

Abstract
Engineered enzyme variants of potato epoxide hydrolase (StEH1) display varying degrees of enrichment of (2R)‐3‐phenylpropane‐1,2‐diol from racemic benzyloxirane. Curiously, the observed increase in the enantiomeric excess of the (R)‐diol is not only a consequence of changes in enantioselectivity for the preferred epoxide enantiomer, but also to changes in the regioselectivity of the epoxide ring opening of (S)‐benzyloxirane. In order to probe the structural origin of these differences in substrate selectivity and catalytic regiopreference, we solved the crystal structures for the evolved StEH1 variants. We used these structures as a starting point for molecular docking studies of the epoxide enantiomers into the respective active sites. Interestingly, despite the simplicity of our docking analysis, the apparent preferred binding modes appear to rationalize the experimentally determined regioselectivities. The analysis also identifies an active site residue (F33) as a potentially important interaction partner, a role that could explain the high conservation of this residue during evolution. Overall, our experimental, structural, and computational studies provide snapshots into the evolution of enantioconvergence in StEH1‐catalyzed epoxide hydrolysis.
Keywords: enantioselectivity, epoxide hydrolysis, evolutionary snapshots, laboratory evolution, protein engineering
Solanum tuberosum epoxide hydrolase 1 (StEH1) is an attractive target for protein engineering, as it has been shown to be susceptible to changes in environment and structure, both of which can affect the enantio‐ and regioselectivity of this enzyme towards a range of epoxide substrates.1, 2, 3 StEH1 catalyzes the hydrolysis of trans‐ and monosubstituted epoxides by a two‐step mechanism.4 This begins with nucleophilic attack of an enzyme‐contributed carboxylate nucleophile (D105) on one of the epoxide carbons, thus forming an alkylenzyme intermediate; the alkylenzyme subsequently decays back to the Michaelis complex, or is hydrolyzed to form the diol product. StEH1 shows a modest preference for the (S)‐enantiomer of benzyloxirane (1, Scheme 1) and complete regiospecificity. The enzyme catalyzes epoxide ring opening exclusively at the least sterically hindered, achiral, carbon (C‐2) with both enantiomers of 1, and this results in retention of stereoconfiguration in the product.
Scheme 1.
Benzyloxirane (1) and 3‐phenylpropane‐1,2‐diol (2).
We have obtained, by ISM‐driven5 directed evolution, enzyme variants that were able to catalyze the formation of the hydrolysis product (2) with enriched optical purity.2, 3 We also isolated laboratory‐evolved variants that catalyze the production of enriched (R)‐2 from racemic 1. The enrichment appears to be a consequence of a combination of changes in both enantio‐ and regiopreference (to different degrees in different enzyme variants). Interestingly, the regioselectivity for C‐2 in the hydrolysis of (R)‐1 was preserved in all variants, but was shifted towards C‐1 with (S)‐1.3 As epoxide ring opening at the asymmetric carbon C‐1 causes inversion of the product's stereoconfiguration, these variants also produced (R)‐2 from (S)‐1. This additional production of (R)‐2 from racemic 1 therefore leads to enantioconvergence, and thus the possibility to exceed the usual 50 % limit of optically pure product (otherwise obtainable by kinetic resolution of a racemic substrate). In the variant displaying the highest level of enrichment (R‐C1B1D33E6), the regiopreference had reverted to that of the wild‐type enzyme, and thus the enantiomeric excess of (R)‐2 was instead attributable to kinetic resolution of the two epoxide enantiomers.
Structure analysis of in vitro evolved enzymes
The mechanisms behind the observed changes in selectivity were initially speculative, based on models of the wild‐type structure with mutations introduced in silico.2 Substitutions W106L and L109Y were proposed to be responsible for the modified regioselectivity in the variant (R‐C1B1) displaying the largest change from the parent enzyme. The side chains of these residues are believed to interact with the substrate. In the wild‐type enzyme, W106 also has a catalytic role, in that its main‐chain amide contributes to an oxyanion hole and stabilizes the tetrahedral intermediate formed during the hydrolytic half‐reaction. The residue at this position (adjacent to the nucleophile) has been shown to influence enzyme selectivity and activity in related epoxide hydrolases,6 thus further supporting involvement here. A role for W106 in substrate recognition has also been suggested based on empirical valence‐bond calculations of (wild‐type) StEH1‐catalyzed hydrolysis of the related styrene oxide. These calculations proposed that the residue stabilizes a certain binding mode through interactions between the indole and the substrate.7 This study also identified a possible link between the preferred binding mode of styrene oxide and the preferred point of epoxide ring opening, hence, to an extent, predicting the stereoconfiguration of the ultimate diol product. The methylene carbon in 1 gives this compound fundamentally different dynamic properties from those of styrene oxide, in which the phenyl ring is directly bonded to one of the epoxide carbons. The larger conformational flexibility severely increases the challenges involved in theoretical predictions of binding modes as well as the amount of sampling required to obtain convergent free energies in empirical valence‐bond or QM/MM free energy calculations. Therefore, resolving the structures of these in‐vitro‐evolved enzymes would provide previously unavailable information about the structural causes of the observed changes in regioselectivity, and also about how an enzyme active‐site structure can adapt to catalyze the transformation of a (new) reaction.
Structures of evolved enzymes
The crystal structures of these laboratory‐evolved variants are presented in Figure 1. All variants were crystallized under similar, but not identical, conditions to those used for wild‐type StEH1;8 this resulted in crystals of the same space group and unit cell dimensions, containing two monomers per asymmetric unit. Pairwise superposition of all monomers yielded RMSDs of 0.14–0.46 Å, with the largest deviations observed for residues 93–96 of a solvent‐exposed loop. This deviation in loop structure is most likely associated with an unintentional additional mutation at position 94 (P94L) introduced during the course of the directed evolution; this occurred first in R‐C1B1 and was inherited by its offspring R‐C1B1D33 and R‐C1B1D33E6. The P94L exchange is not expected to influence StEH1 catalytic parameters because of its distance from the active site (∼25 Å). The details of the R‐C1B1 structure have been described in detail elsewhere.7
Figure 1.
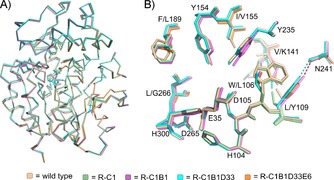
A) Superposition of Cα carbons from the crystal structures of the wild‐type enzyme and evolved variants. B) Key active‐site residues, including those mutated during the directed evolution. Figures were generated in PyMol (v. 1.81) from atomic coordinates in PDB IDs: 2CJP 8 (wild type), 4UHB (R‐C1), 4UFN 7 (R‐C1B1), 4UFP (R‐C1B1D33), and 4UFO (R‐C1B1D33E6). All enzymes were expressed and purified as described.9 Crystallization details and model refinements are in the Supporting Information.
The R‐C1 variant has two point mutations, V141K and I155V. V141 is located at the entrance to the active site, and is not expected to form direct contacts with the tested enantiomers of 1. The mutation to (larger) lysine could affect active‐site accessibility by partially blocking the entrance, but the weakness or lack of electron density for all atoms beyond Cγ indicates high mobility of this side chain and the lack of such an effect. The side chain of I155 is close to H153 and the two catalytic tyrosines, Y154 and Y235 (Figure 1 B). The I155V exchange could cause increased flexibility for the lid tyrosine phenol rings. Although the hydroxy group position of Y235 differs by 1.2 Å between wild‐type StEH1 and R‐C1B1 (Figure 1 B) and the distances between the hydroxyl groups of Y235 and Y154 range from 4.4 Å (R‐C1B1) to 5.0 Å (R‐C1B1D33), there is little support for markedly increased flexibility from the crystallographic temperature factors (Figure S2 in the Supporting Information). The differences are minor in extent and might be attributed to interactions with ligands bound to the active sites (glycerol in the case of R‐C1, and dioxane in R‐C1B1).
In R‐C1B1 and its offspring, there are two additional point mutations, W106L and L109Y. Surprisingly, the exchange of L109 with the considerably bulkier tyrosine causes no structural change in the active site. The main difference between wild‐type StEH1 and R‐C1B1 is the formation of a hydrogen bond between the side chain of N241 and the hydroxyl group of Y109 (Figure 1 B). As a consequence, the Y109 side chain is oriented away from the binding site, thus increasing its volume (Figure 2, Table S1) and weakening rather than strengthening the interactions with the bound substrate. Replacement of the bulky and aromatic hydrophobic side chain of W106 with the smaller leucine side chain also did not result in any significant structural changes in the active site. The only discernible difference from wild‐type StEH1 is a slight movement (0.2–0.3 Å) of the Y235 phenol ring towards L109 in R‐C1, probably as a consequence of structural relaxation of the protein in the absence of the W106 side chain.
Figure 2.
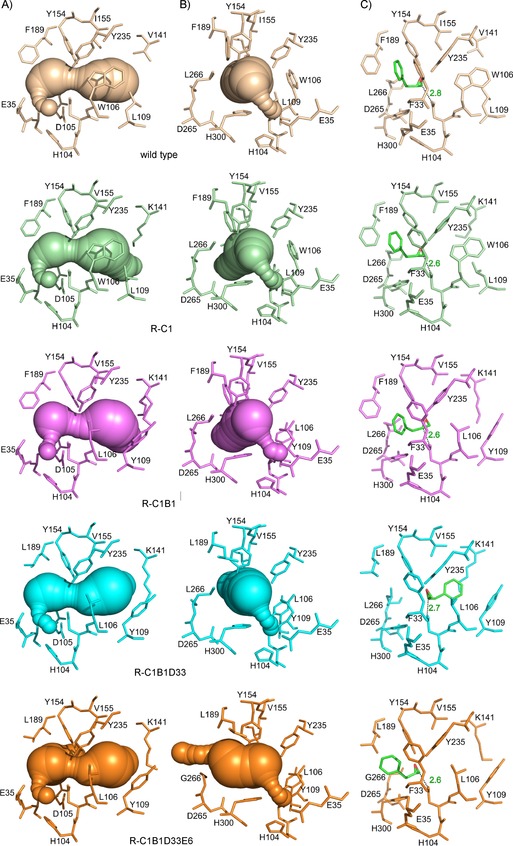
A) Side view (active‐site entrance on right), B) axial view, and C) preferred docking poses of (S)‐1 of wild‐type and variants. Volumes in the crystal structures were calculated with Caver12 (v. 3.01;0.9 Å radius probe). Catalytic residues and those altered by laboratory evolution are shown in stick representation. Water molecules are omitted for clarity. Distances are in ångström, and are drawn between the closest epoxide carbon and D105 Oδ. Images were generated in PyMol (v. 1.8.1; www.pymol.org).
R‐C1B1D33 has one additional substitution, F189L. Again, no significant change in position or conformation of surrounding active‐site residues resulted. F189L increases the volume of the active‐site pocket (Figure 2).
The L266G replacement in R‐C1B1D33E6 potentially increases backbone flexibility, but no backbone shifts or changes in backbone dihedral angles were observed in the crystal structure (representing the most highly evolved StEH1 variant). The side‐chain deletion just increased the active‐site volume, without causing conformational changes of the surrounding residues.
It should be noted that we were unsuccessful in all our attempts to crystallize any of these enzymes with relevant ligands bound to the active site, although a glycerol molecule from the crystallization solution was found in one of the R‐C1 monomers (Figure S1). This triol can be viewed as a product analogue of epoxide (glycidol) hydrolysis and does provide information regarding possible product binding. For further analysis of how the evolved structures acquired the changes in regioselectivity displayed by R‐C1B1 and R‐C1B1D33, we pursued computational modeling approaches.
Predicting substrate binding modes in the active sites
Although the crystallographic data provide insight into how the enzyme structure adapted during laboratory evolution of StEH1 activity, they do not provide insights into how this affects substrate binding in the active site (something which we recently showed to be important for determining StEH1 selectivity towards a different epoxide).7 Hence, and given the success of related approaches on similar systems,10 we performed constrained molecular docking to explore the available binding modes in the StEH1 active site (Table 1). Both enantiomers of 1 were modeled into the active sites of the crystal structure as a starting point. Restraints were set for the enzyme–substrate complexes to maintain hydrogen‐bonding distances between the epoxide ring oxygen and the catalytic tyrosine pair (Y154 and Y235). The preferred binding mode(s) and the distance between the nucleophile (D105) and each of the epoxide carbons were noted and compared with the experimentally determined regioselectivities. In all but one of the cases, we found poses of the docked substrate enantiomers that were in agreement with the observed regioselectivities (Tables 1 and Table S2), if assuming that the epoxide ring‐opening and alkylenzyme formation steers the stereoconfiguration of the final product with this epoxide substrate. The exception was (R)‐1 docked into R‐C1B1. This variant showed a clear preference for ring opening at C‐2, but the distances in the docking poses were similar between the nucleophile and either epoxide carbon (Table 1). Earlier empirical valence‐bond studies on the same enzyme with both trans‐stilbene oxide11 and styrene oxide7 suggested that this is not always the case; an energetically favorable pathway leading to alkylenzyme formation might be (kinetically) trapped due to a comparably higher barrier for hydrolysis. Considering the complexity of the catalysis, our docking results should be viewed mainly as suggestions as to how regioselectivity towards (S)‐1 altered during the trajectory of the laboratory evolution.
Table 1.
Preferred binding modes after docking of (R)‐1 and (S)‐1 into the active sites of wild‐type and evolved StEH1 enzymes.
| (R)‐1 | (S)‐1 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Enzyme | C‐1[a] [%] | r C−1 [b] [Å] | C‐2[a] [%] | r C−2 ]b] [Å] | Mode[c] | C‐1[a] [%] | r C−1 [b] [Å] | C‐2[a] [%] | r C−2 ]b] [Å] | Mode |
| StEH1 (wt) | 5 | 3.6 | 95 | 2.7 | 3 | 7 | 3.0 | 93 | 2.8 | 3 |
| R‐C1 | 3 | 2.8 | 97 | 2.7 | 1′ | 15 | 3.0 | 85 | 2.6 | 3 |
| R‐C1B1 | 2 | 2.8 | 98 | 2.8 | 1′ | 60 | 2.6 | 40 | 3.0 | 1′ |
| R‐C1B1D33 | 2 | 3.0 | 98 | 2.7 | 3 | 62 | 2.7 | 38 | 2.8 | 2 |
[a] Experimental data from ref. 3 [b] Calculated distance between D105 and C‐1/C‐2 of the epoxide ring of 1. [c] Mode 1′ involves interaction of the phenyl ring and H300; Mode 2 directs the phenyl ring of the substrate towards W/L106; in Mode 3, the phenyl substituent is sandwiched between F189 and F33 (Figure 2). Binding Modes 3 (wild type and R‐C1), 1′ (R‐C1B1), and 2 (R‐C1B1D33) provide distances between the nucleophile and the epoxide carbons in line with experimentally determined product configurations. For calculations see the Supporting Information (all docking poses in Table S2).
In the wild‐type enzyme and variant R‐C1, the preferred binding mode places the phenyl substituent of (S)‐1 such that it is sandwiched between the rings of F189 and F33 (Figure 2 C). This binding mode was not observed in earlier studies of monosubstituted epoxides modeled into the active site of StEH1, and is thus termed “Mode 3”. Binding in Mode 3 places the C‐2 carbon of the epoxide (Scheme 1) closest to the Oδ of the D105 nucleophile. The R‐C1B1D33E6 variant displays a similar binding pose, but its enlarged active‐site cavity allows for many different binding configurations, and the relatively poor activity of this variant makes it harder to discern which binding mode is relevant. The R‐C1B1 variant displays a slightly different binding mode, which resembles (but is different from) that found for (S)‐styrene oxide,7 where the epoxide phenyl substituent is directed towards H300 (“Mode 1” in that report). This binding mode is termed “Mode 1′ ”. Binding of the substrate in Mode 1′ shifts the position of the epoxide ring so that the asymmetric C‐1 is closer to the nucleophile. R‐C1B1D33 displays a distinct binding mode, similar to that for styrene oxide binding in Mode 2,7 where the phenyl substituent is directed towards W106. In this variant W106 is substituted by leucine, but the concomitant L109Y substitution provides aromatic edge‐on interactions between the substrate and Y109. Binding in Mode 2 also places C‐1 closer to the nucleophilic carboxylate.
The crystal structure of R‐C1B1D33 was the lowest‐resolution structure (2.95 Å); thus fewer water molecules could be modeled with maintained stringency. This might have affected the docking results. With this in mind, the preferred binding mode of (S)‐1 in this variant (Mode 2) places the phenyl ring of the substrate towards the mutation‐introduced Y109, thus making an edge‐on interaction. The finding that R‐C1B1D33 also displays clear quenching of the tyrosine fluorescence when mixed with (S)‐1 in stopped‐flow experiments (see the Supporting Information) suggests that this interaction could be valid. Also in this binding mode, C‐1 of the epoxide comes closest to the D105 side chain, thus facilitating nucleophilic attack, again in agreement with experimental results of regiopreference in this enzyme.
Conclusion
When comparing the catalytic parameters for the evolved variants to those for wild‐type StEH1, the K M for both enantiomers of 1 increased but k cat for (S)‐1 decreased to a larger extent, thus resulting in lower overall catalytic efficiency with this enantiomer (Table S3). The relatively high K M for the evolved enzymes probably resulted from the initial screening conditions, under which high epoxide concentrations were used, and thus did not select for low values of K M. The active‐site volumes of the variants also gradually grew, compared to the wild‐type enzyme (Figure 2, Table S1), thus further decreasing the possibility of tight binding of the substrate, and possibly contributing to the observed elevated K M values. The rotational flexibility of epoxide 1 is another possible cause of low affinity. We assume that the low binding affinity in the respective Michaelis complexes is responsible for our inability to achieve stability in MD simulations. However, combining the new structural data with the restrained docking results still provided important information about structural changes that lead to enrichment of (R)‐2 during our laboratory evolution. It is interesting that a new epoxide‐binding mode (Mode 3) was preferred for most variants as well as the wild type. This binding mode involves π–π interactions between the epoxide phenyl ring and the side chains of F33 and F189. The involvement of F33 in substrate binding might explain the degree of conservation of this residue during the directed evolution (only exchanges to tyrosine were observed).3 The docking also proposed previously neglected residues as putative targets for future mutagenesis studies (including F191 and F158 interacting with (R)‐1 and Y183 and F301 interacting with (S)‐1). Although we could rationalize the observed regioselectivity from simple docking of the different enantiomers of 1 into the respective active sites, it is important to keep in mind that selectivity is often more complicated than binding and productive orientation of the substrate in its ground state.
In summary, we present the crystal structures of enzyme variants isolated after laboratory evolution; these were selected for enrichment of a defined product enantiomer from a racemic substrate mixture. By combining crystallographic data and molecular simulation with previous experimental studies of this system, we were able to rationalize possible structure–function relationships that have evolved over the course of multiple rounds of mutagenesis and selection. Our work provides an atomic‐level insight into the (engineered) evolution of enantioconvergence in this highly biocatalytically important system.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was supported by the Swedish Research Council (M.W.). The European Research Council provided financial support under the European Community's Seventh Framework Programme (FP7/2007‐2013/ERC Grant Agreement 306474). The authors also acknowledge a Sven and Lilly Lawski scholarship for doctoral studies as well as support from COST Action 1303 Systems Biocatalysis. Finally, we are very grateful for the allocation of computational resources from the Swedish National Infrastructure for Computing (SNIC, Grant Number SNIC2015‐16‐12) and thank the Diamond Light Source and the European Synchrotron Radiation Facility for beam time (proposals mx11171, mx1639), and the staff of beamlines I02, I04 (DLS), and ID23‐2 (ESRF) for assistance with crystal testing and data collection. Access was supported in part by the EU FP7 infrastructure grant BIOSTRUCT‐X (contract no. 283570).
Å. Janfalk Carlsson, P. Bauer, D. Dobritzsch, M. Nilsson, S. C. L. Kamerlin, M. Widersten, ChemBioChem 2016, 17, 1693.
Contributor Information
Dr. Doreen Dobritzsch, Email: doreen.dobritzsch@kemi.uu.se
Dr. S. C. Lynn Kamerlin, Email: kamerlin@icm.uu.se
Prof. Dr. Mikael Widersten, Email: mikael.widersten@kemi.uu.se
References
- 1. Lindberg D., de la Fuente Revenga M., Widersten M., Biochemistry 2010, 49, 2297–2304. [DOI] [PubMed] [Google Scholar]
- 2. Gurell A., Widersten M., ChemBioChem 2010, 11, 1422–1429. [DOI] [PubMed] [Google Scholar]
- 3. Janfalk Carlsson A., Bauer P., Ma H., Widersten M., Biochemistry 2012, 51, 7627–7637. [DOI] [PubMed] [Google Scholar]
- 4. Widersten M., Gurell A., Lindberg D., Biochim. Biophys. Acta Gen. Subj. 2010, 1800, 316–326. [DOI] [PubMed] [Google Scholar]
- 5. Reetz M. T., Prasad S., Carballeira J. D., Gumulya Y., Bocola M., J. Am. Chem. Soc. 2010, 132, 9144–9152. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Laughlin L. T., Tzeng H.-F., Lin S., Armstrong R. N., Biochemistry 1998, 37, 2897–2904; [DOI] [PubMed] [Google Scholar]
- 6b. van Loo B., Lutje Spelberg J. H., Kingma J., Sonke T., Wubbolts M. G., Janssen D. B., Chem. Biol. 2004, 11, 981–990; [DOI] [PubMed] [Google Scholar]
- 6c. Rui L., Cao L., Chen W., Reardon K. F., Wood T. K., Appl. Environ. Microbiol. 2005, 71, 3995–4003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bauer P., Janfalk Carlsson Å., Amrein B. A., Dobritzsch D., Widersten M., Kamerlin S. C. L., Org. Biomol. Chem. 2016, 14, 5639–5651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mowbray S. L., Elfström L. T., Ahlgren K. M., Andersson C. E., Widersten M., Protein Sci. 2006, 15, 1628–1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Elfström L. T., Widersten M., Biochem. J. 2005, 390, 633–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zheng H., Kahakeaw D., Acevado J. P., Reetz M. T., ChemCatChem 2010, 2, 958–961. [Google Scholar]
- 11. Amrein B. A., Bauer P., Duarte F., Janfalk Carlsson Å., Naworyta A., Mowbray S. L., Widersten M., Kamerlin S. C. L., ACS Catal. 2015, 5, 5702–5713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chovancova E., Pavelka A., Benes P., Strnad O., Brezovsky J., Kozlikova B., Gora A., Sustr V., Klvana M., Medek P., Biedermannova L., Sochor J., Damborsky J., PLoS Comput. Biol. 2012, 8, e1002708. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary