

Abstract
Differences in the pathogenesis of microsatellite stable (MSS) sporadic colorectal cancers (CRCs) between left‐sided CRC (LC) and right‐sided CRC (RC) have not been clarified. To identify pathogenesis‐related genomic differences between MSS CRCs within the two locations, we performed a comprehensive molecular analysis using crypt isolation with samples from 92 sporadic CRCs. Microsatellite instability (MSI; high and low/negative) and DNA methylation status (low methylation epigenome; intermediate methylation epigenome [IME] or high methylation epigenome [HME]) were determined using polymerase chain reaction (PCR) microsatellite analysis and PCR‐bisulfite pyrosequencing, respectively. Additionally, mutations in the TP53, KRAS, BRAF and PIK3CA genes were examined using PCR‐bisulfite pyrosequencing (for KRAS and BRAF mutations) or PCR‐single conformation polymorphism (for TP53 and PIK3CA mutations), followed by sequencing of aberrant bands. Finally, a genome‐wide study using a copy number alteration (CNA)‐targeted single nucleotide polymorphism array was performed. Ninety‐two CRCs were classified into 71 MSS and 21 MSI phenotypes. We examined 71 CRCs with the MSS phenotype (LC, 56; RC, 15). Mutations in KRAS were associated with RC with the MSS phenotype, whereas mutations in TP53 were more frequently found in LC with the MSS phenotype. There were significant differences in the frequencies of KRAS and TP53 mutations in the IME between LC and RC with the MSS phenotype. Although CNA gains were associated with LC with the MSS phenotype, CNA losses were not major alterations associated with the MSS phenotype. These findings suggested that the molecular pathogenesis of the MSS phenotype in LC was different from that in RC.
Keywords: colorectal cancer, copy number alteration, microsatellite stable, mutation, tumor location
Short abstract
What's new?
The classification of colorectal cancer (CRC) based on tumor location is simple, comprehensive, and consistent with recent attempts to characterize tumors by pathological and molecular features. Differences in the pathogenesis of microsatellite stable (MSS) sporadic CRCs between left‐sided CRC (LC) and right‐sided CRC (RC) have however not been clarified. Here, the authors found that TP53 mutations are closely associated with the development of LC whereas RC is characterized by KRAS mutations. Using an integrated genome‐wide analysis, they also show significant differences in copy number alterations. The findings suggest a different molecular pathogenesis of the MSS phenotype between LC and RC.
Colorectal cancer (CRC) is a major cause of morbidity and mortality throughout the world.1 It is the third most common cancer worldwide and the fourth most common cause of death.1 Therefore, CRC is one of the most important cancers in the world.1, 2 The prognosis of patients with CRC has improved owing to recent advances in technology, and further improvements in our understanding of the pathological and molecular basis of disease have contributed to potential new approaches for the diagnosis and treatment of CRC. Therefore, it is important to continue to evaluate the role of molecular alterations in colorectal carcinogenesis.
Genomic instability plays an essential role in the development and progression of CRC.3, 4, 5 Based on different forms of genomic instability, sporadic CRC can broadly be divided into two groups: the microsatellite instability (MIN; MSI) type, caused by MLH1 methylation of the promoter region, and the chromosome instability (CIN) type, characterized by genomic instability at the gross chromosomal level.3, 4, 5 The majority of sporadic CRCs (∼90%) are classified into the CIN type, whereas only approximately 10% of sporadic CRCs are classified into the MIN type.3, 4, 5, 6, 7 In addition, the microsatellite stable (MSS) type, which is similar to CIN, is also important in colorectal carcinogenesis. Studies have shown that MSI and MSS are mutually exclusive and are also used as basic molecular classification.3, 7
CRCs that occur proximal (right) or distal (left) to the splenic flexure exhibit differences in their embryologic development, blood supply and clinicopathological findings.8, 9, 10, 11, 12 A previous study showed that classification based on tumor location, that is, left‐sided CRC (LC) and right‐sided CRC (RC), is also useful and essential for understanding colorectal carcinogenesis.8, 9 Although CRC with the MSI‐high phenotype preferentially occurs in the right‐sided colon, it is rarely found in the left‐sided colon.8, 9 However, the MSS phenotype is the main molecular phenotype in both LC and RC.10, 11 Previous reports have shown that the MSS phenotype is found in approximately 60–70% of RC and around 90% of LC.3, 7 To identify the molecular or clinicopathological differences between LC and RC, we must examine the molecular or clinicopathological differences in the MSS phenotype between LC and RC, given that the MSS phenotype is common in CRC.
In the current study, we aimed to validate the molecular profiles of MSS CRC based on tumor location.
Material and Methods
Patients
Tumor specimens were obtained from 92 patients who had undergone colectomy at Iwate Medical University Hospital (Iwate, Japan) between 2013 and 2015. Clinicopathological findings, including age, sex, tumor location, histological classification, lymphatic invasion, venous invasion and tumor stage, were determined according to the Classification of the Japanese Society for Cancer of the Colon and Rectum.13 No preoperative neoadjuvant therapy or radiotherapy was given to any of these patients.
All patients who participated in this study provided written informed consent, and the study was approved by the Ethical Committee of Iwate Medical University.
Crypt isolation method
Crypt isolation from the tumor and normal mucosa was performed in accordance with previously reported methods.14, 15 Briefly, fresh mucosa and tumor samples were minced with a razor into small pieces and then incubated at 37°C for 60 min in calcium‐ and magnesium‐free Hanks’ balanced salt solution (CMF) containing 30 mM ethylenediaminetetraacetic acid (EDTA). The tissue was then stirred in CMF for 30–40 min. The isolated crypts were immediately fixed in 70% ethanol and stored at 4°C until DNA extraction.
The fixed isolated crypts were observed under a dissecting microscope (SZ60; Olympus, Tokyo). Some isolated crypts were routinely processed for histopathological analysis to morphologically confirm their isolated nature. No contamination, such as interstitial cells, was observed in any of our 92 samples.
DNA extraction
DNA from normal and tumor glands was extracted by standard SDS proteinase K treatment. DNA extracted from the samples was resuspended in TE buffer (10 mM Tris‐HCl, 1 mM EDTA [pH 8.0]).
Analysis of MSI
The extracted DNA was amplified by polymerase chain reaction (PCR) with fluorescent dye‐labeled primers targeting five microsatellite loci: BAT25, BAT26, D5S346, D2S123 and D17S250. DNA was detected using a DNA sequencer (ABI PRISM‐310 Genetic Analyzer; ABI), and fragment analyses were carried out with GeneScan software (ABI). MSI status was determined by the presence of additional bands in the PCR product from tumor DNA. MSI‐high was defined as instability in at least two of the five microsatellite loci; MSI‐low was defined as instability in only one locus; and MSS was defined as having no shifted loci according to NCI criteria.16 In the present study, MSI‐low was excluded.
Analysis of KRAS and BRAF mutations
Mutations in KRAS and BRAF genes were examined using a pyrosequencer (Pyromark Q24; Qiagen NV), as previously described.6 The primers used in the present study were described previously.6
Analysis of TP53 and PIK3CA mutations
Mutations in the TP53 gene in exons 5–8 and in the PIK3CA gene in exons 9 and 20 were identified by PCR single‐stranded conformation polymorphism (PCR‐SSCP). The conditions for PCR were described previously.17 SSCP analysis was performed as previously described, with some modifications.17 Briefly, the PCR products (2 μL) were mixed with 10 μL of gel loading solution (9.5% deionized formamide, 20 mM EDTA‐Na, 0.05% xylene cyanol and bromphenol blue) and then denatured at 95°C for 5 min. Nondenaturing 7.5% polyacrylamide gels were used for electrophoresis at 260–300 V for 3–12 h, with the temperature controlled at 22°C using a temperature controller (Resolmax, ATTO Co., Tokyo, Japan). The gels were visualized by silver staining and photographed. The migrated band was removed from the gel, and the DNA was extracted. Suspected mutations obtained by SSCP in the TP53 gene and PIK3CA gene were then confirmed by sequence analysis.17
Pyrosequencing for evaluation of methylation
The DNA methylation status was determined by PCR analysis of bisulfite‐modified genomic DNA (EpiTect Bisulfite Kit; Qiagen) using pyrosequencing for quantitative methylation analysis (Pyromark Q24; Qiagen NV). The primers used in this study were designed previously.6, 18
DNA methylation was quantified using six specific promoters originally described by Yagi and collegues.18, 19 Briefly, after methylation analysis of the first panel of three markers (RUNX3, MINT31 and LOX), tumors with hypermethylated epigenomes (HMEs) were defined as those with at least two methylated markers. The remaining tumors were examined using a second panel of three markers (NEUROG1, ELMO1 and THBD). Tumors with intermediate methylated epigenomes (IMEs) were defined as those with at least two methylated markers, and tumors not classified as having HMEs or IMEs were designated as showing hypomethylated epigenomes (LMEs). The cut‐off value for the mutation assay was 15% mutant alleles, while that for the methylation assay was 30% of tumor cells, as previously reported.6
Copy number alteration analysis
Extracted DNA was adjusted to a concentration of 50 ng/μL. All 92 paired samples were assayed using the Infinium HumanCytoSNP‐12v2.1 BeadChip (Illumina, San Diego, CA), which contains 299,140 single nucleotide polymorphism (SNP) loci, according to the Illumina Infinium HD assay protocol. BeadChips were scanned using iScan (Illumina) and analyzed using GenomeStudio software (v.2011.1; Illumina). The log R ratio (LRR) and B allele frequency (BAF) for each sample were exported from normalized Illumina data using GenomeStudio. Data analysis was carried out using KaryoStudio 1.4.3 (CNV Plugin v3.0.7.0; Illumina) with default parameters. CNAs were classified as described below. In the classification of chromosome copy number alterations by CNA partition algorithms, LRR 0 indicated a normal diploid region, LRR > 0 indicated a copy number gain and LRR < 0 indicated a copy number loss‐of‐heterozygosity (LOH). BAF values ranged from 0 to 1; homozygous SNPs had BAFs near 0 (A‐allele) or 1 (B‐allele), and heterozygous diploid region SNPs had BAFs near 0.5 (AB genotype). Additionally, LRR and BAF data were used to identify regions of hemizygous and copy‐neutral LOH.
Calculation of the length of CNAs on a genome‐wide scale in CRCs
To quantify CNAs on a genome‐wide scale, we calculated the total lengths of CNAs (losses + gains), total length of CNA gain, total length of CNA LOH and total length of CNA‐copy neutral LOH identified by the SNP‐array analysis, as previously described.20 We, therefore, used the total CNA length as an index representing the degree of chromosomal alterations and assessed the relationship between CNA length (total CNA, CNA gain, CNA LOH and CNA copy‐neutral LOH) and LC or RC.
Statistical analysis
Data obtained for histological features, mutations, methylation and CNA status based on each subgroup were analyzed using χ2 tests with Yates’ correction with the aid of Stat Mate‐III software (Atom, Tokyo, Japan). Differences in age distributions between the two groups were analyzed using Mann‐Whitney U tests (PRISM6; GraphPad software, La Jolla, CA). Differences with p‐values of less than 0.05 were considered significant.
Results
Analysis of MSI and its association with clinicopathological findings in LC and RC
Based on the MSI status, 92 tumors were classified into 71 MSS tumors and 21 MSI tumors (MSI‐low, seven cases; MSI‐high, 14 cases). Although the frequency of CRC with the MSS phenotype in women was higher in RC than in LC, there was no significant difference between women and men. The median age of patients with RC was significantly higher than that in patients with LC (p = 0.01). The clinicopathological findings are listed in Table 1.
Table 1.
Clinicopathological features in patients with left‐ and right‐sided colorectal cancers with the microsatellite stable phenotype
| Characteristics | Left‐sided MSS CRCs (%) | Right‐sided MSS CRCs (%) | p‐values |
|---|---|---|---|
| Total | 56 | 15 | |
| Gender | |||
| Male | 33 (58.9) | 5 (33.3) | NS |
| Female | 23 (41.1) | 10 (66.7) | |
| Age | |||
| Median (range) | 68 (39–87) | 76 (63–90) | 0.01 |
| Differentiation | |||
| WDA | 1 (1.8) | 1 (6.7) | NS |
| MDA | 55 (98.2) | 14 (93.3) | |
| Lymphatic invasion | |||
| Negative or low | 52 (92.9) | 15 (100) | NS |
| High | 4 (7.1) | 0 | |
| Venous invasion | |||
| Negative or low | 50 (89.3) | 13 (86.7) | NS |
| High | 6 (10.7) | 2 (13.3) | |
| TNM stage | |||
| I | 9 (16.1) | 2 (13.3) | NS |
| II | 16 (28.5) | 3 (20.0) | |
| III | 23 (41.1) | 7 (46.7) | |
| IV | 8 (14.3) | 3 (20.0) | |
Abbreviations: WDA: well‐differentiated adenocarcinoma; MDA: moderately differentiated adenocarcinoma; Other: mucinous carcinoma or papillary carcinoma. CRCs: colorectal cancer.
Molecular alterations in LC and RC with the MSS phenotype
There was a significant difference in the frequency of TP53 mutations in LC (33/56, 58.9%) compared with that in RC (3/15, 20%; p = 0.006). In addition, a significant difference in the frequency of KRAS mutations was observed between LC (16/56, 28.6%) and RC (11/15, 73.3%; p = 0.001). However, no differences were found in the frequencies of BRAF and PIK3CA mutations between LC and RC. These results are depicted in Table 2‐a.
Table 2.
Frequencies of mutations and DNA methylation statuses in left‐and right‐sided colorectal cancers with the microsatellite stable phenotype
| 2‐a | ||||
|---|---|---|---|---|
| Mutated | Gene | Left‐sided MSS CRCs (%) | Right‐sided MSS CRCs (%) | p‐values |
| Total | 56 | 15 | ||
| TP53 | positive | 33 (58.9) | 3 (20.0) | 0.006 |
| KRAS | positive | 16 (28.6) | 11 (73.3) | 0.001 |
| BRAF | positive | 1 (1.8) | 0 | NS |
| PIK3CA | positive | 5 (8.9) | 1 (6.7) | NS |
| Chi‐square test | ||||
| 2‐b | ||||
|---|---|---|---|---|
| Left‐sided MSS CRCs (%) | Right‐sided MSS CRCs (%) | p‐values | ||
| Total | 56 | 15 | ||
| HME | 3 (5.3) | 0 | NS | |
| IME | 28 (50.0) | 11 (73.3) | ||
| LME | 25 (44.7) | 4 (26.7) | ||
| Chi‐square test | ||||
Abbreviations: CRCs: colorectal cancers; HME: high méthylation phenotype; IME: intermediate methylation phenotype; LME: low methylation phenotype.
Although the HME status was not found in RC (0/15), three of 56 LC (5.3%) showed the HME status (Table 2‐b). IME and LME statuses were common in LC and RC (Table 2‐b).
Differences in mutations in cancer‐related genes between LC and RC based on methylation status
Next, we examined mutations in TP53, KRAS, BRAF and PIK3CA genes based on the methylation status in LC and RC. The frequency of KRAS mutations based on the IME was significantly higher in RC (9/11, 81.1%) than in LC (9/28, 32.1%; p = 0.01). In addition, there was a significant difference in the frequency of TP53 mutations in the IME between LC (2/11, 18.2%) and RC (18/28, 64.3%; p = 0.02). There were no correlations in BRAF (0 in LC and RC) or PIK3CA mutations (3/28 [10.7%] in LC and 1/11 [9.1%] in RC) based on the IME between LC and RC. Moreover, there were no significant differences in the frequencies of TP53 (15/25 [60%] in LC; 1/4 [25%] in RC), KRAS (6/25 [24%] in LC; 2/4 [50%] in RC), BRAF (0/25 [0%] in LC; 0/4 [0%] in RC) and PIK3CA mutations (1/25 [4%] in LC; 0/4 [0%] in RC) based on the LME between LC and RC.
Differences in genomic alterations between LC and RC with the MSS phenotype
The 71 pairs of CRC samples were examined using SNP arrays to detect differences between LC and RC. The CNAs of all chromosomes according to LC and RC are shown in Figure 1. The frequency of CNAs was detected across the entire genome. Genomic CNAs detected in ≧50% of cases are summarized in Table 3. The mean total number of chromosomal aberrations per patient was 447 with an average of 280 gains, ranging from 64 to 568; the mean number of LOHs was 53, ranging from 0 to 184 and the mean number of copy‐neutral LOHs averaged 57, ranging from 0 to 292 in LC. On the other hand, the mean total number of chromosomal aberrations per patient was 219, with an average of 150 gains ranging from 1 to 384; there was an average of 30 LOHs, ranging from 0 to 99 and we found mean number of 42 copy‐neutral LOHs, ranging from 0 to 282, in RC. Regions of gain detected in ≧50% of cases were located at 8p11.1–11.23, 8q, 20p, 20q, 13q, 7p, 7q11–21.11 and 7q21.12‐36.3 (in decreasing order of frequency) in LC and at 13q12.13‐12.3, 20q13.33, 7p21.1–2, 7q31.33, 8q11.21–13.3, 13q12.11–12 and 20q11.21–13.32 in RC. Regions of loss (LOH) detected in ≧50% of cases were found at 8p12‐23.1, 17p11.2‐13.1, 18p and 18q in LC and at 18q21.1–22.3, 18p11.21–32 and 18q11.2‐12.3 in RC. Copy‐neutral LOH was not detected in either LC or RC. These results are summarized in Table 3.
Figure 1.
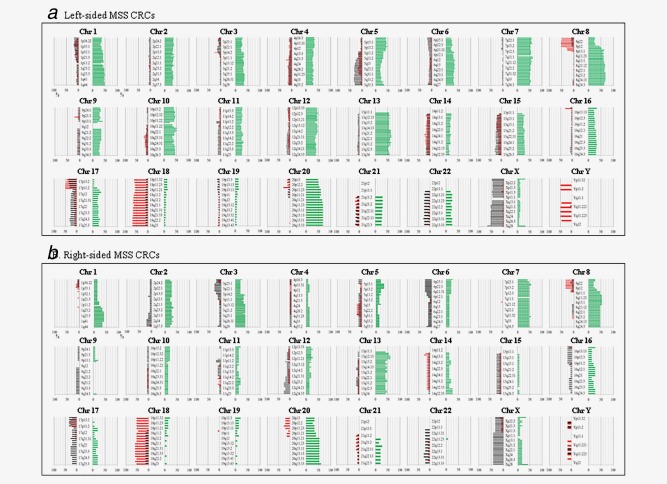
Ideogram of genomic imbalance in 92 colorectal cancers. Chromosomes are ordered from 1 to 22. The colored horizontal lines represent the frequencies of gains, LOHs, and copy‐neutral LOHs. Lines on the left indicate losses (red, LOHs; gray, copy‐neutral LOHs) and those on the right (green) indicate gains. (a). Ideogram of genomic imbalance in left‐sided colorectal cancer. (b). Ideogram of genomic imbalance in right‐sided colorectal cancer.
Table 3.
Comparison of regions frequently harboring CNAs in left‐ and right‐sided colorectal cancers with the micro satellite stable phenotype
| Chromosomal regions | Left‐sided MSS CRCs n = 56 (%) | Chromosomal regions | Right‐sided MSS CRCs n = 15 (%) |
|---|---|---|---|
| Gain | Gain | ||
| 8p11.1‐11.23 | 28–35(50.0–62.5) | 13q12.13‐12.3 | 9(60.0) |
| 8q | 31–38(55.3–67.8) | 20q13.33 | 9(60.0) |
| 20p | 28–33(50.0–58.9) | 7p21.1‐2 | 8(53.3) |
| 20q | 36–38(64.2–67.8) | 7q31.33 | 8(53.3) |
| 13q | 29–36(51.7–64.2) | 8q11.21‐13.3 | 8(53.3) |
| 7p | 29–33(51.7–58.9) | 13q12.11‐12 | 8(53.3) |
| 7q11‐21.11 | 33–35(58.9–62.5) | 20q11.21‐13.32 | 8(53.3) |
| 7q21.12‐36.3 | 28–31 (50–55.9) | ||
| LOH | LOH | ||
| 8p 12‐23.1 | 28–29(50.0–51.7) | 18q21.1‐22.3 | 9(60.0) |
| 17p11.2‐13.1 | 28(50.0) | 18q11.2‐12.3 | 8(53.8) |
| 18p | 32–34 (57.1–60.7) | 18p11.21‐32 | 8(53.8) |
| l8q | 33–38(58.9–67.8) | ||
| Copy‐neutral LOH | Copy‐neutral LOH | ||
| None | None |
Abbreviations: CRCs: colorectal cancers; LOH: loss of heterozygosity.
Genomic differences between LC and RC with the MSS phenotype
Next, we examined differences in CNAs between LC and RC, as shown in Table 4. Regions of gain detected in >30% of cases were selected for comparison of LC with RC. Significant differences in gains between LC and RC were found at 10q21.1, 10q21.3, 12q14.1, 12q14.2, 9q21.11, 9q21.12, 9q21.33, 9q22.1, 9q22.31 and 9q22.32 (Table 4). We also examined regions of loss found in ≧20% of cases of LC and RC. Significant differences in the frequencies of copy‐neutral LOH between LC and RC were found at 3p24.2, 3p24.3, 16p12.1 and 16p13.2 (Table 4). However, no differences in the frequency of LOH were found between LC and RC.
Table 4.
Significant differences in the frequencies of CNAs between left‐ and right‐sided colorectal cancers with the microsatellite stable phenotype.
| Chromosomal regions | Left‐sided MSS CRCs n = 56 (%) | Right‐sided MSS CRCs n = 15 (%) | p‐values |
|---|---|---|---|
| Gain | |||
| 10q21.1 | 27(48.2) | 2(13.3) | 0.03 |
| 10q21.3 | 27(48.2) | 2(13.3) | 0.03 |
| 12q14.1 | 27(48.2) | 2(13.3) | 0.03 |
| 12q14.2 | 27(48.2) | 2(13.3) | 0.03 |
| 9q21.11 | 17(30.3) | 0 | 0.04 |
| 9q21.12 | 17(30.3) | 0 | 0.04 |
| 9q21.33 | 17(30.3) | 0 | 0.04 |
| 9q22.1 | 17(30.3) | 0 | 0.04 |
| 9q22.31 | 17(30.3) | 0 | 0.04 |
| 9q22.32 | 17(30.3) | 0 | 0.04 |
| LOH | None | ||
| Copy‐neutral LOH | |||
| 3p24.2 | 3(5.3) | 5(33.3) | 0.01 |
| 3p24.3 | 4(7.1) | 5(33.3) | 0.02 |
| 16p12.1 | 1 (1.7) | 3(20.0) | 0.04 |
| 16p13.2 | 1(1.7) | 3 (20.0) | 0.04 |
Abbreviations: CRCs: colorectal cancers; LOH: loss of heterozygosity χ2 test.
Association of the length of CNAs on the genome‐wide scale in LC and RC with the MSS phenotype
Overall, the total length of CNAs was longer in LC than in RC (Fig. 2; p < 0.03). We analyzed genomic losses (LOH and copy‐neutral LOH) and gains separately. The total length of CNA gains was significantly longer in LC than in RC (Fig. 2; p < 0.02). No significant differences in the total length of losses (LOH and copy‐neutral LOH) were found between LC and RC.
Figure 2.
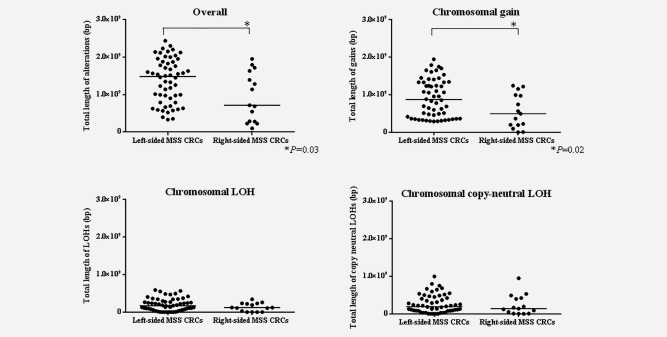
Comparison of the total lengths of abnormal regions in CNAs of each subject for left‐ and right‐sided colorectal cancer with the microsatellite stable phenotype.
Associations of clinicopathological or molecular profiles of rectal cancers with LC and LC excluding rectal cancer
No differences were observed in clinicopathological features between LC (excluding rectal cancer) and rectal cancer. There were no differences in the frequencies of mutations (KRAS, BRAF, TP53 and PIK3CA) between LC (excluding rectal cancer) and rectal cancer. In addition, no differences were observed in the methylation status (HME, IME and LME). Genomic CNAs detected in ≧50% of cases are summarized in Supporting Information Table 1. In rectal cancer, the mean total number of chromosomal aberrations per patient was 387 with an average of 202 gains, ranging from 64 to 511; the mean number of LOHs was 66, ranging from 0 to 292, and the mean number of copy‐neutral LOHs averaged 49, ranging from 0 to 292. In contrast, in LC excluding rectal cancer, the mean total number of chromosomal aberrations per patient was 475, with an average of 338 gains ranging from 100 to 568; there was an average of 46 LOHs, ranging from 0 to 184, and we found a mean number of 78 copy‐neutral LOHs, ranging from 0 to 244. Regions of gain detected in ≧50% of cases were located at 7p, 7q11–21.11, 8p, 8q, 13q, 20p and 20q (in decreasing order of frequency) in rectal cancers and at 7p, 7q11–21.11, 8p, 8q, 13q, 20p and 20q in LC excluding rectal cancers. Regions of loss (LOH) detected in ≧50% of cases were found at 8p12‐23.1, 18q and 18p in rectal cancers and at 8p12‐23.1, 18p and 18q in LC excluding rectal cancers. Copy‐neutral LOH was not detected in either rectal cancers or LC excluding rectal cancers. Regions of gain detected in >30% of cases were not selected for comparisons of rectal cancers with LC excluding rectal cancers.
Finally, representative features of isolated glands are shown in Supporting Information Figure 1.
Discussion
Multiple studies have shown that the molecular characteristics of CRC vary according to anatomical sites, including LC and RC; therefore, researchers have focused on the potential etiologies of these two types of CRC.8, 9, 21, 22 Differences between LC and RC have been found. For example, RC is more likely to have mucinous histology, to be diploid, and to exhibit a high frequency of MSI and CpG island methylation.7, 9, 12 whereas LC often exhibits a differentiated histology, have chromosomal instability, and are more often aneuploidy.7, 9, 11 Classification based on tumor location is simple, comprehensive and consistent with recent attempts to characterize tumors by pathological and molecular features.8, 9 However, for validation of the distinct molecular profiles of CRC based on tumor location, it is necessary to identify differences in molecular alterations between LC and RC with the MSS phenotype. It is unclear whether these biological differences translate into meaningful differences in colorectal carcinogenesis. In the present study, the clinical and research value of this simple approach based on tumor location and tumor classification was evaluated.
The TP53 gene is one of the most frequently mutated genes in human cancers.4, 5, 23, 24 Approximately half of all cancers have mutated TP53.4, 5 In addition, mutation of the TP53 gene is found in 60–80% of CRCs.4, 5, 7 Mutation of the TP53 gene is a representative event during the progression of CRC.4, 5, 23, 24 Recent studies have shown that mutation of TP53 is observed more frequently in LC than in RC.9, 25 These findings are consistent with our study. In the present study, although the MSS type in LC was characterized by TP53 mutations, TP53 mutations in RC with the MSS type were only infrequently detected. TP53 mutations are generally believed to occur at the time of transition from adenoma to cancer, suggesting that TP53 gene mutations accumulate within the cancer tissue during cancer progression.6, 7 These findings support the notion that the genetic mechanisms defining the conversion of adenoma into carcinoma may differ between RC and LC.
KRAS mutations are closely associated with progression from low‐grade adenoma to high‐grade adenoma.4, 6, 7, 23 Therefore, mutation of KRAS is thought to be a critical genetic event in colorectal carcinogenesis.4, 6, 7, 23 KRAS mutations have recently emerged as a major predictor of resistance to epidermal growth factor (EGF) receptor (EGFR) inhibitors.26 The EGF ligand activates a signaling cascade via 2 pathways, that is, the RAS‐RAF‐mitogen‐activated protein kinase (MAPK) and phosphoinositide 3‐kinase (PI3K)‐AKT pathways, suggesting that KRAS mutations may be a key molecular factor mediating the efficacy of EGFR monoclonal antibody therapy in CRC.26, 27 Approximately 30–50% of colorectal tumors are known to harbor mutated KRAS, indicating that up to 50% of patients with CRC may benefit from specific treatment strategies based on KRAS status.23, 26, 27 In the present study, the frequency of KRAS mutations was found to be significantly higher in RC than in LC. Although previous reports are consistent with our data,28, 29 the finding that KRAS mutations preferentially occur in RC with the MSS phenotype has not been previously demonstrated. In addition, this finding also suggested that patients having LC with the MSS phenotype may benefit from EGFR antibody therapy, whereas those having RC with the MSS phenotype may not benefit. This is an interesting finding and suggests that the strategy for molecular‐targeted therapy in patients with the MSS phenotype may differ among those with LC and RC.
DNA methylation plays a major role in colorectal carcinogenesis.30, 31 DNA methylation can be classified into three types (HME, IME and LME) according to the 2‐panel method established by Yagi and Kaneda.18, 19 HME corresponds to CIMP‐high, as proposed by Toyota et al.18, 19, 30, 31 Most sporadic MSI colon tumors (MSI‐high tumors) are CIMP positive, whereas CIMP is uncommon in MSS cancer that exhibits genomic instability at the chromosomal level, indicative of distinct underlying molecular processes.18, 19, 31 Thus, CIMP is significantly more frequent in tumors of sporadic MSI cancers and is associated with BRAF mutations.4, 5, 12 In the present study, our results showed that although IME and LME were commonly found in LC and RC with the MSS phenotype, HME was rarely observed in MSS‐type CRCs. Kaneda et al. found that methylation was sufficiently accumulated at the adenoma stage to develop an epigenotype, suggesting that accumulation of aberrant promoter methylation was mostly complete at the adenoma stage.18, 19 This finding suggests that MSS‐type CRC has an epigenotype similar to that of early precursor lesions, irrespective of tumor location.6, 18, 19
Recent studies have shown that IME with KRAS mutation is closely associated with tumor survival.18, 19 In the present study, a significant difference was found in the frequency of KRAS mutation in the intermediate methylation phenotype between LC and RC with the MSS phenotype. These results support the finding that patients with RC have a significantly worse prognosis than those with LC in terms of overall survival.10 In addition this finding may suggest that RC should be treated distinctively from LC. In the present study, the frequency of TP53 mutation in the IME of LC was significantly higher than that of RC. This finding is interesting and is based on the possibility that TP53 mutation in LC is associated with a specific methylation status (i.e., IME in the present study). However, the relevance of this finding to colorectal carcinogenesis is unknown. Further studies are needed to investigate the association between methylation status and specific mutations.
To the best of our knowledge, no other studies have reported a comprehensive genomic analysis of MSS phenotype based on tumor location. The key finding of this study was the discovery of a potential genomic stratification marker in LC and RC with the MSS phenotype, that is, gains at 10q21.1–21.3, 12q14.1–14.2 and 9q21–22 and losses at 3p24.2–24.3 and 16p12.1–13.2. These findings suggested that gains at 10q21.1–21.3, 12q14.1–14.2 and 9q21–22 were closely associated with the development of LC with the MSS phenotype, whereas losses (copy‐neutral LOH) at 3p24.2–24.3 and 16p12.1–13.2 may characterize the progression of RC with the MSS phenotype. Although we identified genomic alterations associated with tumor location, additional research is needed to understand the molecular differences between RC and LC.
Numerous genes have been mapped to the regions of gains at 10q21.1–21.3, 12q14.1–14.2 and 9q21–22 and losses at 3p24.2–24.3 and 16p12.1–13.2 found in this study. However, many genes in these regions are not eligible as candidate genes with major roles in colorectal carcinogenesis. Of the many candidate genes, we identified cyclin‐dependent kinase (CDK) 4/6 on 12q14.1–14.2,32, 33 death‐associated protein kinase 1 (DAPK1) on 9q21–22,34 retinoic acid receptor (RAR) β235 and topoisomerase II beta (TOP2B) on 3p2436 as representative genes contributing to tumor development. Of these candidate genes, we selected CDK4/6 and RARβ2 as genes involved in colorectal tumorigenesis. Thus, further studies are needed to elucidate the roles of these two genes in colorectal carcinogenesis.
CIN is observed in approximately 80–90% of sporadic CRCs4, 5, 7 and is characterized by an accelerated rate of gains or losses in whole or large portions of chromosomes, resulting in karyotypic variability.11 CIN causes imbalances in chromosome numbers (aneuploidy), sub‐chromosomal genomic amplifications and a high frequency of LOH.11 In this study, we aimed to analyze the association of CNAs with tumor location in CRC and to understand the role of CNAs in sporadic CRC development according to tumor location. We examined the total length of CNAs to identify genomic damage in LC and RC with the MSS phenotype. Notably, although total length of loss (LOH or copy‐neutral LOH) of CNAs did not differ among LC and RC with the MSS phenotype, the total lengths of overall CNAs and gain of CNAs were significantly longer in LC with the MSS phenotype than in RC with the MSS phenotype. This finding suggested that cellular genomic destabilization was enhanced to a greater degree in LC than in RC, indicating that CIN in cancer cells was caused by TP53 mutations.37 These findings provide important insights into the pathogenesis of CRC with the MSS phenotype.
Rectal cancers have been reported to be distinct from other distal cancers.38, 39 The few studies that have addressed the molecular and/or biological differences between the two diseases (rectal cancers and other distal LC) have focused on a single marker, such as KRAS, BRAF, MSI or LOH.38, 39 In the present study, however, we found no differences in clinical or molecular findings between rectal cancer and other distal cancers. Our study was a small‐sized study and was different from previous studies in that we performed a comprehensive molecular analysis. To identify differences in clinical and molecular findings between the two cancers, more detailed studies should be performed using a larger cohort in the near future.
In conclusion, we examined differences in genetic alterations between LC and RC with the MSS phenotype. Although TP53 mutations were closely associated with the development of LC with the MSS phenotype, RC with the MSS phenotype was characterized by KRAS mutations. IME and LME statuses were commonly observed in CRC with the MSS phenotype, regardless of the location. An integrated genome‐wide analysis showed that there were significant differences (total lengths of gains and overall CNAs) in CNAs between LC and RC with the MSS phenotype. Thus, significant differences in chromosomal regions were detected between LC and RC; however, further studies are needed to confirm these results. Moreover, although our study included a small number of patients, our findings provide important insights into the molecular profiles of CRC according to tumor location and improve our understanding of colorectal carcinogenesis. In addition, although a second cohort is needed to validate the present results, we believe that our results will improve our understanding of colorectal carcinogenesis.
Supporting information
Supporting Information Figure 1
Supporting Information Table 1
Acknowledgements
The authors gratefully acknowledge the technical assistance of Ms. E. Sugawara and Mr. T. Kasai. They also thank members of the Department of Molecular Diagnostic Pathology, Iwate Medical University for their support. The authors have no conflicts of interest to declare.
References
- 1. Haggar FA, Boushey RP. Colorectal cancer epidemiology: incidence, mortality, survival, and risk factors. Clin Colon Rectal Surg 2009;22:191–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Herszényi L, Tulassay Z. Epidemiology of gastrointestinal and liver tumors. Eur Rev Med Pharmacol Sci 2010;14:249–58. [PubMed] [Google Scholar]
- 3. Lengauer C, Kinzler KW, Vogelstein B. Genetic instability in colorectal cancers. Nature 1997;386:623–27. [DOI] [PubMed] [Google Scholar]
- 4. Ogino S, Goel A. Molecular classification and correlates in colorectal cancer. J Mol Diagn 2008;10:13–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012;487:330–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yamamoto E, Suzuki H, Yamano HO, et al. Molecular dissection of premalignant colorectal lesions reveals early onset of the CpG island methylator phenotype. Am J Pathol 2012;181:1847–61. [DOI] [PubMed] [Google Scholar]
- 7. Jass JR, Whitehall VL, Young J, et al. Emerging concepts in colorectal neoplasia. Gastroenterology 2002;123:862–76. [DOI] [PubMed] [Google Scholar]
- 8. Iacopetta B. Are there two sides to colorectal cancer? Int J Cancer 2002;101:403–8. [DOI] [PubMed] [Google Scholar]
- 9. Sugai T, Habano W, Jiao Y‐F, et al. Analysis of molecular alterations in left‐ and right‐sided colorectal carcinomas reveals distinct pathways of carcinogenesis: proposal for new molecular profile of colorectal carcinomas. J Mol Diagn 2006;8:193–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yahagi M, Okabayashi K, Hasegawa H, et al. The worse prognosis of right‐sided compared with left‐sided colon cancers: a systematic review and meta‐analysis. J Gastrointest Surg 2016;20:648–55. [DOI] [PubMed] [Google Scholar]
- 11. Pino MS, Chung DC. The chromosomal instability pathway in colon cancer. Gastroenterology 2010;138:2059–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Leggett B, Whitehall V. Role of the serrated pathway in colorectal cancer pathogenesis. Gastroenterology 2010;138:2088–100. [DOI] [PubMed] [Google Scholar]
- 13. Japanese Society for Cancer of the Colon and Rectum . Japanese classification of colorectal carcinoma, Second English Edition Tokyo: Kanehara Co, 2009. 30–63. [Google Scholar]
- 14. Arai T, Kino I. Morphometrical and cell kinetic studies of normal human colorectal mucosa: comparison between the proximal and the distal large intestine. Acta Pathol Jpn 1989;39:725–30. [DOI] [PubMed] [Google Scholar]
- 15. Nakamura S, Goto J, Kitayama M, et al. Application of the crypt‐isolation technique to flow‐cytometric analysis of DNA content in colorectal neoplasms. Gastroenterology 1994;106:100–7. [DOI] [PubMed] [Google Scholar]
- 16. Boland CR, Thibodeau SN, Hamilton SR, et al. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res 1998;58:5248–57. [PubMed] [Google Scholar]
- 17. Sugai T, Habano W, Nakamura S, et al. A unique method for mutation analysis of tumor suppressor genes in colorectal carcinomas using a crypt isolation technique. Arch Pathol Lab Med 2000;124:382–6. [DOI] [PubMed] [Google Scholar]
- 18. Yagi K, Takahashi H, Akagi K, et al. Intermediate methylation epigenotype and its correlation to KRAS mutation in conventional colorectal adenoma. Am J Pathol 2012;180:616–25. [DOI] [PubMed] [Google Scholar]
- 19. Kaneda A, Yagi K. Two groups of DNA methylation markers to classify colorectal cancer into three epigenotypes. Cancer Sci 2011;102:18–24. [DOI] [PubMed] [Google Scholar]
- 20. Sawada T, Yamamoto E, Suzuki H, et al. Association between genomic alterations and metastatic behavior of colorectal cancer identified by array‐based comparative genomic hybridization. Genes Chromosomes Cancer 2013;52:140–9. [DOI] [PubMed] [Google Scholar]
- 21. Richman S, Adlard J. Left and right sided large bowel cancer. BMJ 2002;324:931–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Weiss JM, Pfau PR, O'Connor ES, et al. Mortality by stage for right‐ versus left‐sided colon cancer: analysis of surveillance, epidemiology, and end results–Medicare data. J Clin Oncol 2011;29:4401–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sugai T, Habano W, Uesugi N, et al. Molecular validation of the modified Vienna classification of colorectal tumors. J Mol Diagn 2002;4:191–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Russo A, Bazan V, Iacopetta B, et al. The TP53 colorectal cancer international collaborative study on the prognostic and predictive significance of p53 mutation: influence of tumor site, type of mutation, and adjuvant treatment. J Clin Oncol 2005;23:7518–28. [DOI] [PubMed] [Google Scholar]
- 25. Park SY, Lee HS, Choe G, et al. Clinicopathological characteristics, microsatellite instability, and expression of mucin core proteins and p53 in colorectal mucinous adenocarcinomas in relation to location. Virchows Arch 2006;449:40–7. [DOI] [PubMed] [Google Scholar]
- 26. Wheeler DL, Dunn EF, Harari PM. Understanding resistance to EGFR inhibitors‐impact on future treatment strategies. Nat Rev Clin Oncol 2010;7:493–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Siddiqui AD, Piperdi B. KRAS mutation in colon cancer: a marker of resistance to EGFR‐I therapy. Ann Surg Oncol 2010;17:1168–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bleeker WA, Hayes VM, Karrenbeld A, et al. Impact of KRAS and TP53 mutations on survival in patients with left‐ and right‐sided Dukes' C colon cancer. Am J Gastroenterol 2000;95:2953–7. [DOI] [PubMed] [Google Scholar]
- 29. Brulé SY, Jonker DJ, Karapetis CS, et al. Location of colon cancer (right‐sided versus left‐sided) as a prognostic factor and a predictor of benefit from cetuximab in NCIC CO.17. Eur J Cancer 2015;51:1405–14. [DOI] [PubMed] [Google Scholar]
- 30. Toyota M, Ahuja N, Ohe‐Toyota M, et al. CpG island methylator phenotype in colorectal cancer. Proc Natl Acad Sci USA 1999;96:8681–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shen L, Toyota M, Kondo Y, et al. Integrated genetic and epigenetic analysis identifies three different subclasses of colon cancer. Proc Natl Acad Sci USA 2007;104:18654–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ziemke EK, Dosch JS, Maust JD, et al. Sensitivity of KRAS‐mutant colorectal cancers to combination therapy that cotargets MEK and CDK4/6. Clin Cancer Res 2016;22:405–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kato S, Schwaederle M, Daniels GA, et al. Cyclin‐dependent kinase pathway aberrations in diverse malignancies: clinical and molecular characteristics. Cell Cycle 2015;14:1252–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Murria R, Palanca S, de Juan I, et al. Methylation of tumor suppressor genes is related with copy number aberrations in breast cancer. Am J Cancer Res 2014;5:375–85. [PMC free article] [PubMed] [Google Scholar]
- 35. Perraud A, Nouaille M, Akil H, et al. Retinoid acid receptors in human colorectal cancer: an unexpected link with patient outcome. Exp Ther Med 2011;2:491–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhang YJ, Li AJ, Han Y, et al. Inhibition of Girdin enhances chemosensitivity of colorectal cancer cells to oxaliplatin. World J Gastroenterol 2014;20:8229–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Raskov H, Pommergaard HC, Burcharth J, et al. Colorectal carcinogenesis–update and perspectives. World J Gastroenterol 2014;20:18151–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yamauchi M, Morikawa T, Kuchiba A, et al. Assessment of colorectal cancer molecular features along bowel subsites challenges the conception of distinct dichotomy of proximal versus distal colorectum. Gut 2012;61:847–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Minoo P, Zlobec I, Peterson M, et al. Characterization of rectal, proximal and distal colon cancers based on clinicopathological, molecular and protein profiles. Int J Oncol 2010;37:707–18. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information Figure 1
Supporting Information Table 1