined with a cytotoxic that met the inclusion criteria was 372 (24,326 patients; 248 drug combinations; Table 1, Supporting Information Table 1). The targeted and cytotoxic agents were FDA approved by the time of the current analysis for the majority of studies (N = 294 of 372 studies for the targeted agent; N = 362 of 372 studies for the cytotoxic agent). The targeted agent was an antibody in 121 studies and a small molecule inhibitor in 251 studies. The median (range) for the combined dose was 180% (41–200%) of the additive FDA/RP2D/MTD dose.
Table 1.
Two drug combinations including a cytotoxic and targeted agent reported over 4 years (Phase I–III studies on PubMed January 1, 2010 to December 31, 2013)
| Cytotoxic agent plus targeted agenta | |
|---|---|
| Number of studies | 372 |
| Number of drug combinations | 248 |
| Number of patients | 24,326 |
| Median (range) additive dose percentage | 180 (41–200) |
| Number (%) of studies where ≥ one drug dose percentage was 100% | 310 (83) |
| Number (%) of drug combinations where ≥ one drug dose percentage was 100% | 210 (85) |
| Number (%) of patients where ≥ one drug dose percentage was 100% | 21,492 (88) |
| Median (range) percentile for second drug when one drug dose percentage was 100% | 90 (16–100) |
| Number (%) of studies where targeted agent dose percentage was 100% | 203 (55) |
| Number (%) of drug combinations where targeted agent dose percentage was 100% | 137 (55) |
| Number (%) of patients where targeted agent dose percentage was 100% | 14,860 (61) |
| Median (range) percentile for cytotoxic drug when targeted agent dose percentage was 100% | 100 (36–100) |
| Number (%) of studies where cytotoxic agent dose percentage was 100% | 248 (67) |
| Number (%) of drug combinations where cytotoxic agent dose percentage was 100% | 171 (69) |
| Number (%) of patients where cytotoxic agent dose percentage was 100% | 17,635 (72) |
| Median (range) percentile for targeted drug when cytotoxic agent dose percentage was 100% | 100 (17–100) |
| Number (%) of studies where each drug's dose percentage was 100% (e.g. additive dose percentage2 = 200%) | 141 (38) |
| Number (%) of drug combinations where each drug's dose percentage was 100% (e.g. additive dose percentage = 200%) | 97 (39) |
| Number (%) of patients where each drug's dose percentage was 100% (e.g. additive dose percentage = 200%) | 11,003 (45) |
| Number (%) of studies where additive dose percentage was >100%, with no single drug given at 100% | 37 (10) |
| In studies where additive dose percentage was >100%, with no single drug given at 100%, median (range) of additive dose percentage | 135 (102–175) |
| Number (%) of studies where additive dose percentage was ≤100% and safe dose was found | 14 (4) |
| In studies where additive dose percentage was ≤100% and safe dose was found, median (range) of additive dose percentage | 85 (41–100) |
| Number (%) of studies where additive dose percentage was ≤100% and safe dose was not found | 3 (1) |
| In studies where additive dose percentage was ≤100% and safe dose was not found, median (range) of additive dose percentage studied | 100 (92–100) |
| Number (%) of studies where additive dose percentage was >100% and safe dose was not found | 8 (2) |
| In studies where additive dose percentage was >100% and safe dose was not found, median (range) of additive dose percentage given | 181 (101–200) |
Excludes hormonal modulators and immunotherapy. 2Additive dose percentage = [(safe dose of drug A in combination/dose of drug A as single agent at FDA‐approved dose or RP2D or MTD) × 100] + [(safe dose of drug B in combination/dose of drug B as single agent at FDA‐approved dose or RP2D or MTD) × 100].
One drug at 100% dose percentage of the FDA/RP2D/MTD dose
Three hundred ten studies (including 210 drug combinations) were published (N = 21,492 patients) in which one targeted agent and one cytotoxic were administered and one of the agents was given at full (100%) dose (Supporting Information Refs. S1–S294; Table 2). These included 92 Phase I studies (N = 2,543 patients), 200 Phase II or III studies (N = 17,987 patients) and 18 Phase I/II combined studies (N = 962 patients). (In the Phase I studies, only a subset of patients were treated at the FDA/RP2D/MTD dose.) In total, in 141 studies (45% of the 310 studies where either the cytotoxic or the targeted agent was at 100% of the FDA/RP2D/MTD dose; N = 97 drug combinations), the other drug was also administered at 100% (N = 11,003 patients received both drugs at 100% dose).
Table 2.
Summary of two drugs (targeted and cytotoxic) in combination
| Second drug at 100% dose percentage of FDA/RP2D/MTD | Lowest additive dose percentage of the combination | |
|---|---|---|
| One drug at 100% dose percentage of FDA/RP2D/MTD |
45% of studies (141/310) Note: 141 of the 372 total studies (38%) administered each drug at 100% dose |
117%a (cytotoxic agent 100%) 136%2 (targeted agent 100%) |
| Cytotoxic at 100% dose percentage of FDA/RP2D/MTD (N = 248 studies) | 57% (141/248) | 117%a |
| Targeted agent at 100% dose percentage of FDA/RP2D/MTD (N = 203 studies) | 69% (141/203) | 136%2 |
| No single drug at 100% of the FDA/RP2D/MTD (N = 51 studies)3 | Not applicable |
41%4
82%5 (without HDAC or PARP inhibitors) 97%6 (with antibody as targeted agent) |
Lowest dose percentage was 17% (targeted) plus 100% (cytotoxic): vorinostat (HDAC inhibitor) plus vinorelbine (Supporting Information Ref. S291); atrasentan (endothelin A receptor antagonist) and pegylated liposomal doxorubicin or docetaxel (Supporting Information Refs. S292–S293). However, cenersen (antisense against TP53) was given at 16% and idarubicin was at 100%, but dose was not chosen based on toxicity (Supporting Information Ref. S294). 2Combretastatin A4 phosphate (tubulin binder) was at 36% and bevacizumab was at 100% (Supporting Information Ref. S188). 3In the 51 studies where no single drug was given at 100% dose percentage, the most common grade 3 or 4 toxicities were as follows: (i) neutropenia (N = 24 (47%) studies); (ii) thrombocytopenia (N = 8 (16%) studies); (iii) anemia (N = 5 (10%) studies). For studies (N = 7) that included HDAC or PARP inhibitors, the most common toxicities leading to lowered doses were neutropenia and thrombocytopenia (57% and 29% of studies). 4Topotecan and veliparib (PARP inhibitor) (Supporting Information Ref. S345). 5Chlorambucil was at 46% and imatinib was at 36% (Supporting Information Ref. S339). 6Fludarabine was at 72% and alemtuzumab was at 25% (Supporting Information Refs. S336–S337).
Abbreviations: FDA: Food and Drug Administration‐approved dose; HDAC: histone deacetylase; MTD: maximum tolerable dose; PARP: poly‐ADP ribose polymerase; RP2D: recommended Phase 2 dose.
For combinations involving an antibody as the targeted agent, 69 out of 121 studies (57%; 37 out of 68 drug combinations) could give both agents at 100% of the dose. For combinations involving a small molecule inhibitor as the targeted agent, only 76 out of 251 studies (30%; 62 out of 180 drug combinations) could give both agents at 100% of the single agent dose.
Targeted agent at 100% dose percentage of the FDA/RP2D/MTD dose
Two hundred three studies (including 137 drug combinations) were published (N = 14,860 patients) in which one targeted agent and one cytotoxic were administered and the targeted agent was given at full (100%) dose (Supporting Information Refs. S1–S188; Table 2). The median dose percentage for the cytotoxic agent was at 100% of the FDA/RP2D/MTD dose (range, 36–100%). The median (range) for the additive dose percentage was 200% (136–200%) of the additive FDA/RP2D/MTD dose. The lowest safe additive dose percentage was 136%; this was for bevacizumab (an antiangiogenesis antibody) and combretastatin A4 phosphate (a class of natural phenols that binds tubulin which despite its name is unrelated to statins; Supporting Information Refs. S188).
Cytotoxic agent at 100% dose percentage of the FDA/RP2D/MTD dose
Two hundred forty eight studies (including 171 drug combinations) were published (N = 17,635 patients) in which one targeted agent and one cytotoxic were administered and the cytotoxic was given at full (100%) dose (Supporting Information Refs. S1–S132 and S189–S294; Table 2). The median dose percentage for the targeted agent was at 100% of the FDA/RP2D/MTD (range, 17–100%). The median (range) for the additive dose percentage was 200% (117–200%) of the additive FDA/RP2D/MTD dose. The lowest safe additive dose percentage was 117%, which was given for: vorinostat [histone deacetylase (HDAC) inhibitor] plus vinorelbine (Supporting Information Ref. S291); atrasentan (endothelin A receptor antagonist) and pegylated liposomal doxorubicin or docetaxel (Supporting Information Refs. S292–S293; Table 2). While cenersen (antisense against TP53) was given at 16% and idarubicin was at 100%, this dose was not chosen based on toxicity (Supporting Information Ref. S294).
Additive dose percentage ≥100%, but no single drug at 100% of the FDA/RP2D/MTD dose
There were 37 studies (N = 28 drug combinations) where the additive dose percentage was >100%, but no single drug was at 100% of the FDA/RP2D/MTD dose due to toxicity of higher doses (Supporting Information Refs. S295–S331). The lowest safe additive dose of the combination was 102% which was required for rituximab and cladribine (Supporting Information Ref. S331; 33% and 69% of the dose percentage, respectively; Table 2).
Additive dose percentage ≤ 100%
Fourteen studies (everolimus and capecitabine, Supporting Information Ref. S332; gemtuzumab oogomycin and clofarabine, Supporting Information Ref. S333; obatoclax mesylate and topotecan, Supporting Information Refs. S334–S335; alemtuzumab and fludarabine, Supporting Information Refs. S336–S337; bryostatin‐1 and cisplatin, Supporting Information Ref. S338; imatinib and chlorambucil, Supporting Information Ref. S339; veliparib and cyclophosphamide, Supporting Information Ref. S340; vorinostat and decitabine, Supporting Information Ref. S341; olaparib and topotecan, Supporting Information Ref. S342; olaparib and dacarbazine, Supporting Information Ref. S343; panobinostat and melphalan, Supporting Information Ref. S344 and veliparib and topotecan, Supporting Information Ref. S345) were published, where one agent was targeted and one was a cytotoxic and the additive dose percentage was ≤100%. The dose for capecitabine and everolimus was 50% and 50% of the FDA/RP2D/MTD dose of each drug, respectively; clofarabine and gemtuzumab oogomycin, 50% and 50%, respectively; for topotecan and obatoclax mesylate, 83% and 17% in both studies, respectively; for fludarabine and alemtuzumab, 72% and 25% in both studies, respectively; cisplatin and bryostatin‐1, 67% and 25%, respectively; for chlorambucil and imatinib, 46% and 36%, respectively; for cyclophosphamide and veliparib, 70% and 8%, respectively; for decitabine and vorinostat, 50% and 25%, respectively; for topotecan and olaparib, 40% and 25%, respectively; for dacarbazine and olaparib, 48% and 8%, respectively; for panobinostat and melphalan 38% and 15%, respectively; and for topotecan and veliparib, 40% and 1%, respectively (additive dose = 100%, 100%, 100%, 97%, 92%, 82%, 78%, 75%, 65%, 56%, 53%, 41% of the combined FDA/RP2D/MTD dose). In conclusion, if a HDAC or poly‐ADP ribose polymerase (PARP) inhibitor is used, the lowest additive safe dose percentage is 41%. If neither of these drugs is included, the lowest safe additive dose percentage is 82% (imatinib and chlorambucil). If the combination involved an antibody as the targeted agent, the lowest safe additive dose percentage was 97% (alemtuzumab and fludarabine; Table 2).
Targeted and cytotoxic combinations where the safety of the combination dose was unacceptable
There were eight studies published where the additive dose was >100% and the studies did not find an acceptable dose [pazopanib and pemetrexed, Supporting Information Ref. S346 (both drugs at 100%); gefitinib and docetaxel, Supporting Information Ref. S347 (both drugs at 100%); dasatinib and ixabepilone, Supporting Information Ref. S348 (both drugs at 100%), axitinib and docetaxel, Supporting Information Ref. S132 (both drugs at 100%); bendamustine and erlotinib, Supporting Information Ref. S349 [100% and 61%, respectively (additive 161%)]; vinflunine and pazopanib, Supporting Information Ref. S350 [80% and 25%, respectively (additive 105%)]; vinflunine and erlotinib, Supporting Information Ref. S351 [71% and 33%, respectively (additive 104%)]; and topotecan and lenalidomide, Supporting Information Ref. S352 [83% and 18%, respectively (additive 101%)]. Three studies docetaxel and vorinostat, Supporting Information Ref. S353 [83% and 17% dose percentage, respectively (additive = 100%)]; PR‐104 and sorafenib, Supporting Information Ref. S354 [50% and 50%, respectively (additive 100%)]; 5‐fluorouracil and vorinostat, Supporting Information Ref. S355 [56% and 36%, respectively (additive = 92%)] were published where the lowest dose level tried was ≤100% and there was not an acceptable safety profile. However, lower doses were not attempted.
HDAC and PARP inhibitor combinations
Combinations with HDAC or PARP inhibitors often required significant dose reductions of both agents and resulted in the lowest additive dose percentages seen in the study. Panobinostat and melphalan were reduced to 38% and 15%, Supporting Information Ref. S344, topotecan and veliparib were reduced to 40% and 1%, Supporting Information Ref. S345, dacarbazine and olaparib were reduced to 48% and 8%, Supporting Information Ref. S343, topotecan and olaparib, Supporting Information Ref. S342 were administered at 40% and 25%, decitabine and vorinostat, Supporting Information Ref. S341 were administered at 50% and 25%, and cyclophosphamide and veliparib, Supporting Information Ref. S340 were administered at 70% and 8% of the FDA/RP2D/MTD dose, respectively due to toxicities at the higher doses. Because some of these combinations involved topotecan, which is a notoriously difficult agent to give at full dose, we also examined the overall impact of topotecan. When topotecan was given with drugs other than HDAC or PARP inhibitors (N = 8 studies); the lowest safe additive dose percentage was 100%; when PARP inhibitors were given with a drug other than topotecan, the lowest additive dose percentage was 56% (dacarbazine and olaparib).
Discussion
Combining targeted agents with cytotoxic chemotherapy represents a potential approach to improve therapy in metastatic carcinomas. In many cases, toxicity is nonoverlapping between cytotoxic drugs and targeted agents especially with antibody therapeutics, and these combinations can theoretically modulate chemoresistance pathways to promote apoptosis in cancer cells without increasing toxicity. Bevacizumab, rituximab and trastuzumab have been combined with cytotoxic chemotherapy with improved survival and tolerable side effects for colon cancer, non‐Hodgkin's lymphoma and breast cancer, respectively.10, 11, 12, 13 While classical cytotoxic combination chemotherapy dosing was previously limited by significant toxicity necessitating conservative initial dosing, standard care for nononcology patients or for individuals with cancer and multiple medical issues routinely involves combining medications based on established algorithms. Indeed, the average patient with advanced cancer is on a median of eight drugs for other health problems prior to starting treatment for malignancy.8 Although combining targeted therapies with cytotoxic agents can theoretically have fewer side effects than cytotoxic combinations, a process to calculate initial safe doses either within a clinical trial or in practice would be of benefit. Furthermore, given the molecular heterogeneity of metastatic carcinomas, an optimized precision medicine strategy is required for effective treatment and will necessitate the use of such combinations.
The current study aimed to determine the lowest safe starting doses required for novel two‐drug combinations involving a targeted agent and a cytotoxic agent. We assessed Phase I–III clinical trials over a 4‐year span (N = 24,326 patients). While all studies were able to define safe starting doses if the additive dose percentage of the combination was sufficiently lowered, only 38% of studies could administer both agents at 100% of the FDA‐approved/RP2D/MTD dose. Subset analyses demonstrated significantly fewer dose reductions with antibody combinations where 57% of studies could administer both agents at 100% of the dose percentage as compared to 30% with small molecule inhibitors as the targeted agent. Combinations involving HDAC or PARP inhibitors often resulted in significantly compromised doses, with the lowest additive dose percentage of the study occurring with veliparib and topotecan (additive dose percentage = 41%; Supporting Information Ref. S345). PARP inhibitors are generally well tolerated as single agents when used for BRCA‐mutated patients, but otherwise have limited single agent activity necessitating combination therapy approaches.14 Although PARP and HDAC inhibitors were classified as targeted therapies, they damage DNA by affecting DNA expression through acetylation/deactylation of histones15 and inhibiting repair of single‐strand DNA breaks, respectively,16 and thus can potentiate the effects of cytotoxic chemotherapy, perhaps leading to significant toxicity. As such, they can be considered to have an overlapping target with cytotoxic chemotherapeutic agents. In the absence of HDAC and PARP inhibitors, the lowest safe additive dose was 82% (imatinib and chlorambucil; Supporting Information Ref. S339) and 97% when an antibody was the targeted agent (alemtuzumab and fludarabine; Supporting Information Refs. S336–S337; Table 2).
In certain settings, the dose of either the cytotoxic or the targeted agent may need to be held at 100% of the FDA‐approved/RP2D/MTD dose. We therefore also analyzed safe starting doses in these situations (Table 2). If the cytotoxic was given at 100% dose percentage, the lowest safe starting dose of the targeted agent was 17% of the FDA‐approved/RP2D/MTD dose. If the targeted compound was given at 100% dose percentage, the lowest safe starting dose of the cytotoxic agent was 36% of the FDA‐approved/RP2D/MTD dose.
We recently described combination therapy with two targeted agents from adult oncology clinical trials published between January 1, 2010, through December 31, 2013, and found that in 144 studies (N = 8,568 patients; 95 combinations), the majority of combinations could have both drugs administered at 100% of the single agent dose percentage. The lowest safe additive dose percentage was 60% if targets and/or class of drugs overlapped, or in the presence of mTor inhibitors, but was otherwise 143%.17 In contrast, the current study which combines a targeted agent with a cytotoxic agent found significantly more toxicity with more substantial dose reductions seen.
The current study attempted to provide a rational starting point for dosing de novo drug combinations involving a targeted agent and a cytotoxic agent for clinical trials or in practice. Toxicity has been a primary concern for oncology therapeutics given narrow therapeutic windows for classic cytotoxic chemotherapy, thus the current study focused on determining safe starting doses to minimize toxicity. A prior study of 55 clinical trials sponsored by a single entity suggested that patients on Phase I trials had better response rates and overall survival on higher doses,18 while a single institution study of 24 clinical trials from various sponsors demonstrated that patients who received lower drug doses did not fare worse than those on higher doses.19 Although it is unclear if dose reductions in the setting of administration of multiple agents will alter efficacy, in the absence of significant toxicity, intrapatient dose escalation to allow for improved efficacy can occur if needed. An alternative strategy to maximize efficacy would consider starting most agents at 100% of the dose percentage and then de‐escalating the dose in the setting of toxicity. However, given that only 38% of the studies could administer both agents at 100% of the FDA‐approved/RP2D/MTD dose, it is likely that this strategy would result in significant toxicity for the majority of patients treated.
The current study has several limitations. It was restricted to two‐drug combinations of cytotoxic and targeted agents in adult patients without organ dysfunction. It excluded patients with renal or hepatic failure, children and elderly patients all of whom would likely require dosing modifications due to alterations in organ function based on the metabolism of the therapeutic. The study also assumed that all patients enrolled in the trials were at similar risk of adverse events given that organ dysfunction and elderly patients were excluded, but differences in performance status and baseline entry criterion across the trials may have contributed to varying toxicities between studies. Drug–drug interactions and effects on metabolic proteins leading to changes in the pharmacology of the therapeutics may have also led to lower safe dose levels. The study did not consider individual pharmacology, metabolism, or absorption, but these factors are generally not evaluated when dosing individual patients even with approved drugs. The exposures of targeted agents may have varied across trials with differing chemotherapeutics which may have led to changes in toxicity and lowered the additive dose percentage; however, this information was not consistently available in the studies. While the study represented a large number of patients, some clinical trials with significant toxicity may never have been published, which could result in publication bias, and thus the observations of the current study may not be representative of all possible drug combinations. The effect of lower doses or reduced starting doses prior to individual dose escalation on efficacy was not analyzed in the current study. The study also included Phase I–III trials that had different objectives, and thereby represented a heterogeneous data set. While the current study attempts to address safe starting doses, it did not analyze dose escalation schemes, since these have been discussed elsewhere.20, 21 While in general lower doses given in combination were necessitated by toxicity, in some studies the investigators held one drug at a preconceived dosing level, which may have led to lower additive dose percentages.
In conclusion, the basis for starting drugs in Phase I trials has traditionally been one‐tenth of the LD10 in mice (the dose that results in 10% lethality) or one‐third of the toxic dose low (TDL; the lowest dose that produces a toxic effect in a species over a certain exposure period) in larger species. However in combination studies, the method of selection of starting dose is very unclear. Options include fixing one drug dose and escalating the other or stepwise escalation of both drugs. The current study suggests that in adults with intact organ function, a targeted and cytotoxic dual drug combination will often require dose reductions for toxicity. The lowest safe starting additive dose percentage is about 40% if PARP or HDAC inhibitors are included in the combination. In the absence of PARP and HDAC inhibitors, the lowest safe additive dose percentage is around 80% and increases to 100% if an antibody is included in the combination. A conservative dosing approach to avoid toxicity would thus start a novel targeted and cytotoxic combination at 40% and 40% of each drug's respective FDA/RP2D/MTD dose. When a PARP or HDAC inhibitor is the targeted agent, the starting dose will decrease to 20% and 20%, while if an antibody is used it will increase to 50% and 50% of the respective FDA/RP2D/MTD doses. It is important to keep in mind that 38% of patients could tolerate full doses of each drug, and the median additive dose percentage was 180%, suggesting that intrapatient dose escalation may permit higher doses in many individuals. These results provide information on the lowest safe doses observed in prior studies, which can help inform decisions for safe starting doses of de novo two‐agent drug combinations with a cytotoxic and targeted agent in clinical trials and practice.
Supporting information
Supporting Information
Supporting Information Table 1.
Acknowledgements
This study was funded in part by the Joan and Irwin Jacobs philanthropic fund. Dr. Kurzrock receives research funds from Genentech, Pfizer, Foundation Medicine, Guardant, Sequenom and Merck Serono, as well as consultant/advisory board fees from Sequenom, Actuate Therapeutics and XBiotech and has an ownership interest in Novena Inc, and Curematch Inc.
References
- 1. Kurzrock R, Giles FJ. Precision oncology for patients with advanced cancer: the challenges of malignant snowflakes. Cell Cycle 2015;14:2219–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gerlinger M, Rowan AJ, Horswell S, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med 2012;366:883–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kandoth C, McLellan MD, Vandin F, et al. Mutational landscape and significance across 12 major cancer types. Nature 2013;502:333–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wheler JJ, Parker BA, Lee JJ, et al. Unique molecular signatures as a hallmark of patients with metastatic breast cancer: implications for current treatment paradigms. Oncotarget 2014;5:2349–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wheler J, Lee JJ, Kurzrock R. Unique molecular landscapes in cancer: implications for individualized, curated drug combinations. Cancer Res 2014;74:7181–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tsimberidou AM, Iskander NG, Hong DS, et al. Personalized medicine in a phase I clinical trials program: the MD Anderson Cancer Center initiative. Clin Cancer Res 2012;18:6373–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. DeVita VT Jr., Chu E. A history of cancer chemotherapy. Cancer Res 2008;68:8643–53. [DOI] [PubMed] [Google Scholar]
- 8. Borad MJ, Curtis KK, Babiker HM, et al. The impact of concomitant medication use on patient eligibility for phase I cancer clinical trials. J Cancer 2012;3:345–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Liu S, Kurzrock R. Toxicity of targeted therapy: implications for response and impact of genetic polymorphisms. Cancer Treat Rev 2014;40:883–91. [DOI] [PubMed] [Google Scholar]
- 10. Averbuch SD, Wolf MK, El‐Rayes BF, et al. Clinical development issues In: Prendergast GC, eds. Molecular cancer therapeutics: strategies for drug discovery and development. Hoboken, NJ: Wiley‐LSS, 2004. 287–306. [Google Scholar]
- 11. Hurwitz H, Fehrenbacher L, Novotny W, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med 2004;350:2335–42. [DOI] [PubMed] [Google Scholar]
- 12. Sehn LH, Donaldson J, Chhanabhai M, et al. Introduction of combined CHOP plus rituximab therapy dramatically improved outcome of diffuse large B‐cell lymphoma in British Columbia. J Clin Oncol 2005;23:5027–33. [DOI] [PubMed] [Google Scholar]
- 13. Baselga J. Herceptin alone or in combination with chemotherapy in the treatment of HER2‐positive metastatic breast cancer: pivotal trials. Oncology 2001;61: 14–21. [DOI] [PubMed] [Google Scholar]
- 14. Drew Y, Plummer R. PARP inhibitors in cancer therapy: two modes of attack on the cancer cell widening the clinical applications. Drug Resist Updates 2009;12:153 [DOI] [PubMed] [Google Scholar]
- 15. Xu WS, Parmigiani RB, Marks PA. Histone deacetylase inhibitors: molecular mechanisms of action. Oncogene 2007;26:5541–52. [DOI] [PubMed] [Google Scholar]
- 16. Underhill C, Toulmonde M, Bonnefoi H. A review of PARP inhibitors: from bench to bedside. Ann Oncol 2011;22:268–79. [DOI] [PubMed] [Google Scholar]
- 17. Liu S, Nikanjam M, Kurzrock R. Dosing de novo combinations of two targeted drugs: towards a customized precision medicine approach to advanced cancers. Oncotarget 2016;7:11310–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gupta S, Hunsberger S, Boerner SA, et al. Meta‐analysis of the relationship between dose and benefit in phase I targeted agent trials. J Natl Cancer Inst 2012;104:1860–6. [DOI] [PubMed] [Google Scholar]
- 19. Jain RK, Lee JJ, Hong D, et al. Phase I oncology studies: evidence that in the era of targeted therapies patients on lower doses do not fare worse. Clin Cancer Res 2010;16:1289–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hamberg P, Verweij J. Phase I drug combination trial design: walking the tightrope. J Clin Oncol 2009;27:4441–3. [DOI] [PubMed] [Google Scholar]
- 21. Hamberg P, Ratain MJ, Lesaffre E, et al. Dose‐escalation models for combination phase I trials in oncology. Eur J Cancer 2010;46:2870–8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Supporting Information Table 1.