

Summary
The components and reactions of the fibrinolysis system are well understood. The pathway has fewer reactants and interactions than coagulation, but the generation of a complete quantitative model is complicated by the need to work at the solid‐liquid interface of fibrin. Diagnostic tools to detect disease states due to malfunctions in the fibrinolysis pathway are also not so well developed as is the case with coagulation. However, there are clearly a number of inherited or acquired pathologies where hyperfibrinolysis is a serious, potentially life‐threatening problem and a number of antifibrinolytc drugs are available to treat hyperfibrinolysis. These topics will be covered in the following review.
Keywords: fibrinolysis, acute promyelocytic leukaemia, antiplasmin, bleeding disorders, plasminogen activator inhibitor type 1
The biochemistry of fibrinolysis
The basic mechanisms and regulation of fibrinolysis have been reviewed recently (Longstaff & Kolev, 2015) and will not be covered in detail. In summary, fibrinolysis is readily considered as two consecutive steps: (i) the generation of plasmin by plasminogen activators, and (ii) the digestion of fibrin by plasmin. The whole system is kept in check by a system of protease inhibitors and other regulatory mechanisms. Some key reactions and structural background are highlighted in Fig 1.
Figure 1.
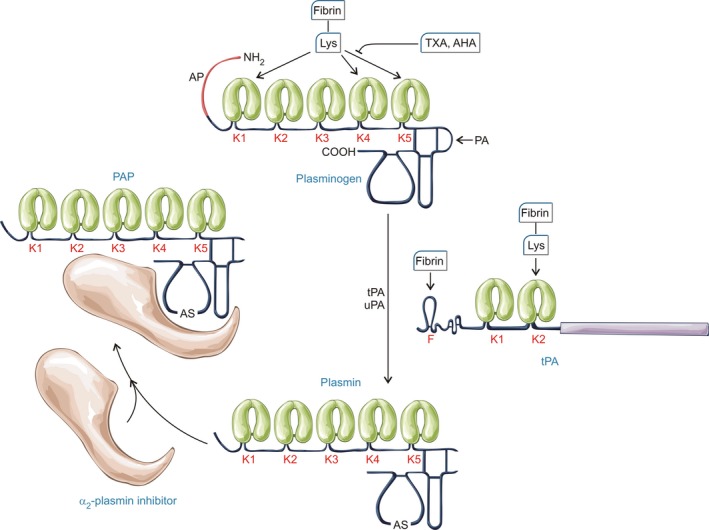
Plasminogen generation and inhibition. Activation of the zymogen plasminogen to the serine protease plasmin is generally a simple peptide bond cleavage (at Arg561) catalysed by tissue‐type plasminogen activator (tPA) or urokinase‐type plasminogen activator (uPA). However, the underlying mechanisms of these two plasminogen activators are inherently different, which is important when considering their regulation. uPA is generated from a zymogen, single chain precursor, scuPA, whereas tPA is constitutively active in single chain and two chain forms (Thelwell & Longstaff, 2007). tPA activity is poor in free solution but stimulation is achieved via a co‐localization mechanism relying on the binding of plasminogen and tPA in close proximity on the fibrin surface (Horrevoets et al, 1997). Plasminogen binding is accomplished through kringle (K) domains, of which there are 5 with different affinities and specificities although K1, K4 and K5 seem to be most important for binding to lysine residues in fibrin. The primary inhibitor of tPA and uPA is the serpin, plasminogen activator inhibitor 1 (PAI‐1). PAI‐1 reacts rapidly with both target enzymes, although the rate is modulated by the presence of fibrinogen and fibrin (Thelwell & Longstaff, 2007). Similarly, α2‐plasmin inhibitor (α2‐PI also known as α2‐antiplasmin) reacts very rapidly with plasmin in free solution but the rate is significantly compromised in the presence of fibrin and lysine analogues. AP, activation peptide; PAP, plasmin‐antiplasmin complex; AS, active site; AHA, 6‐aminohexanoic acid; TXA, tranexamic acid.
It is important to remember that fibrin is not a passive target in fibrinolysis but plays an important regulatory role through the binding of reactants and focussing plasmin activity at cleavage sites (Varju et al, 2014). During the course of fibrin degradation, plasmin proteolysis generates C‐terminal lysines, which provide new binding sites, primarily for plasminogen and plasmin (Silva et al, 2012) and these nascent binding sites constitute an important positive feedback mechanism to accelerate fibrin degradation. Binding to lysine residues in fibrin can be blocked by soluble fibrin analogues, such as aminohexanoic acid (AHA, also known as epsilon aminocaproic acid, EACA) or tranexamic acid (TXA), which are discussed below. Tissue‐type plasminogen activator (tPA)‐fibrin binding can occur through lysine interactions with its kringle 2 domain, but the finger domain with different specificity is dominant (Longstaff et al, 2011; Silva et al, 2012). Urokinase‐type plasminogen activator (uPA) does not bind to fibrin, and is more sensitive to the conformation of plasminogen, which is affected by lysine residues in fibrin and free lysine analogues in solution (Sinniger et al, 1999; Silva et al, 2012). Recent crystal structures of plasminogen have greatly extended our understanding of the intra‐molecular interactions behind these structural changes, providing detailed molecular models to explain how plasminogen structure can regulate its activation (Xue et al, 2012; Law et al, 2013). Cell surface plasminogen activation shares some similarities with fibrin‐bound plasminogen activation, however this topic is beyond the scope of the current review. The particular example of enhanced cell surface plasminogen activation in acute promyelocytic leukaemia is covered below.
Inhibitors of fibrinolysis
As in the case of coagulation, regulation of fibrinolysis involves dedicated serpin protein inhibitors, operating at the level of plasminogen activation and plasmin action, outlined in Figs 1 and 2. A quite different mode of inhibition of fibrinolysis is accomplished by thrombin activatable fibrinolysis inhibitor (TAFIa, also termed CPB2, CPU). TAFI (proCPU), the zymogen form, is activated by thrombin or plasmin, but primarily by thrombin/thrombomodulin to TAFIa, a zinc‐dependent carboxypeptidase that removes C‐terminal lysine (and arginine) residues from fibrin thereby reducing the number of binding sites available for plasminogen and plasmin. The clinical consequences of disturbances in levels of α2 plasmin inhibitor (α2‐PI), plasminogen activator inhibitor type 1 (PAI‐1, also termed SERPINE1) and TAFI are discussed below. The gross structure of fibrin clots, including fibrin fibre thickness and branching, cellular components and other associated blood borne molecules, also modify clot physical and biochemical properties and susceptibility to lysis (Bridge et al, 2014). Hence the rate of fibrin clot breakdown is partly determined at the clot formation stage and has a bearing on the presentation of bleeding complications, as described in the section on secondary hyperfibrinolysis below.
Figure 2.
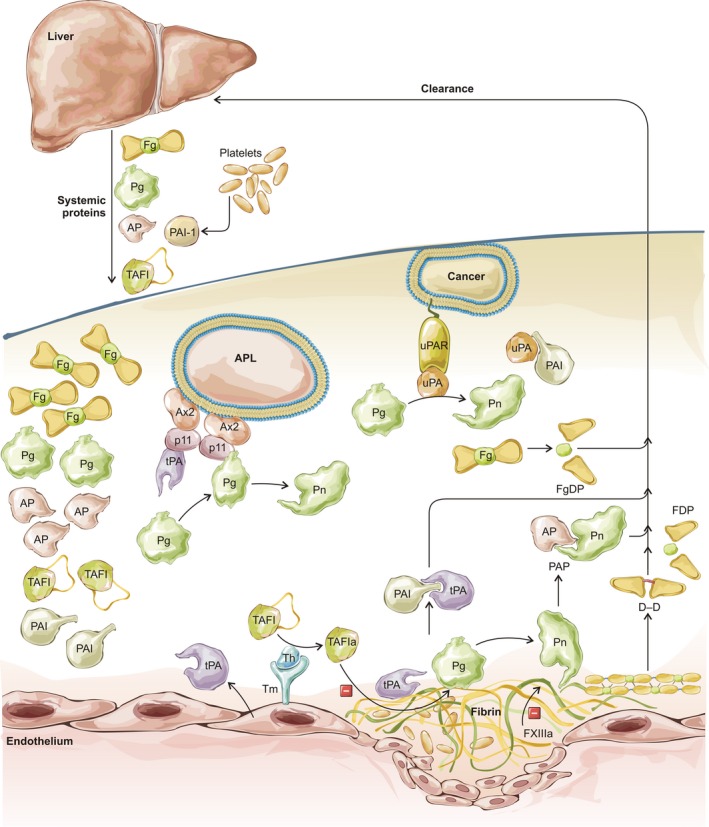
Overview of fibrinolysis including significant reactions, markers and points where disturbance may lead to clinical bleeding. Liver synthesises and releases fibrinogen (Fg), plasminogen (Pg), α2‐plasmin inhibitor (AP), thrombin activatable fibrinolysis inhibitor (TAFI) into the blood and takes up circulating fibrinogen and fibrin degradation products (FgDP, FDP, D‐D), as well as the complexes of proteases and their inhibitors (AP, plasminogen activator inhibitor 1, PAI). Fibrin and cells (myeloid precursor cells in acute promyelocytic leukaemia, APL, or malignant cells in cancer) provide a template for binding of plasminogen activators (tissue‐type, tPA and urokinase‐type, uPA) and plasminogen for more efficient generation of plasmin (Pn). Cell surface activation complexes are assembled on annexin 2 (Ax2) and S100A10 (p11) tetramers in APL or uPA receptors (uPAR). Thrombin (Th) in complex with thrombomodulin (Tm) activates TAFI (TAFIa), which eliminates plasminogen binding sites in fibrin. Activated factor XIII (FXIIIa) confers fibrinolytic resistance through modification of fibrin structure and crosslinking of AP to fibrin.
Recent work on highly charged polymers including polyphosphate from platelets, DNA and histones from neutrophils, and exogenous heparinoids, provide interesting new insights into factors regulating clotting and lysis (Smith & Morrissey, 2008; Longstaff et al, 2016). A diagrammatic representation of components and interactions of the fibrinolysis pathway relevant to this review is presented in Fig 2.
Diagnostic tools to measure disturbed fibrinolysis
Detecting and measuring fibrinolysis is less standardized than is the case with coagulation where batteries of tests are routinely performed using high throughput automated analysers and standardized reagents to diagnose coagulation defects or monitor anticoagulation therapy, for example. Ongoing fibrinolysis can be detected by fluctuations in fibrinolytic components, such as plasmin‐α2‐PI complexes, D‐dimer formation and depletion of circulating plasminogen and fibrinogen. Other fibrinolysis proteins, such as antigen levels of PAI‐1, tPA and TAFI, or activity of PAI‐1 or TAFIa are also used as markers of the state of the fibrinolysis system. Data on individual haemostasis components do not give a complete picture of the fibrinolytic system and global assays are an alternative option. Global assays of haemostasis is a huge topic and the subject of several recent reviews [e.g. (van Geffen & van Heerde, 2012)]. Again, coagulation assays dominate, from simple time courses of clotting of plasma or whole blood to more sophisticated thrombin generation tests where thrombin activity is monitored by following hydrolysis of chromogenic or fluorogenic substrates after coagulation is triggered in re‐calcified plasma. There is no equivalent system for directly measuring plasmin evolution during activation of the fibrinolytic system, but there are several variations of clot lysis assays which rely on turbidity changes as clots are formed and lysed (He et al, 2007; van Geffen & van Heerde, 2012). Parameters derived from such time courses are used to assess fibrinolytic capacity of test plasma, but this is not achieved under normal circumstances, on a reasonable timescale, without the addition of a plasminogen activator, usually tPA. Thus it is not possible to get information on the intrinsic activity of tPA or subtle changes in the balance with PAI‐1. Another approach, which has been around for many years, is the Euglobulin Clot Lysis Time (ECLT). In this case, a plasma fraction is precipitated by acid treatment then re‐dissolved and this ‘euglobulin fraction’ is assessed for fibrinolytic capacity. Although additional plasminogen activator is unnecessary, the euglobulin fraction contains a different mix of proteins than plasma, including 60–70% of fibrinogen and plasminogen and 90% of tPA, but is depleted of inhibitors PAI‐1 and TAFI (at around 40%) and, most significantly, <10% α2‐PI (Smith et al, 2003) (these values may vary with the precise methods used to precipitate the euglobulin fraction). The development of whole blood assays is potentially very advantageous from the point of view of including all plasma components and also cells, most importantly platelets, which in addition to modifying clot structure during clot retraction (Tutwiler et al, 2016), are a source of PAI‐1. The most obvious problem when using whole blood is the handicap of not easily being able to use optical methods. One way around this has been developed by Rijken et al (2012) who employed enzyme‐linked immunosorbent assay‐based methods to detect fibrin degradation products in clotted‐lysing blood samples, while using aprotinin‐treated parallel samples as baseline (aprotinin is a potent plasmin inhibitor) (Rijken et al, 2012). A different approach to monitoring clot development, stabilization and lysis relies on viscoelastic techniques, known as thromboelastography (commercially available as TEG) or rotational thromboelastometry (ROTEM). These methods commonly employ re‐calcified whole blood with or without additional factors depending on the test or component(s) of interest (Luddington, 2005). The basic technique is old but publications citing TEG or ROTEM in their abstracts have increased around 20‐fold since 2000 and they have been applied to investigate haemostasis in research and clinical studies, including monitoring coagulation factor replacement therapy in haemophilia, or during surgery, and in trauma and sepsis. Besides the advantages of using whole blood, these methods can be used in a near‐patient context and generate rapid results to assist clinical decision‐making. On the downside, the methods are not well standardized and interpretation of the many readouts available from the profiles generated by TEG and ROTEM is not always simple. However, these are methods able to identify hyperfibrinolysis.
Bleeding tendencies based on abnormal fibrinolysis
Clinical bleeding in the absence of overt mechanical injury is always the outcome of impaired haemostatic balance stemming either from inadequate pro‐coagulant mechanisms or enhanced fibrinolysis. In certain cases, such as the lack of a specific inhibitor, the causative relationship between the fibrinolytic abnormality and the laboratory findings or clinical symptoms of the bleeding patient is straightforward. In other circumstances, defective clotting and abnormal fibrin may lead to enhanced fibrinolysis but there is no routinely used standardized laboratory assay (or a panel of assays) that can clearly identify this relative hyperfibrinolysis. In order to emphasize the variable role of fibrinolysis in clinical bleeding, in the following discussion we make a distinction between primary hyperfibrinolysis (absolute quantitative or qualitative abnormalities of proteins directly involved in the lytic process) and secondary hyperfibrinolysis (conditions of imbalanced excessive activation of structurally normal fibrinolytic enzymes or enhanced susceptibility of fibrin to proteolysis). Both types of haemorrhagic trends can be traced back to inherited or acquired disease states. The distinction of primary and secondary hyperfibrinolysis introduced in this review, and summarized in Table 1, is helpful from therapeutic point of view, because it emphasizes what should be the primary target of treatment (the fibrinolytic system or the underlying disease state). In addition to the hyperfibrinolytic states discussed in detail below, Table 1 presents several less prevalent conditions with a selected key reference for each one.
Table 1.
Haemorrhagic conditions related to hyperfibrinolysis
| Class | Disease/condition | Mechanistic features | Effect of antifibrinolytics | Reference |
|---|---|---|---|---|
| Inherited primary hyperfibrinolysis | α2‐PI deficiency | Uncontrolled excessive plasmin activity | Major therapeutic modality | See text |
| PAI‐1 deficiency | Uncontrolled excessive plasminogen activation | Major therapeutic modality | See text | |
| Quebec platelet disorder | Overexpression of uPA in platelets and excessive plasminogen activation | Major therapeutic modality | Blavignac et al (2011) | |
| Acquired primary hyperfibrinolysis | End‐stage liver cirrhosis | Reduced levels of α2‐PI and TAFI, impaired hepatic clearance of tPA | Therapeutic benefit, if hyperfibrinolysis is supported by laboratory findings | See text |
| Acute promyelocytic leukaemia | Overexpression of the tPA‐cofactor (S100A10)2‐(annexin A2)2 | Beneficial as adjuvant to the basal ATRA therapy with potential thrombotic side‐effects | See text | |
| Inherited secondary hyperfibrinolysis | Haemophilia | Enhanced lytic susceptibility of fibrin structure, impaired TAFI activation | Systemic and local administration as adjuvant to substitution therapy | See text |
| FXIII deficiency | Enhanced lytic susceptibility of fibrin structure, impaired α2‐PI crosslinking to fibrin | Isolated case reports for beneficial effects as adjuvant to substitution therapy | See text | |
| Dysfibinogenaemias | Abnormal fibrin structure, more susceptible to lysis | Beneficial in cases of mild bleeding and menorrhagia | Casini et al (2015) | |
| Acquired secondary hyperfibrinolysis | Trauma | DIC | Beneficial within 3 h after the injury | See text |
| Thrombolytic therapy | Iatrogenic side effect | Beneficial effect of acute infusion | de Bono and More (1993) | |
| Cardiopulmonary bypass | Excessive plasmin generation secondary to activation of coagulation | Beneficial administration according to specific perioperative protocols | Edmunds (2010) | |
| Systemic amyloidosis | DIC | Beneficial effect in isolated case reports | Colucci et al (2009) | |
| Malignant prostatic and other solid tumours | DIC | Beneficial effect in isolated case reports | Hyman et al (2011) | |
| Placenta disorders (placenta accreta) | Release of plasminogen activators from the uterine and placenta | Efficient control of hyperfibrinolysis | Schroder et al (2015) |
α2‐PI, α2 plasmin inhibitor; DIC, disseminated intravascular coagulation; FXIII, factor XIII; PAI‐1, plasminogen activator inhibitor type 1; TAFI, thrombin activatable fibrinolysis inhibitor; tPA, tissue‐type plasminogen activator; uPA, urokinase‐type plasminogen activator.
Inherited primary hyperfibrinolysis
Based on their role in the control of protease activity, a haemorrhagic phenotype can be predicted in cases of failure of the physiological inhibitors of plasmin and plasminogen activators. Congenital deficiencies of α2‐PI and PAI‐1 have been reported in a few patients [e.g. (Maino et al, 2008; Mehta & Shapiro, 2008)], but the lack of routine laboratory screening means the real prevalence of these inherited bleeding conditions is unknown.
Depending on the nature of the mutation, the molecular defect in the structure of α2‐PI can result in either lower plasma concentrations or decreased inhibiting potential despite normal α2‐PI antigen levels in blood. For example, the mutant α2‐PI characterized by Miura and Aoki (1990) is elongated, with 166 amino acids and consequent misfolding of the protein; only a minor fraction of the mutant α2‐PI is secreted in the blood (Miura & Aoki, 1990). An α2‐PI variant from a Dutch family (α2‐antiplasmin Enschede) was found to be associated with bleeding in homozygous individuals. An alanine insertion near the reactive centre converts the protein from an inhibitor to a substrate of plasmin (Rijken et al, 1988). Thus, α2‐PI antigen determination in plasma shows normal values, but activity measurements indicate lack of inhibitor. In young patients with homozygous α2‐PI deficiency, haematomas may develop rapidly after minor injuries and persist for days, and apparently unprovoked intracranial and joint bleedings occur (Kluft et al, 1982). The phenotype of heterozygous α2‐PI deficiency is expressed as a bleeding tendency with variable severity following surgery, trauma, venepuncture or tooth extraction (Kluft et al, 1982). However, many heterozygous subjects have no such bleeds (Aoki et al, 1979).
PAI‐1 deficiency has also been observed and complete loss of inhibitory activity in homozygous individuals has been classified as a moderate bleeding disorder (Fay et al, 1997). Bleeding has been observed in response to trauma or surgery but was successfully treated using antifibrinolytic lysine analogues. A single PAI‐1 mutation has been well‐characterized in a family including 7 homozygous and 19 heterozygous members. In this example, a dinucleotide insertion in exon 4 of the gene encoding PAI‐1 (SERPINE1) causes a frameshift and a premature stop codon (Fay et al, 1992) and the shorter mutant molecule is degraded prior to secretion.
Acquired primary hyperfibrinolysis
More than 100 years ago Goodpasture presented elegant evidence that the haemorrhagic trend in end‐stage chronic alcoholic liver disease develops against the background of relatively normal blood coagulation capacity (normal blood and plasma clotting) (Goodpasture, 1914). However, liver disease patients may develop bleeding or venous thrombotic complications (e.g. reviewed in Roberts et al, 2010), as many clotting factors, plasminogen and inhibitors are synthesized in the liver, leading to a complex coagulopathy. So, while reduced levels of circulating plasminogen are antifibrinolytic, reduced levels of α2‐PI and TAFI will be profibrinolytic, as will impaired hepatic clearance of tPA. Thus, it has been argued there is a rebalancing of the haemostatic system, which may be driven towards a hypercoagulable state or hyperfibrinolysis, and the balance may be precarious (Lisman, 2012). In practice hyperfibrinolysis was correlated with the degree of cirrhosis in some studies (Colucci et al, 2003; Rijken et al, 2012), but not all (Lisman et al, 2001). Rijken et al (2012) found that the majority of cirrhosis patients (60%) showed accelerated clot lysis after paying particular attention to assay methodology using undiluted whole blood and avoiding the need to add tPA. uPA may also be affected and contribute to increased fibrinolysis (Booth et al, 1984). Standardized laboratory testing and large scale prospective studies are needed to substantiate the administration of antifibrinolytics to combat hyperfibrinolysis in liver disease (Gunawan & Runyon, 2006).
Acute promyelocytic leukaemia
Acute promyelocytic leukaemia (APL) is characterized by a high rate of life‐threatening haemorrhagic events related to hyperfibrinolysis. Despite the normal plasma levels of TAFI antigen, low TAFI activity has been reported in APL patients probably due to excessive inactivation of TAFI by plasmin (Meijers et al, 2000). The primary cause of the bleeding complications appears to be a unique pathway of enhanced plasminogen activation. A single genetic defect has been identified in APL, a translocation between chromosomes 15 and 17 resulting in fusion of the retinoic acid receptor‐α (RAR‐α) gene (RARA) and the promyelocytic leukaemia protein gene (PML). The expressed PML‐RAR‐α fusion protein arrests the differentiation of the myeloid precursor cells and prolongs their survival with consequent APL (Grignani et al, 1993). At the same time, the PML‐RAR‐α enhances the expression of S100A10 (p11), a member of the S100 family of calcium‐binding proteins (O'Connell et al, 2011). S100A10 forms a heterotetrameric (S100A10)2‐(annexin A2)2 complex on the surface of various cells, which through its C‐terminal lysine binds plasminogen, tPA and plasmin (MacLeod et al, 2003). In this way, the (S100A10)2‐(annexin A2)2 complex provides a template for plasminogen activation on the cell surface and protects plasmin against plasma inhibitors in a similar way to fibrin. These S100A10‐related pro‐fibrinolytic effects are consistent with the bleeding profile in APL. It is noteworthy that annexin A2 is also induced by PML‐RAR‐α (Liu et al, 2011) and following proteolytic cleavage (but not in native form) it binds plasminogen and tPA (Das et al, 2007). However, because such a cleaved form has not been demonstrated in vivo, annexin A2 appears to be important as a complex‐forming subunit in the heterotetrameric template rather than a direct cofactor in plasminogen activation. An established therapeutic modality in APL is all‐trans retinoic acid (ATRA), which induces differentiation of the leukaemic promyelocytes concomitant with improvement of the haemorrhagic symptoms (Dombret et al, 1993). ATRA treatment reverses the S100A10 and annexin A2 induction in PML‐RAR‐α positive cells and suppresses plasmin generation on their surface (O'Connell et al, 2011). Beneficial therapeutic effects can be expected from adjuvant antifibrinolytic treatment in cases with definite laboratory signs of hyperfibrinolysis, as evidenced by the effect of AHA in APL patients with a more than 50% reduction in the plasma α2‐PI level, which is used as a marker of excessive plasmin generation (Wassenaar et al, 2008). An additional factor for the pro‐fibrinolytic state in APL could be the release of neutrophil elastase from the leukaemic promyelocytes (Oudijk et al, 2000). Despite its lower catalytic efficiency in the degradation of fibrin compared to plasmin, elastase can promote fibrinolysis, because it converts plasminogen to miniplasminogen, a zymogen that is more readily activated, and which inactivates α2‐PI (Machovich & Owen, 1990). Taken together, the evidence justifies the classification of the haemostatic imbalance in APL as primary hyperfibrinolysis, and this conclusion is supported by the typical laboratory findings in the blood of APL patients (normal prothrombin and activated partial thromboplastin time, normal antithrombin level, low fibrinogen concentration in combination with elevated fibrin and fibrinogen degradation products) (Oudijk et al, 2000).
Inherited secondary hyperfibrinolysis
The term secondary hyperfibrinolysis can be applied for pathological situations when the members of the fibrinolytic system have normal structure and availability, but they either act on fibrin that is more susceptible to lysis or their hyperfunction is provoked in response to overt abnormal systemic blood clotting. Both inherited and acquired conditions can result in such alterations of fibrinolytic activity.
Haemophilia
Haemophilia A, B and C are rare congenital deficiencies of coagulation factors VIII, IX and XI, respectively, that are accompanied by devastating bleeding episodes, often unprovoked in severe cases. Because the culprit factors participate in the positive feedback circuit of the blood coagulation cascade mediated by the intrinsic tenase (FVIIIa as cofactor, FIXa as an executioner protease, FXIa as an activator), their deficiency causes a striking delay in thrombin generation in the propagation phase of whole blood clotting, but not at its initiation (Cawthern et al, 1998). Thus, significant fibrin is formed at clot time of whole haemophilic blood, suggesting that bleeding is due to clot instability rather than ab ovo fibrin deficiency. This concept is supported also by the elevated D‐dimer concentration in the circulation of haemophilic patients (Grunewald et al, 2002). In view of the effects of thrombin concentration on fibrin structure, mentioned in the Introduction, it is not surprising that fibrin architecture is altered in haemophilic plasma clots. Fibrin formed in haemophilic plasmas shows thicker fibres, larger pores, lower density and increased permeability compared to normal plasma [e.g. (Antovic et al, 2014; Wolberg et al, 2005; Zucker et al, 2014)]. These structural characteristics of the haemophilic clots augment their susceptibility to tPA‐mediated lysis through improved permeation of the activator, higher potency of the fibrin structure as a template for plasminogen activation and more efficient pattern of plasmin digestion. The low‐grade thrombin generation in haemophilia results in a delay in the activation of TAFI, thereby contributing to the instability of the haemophilic clots owing to the downregulation of an essential negative feedback mechanism (Broze & Higuchi, 1996). Supplementation of haemophilic plasma with TAFI (Mosnier et al, 2001) or soluble thrombomodulin (to stimulate TAFIa formation by thrombin‐thrombomodulin complex) (Foley & Nesheim, 2009), corrects this defect through enhancement of TAFI activation and consequent suppression of fibrinolysis. However, the lack of elevation in the circulating plasmin‐α2‐PI complexes in the plasma of bleeding haemophilic patients (Grunewald et al, 2002) suggests that in vivo the altered fibrin structure is a more important causative factor for overactive fibrinolysis in haemophilia than the enhancement of plasminogen activation. Furthermore, the reduced rate of thrombin generation in haemophilia delays the activation of plasma FXIII (Brummel‐Ziedins et al, 2009), which is an independent factor for clot instability, as discussed in more detail below. Although the exact mechanistic contribution of hyperfibrinolysis to the haemorrhagic phenotype of haemophilia was essentially unknown, its causative role was appreciated long ago as reflected in the positive therapeutic experience with systemic and local administration of antifibrinolytics (TXA, AHA) for prevention of bleeding in haemophiliacs at risk (Forbes et al, 1972).
FXIII deficiency
A different type of abnormal fibrin structure originates from FXIII deficiency, where both inherited and acquired deficiency states occur with low prevalence. The earliest clinical manifestation of congenital FXIII deficiency is delayed umbilical bleeding, and recurrent subcutaneous and intramuscular haematomas occur later. Life‐threatening intracranial bleeding is often provoked by minor traumas (Anwar & Miloszewski, 1999). Most cases of inherited FXIII deficiency are caused by defects in the gene encoding the catalytic A subunit and a broad array of mutations have been described (missense and nonsense mutations, splice site defects, insertions and deletions). Only a small number of cases of congenital B subunit defects with mild bleeding symptoms have been described, the loss of the carrier B subunit resulting in low plasma FXIII levels (whilst platelet FXIII is preserved). The most common causes of acquired FXIII deficiency are consumptive depletion (e.g. following major surgery) and auto‐antibodies in systemic lupus erythematosus that block the activation, the catalytic activity or the fibrin‐binding of subunit A (Souri et al, 2015); or bind to structural domains of subunit B with consequent loss of stable plasma FXIII (Ajzner et al, 2009). Susceptibility of tPA‐catalysed fibrin lysis may be explained by reduced cross‐linking and altered clot structure in the absence of FXIII (Hethershaw et al, 2014). However, in a plasma environment under flow, the major determinant of the lytic resistance conferred by FXIII appears to be the crosslinking of α2‐PI to fibrin (Fraser et al, 2011). The plasma and whole blood thromboelastograms of 10 patients with congenital FXIII A subunit deficiency evidenced accelerated lysis that could be corrected with recombinant FXIII replacement therapy (Lovejoy et al, 2006). Despite convincing evidence for the role of hyperfibrinolysis in the pathogenesis of bleeding, only isolated case reports show the potential benefits of antifibrinolytics, for example oral TXA in acquired FXIII deficiency (Janning et al, 2013).
Acquired secondary hyperfibrinolysis
Disseminated intravascular coagulation (DIC)
In contrast to the rare acquired FXIII deficiency, DIC is a common severe complication of systemic infection (sepsis) and extensive tissue destruction (major trauma). DIC is defined as ‘an acquired syndrome characterized by the intravascular activation of coagulation with loss of localization arising from different causes. It can originate from and cause damage to the microvasculature, which if sufficiently severe, can produce organ dysfunction’ (Taylor et al, 2001). Innate immune mechanisms are activated by pathogen‐associated molecular patterns (PAMPs) in sepsis or altered self‐tissue damage‐associated molecular patterns (DAMPs) [reviewed by (Manson et al, 2012)] that provoke pro‐thrombotic mechanisms in the microvasculature of the injured tissue to restrict the propagation of the damaging agent. Recruited monocytes express tissue factor, which initiates the extrinsic pathway of blood coagulation, whereas neutrophils release neutrophil extracellular traps (NETs). Histones, the most abundant proteins in NETs, induce activation of platelets (Fuchs et al, 2011) and endothelial cells (Saffarzadeh et al, 2012) and inhibit the activated protein C‐mediated negative feedback of the coagulation cascade (Ammollo et al, 2011). If the impaired anticoagulant pathways cannot localize the pro‐thrombotic factors to the primary injury site, overt multi‐organ microthrombosis develops that is accompanied by activation of fibrinolysis. The outcome of simultaneous systemic activation of coagulation and fibrinolysis is consumptive coagulopathy, the laboratory signs of which provide the diagnostic criteria: thrombocytopenia, prolonged prothrombin time, low fibrinogen and high circulating levels of fibrin degradation products (FDP) (Taylor et al, 2001). Depending on the nature of the provoking factor and the stage of the disease, the phenotype of DIC can be either thrombotic or haemorrhagic. Strong evidence supports the primary role of hyperfibrinolysis in the early stage of trauma‐associated DIC presenting typically as bleeding and requirement for massive transfusion. Excessively elevated levels of D‐dimers in patients with major trauma are documented at their hospital admission (Brohi et al, 2008; Sawamura et al, 2009) and thromboelastography has confirmed a hyperfibrinolytic state (Kashuk et al, 2010) or identified enhanced fibrinolysis even in cases when FDP levels are normal (Perouansky et al, 1999). The pattern of fibrinolytic and anticoagulant markers in trauma patients (elevated D‐dimer, tPA and soluble thrombomodulin, low protein C and normal PAI‐1 levels) (Brohi et al, 2008) is consistent with a pathomechanism initiated by hypoperfusion‐induced endothelial damage in posttraumatic shock. Hypoxia is known to induce tPA release from endothelial cells, whereas thrombomodulin detached from the damaged endothelium promotes the activation of protein C, which neutralizes PAI‐1 (Sakata et al, 1986). The imbalance of tPA and its inhibitor results in early hyperfibrinolysis in patients with major traumatic injury, as discussed in more detail below.
Iatrogenic hyperfibrinolysis: thrombolytic therapy and bleeding
Thrombolytic therapy for the removal of coronary thrombi began with first generation plasminogen activators, streptokinase and uPA. First generation thrombolytics were characterized by systemic plasminogen activation and bleeding side effects, in particular serious or fatal cerebral haemorrhage was a problem (Verstraete, 2000). Uncontrolled plasmin generation is known to cause depletion of important haemostatic proteins, including fibrinogen, FV, FVIII and α2‐PI, promoting bleeding (Rick & Krizek, 1986; Lee & Mann, 1989; Nogami et al, 2007). Subsequent development of more advanced, 2nd and 3rd generation thrombolytics (e.g. Alteplase, Reteplase and Tenecteplase,) was spurred on by the notion that improved fibrin specificity would localize plasmin activity and reduce bleeding side effects. Unfortunately the holy grail of finding a therapeutic agent able to leave existing stable haemostatic plugs intact while targeting new unwanted thrombus has remained elusive, and significant intracranial haemorrhage is observed in around 1% of patients over a range of treatment regimens (Marder & Stewart, 2002). Indeed, the debate on risk/benefit carries on over treatment for stroke using tPA (Alteplase), currently the only licensed therapy in the US and Europe. Despite Alteplase being licensed since the mid 1980s there are still periodic reviews, though the conclusions and ongoing work still favour treatment for stroke within 4·5 h in most countries (Medicines and Healthcare products Regulatory Agency 2015). The newer thrombolytic drugs have failed to impact on bleeding risk associated with thrombolytic therapy and significant future developments are doubtful.
Antifibrinolytics
The goal of antifibrinolytics is to reduce plasmin activity, which may be achieved at the level of plasminogen activation or direct inhibition of plasmin. Both mechanisms are illustrated by the action of the most well known therapeutic antifibrinolytics, aprotinin and the lysine analogues TXA and AHA. Early evidence of the effectiveness of aprotinin (Trasylol®) in reducing blood loss and the requirement for replacement units was presented (Royston et al, 1987), with some speculation on its mechanism of action. Plasmin inhibition was the main focus, but possibly acting by sparing platelet loss and/or preserving von Willebrand factor‐platelet interactions. Aprotinin was used successfully in cardiopulmonary bypass surgery and other types of vascular, thoracic and orthopaedic surgery (e.g. reviewed by Henry et al, 2011). However, by 2008, safety concerns suggesting increased risk of renal failure, myocardial infarction and stroke led to the revocation of licences in Europe and the US, though the validity of the analysis behind these decisions is still debated (McMullan & Alston, 2013). Infusion of aprotinin was designed to achieve a circulating concentration of 3‐4 μM inhibitor, whereas the inhibition constant (Ki) for aprotinin as an active site inhibitor of plasmin is 0·4 nmol/l or 2 nmol/l in the presence of fibrin (Longstaff, 1994). This apparent discrepancy between Ki and therapeutic level resulted in proposals for other protease targets, such as kallikrein. However, plasmin remains the most likely target and high concentrations of aprotinin are needed to deal with potentially high concentrations of plasmin (the circulating concentration of plasminogen is around 2 μmol/l).
Lysine analogues
Lysine analogues TXA and AHA have been known for many years as inhibitors of fibrinolysis (reviewed by Roberts, 2015). TXA may be viewed as a more chemically rigid version of AHA, fixed by a ring structure into a less flexible, more active, conformation, with high kringle‐binding affinity. Lysine analogues bind to kringle domains to block fibrin interactions and, as a general rule, TXA demonstrates around 6‐ to 10‐fold higher affinity for lysine binding sites than AHA (McCormack, 2012). TXA is the more widely used therapeutic antifibrinolytic, but both lysine analogues and aprotinin are all effective at reducing blood loss in surgery, to a similar extent (Henry et al, 2011). It is important to remember that aprotinin and lysine analogues used in elective surgery are administered before any blood loss begins, a more controlled situation than dealing with trauma patients. Trauma‐induced coagulopathy involves diverse pathways: coagulation factor consumption, DIC, activated protein C and FXIa, as well haemodilution, hypothermia and acidosis (Mann & Freeman, 2015). Some patients demonstrate a syndrome of hyperfibrinolysis, identified and characterized by reduced inhibitors, α2‐PI, TAFI, PAI‐1, and FXIII, in addition to increased tPA, uPA and elastase (Mann & Freeman, 2015). Hyperfibrinolysis can be detected in some cases by thromboelastography, and is observed to be a bad prognostic indicator on admission to the emergency department (Brohi et al, 2008). However, ROTEM and TEG are relatively insensitive techniques and ongoing fibrinolysis may be better assessed by measuring raised plasmin‐α2‐PI complexes or D‐dimer (Raza et al, 2013). The CRASH‐2 trial (Shakur et al, 2010) (20 211 patients from 270 hospitals in 40 countries) demonstrated the successful application of TXA when given to trauma patients within 8 h. Importantly, there were no indications of increased risk of vascular occlusion in the treated group, including myocardial infarction, stroke or pulmonary embolism. This is important as it has been shown that failure to prevent severe traumatic bleeding is associated with increased vascular occlusive events (Pealing et al, 2012). A subsequent detailed reanalysis of the original CRASH‐2 trial data (Roberts et al, 2011) revealed TXA was only effective at preventing death due to bleeding when given quickly. In the group treated <3 h the relative risk of death from bleeding was 0·68 (95% confidence interval 0·57–0·82) while after 3 h it was 1·44 (1·12–1·84). With this in mind, and noting the excellent safety profile of TXA (no increased risk of thrombosis) it has been forcefully argued that any delay in giving TXA while waiting for test results to confirm ongoing fibrinolysis (by thromboelastography for example) will result in thousands of unnecessary deaths (Shakur et al, 2012). European guidelines to maximize the benefit of TXA recommend aggressive treatment for patients who are bleeding or at risk of bleeding, with the initial infusion over 10 min, immediately, on first encounter with the patient (Spahn et al, 2013). Nevertheless, the mechanism underlying the 3 h time window and the missing detail on exactly how TXA and plasmin are associated with increasing risk of death in trauma may leave some room for doubt and hesitation in the universal uptake of TXA as an immediate first line treatment.
Lysine analogues: mechanism of action
It is worth exploring the possible mechanisms of action of TXA and probe what basic research might tell us to explain under what circumstances TXA could be profibrinolytic and hence ineffective (or dangerous) >3 h after trauma. The circulating concentration of TXA used in surgery and the target when treating trauma is around 200 μmol/l (Fiechtner et al, 2001), easily within the range to prevent tPA activation of plasminogen on fibrin [with an 50% inhibitory concentration of <50 μmol/l (Silva et al, 2012), Fig 3A], suggesting the most likely antifibrinolytic mechanism. A much higher TXA concentration would be needed to block the plasmin active site, Ki = 16 mmol/l (Christensen, 1978). However, TXA can also be profibrinolytic. Above 100 μmol/l, TXA can bind to a plasminogen low affinity kringle and induce an open conformation that is more activatable by uPA (but not tPA), with or without fibrin present (Sinniger et al, 1999; Silva et al, 2012), as shown in Fig 3A. Furthermore, once plasmin is generated, inhibition by α2‐PI is normally rapid in the absence of fibrin (Thelwell & Longstaff, 2007), but plasmin is protected by TXA in the range 10–500 μmol/l (Longstaff & Gaffney, 1991). Another profibrinolytic mechanism has been ascribed to improved diffusion of plasmin through fibrin clots due to reduced binding to C‐terminal lysines. Thus, fibrinolysis was enhanced when plasmin was added to fibrin treated with carboxypepetidase (which removes C‐terminal lysines) (Kovacs et al, 2014) or in the presence of AHA (Varju et al, 2014), as shown in Fig 3B. The concentration of AHA used in (Varju et al, 2014) was 1 mmol/l, so applying the 6‐ to 10‐fold rule (McCormack, 2012), a therapeutic concentration of 100–150 μmol/l TXA may have the same pro‐fibrinolytic effect. Hence, profibrinolytic effects of TXA are possible under circumstances where uPA develops a significant role as activator; or when sufficient free plasmin has been generated and TXA protects plasmin from inhibition by α2‐PI; or where TXA enhances plasmin diffusion and degradation of fibrin. Further work is needed to explore if these hypotheses account for the failure of TXA to prevent bleeding when given >3 h after trauma. However, recent work using a mouse model to study delayed intracranial haemorrhage after traumatic brain injury supports a major role for enhanced fibrinolysis, and the evolution of uPA activity in particular (Hijazi et al, 2015). It was shown that after injury, a spike of tPA appears in the cerebrospinal fluid, which then rapidly declines, while uPA release is slower but predominates after 3–4 h. Significantly, using knockout mice, these authors also demonstrated the ability of TXA to further stimulate fibrinolysis when uPA is the plasminogen activator in the absence of tPA, in this model.
Figure 3.
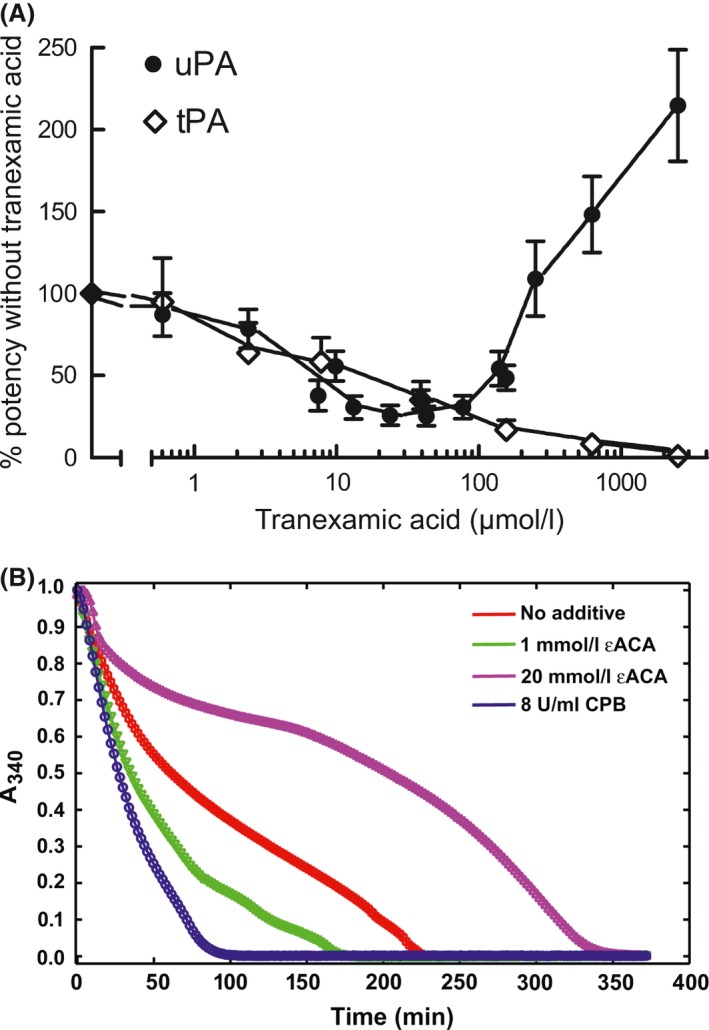
Inhibition or stimulation of fibrinolysis modulated by lysine binding site interactions. Both figures show fibrin clot lysis data from purified systems using clots formed by mixing thrombin and fibrinogen. Figure 3A shows clear inhibition by tranexamic acid (TXA) of tissue‐type plasminogen activator (tPA) activity in fibrinolysis assays (open symbols) but stimulation by TXA > 100 μmol/l when urokinase‐type plasminogen activator (uPA) is the activator (solid circles) (from (Silva et al, 2012), with permission). In Fig 3B, clot lysis achieved by adding plasmin to a preformed fibrin clot is stimulated by low concentrations of 6‐aminohexanoate (AHA) or by carboxypeptidase B (CPB) removal of C‐terminal lysines in fibrin. Reprinted with permission from: Varju et al (2014). Copyright 2014 American Chemical Society.
Conclusions and directions for future work
High throughput or rapid screening for disturbances of fibrinolysis is difficult and currently available approaches are poorly standardized. This may explain in part the scarcity of data associating defects in fibrinolysis with disease states, compared with the situation for coagulation. However, genome studies, results with knockout mice, and low numbers of affected individuals, all suggest the fibrinolysis system is robust with inbuilt redundancy. A number of inherited bleeding conditions and some acquired diseases are clearly linked to hyperfibrinolysis and may be treated with antifibrinolytics, and there is a history of successful application of the lysine analogues AHA and TXA. These simple drugs have proved useful in reducing blood loss across a range of situations, including some inherited and acquired haemostasis disorders, surgery and trauma. Further work directed towards a complete understanding of the mechanism of action of antifibrinolytics in different circumstances, or understanding why they sometimes fail, may extend their safe use more widely. An important additional line of future work is the development of standardized diagnostic tests for identifying the primary role of hyperfibrinolysis in haemorrhagic states, which would improve clinical decision making.
Acknowledgements
Both authors contributed equally to the writing of this review. This review was written while KK was receiving support from the Hungarian Scientific Research Fund OTKA 112612. The authors are grateful to Gergely Ángyán for the preparation of the graphical illustrations.
References
- Ajzner, E. , Schlammadinger, A. , Kerenyi, A. , Bereczky, Z. , Katona, E. , Haramura, G. , Boda, Z. & Muszbek, L. (2009) Severe bleeding complications caused by an autoantibody against the B subunit of plasma factor XIII: a novel form of acquired factor XIII deficiency. Blood, 113, 723–725. [DOI] [PubMed] [Google Scholar]
- Ammollo, C.T. , Semeraro, F. , Xu, J. , Esmon, N.L. & Esmon, C.T. (2011) Extracellular histones increase plasma thrombin generation by impairing thrombomodulin‐dependent protein C activation. Journal of Thrombosis and Haemostasis, 9, 1795–1803. [DOI] [PubMed] [Google Scholar]
- Antovic, A. , Mikovic, D. , Elezovic, I. , Zabczyk, M. , Hutenby, K. & Antovic, J.P. (2014) Improvement of fibrin clot structure after factor VIII injection in haemophilia A patients treated on demand. Thrombosis and Haemostasis, 111, 656–661. [DOI] [PubMed] [Google Scholar]
- Anwar, R. & Miloszewski, K.J. (1999) Factor XIII deficiency. British Journal of Haematology, 107, 468–484. [DOI] [PubMed] [Google Scholar]
- Aoki, N. , Saito, H. , Kamiya, T. , Koie, K. , Sakata, Y. & Kobakura, M. (1979) Congenital deficiency of alpha 2‐plasmin inhibitor associated with severe hemorrhagic tendency. J Clin Invest, 63, 877–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blavignac, J. , Bunimov, N. , Rivard, G.E. & Hayward, C.P. (2011) Quebec platelet disorder: update on pathogenesis, diagnosis, and treatment. Seminars in Thrombosis and Hemostasis, 37, 713–720. [DOI] [PubMed] [Google Scholar]
- de Bono, D.P. & More, R.S. (1993) Prevention and management of bleeding complications after thrombolysis. International Journal of Cardiology, 38, 1–6. [DOI] [PubMed] [Google Scholar]
- Booth, N.A. , Anderson, J.A. & Bennett, B. (1984) Plasminogen activators in alcoholic cirrhosis: demonstration of increased tissue type and urokinase type activator. Journal of Clinical Pathology, 37, 772–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridge, K.I. , Philippou, H. & Ariens, R. (2014) Clot properties and cardiovascular disease. Thrombosis and Haemostasis, 112, 901–908. [DOI] [PubMed] [Google Scholar]
- Brohi, K. , Cohen, M.J. , Ganter, M.T. , Schultz, M.J. , Levi, M. , Mackersie, R.C. & Pittet, J.F. (2008) Acute coagulopathy of trauma: hypoperfusion induces systemic anticoagulation and hyperfibrinolysis. Journal of Trauma, 64, 1211–1217; discussion 1217. [DOI] [PubMed] [Google Scholar]
- Broze, G.J. & Higuchi, D.A. (1996) Coagulation‐dependent inhibition of fibrinolysis: role of carboxypeptidase‐U and the premature lysis of clots from hemophilic plasma. Blood, 88, 3815–3823. [PubMed] [Google Scholar]
- Brummel‐Ziedins, K.E. , Branda, R.F. , Butenas, S. & Mann, K.G. (2009) Discordant fibrin formation in hemophilia. Journal of Thrombosis and Haemostasis, 7, 825–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casini, A. , Neerman‐Arbez, M. , Ariens, R.A. & de Moerloose, P. (2015) Dysfibrinogenemia: from molecular anomalies to clinical manifestations and management. Journal of Thrombosis and Haemostasis, 13, 909–919. [DOI] [PubMed] [Google Scholar]
- Cawthern, K.M. , van‘t Veer, C. , Lock, J.B. , DiLorenzo, M.E. , Branda, R.F. & Mann, K.G. (1998) Blood coagulation in hemophilia A and hemophilia C. Blood, 91, 4581–4592. [PubMed] [Google Scholar]
- Christensen, U. (1978) Allosteric effects of some antifibrinolytic amino acids on the catalytic activity of human plasmin. Biochimica et Biophysica Acta, 526, 194–201. [DOI] [PubMed] [Google Scholar]
- Colucci, M. , Binetti, B.M. , Branca, M.G. , Clerici, C. , Morelli, A. , Semeraro, N. & Gresele, P. (2003) Deficiency of thrombin activatable fibrinolysis inhibitor in cirrhosis is associated with increased plasma fibrinolysis. Hepatology, 38, 230–237. [DOI] [PubMed] [Google Scholar]
- Colucci, G. , Alberio, L. , Jahns, M. , Keller, P. , Steiner, S. , Rusges‐Wolter, I. & Lammle, B. (2009) Effective therapy with tranexamic acid in a case of chronic disseminated intravascular coagulation with acquired alpha2‐antiplasmin deficiency associated with AL amyloidosis. Thrombosis and Haemostasis, 102, 1285–1287. [DOI] [PubMed] [Google Scholar]
- Das, R. , Burke, T. & Plow, E.F. (2007) Histone H2B as a functionally important plasminogen receptor on macrophages. Blood, 110, 3763–3772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dombret, H. , Scrobohaci, M.L. , Ghorra, P. , Zini, J.M. , Daniel, M.T. , Castaigne, S. & Degos, L. (1993) Coagulation disorders associated with acute promyelocytic leukemia: corrective effect of all‐trans retinoic acid treatment. Leukemia, 7, 2–9. [PubMed] [Google Scholar]
- Edmunds, L.H. Jr (2010) Managing fibrinolysis without aprotinin. Annals of Thoracic Surgery, 89, 324–331. [DOI] [PubMed] [Google Scholar]
- Fay, W.P. , Shapiro, A.D. , Shih, J.L. , Schleef, R.R. & Ginsburg, D. (1992) Brief report: complete deficiency of plasminogen‐activator inhibitor type 1 due to a frame‐shift mutation. New England Journal of Medicine, 327, 1729–1733. [DOI] [PubMed] [Google Scholar]
- Fay, W.P. , Parker, A.C. , Condrey, L.R. & Shapiro, A.D. (1997) Human plasminogen activator inhibitor‐1 (PAI‐1) deficiency: characterization of a large kindred with a null mutation in the PAI‐1 gene. Blood, 90, 204–208. [PubMed] [Google Scholar]
- Fiechtner, B.K. , Nuttall, G.A. , Johnson, M.E. , Dong, Y. , Sujirattanawimol, N. , Oliver, W.C. Jr , Sarpal, R.S. , Oyen, L.J. & Ereth, M.H. (2001) Plasma tranexamic acid concentrations during cardiopulmonary bypass. Anesthesia and Analgesia, 92, 1131–1136. [DOI] [PubMed] [Google Scholar]
- Foley, J.H. & Nesheim, M.E. (2009) Soluble thrombomodulin partially corrects the premature lysis defect in FVIII‐deficient plasma by stimulating the activation of thrombin activatable fibrinolysis inhibitor. Journal of Thrombosis and Haemostasis, 7, 453–459. [DOI] [PubMed] [Google Scholar]
- Forbes, C.D. , Barr, R.D. , Reid, G. , Thomson, C. , Prentice, C.R. , McNicol, G.P. & Douglas, A.S. (1972) Tranexamic acid in control of haemorrhage after dental extraction in haemophilia and Christmas disease. British Medical Journal, 2, 311–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser, S.R. , Booth, N.A. & Mutch, N.J. (2011) The antifibrinolytic function of factor XIII is exclusively expressed through alpha(2)‐antiplasmin cross‐linking. Blood, 117, 6371–6374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs, T.A. , Bhandari, A.A. & Wagner, D.D. (2011) Histones induce rapid and profound thrombocytopenia in mice. Blood, 118, 3708–3714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Geffen, M. & van Heerde, W.L. (2012) Global haemostasis assays, from bench to bedside. Thrombosis Research, 129, 681–687. [DOI] [PubMed] [Google Scholar]
- Goodpasture, E.W. (1914) Fibrinolysis and chronic hepatic insufficiency. Bulletin of the Johns Hopkins Hospital, 25, 330–336. [Google Scholar]
- Grignani, F. , Ferrucci, P.F. , Testa, U. , Talamo, G. , Fagioli, M. , Alcalay, M. , Mencarelli, A. , Grignani, F. , Peschle, C. , Nicoletti, I. & Pelicci, P.G. (1993) The acute promyelocytic leukemia‐specific PML‐RAR alpha fusion protein inhibits differentiation and promotes survival of myeloid precursor cells. Cell, 74, 423–431. [DOI] [PubMed] [Google Scholar]
- Grunewald, M. , Siegemund, A. , Grunewald, A. , Konegan, A. , Koksch, M. & Griesshammer, M. (2002) Paradoxical hyperfibrinolysis is associated with a more intensely haemorrhagic phenotype in severe congenital haemophilia. Haemophilia, 8, 768–775. [DOI] [PubMed] [Google Scholar]
- Gunawan, B. & Runyon, B. (2006) The efficacy and safety of epsilon‐aminocaproic acid treatment in patients with cirrhosis and hyperfibrinolysis. Alimentary Pharmacology & Therapeutics, 23, 115–120. [DOI] [PubMed] [Google Scholar]
- He, S. , Zhu, K. , Skeppholm, M. , Vedin, J. , Svensson, J. , Egberg, N. , Blombäck, M. & Wallen, H. (2007) A global assay of haemostasis which uses recombinant tissue factor and tissue‐type plasminogen activator to measure the rate of fibrin formation and fibrin degradation in plasma. Thrombosis and Haemostasis, 98, 871–882. [PubMed] [Google Scholar]
- Henry, D.A. , Carless, P.A. , Moxey, A.J. , O'Connell, D. , Stokes, B.J. , Fergusson, D.A. & Ker, K. (2011) Anti‐fibrinolytic use for minimising perioperative allogeneic blood transfusion. Cochrane Database Systematic Review, Art. No. CD001886. [DOI] [PubMed] [Google Scholar]
- Hethershaw, E.L. , Cilia La Corte, A.L. , Duval, C. , Ali, M. , Grant, P.J. , Ariëns, R.A.S. & Philippou, H. (2014) The effect of blood coagulation factor XIII on fibrin clot structure and fibrinolysis. Journal of Thrombosis and Haemostasis: JTH, 12, 197–205. [DOI] [PubMed] [Google Scholar]
- Hijazi, N. , Abu Fanne, R. , Abramovitch, R. , Yarovoi, S. , Higazi, M. , Abdeen, S. , Basheer, M. , Maraga, E. , Cines, D.B. & Higazi, A.A. (2015) Endogenous plasminogen activators mediate progressive intracerebral hemorrhage after traumatic brain injury in mice. Blood, 125, 2558–2567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horrevoets, A.J.G. , Pannekoek, H. & Nesheim, M.E. (1997) A steady‐state template model that describes the kinetics of fibrin‐ stimulated. Journal of Biological Chemistry, 272, 2183–2191. [DOI] [PubMed] [Google Scholar]
- Hyman, D.M. , Soff, G.A. & Kampel, L.J. (2011) Disseminated intravascular coagulation with excessive fibrinolysis in prostate cancer: a case series and review of the literature. Oncology, 81, 119–125. [DOI] [PubMed] [Google Scholar]
- Janning, M. , Holstein, K. , Spath, B. , Schnabel, C. , Bannas, P. , Bokemeyer, C. & Langer, F. (2013) Relevant bleeding diathesis due to acquired factor XIII deficiency. Hamostaseologie, 33, S50–S54. [PubMed] [Google Scholar]
- Kashuk, J.L. , Moore, E.E. , Sawyer, M. , Wohlauer, M. , Pezold, M. , Barnett, C. , Biffl, W.L. , Burlew, C.C. , Johnson, J.L. & Sauaia, A. (2010) Primary fibrinolysis is integral in the pathogenesis of the acute coagulopathy of trauma. Annals of Surgery, 252, 434–442; discussion 443‐434. [DOI] [PubMed] [Google Scholar]
- Kluft, C. , Vellenga, E. , Brommer, E.J. & Wijngaards, G. (1982) A familial hemorrhagic diathesis in a Dutch family: an inherited deficiency of alpha 2‐antiplasmin. Blood, 59, 1169–1180. [PubMed] [Google Scholar]
- Kovacs, A. , Szabo, L. , Longstaff, C. , Tenekedjiev, K. , Machovich, R. & Kolev, K. (2014) Ambivalent roles of carboxypeptidase B in the lytic susceptibility of fibrin. Thrombosis Research, 133, 80–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law, R.H. , Abu‐Ssaydeh, D. & Whisstock, J.C. (2013) New insights into the structure and function of the plasminogen/plasmin system. Current Opinion in Structural Biology, 23, 836–841. [DOI] [PubMed] [Google Scholar]
- Lee, C.D. & Mann, K.G. (1989) Activation/inactivation of human factor V by plasmin. Blood, 73, 185–190. [PubMed] [Google Scholar]
- Lisman, T. (2012) Fibrinolysis in liver disease. Journal of Thrombosis and Haemostasis, 10, e25. [Google Scholar]
- Lisman, T. , Leebeek, F.W. , Mosnier, L.O. , Bouma, B.N. , Meijers, J.C. , Janssen, H.L. , Nieuwenhuis, H.K. & De Groot, P.G. (2001) Thrombin‐activatable fibrinolysis inhibitor deficiency in cirrhosis is not associated with increased plasma fibrinolysis. Gastroenterology, 121, 131–139. [DOI] [PubMed] [Google Scholar]
- Liu, Y. , Wang, Z. , Jiang, M. , Dai, L. , Zhang, W. , Wu, D. & Ruan, C. (2011) The expression of annexin II and its role in the fibrinolytic activity in acute promyelocytic leukemia. Leukemia Research, 35, 879–884. [DOI] [PubMed] [Google Scholar]
- Longstaff, C. (1994) Studies on the mechanisms of action of aprotinin and tranexamic acid as plasmin inhibitors and antifibrinolytic agents. Blood Coagulation & Fibrinolysis, 5, 537–542. [PubMed] [Google Scholar]
- Longstaff, C. & Gaffney, P.J. (1991) Serpin‐serine protease binding kinetics: alpha 2‐antiplasmin as a model inhibitor. Biochemistry, 30, 979–986. [DOI] [PubMed] [Google Scholar]
- Longstaff, C. & Kolev, K. (2015) Basic mechanisms and regulation of fibrinolysis. Journal of Thrombosis and Haemostasis, 13, S98–S105. [DOI] [PubMed] [Google Scholar]
- Longstaff, C. , Thelwell, C. , Williams, S.C. , Silva, M.M. , Szabo, L. & Kolev, K. (2011) The interplay between tissue plasminogen activator domains and fibrin structures in the regulation of fibrinolysis: kinetic and microscopic studies. Blood, 117, 661–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longstaff, C. , Hogwood, J. , Gray, E. , Komorowicz, E. , Varju, I. , Varga, Z. & Kolev, K. (2016) Neutralisation of the anti‐coagulant effects of heparin by histones in blood plasma and purified systems. Thrombosis and Haemostasis, 115, 591–599. [DOI] [PubMed] [Google Scholar]
- Lovejoy, A.E. , Reynolds, T.C. , Visich, J.E. , Butine, M.D. , Young, G. , Belvedere, M.A. , Blain, R.C. , Pederson, S.M. , Ishak, L.M. & Nugent, D.J. (2006) Safety and pharmacokinetics of recombinant factor XIII‐A2 administration in patients with congenital factor XIII deficiency. Blood, 108, 57–62. [DOI] [PubMed] [Google Scholar]
- Luddington, R.J. (2005) Thrombelastography/thromboelastometry. Clinical and Laboratory Haematology, 27, 81–90. [DOI] [PubMed] [Google Scholar]
- Machovich, R. & Owen, W.G. (1990) The elastase‐mediated pathway of fibrinolysis. Blood Coagulation & Fibrinolysis, 1, 79–90. [DOI] [PubMed] [Google Scholar]
- MacLeod, T.J. , Kwon, M. , Filipenko, N.R. & Waisman, D.M. (2003) Phospholipid‐associated annexin A2‐S100A10 heterotetramer and its subunits: characterization of the interaction with tissue plasminogen activator, plasminogen, and plasmin. Journal of Biological Chemistry, 278, 25577–25584. [DOI] [PubMed] [Google Scholar]
- Maino, A. , Garagiola, I. , Artoni, A. , Al‐Humood, S. & Peyvandi, F. (2008) A novel mutation of alpha2‐plasmin inhibitor gene causes an inherited deficiency and a bleeding tendency. Haemophilia, 14, 166. [DOI] [PubMed] [Google Scholar]
- Mann, K.G. & Freeman, K. (2015) TACTIC: Trans‐Agency Consortium for Trauma‐Induced Coagulopathy. Journal of Thrombosis and Haemostasis, 13, S63–S71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manson, J. , Thiemermann, C. & Brohi, K. (2012) Trauma alarmins as activators of damage‐induced inflammation. British Journal of Surgery, 99, 12–20. [DOI] [PubMed] [Google Scholar]
- Marder, V.J. & Stewart, D. (2002) Towards safer thrombolytic therapy. Seminars in Hematology, 39, 206–216. [DOI] [PubMed] [Google Scholar]
- McCormack, P.L. (2012) Tranexamic acid: a review of its use in the treatment of hyperfibrinolysis. Drugs, 72, 585–617. [DOI] [PubMed] [Google Scholar]
- McMullan, V. & Alston, R.P. (2013) III. Aprotinin and cardiac surgery: a sorry tale of evidence misused. British Journal of Anaesthesia, 110, 675–678. [DOI] [PubMed] [Google Scholar]
- Medicines and Healthcare products Regulatory Agency . (2015) Alteplase for treatment of acute ischaemic stroke: independent review. The review of alteplase in the treatment of acute ischaemic stroke by the expert working group of the Commssion on Human Medicines. https://www.gov.uk/government/publications/alteplase-for-treatment-of-acute-ischaemic-stroke-independent-review
- Mehta, R. & Shapiro, A.D. (2008) Plasminogen activator inhibitor type 1 deficiency. Haemophilia, 14, 1255–1260. [DOI] [PubMed] [Google Scholar]
- Meijers, J.C. , Oudijk, E.J. , Mosnier, L.O. , Bos, R. , Bouma, B.N. , Nieuwenhuis, H.K. & Fijnheer, R. (2000) Reduced activity of TAFI (thrombin‐activatable fibrinolysis inhibitor) in acute promyelocytic leukaemia. British Journal of Haematology, 108, 518–523. [DOI] [PubMed] [Google Scholar]
- Miura, O. & Aoki, N. (1990) Impaired secretion of mutant alpha 2‐plasmin inhibitor (alpha 2 PI‐Nara) from COS‐7 and HepG2 cells: molecular and cellular basis for hereditary deficiency of alpha 2‐plasmin inhibitor. Blood, 75, 1092–1096. [PubMed] [Google Scholar]
- Mosnier, L.O. , Lisman, T. , van den Berg, H.M. , Nieuwenhuis, H.K. , Meijers, J.C. & Bouma, B.N. (2001) The defective down regulation of fibrinolysis in haemophilia A can be restored by increasing the TAFI plasma concentration. Thrombosis and Haemostasis, 86, 1035–1039. [PubMed] [Google Scholar]
- Nogami, K. , Shima, M. , Matsumoto, T. , Nishiya, K. , Tanaka, I. & Yoshioka, A. (2007) Mechanisms of plasmin‐catalyzed inactivation of factor VIII: a crucial role for proteolytic cleavage at Arg336 responsible for plasmin‐catalyzed factor VIII inactivation. Journal of Biological Chemistry, 282, 5287–5295. [DOI] [PubMed] [Google Scholar]
- O'Connell, P.A. , Madureira, P.A. , Berman, J.N. , Liwski, R.S. & Waisman, D.M. (2011) Regulation of S100A10 by the PML‐RAR‐alpha oncoprotein. Blood, 117, 4095–4105. [DOI] [PubMed] [Google Scholar]
- Oudijk, E.J. , Nieuwenhuis, H.K. , Bos, R. & Fijnheer, R. (2000) Elastase mediated fibrinolysis in acute promyelocytic leukemia. Thrombosis and Haemostasis, 83, 906–908. [PubMed] [Google Scholar]
- Pealing, L. , Perel, P. , Prieto‐Merino, D. , Roberts, I. & Collaborators, C.‐T. (2012) Risk factors for vascular occlusive events and death due to bleeding in trauma patients; an analysis of the CRASH‐2 cohort. PLoS ONE, 7, e50603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perouansky, M. , Oppenheim, A. , Sprung, C.L. , Eidelman, L.A. & Pizov, R. (1999) Effect of haemofiltration on pathological fibrinolysis due to severe sepsis: a case report. Resuscitation, 40, 53–56. [DOI] [PubMed] [Google Scholar]
- Raza, I. , Davenport, R. , Rourke, C. , Platton, S. , Manson, J. , Spoors, C. , Khan, S. , De'Ath, H.D. , Allard, S. , Hart, D.P. , Pasi, K.J. , Hunt, B.J. , Stanworth, S. , MacCallum, P.K. & Brohi, K. (2013) The incidence and magnitude of fibrinolytic activation in trauma patients. Journal of Thrombosis and Haemostasis, 11, 307–314. [DOI] [PubMed] [Google Scholar]
- Rick, M.E. & Krizek, D.M. (1986) Platelets modulate the proteolysis of factor VIII: C protein by plasmin. Blood, 67, 1649–1654. [PubMed] [Google Scholar]
- Rijken, D.C. , Groeneveld, E. , Kluft, C. & Nieuwenhuis, H.K. (1988) Alpha 2‐antiplasmin Enschede is not an inhibitor, but a substrate, of plasmin. Biochem J, 255, 609–615. [PMC free article] [PubMed] [Google Scholar]
- Rijken, D.C. , Kock, E.L. , Guimarães, A.H.C. , Talens, S. , Murad, S.D. , Janssen, H.L.A. & Leebeek, F.W.G. (2012) Evidence for an enhanced fibrinolytic capacity in cirrhosis as measured with two different global fibrinolysis tests. Journal of Thrombosis and Haemostasis, 10, 2116–2122. [DOI] [PubMed] [Google Scholar]
- Roberts, I. (2015) Tranexamic acid in trauma: how should we use it? Journal of Thrombosis and Haemostasis, 13, S195–S199. [DOI] [PubMed] [Google Scholar]
- Roberts, L.N. , Patel, R.K. & Arya, R. (2010) Haemostasis and thrombosis in liver disease. British Journal of Haematology, 148, 507–521. [DOI] [PubMed] [Google Scholar]
- Roberts, I. , Shakur, H. , Afolabi, A. , Brohi, K. , Coats, T. , Dewan, Y. , Gando, S. , Guyatt, G. , Hunt, B.J. , Morales, C. , Perel, P. , Prieto‐Merino, D. & Woolley, T. (2011) The importance of early treatment with tranexamic acid in bleeding trauma patients: an exploratory analysis of the CRASH‐2 randomised controlled trial. Lancet, 377, 1096–1101, 1101 e1091‐1092. [DOI] [PubMed] [Google Scholar]
- Royston, D. , Bidstrup, B.P. , Taylor, K.M. & Sapsford, R.N. (1987) Effect of aprotinin on need for blood transfusion after repeat open‐heart surgery. Lancet, 2, 1289–1291. [DOI] [PubMed] [Google Scholar]
- Saffarzadeh, M. , Juenemann, C. , Queisser, M.A. , Lochnit, G. , Barreto, G. , Galuska, S.P. , Lohmeyer, J. & Preissner, K.T. (2012) Neutrophil extracellular traps directly induce epithelial and endothelial cell death: a predominant role of histones. PLoS ONE, 7, e32366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakata, Y. , Loskutoff, D.J. , Gladson, C.L. , Hekman, C.M. & Griffin, J.H. (1986) Mechanism of protein C‐dependent clot lysis: role of plasminogen activator inhibitor. Blood, 68, 1218–1223. [PubMed] [Google Scholar]
- Sawamura, A. , Hayakawa, M. , Gando, S. , Kubota, N. , Sugano, M. , Wada, T. & Katabami, K. (2009) Disseminated intravascular coagulation with a fibrinolytic phenotype at an early phase of trauma predicts mortality. Thrombosis Research, 124, 608–613. [DOI] [PubMed] [Google Scholar]
- Schroder, L. , Potzsch, B. , Ruhl, H. , Gembruch, U. & Merz, W.M. (2015) Tranexamic acid for hyperfibrinolytic hemorrhage during conservative management of Placenta Percreta. Obstetrics and Gynecology, 126, 1012–1015. [DOI] [PubMed] [Google Scholar]
- Shakur, H. , Roberts, I. , Bautista, R. , Caballero, J. , Coats, T. , Dewan, Y. , El‐Sayed, H. , Gogichaishvili, T. , Gupta, S. , Herrera, J. , Hunt, B. , Iribhogbe, P. , Izurieta, M. , Khamis, H. , Komolafe, E. , Marrero, M.A. , Mejia‐Mantilla, J. , Miranda, J. , Morales, C. , Olaomi, O. , Olldashi, F. , Perel, P. , Peto, R. , Ramana, P.V. , Ravi, R.R. & Yutthakasemsunt, S. (2010) Effects of tranexamic acid on death, vascular occlusive events, and blood transfusion in trauma patients with significant haemorrhage (CRASH‐2): a randomised, placebo‐controlled trial. Lancet, 376, 23–32. [DOI] [PubMed] [Google Scholar]
- Shakur, H. , Roberts, I. , Piot, P. , Horton, R. , Krug, E. & Mersch, J. (2012) A promise to save 100,000 trauma patients. Lancet, 380, 2062–2063. [DOI] [PubMed] [Google Scholar]
- Silva, M.M. , Thelwell, C. , Williams, S.C. & Longstaff, C. (2012) Regulation of fibrinolysis by C‐terminal lysines operates through plasminogen and plasmin but not tissue plasminogen activator (tPA). Journal of Thrombosis and Haemostasis, 10, 2354–2360. [DOI] [PubMed] [Google Scholar]
- Sinniger, V. , Merton, R.E. , Fabregas, P. , Felez, J. & Longstaff, C. (1999) Regulation of tissue plasminogen activator activity by cells. Domains responsible for binding and mechanism of stimulation. Journal of Biological Chemistry, 274, 12414–12422. [DOI] [PubMed] [Google Scholar]
- Smith, S.A. & Morrissey, J.H. (2008) Polyphosphate enhances fibrin clot structure. Blood, 112, 2810–2816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, A.A. , Jacobson, L.J. , Miller, B.I. , Hathaway, W.E. & Manco‐Johnson, M.J. (2003) A new euglobulin clot lysis assay for global fibrinolysis. Thrombosis Research, 112, 329–337. [DOI] [PubMed] [Google Scholar]
- Souri, M. , Osaki, T. & Ichinose, A. (2015) Anti‐factor XIII A subunit (FXIII‐A) autoantibodies block FXIII‐A2 B2 assembly and steal FXIII‐A from native FXIII‐A2 B2. Journal of Thrombosis and Haemostasis, 13, 802–814. [DOI] [PubMed] [Google Scholar]
- Spahn, D.R. , Bouillon, B. , Cerny, V. , Coats, T.J. , Duranteau, J. , Fernandez‐Mondejar, E. , Filipescu, D. , Hunt, B.J. , Komadina, R. , Nardi, G. , Neugebauer, E. , Ozier, Y. , Riddez, L. , Schultz, A. , Vincent, J.L. & Rossaint, R. (2013) Management of bleeding and coagulopathy following major trauma: an updated European guideline. Critical Care, 17, R76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor, F.B. Jr , Toh, C.H. , Hoots, W.K. , Wada, H. & Levi, M. ; Scientific Subcommittee on Disseminated Intravascular Coagulation of the International Society on, Thrombosis and Haemostasis . (2001) Towards definition, clinical and laboratory criteria, and a scoring system for disseminated intravascular coagulation. Thrombosis and Haemostasis, 86, 1327–1330. [PubMed] [Google Scholar]
- Thelwell, C. & Longstaff, C. (2007) The regulation by fibrinogen and fibrin of tissue plasminogen activator kinetics and inhibition by plasminogen activator inhibitor 1. Journal of Thrombosis and Haemostasis, 5, 804–811. [DOI] [PubMed] [Google Scholar]
- Tutwiler, V. , Litvinov, R.I. , Lozhkin, A.P. , Peshkova, A.D. , Lebedeva, T. , Ataullakhanov, F.I. , Spiller, K.L. , Cines, D.B. & Weisel, J.W. (2016) Kinetics and mechanics of clot contraction are governed by the molecular and cellular composition of the blood. Blood, 127, 149–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varju, I. , Tenekedjiev, K. , Keresztes, Z. , Pap, A.E. , Szabo, L. , Thelwell, C. , Longstaff, C. , Machovich, R. & Kolev, K. (2014) Fractal kinetic behavior of plasmin on the surface of fibrin meshwork. Biochemistry, 53, 6348–6356. [DOI] [PubMed] [Google Scholar]
- Verstraete, M. (2000) Third‐generation thrombolytic drugs. American Journal of Medicine, 109, 52–58. [DOI] [PubMed] [Google Scholar]
- Wassenaar, T. , Black, J. , Kahl, B. , Schwartz, B. , Longo, W. , Mosher, D. & Williams, E. (2008) Acute promyelocytic leukaemia and acquired alpha‐2‐plasmin inhibitor deficiency: a retrospective look at the use of epsilon‐aminocaproic acid (Amicar) in 30 patients. Hematological Oncology, 26, 241–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolberg, A.S. , Allen, G.A. , Monroe, D.M. , Hedner, U. , Roberts, H.R. & Hoffman, M. (2005) High dose factor VIIa improves clot structure and stability in a model of haemophilia B. British Journal of Haematology, 131, 645–655. [DOI] [PubMed] [Google Scholar]
- Xue, Y. , Bodin, C. & Olsson, K. (2012) Crystal structure of the native plasminogen reveals an activation‐resistant compact conformation. Journal of Thrombosis and Haemostasis, 10, 1385–1396. [DOI] [PubMed] [Google Scholar]
- Zucker, M. , Seligsohn, U. , Salomon, O. & Wolberg, A.S. (2014) Abnormal plasma clot structure and stability distinguish bleeding risk in patients with severe factor XI deficiency. Journal of Thrombosis and Haemostasis, 12, 1121–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]