

Abstract
Background:
Baicalin is the main bioactive constitute of the dried roots of Scutellaria baicalensis and possesses various biological activities. However, the poor water solubility and low oral bioavailability limit its efficacy.
Objective:
The present study was conducted to enhance the dissolution and oral bioavailability of baicalin (BA) through a novel mesoporous carbon nanopowder (MCN) drug carrier.
Materials and Methods:
Solid dispersions (SDs) of BA with MCN were prepared using a solvent evaporation method. The physical state of the formulations was investigated using SEM, differential scanning calorimetry (DSC) and powder X-ray diffraction (XRD). The pharmaceutical performance of pure BA, physical mixture (PM) and SDs was evaluated by performing an in-vitro dissolution test. The pharmacokinetic studies were conducted in SD rats and the analysis of the biological samples was performed on an Acquity UPLC–MS system. The intestinal and renal toxicity test of MCN was also evaluated.
Results:
The drug release profile indicated that the BA dissolution rate from SDs with a BA/MCN ratio of 1:6 greatly increased in comparison with that of the pure crystalline drug. Furthermore, a pharmacokinetic analysis in rats showed that the BA area under the concentration–time curve for SDs of MCN/BA was 1.83 times larger than that of pure BA. In comparison with the pure drug, the MCN–BA system significantly shortened the time to Tmax and generated higher Cmax. There was no intestinal and renal toxicity of MCN.
Conclusion:
These results indicated that the oral bioavailability of BA was remarkably improved by the MCN carrier. Additionally, intestinal toxicity test showed that MCN produced no toxicity in the gastrointestinal tract. Our results show that MCN-based SDs could be used to enhance the bioavailability of drugs with poor water solubility.
SUMMARY
The drug release profile indicated that the BA dissolution rate from SDs with a BA/MCN ratio of 1:6 greatly increased in comparison with that of the pure crystalline drug.
Furthermore, a pharmacokinetic analysis in rats showed that the BA area under the concentration–time curve for SDs of MCN/BA was 1.83 times larger than that of pure BA.
In comparison with the pure drug, the MCN–BA system significantly shortened the time to Tmax and generated higher Cmax
Abbreviations used: BA: baicalin, MCN: mesoporous carbon nanopowder, SDs: solid dispersions, SEM: scanning electron microscopy, DSC: differential scanning calorimetry, XRD: powder X-ray diffraction, HPLC: high-performance liquid chromatography, PM: physical mixture, S.D.: standard deviation, ANOVA: analysis of variance, RSD: relative standard deviation, ESI: electrospray ionization, IS: internal standard, MRM: multiple reaction monitoring
Keywords: Baicalin, mesoporous carbon nanopowder, solid dispersion, dissolution, oral bioavailability
INTRODUCTION
Baicalin [BA, Figure 1], a type of flavonoids, is the main bioactive constitute of the dried roots of Scutellaria baicalensis. BA has been widely used in a number of therapeutic applications because of its antibacterial, anti-inflammatory, antioxidant and anticancer effects.[1,2,3] However, BA has poor water solubility (91 μg/mL) and low oral bioavailability (2.2 ± 0.2% absolute bioavailability after oral administration in rats),[4,5,6] which could limit its efficacy. For highly lipophilic drugs, the rate and degree of absorption are usually restricted and defined by the dissolution process,[7,8] which necessitates the development of new oral delivery formulations or technologies by the pharmaceutical industry to improve bioavailability and efficacy.
Figure 1.
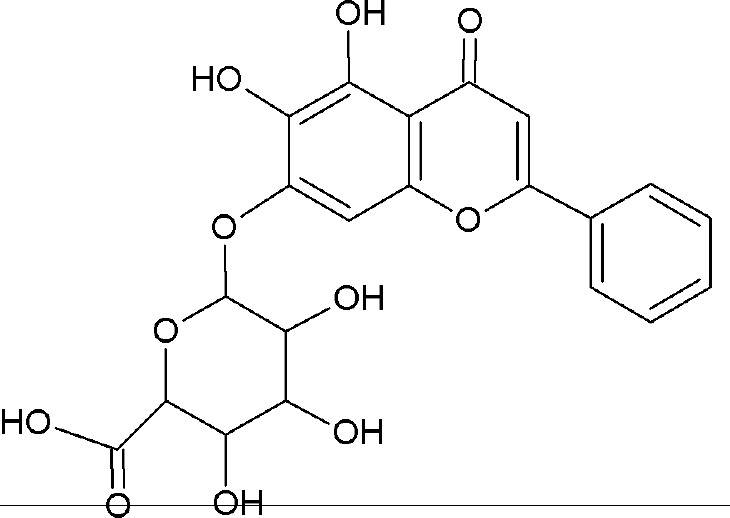
Chemical structure of BA.
Drugs with low aqueous solubility exhibit low oral bioavailability due to insufficient dissolution during their passage through the gastrointestinal tract. By improving the drug release profile of such drugs, their bioavailability can be enhanced while their side effects are reduced. Solid dispersions (SDs) are one of the most successful and widely used strategies to improve the dissolution rate and solubility of poorly soluble drugs.[9,11,12] SDs have advantageous properties, such as small particle size, high wettability and porosity, and a non-crystalline structure.[13,14,15] Moreover, inorganic porous materials, such as mesoporous silica,[16] have been used in SDs. Several reports have shown that porous SDs carriers can improve the dissolution profiles of water-insoluble drugs.
In recent years, significant research interest has been focused on the development of mesoporous carbon materials with a uniform pore structure.[17] Mesoporous carbon nano (MCN) powder is a porous material with a high specific surface area, large pore volume, high thermal stability, strong adsorption capacity, and good biocompatibility. The nano cavities of MCN can change the crystalline state of a drug into an amorphous one, effectively limit drug re-crystallization and significantly reduce the particle size of amorphous drugs.[18,19,21] Mesoporous carbon materials have been successfully applied in oral drug delivery systems;[22,22,24] however, to the best of our knowledge, there are no reports on the use of MCN for oral drug delivery system both in vitro and in vivo, and the influence of the particular properties of MCN on its suitability for use as a vehicle for insoluble drugs in SDs requires further examination. Therefore, we prepared a novel MCN–BA SDs to improve the dissolution rate and oral bioavailability of BA. The molecular state and release behavior of the drug in the system were comprehensively studied by SEM, DSC, and powder XRD. In-vivo pharmacokinetic studies were conducted to confirm the increase in BA oral bioavailability due to its incorporation into the SDs. In addition, the intestinal toxicity tests were carried out to investigate whether MCN produced toxicity in the gastrointestinal tract. Figure 1 Chemical structure of BA.
MATERIALS AND METHODS
Chemicals and drugs
BA (purity 90%) and daidzein (purity 98%) were obtained from Sigma–Aldrich Chemical Co. (St. Louis, MO, USA). MCN (average pore diameter, 64 Å; morphology, almost spherical; pore total volume, 0.342 cm3/g; specific surface area, 150–250 m2/g) was purchased from Beijing AnWeiAn Lab Equipment Co., Ltd. (Beijing, People's Republic of China). Methanol was high-performance liquid chromatography (HPLC) grade and all the other reagents and chemicals were of analytical grade. Deionized water was purified using a Milli-Q water purification system (Millipore, Bedford, MA, USA).
Preparation of solid dispersions and physical mixture systems
The SDs of BA with MCN were prepared using a solvent evaporation method. First, ethanol was added to BA in a beaker to produce a BA solution with a concentration of 2 mg·mL-1. The appropriate amount of MCN was gradually added to the drug solution to produce solutions with mass ratios of BA to MCN of 1:2, 1:4, 1:6, and 1:8. Then, each mixture was ultrasonicated for 15 min to uniformly disperse the MCN particles. After sonication, the samples required gentle stirring for 12 h to achieve adsorption equilibrium. To prevent the loss of ethanol, the container was sealed during the drug loading procedure. Finally, the solvent was evaporated to dryness in a rotary evaporator at room temperature. The physical mixture (PM) of BA and MCN was obtained by mixing BA with MCN at a ratio of 1:6 (w/w), followed by grinding the mixture in a mortar until it was homogeneous.
Dissolution test
In-vitro dissolution test
The pharmaceutical performance of pure BA, PM and SDs was evaluated by performing an in-vitro dissolution test. The test was performed according to the USP 24 method 2 (paddle method) using a BCZ-8A intellectualized dissolution apparatus (Tianjin University Exact Apparatus Co., Ltd., Tianjin, China). The test was carried out at 37°C with constant stirring at 50 rpm for 2 h. Samples equivalent to 20 mg BA were added to the dissolution medium (900 mL of distilled water containing 0.5% sodium dodecyl sulfate). Samples of 5 mL were withdrawn periodically and replaced with fresh and temperature-equilibrated dissolution medium. The obtained samples were filtered through a 0.45-μm membrane and analyzed using the HPLC methods as described below. The in-vitro dissolution test was performed in triplicate, and the results were expressed as mean ± standard deviation. Statistical significance was assessed using one-way analysis of variance (ANOVA) with SPSS software (version 13.0; IBM Corp., Armonk, NY, USA). P-values less than 0.05 were considered significant.
High-performance liquid chromatography analysis of baicalin
The concentration of BA in the dissolution medium was measured using an HPLC system (Waters 600 Controller, Waters717 plus Auto sampler, Waters Empower data processing software; Waters Corp., Milford, MA, USA) with UV detection at 280 nm. Separation was performed at 30°C on on Agilent ZORBAX SB-C18 column (250 mm × 4.6 mm, 5 μm) with a mobile phase of methanol and 0.3% phosphoric acid (47:53, v/v) at a flow rate of 1.0 mL·min-1. The samples were filtered through 0.45-μm membrane filters before analysis, and the injection volume was 20 μL. The linearity of the method was studied in the concentration range of 0.236–23.600 μg·mL-1 (r = 0.9997). The relative standard deviation (RSD) was below 1.5% for intraday and interday precision. The recovery rates for BA were in the range of 97–103%, with RSD less than 3%.
Characterization of baicalin solid dispersion
SEM
The particle size and surface morphology of the BA, MCN, PM, and SDs were characterized using SEM (S-3000N microscope; Hitachi, Tokyo, Japan). Before the observation, the samples were adhered to metal stubs by double-sided adhesive tape and covered with a thin layer of gold.
DSC
The thermographs of the BA, MCN, PM, and SDs were obtained using a DSC 204A/G Phoenix® instrument (Netzsch, Germany). All samples were dried at 40°C for 24 h to remove the free water. The powder (5–8 mg) was hermetically sealed in an aluminum pan and heated from 20°C to 550°C at a rate of 10°C·min-1. All measurements were conducted under nitrogen (50 mL·minute-1 flow rate).
Powder XRD analysis
The physical state of the BA, MCN, PM, and SDs was evaluated using an XRD system (D8-Advance; Bruker, Karlsruhe, Germany), and the data was collected using primary mono chromated radiation (Cu Kα1, λ = 1.5406 Å). The diffraction patterns were recorded at a scanning speed of 6°C/min with a 2θ angular extent of 0–70°C.
Pharmacokinetic experiments
Animals
The pharmacokinetic studies were conducted in Sprague-Dawley rats (220 ± 20 g, half male and half female), which were purchased from Shanghai SLAC Laboratory Animal Co., Ltd. (Shanghai, China). The rats were housed in an animal room at an ambient temperature of 22–24°C and a relative humidity of 60%. Before the experiments, the animals were fasted 12 h with free access to tap water. Permission for the animal studies was obtained from the animal ethics committee of Jiangsu Provincial Academy of Chinese Medicine.
Chromatographic and mass spectrometry conditions
The analysis of the biological samples was performed on an Acquity UPLC/TQ-2000 system (Waters Corp., Milford, MA, USA) equipped with a conditioned autosampler at 4°C. An Acquity UPLC BEH C18 column (i.d., 1.7 μm; 2.1 mm × 50 mm; Waters, Milford, MA, USA) was used to separate the samples. The mobile phase consisted of methanol (solvent A) and 0.1% formic acid in water (solvent B) at a flow rate of 0.2 mL/min. The optimal isocratic elution was 60% A/40% B. The volume of 2 μL was injected into system for analysis. Mass spectrometric condition: Mass spectra were recorded on a quadrupole TQ-2000 mass spectrometer (Waters Corporation, Milford, MA, USA) equipped with an electrospray ionization (ESI) interface. Baicalin and daidzein (internal standard, IS) were both detected in the positive ionization mode. The source temperature and desolvation temperature were set at 120 and 350°C, while desolvation gas (nitrogen), cone gas (nitrogen) and collision gas (argon) flow were 650 L/h, 50 L/h and 0.15 mL/min, respectively. The capillary voltage and cone voltage were set at 3000 V and 40 V, respectively. The multiple reaction monitoring (MRM) model, with transitions m/z 447 ulm/z 271 for baicalin and m/z 255 lim/z 199 for the IS, was applied to quantitatively analyze the samples. The mass range was scanned from 100 to 700 m/z. The data was processed by MassLynx (version 4.1; Waters).
Pharmacokinetic study
Twenty-four rats were selected for experiments to evaluate the pharmacokinetic characteristics of the BA. The rats were randomly divided into three groups (n = 8 rats in each group) for administration of the BA, PM, and SDs (1:6, w/w) at a dose equivalent to 100 mg/kg of BA.[25] Blood samples (500 μL) were collected from the orbital sinus into heparinized centrifuge tubes at 0.083, 0.25, 0.5, 0.75, 1, 1.5, 2, 4, 6, 8, 10, 12, and 24 h after dosing. The blood samples were centrifuged at 3000 rpm for 10 min at 4°C to separate the plasma, and the supernatants were stored at 70°C for later analysis.
Sample preparation and ultraperformance liquid chromatography-mass spectrometry (UPLC-MS) analysis
The plasma samples were prepared as follows. Briefly, 200 μL of plasma was partitioned with 800 μL of methanol (containing 1.0 μg/mL of daidzein as an IS). The mixture was vortexed for 5 min to extract BA and daidzein, followed by centrifugation at 12,000 rpm for 10 min. The precipitate containing the proteins and mesoporous carbon nanomaterial was discarded. The methanol layer was evaporated to dryness at 40°C under N2 gas. The residue was reconstituted with 200 μL of the mobile phase (methanol/0.1% formic acid water; 60:40), vortexed, and centrifuged at 12 000 rpm for 10 min. Finally, 2 μL of the solution was subjected to the chromatographic system for analysis.
Data presentation and analysis
DAS software program version 3.0 (Chinese Pharmacology Society, Beijing, China) was used to analyze the plasma BA concentration-time curve. The main pharmacokinetic parameters, (area under the drug concentration-time (AUC0re and AUC0an), peak plasma concentration (Cmax), time to peak concentration (Tmax), and elimination half-life (t1/2)) were investigated using a non-compartmental model. All results were expressed as mean cstandard deviation. P-values were determined using SPSS software (version 13; IBM Corp., Armonk, NY, USA).
Intestinal and renal toxicity test of MCN
Thirty-two male Sprague–Dawley rats (220 ± 20 g) were divided into four groups (n = 8 rats in each group) to evaluate the intestinal and renal toxicity of MCN. MCN, PM, and SDs (BA/MCN ratio of 1:6) at a dose equivalent to 100 mg/kg of BA were orally administered to appropriate group, while the fourth group received the same volume of physiological saline. Two weeks after administration, the jejunum and kidney was removed and washed thoroughly with physiological saline. The jejunum and kidney samples were preserved in 10% formalin, embedded in paraffin and stained with hematoxylin–eosin (HE). All specimens were visualized and photographed under a microscope using a camera system (Olympus LK2, Tokyo, Japan).
Stability study
The physical stability test of SDs was tested under accelerated conditions (40°C and 75% RH) for 6 months. Changes in drug content and in-vitro dissolution of SDs were assessed regularly.
RESULTS AND DISCUSSION
In-vitro drug dissolution behavior
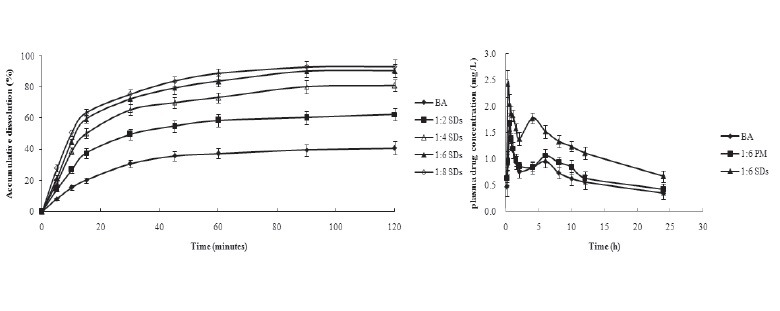
Dissolution tests were performed for pure BA, PM and SDs of BA with MCN samples with different drug/carrier ratios [Figure 2]. The PM showed an improvement on drug dissolution, but there was no significant difference between pure BA and PM. In comparison with pure crystalline BA, the SDs exhibited greater dissolution of BA. The raw drug showed minimal dissolution within a 2-h period, and the maximum accumulated amount of drug was about 30% of the initially applied amount. In contrast, the accumulative dissolutions from the BA–MCN samples prepared with drug/carrier ratios of 1:2, 1:4, 1:6, and 1:8 (w/w) were 62.4%, 81.2%, 90.7% and 93.3%, respectively, and the dissolution rate of the BA–MCN samples was much faster than that of raw BA. Dissolution might have been enhanced because of the amorphous state of the BA in the SDs samples, which increased water solubility.[8,26] Drug release is usually enhanced by utilizing non-crystalline drug forms, because no energy is required to disrupt the crystal lattice during the dissolution process. Moreover, dissolution was clearly associated with the ratio of drug to MCN carrier.[16] The accumulative dissolution of BA from the MCN–BA system at drug/carrier ratios of 1:6 and 1:8 was similar. Therefore, the SDs with a BA/MCN ratio of 1:6 was selected for further study.
Figure 2.
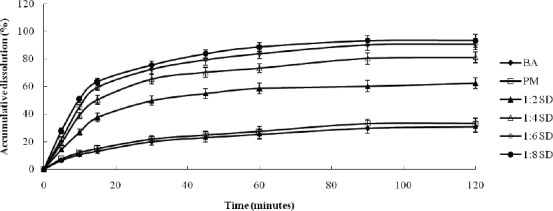
The dissolution profiles of BA and SDs prepared using different ratios of BA/MCN (1:2, 1:4, 1:6, and 1:8).
Characterization of BA solid dispersion
SEM
The scanning electron micrographs of BA, MCN, the BA/MCN PM, and the SDs with a BA/MCN ratio of 1:6 are shown in [Figure 3]. The BA particles consisted of micro particles and large crystals (flat broken needles of different sizes). The porous structure of the MCN carrier, which enhanced drug loading and release, was clearly observed.[20] Additionally, the nanocavity effectively blocked drug re-crystallization and remarkably decreased the particle size of the non-crystalline drug.[27] The features of BA and MCN were visible in the PM. In contrast, BA was not observed separately in the SDs, suggesting that BA dispersed uniformly into the carrier.
Figure 3.

Scanning electron microscopy of pure BA (A), MCN (B), PM (C) and SDs prepared at a BA/MCN ratio of 1:6 (D).
DSC
The thermal behaviors of BA, MCN, the BA/MCN PM, and the SDs with a BA/MCN ratio of 1:6 were recorded. When BA was present in crystalline form, a single depression was seen at its melting point. No melting peak was observed when the drug was in an amorphous state.[28] The DSC thermograms of raw BA and the PM, which possessed a single endothermic melting peak at approximately 223°C, are shown in [Figure 4]. These results were in agreement with the results reported by Yan et al.[29] However, no trace of an endothermic peak at 223°C was identified in the DSC curve for the tested SDs. The absence of phase transitions was evidence that the BA in the SDs was molecularly dispersed and might have been in an amorphous form, which facilitated dissolution.
Figure 4.
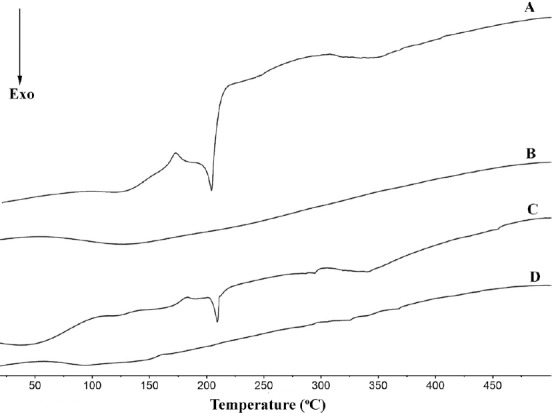
Differential scanning calorimetry curves of pure BA (A), MCN (B), PM (C) and SDs prepared at a BA/MCN ratio of 1:6 (D).
XRD
XRD was performed to determine whether a crystalline BA phase could be detected after loading the drug into MCN. The diffractograms of pure BA, MCN, the BA/MCN PM, and the SDs with a BA/MCN ratio of 1:6 are shown in [Figure 5]. The XRD analysis was generally consistent with the DSC results. The diffraction pattern of pure BA showed numerous peaks, indicating a highly crystalline state. No diffraction peaks were observed for MCN, suggesting an amorphous state. For the BA/MCN PM, prominent and characteristic diffraction peaks of BA were observed, which confirmed that MCN had no effect on the drug crystalline transition of BA. As expected, no crystalline BA was detected in the SDs sample, confirming the non-crystalline state of the incorporated drug; the adsorbed BA molecules did not show their original crystal state, but instead presented an amorphous state. Utilization of amorphous BA could significantly enhance the dissolution of oral BA formulations, leading to a marked improvement in oral bioavailability.
Figure 5.
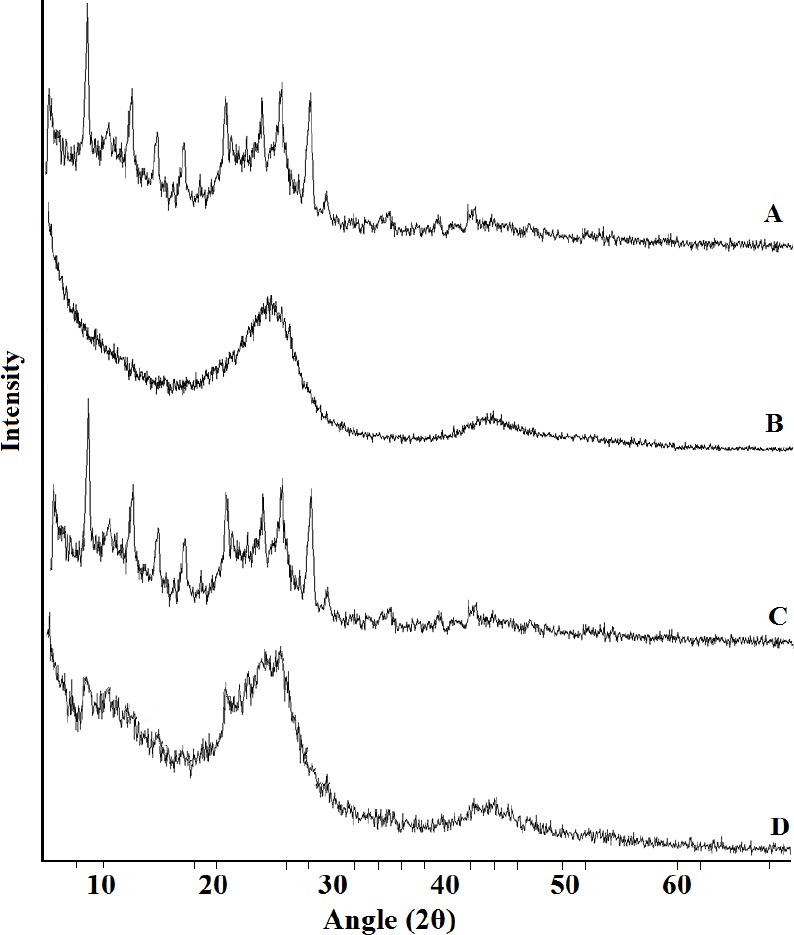
X-ray diffraction. (A) BA, (B) MCN, (C) PM, and (D) SDs.
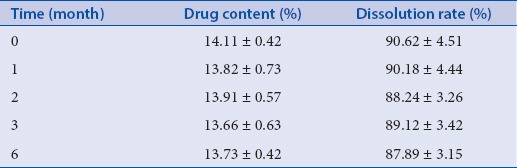
Accelerated stability test
To allow widespread use, SDs should have good physical stability. However, it is difficult to maintain an amorphous drug state (a state of flux) stable for a long time. Therefore, the stability of amorphous BA in the SDs was examined. As shown in [Table 1], the drug content and dissolution rate were changed little after 1, 2, 3, and 6 months. These findings indicated that MCN dispersed and inhibited aggregation and re-crystallization of amorphous BA in SDs.
Table 1.
Stability test of SDs (n = 3, ± standard deviation)
In-vivo absorption study
The concentration of BA in plasma samples was analyzed by performing HPLC/MSMS as described above, and daidzein was used as the IS. The calibration curve showed a good linear relationship at concentrations between 0.01 mg·L-1 and 10.00 mg·L-1 (r = 0.9992). The limit of quantification was 0.01 mg·L-1. The between- and within-day precisions were 6.24% and 5.36%, respectively. The relative recoveries of BA at high, middle, and low concentrations were 102.35% ± 13.21%, 99.47% ± 11.21%, and 95.65% ± 6.98%, respectively. The RSD values were all less than 15%, which were within the acceptable range of the guidelines for bio-analytical methods.
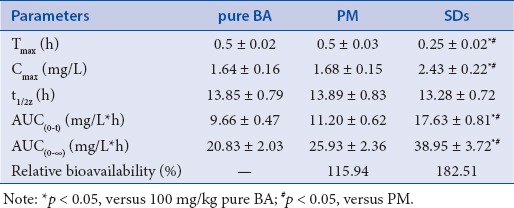
The in-vitro dissolution experiments confirmed that the dissolution of SDs was increased. To evaluate the effect of MCN carrier in vivo, the rats received the BA suspension, the SDs (1:6 drug/carrier ratio), or the PM by intra gastric administration. The mean plasma concentration-time profiles are shown in [Figure 6], and the major pharmacokinetic parameters are listed in [Table 2]. The Cmax of the SDs (2.43 mg/L) was significantly greater than that of pure BA (1.64 mg/L) or the PM (1.68 mg/L) (p < 0.05). The Tmax value of the SDs was 0.25 h, which was remarkably earlier than that of pure BA and the PM (p < 0.05), which both showed a Tmax of 0.5 h. In comparison with pure BA and the PM, the SDs system showed a significant improvement in AUC0–t value. The BA plasma levels following oral administration of pure BA or the PM were very low, with a AUC0–t of 9.66 mg/L.h and 11.20 mg/L.h, respectively. In contrast, the AUC0–t value of the SDs system reached 17.63 mg/L.h. These results indicated that the SDs enhanced the relative oral bio-availability of BA by approximately 182.51%.
Figure 6.
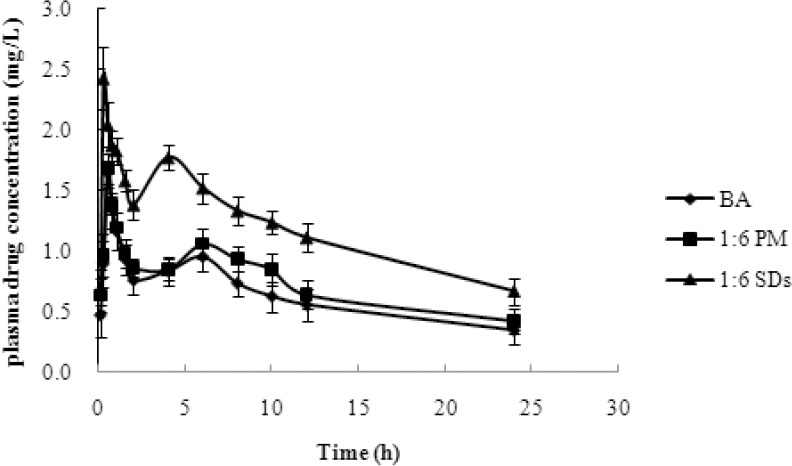
The plasma concentration–time profile of BA after oral administration of BA, PMs and SDs to rats, at a dose of 100 mg/kg. The data are presented as mean ± standard deviation (n = 6).
Table 2.
The mean pharmacokinetic parameters of BA in rats after oral administration of BA, PM (BA/MCN ratio of 1:6) and SDs (BA/MCN ratio of 1:6) in a dose of 100 mg/kg (n = 6, ± standard deviation)
The pharmacokinetic characteristics of BA were in good agreement with the results of the in-vitro dissolution experiment. The mean plasma concentration-time profiles demonstrated a prominent enhancement in oral BA absorption when the SDs formulation utilized MCN as a drug carrier. The nano-scale pores and highly dispersed drug particles remarkably increased the surface area of BA, which accelerated its dissolution from the BA/MCN system into the gastrointestinal tract. Therefore, the oral absorption of BA was promoted by the MCN SDs.[30,31,32] Additionally, the bioavailability of BA from the PM was slightly greater than that of pure BA, perhaps due to the promotion of absorption by MCN.
Intestinal and renal toxicity test of MCN
The use of MCN as a drug carrier should not cause any toxicity. Therefore, gastrointestinal and renal toxicity following oral administration was investigated. As shown in [Figure 7A], no degeneration, necrosis, interstitial congestion, edema, or inflammatory cell infiltration were produced in the mucosa, sub mucosa, muscularis externa, or serosa after exposure to MCN, the BA/MCN PM, or the SDs (1:6 drug/carrier ratio) for 2 weeks. The intestinal mucosal structure was not damaged, and goblet cells were unchanged in the control group. No significant differences in intestinal mucosal structure were observed among the 3 treated groups. It was easy to see that complete membrane, clear layer of cortex and medulla, no atrophy and necrosis of glomerular, no congestion and inflammatory cell infiltration in the renal tubules of control group. Importantly, the pathology report of rats treated with MCN, PM, or SDs was almost the same as the control group [Figure 7B]. Therefore, MCN produced no significant side-effects, and toxicity of the gastrointestinal tract mucosa and kidney was not observed.
Figure 7.
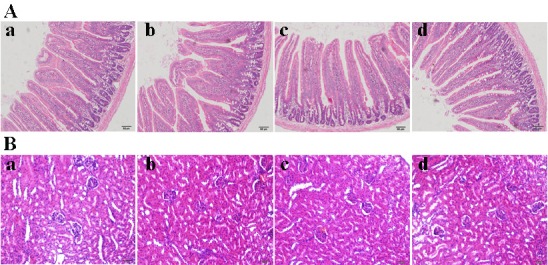
Intestinal and renal toxicity test of MCN. (A) Influences of MCN, PM, and SDs treatment on intestinal mucosal structures of rats, (a) control group, (b) MCN group, (c) PM group, and (d) SDs group. (B) Influences of MCN, PM, and SDs treatment on renal mucosal structures of rats, (a) control group, (b) MCN group, (c) PM group, and (d) SDs group.
CONCLUSION
In this study, the utility of a novel MCN to improve the solubility of poorly water-soluble drugs was assessed using an SDs formulation. BA was loaded into the mesoporous carbon carrier to study its physical state, in-vitro dissolution/release and in-vivo pharmacokinetics. SEM, XRD and DSC investigations confirmed that BA was successfully loaded into the pores of the mesoporous carbon carrier in an amorphous state. In the in-vitro drug dissolution and release tests, MCN could remarkably improved the dissolution rate of BA in comparison with that of pure BA. The pharmacokinetic studies demonstrated the capability of the MCN material to enhance the drug dissolution and bioavailability in the gastrointestinal tract. Thus, MCN appears to be a promising candidate as an SDs carrier that can provide rapid and efficient release of drugs with poor water solubility.
Financial support and sponsorship
Nil
Conflicts of interest
There are no conflicts of interest.
ABOUT AUTHOR

Dr. Xiaobin Jia
Dr. Xiaobin Jia, is a professor in the key laboratory of new drug delivery system of Chinese Materia Medica, Jiangsu provincial academy of Chinese Medicine, 100 shizi road, Nanjing, P.R.China 210028. His main research field is the material basis of Traditional Chinese Medicine.
Acknowledgement
This work was supported by the Natural Science Foundation of China (No. 81403121).
REFERENCES
- 1.Sun J, Li L, Wu J, Liu B, Gong W, Lv Y, et al. Effects of baicalin on airway remodeling in asthmatic mice. Planta Med. 2013;79:199–06. doi: 10.1055/s-0032-1328197. [DOI] [PubMed] [Google Scholar]
- 2.Kim AR, Kim SN, Jung IK, Kim HH, Park YH, Park WS, et al. The inhibitory effect of Scutellaria baicalensis extract and its active compound, baicalin, on the translocation of the androgen receptor with implications for preventing androgenetic alopecia. Planta Med. 2014;80:153–8. doi: 10.1055/s-0033-1360300. [DOI] [PubMed] [Google Scholar]
- 3.Cui L, Feng L, Zhang ZH, Jia XB. The anti-inflammation effect of baicalin on experimental colitis through inhibiting TLR4/NF-κB pathway activation. Int Immunopharmacol. 2014;23:294–03. doi: 10.1016/j.intimp.2014.09.005. [DOI] [PubMed] [Google Scholar]
- 4.Srinivas NR. Baicalin, an emerging multi-therapeutic agent: pharmacodynamics, pharmacokinetics, and considerations from drug development perspectives. Xenobiotica. 2010;40:357–67. doi: 10.3109/00498251003663724. [DOI] [PubMed] [Google Scholar]
- 5.Xing J, Chen X, Zhong D. Absorption and enterohepatic circulation of baicalin in rats. Life Sci. 2005;78:140–6. doi: 10.1016/j.lfs.2005.04.072. [DOI] [PubMed] [Google Scholar]
- 6.Zhou X, Liu D, Liu H, Yang Q, Yao K, Wang X. Effect of low molecular weight chitosans on drug permeation through mouse skin: 1. Transdermal delivery of baicalin. J Pharm Sci. 2010;99:2991–8. doi: 10.1002/jps.22063. [DOI] [PubMed] [Google Scholar]
- 7.Kesisoglou F, Panmai S, Wu Y. Nanosizing-oral formulation development and biopharmaceutical evaluation. Adv Drug Deliv Rev. 2007;59:631–44. doi: 10.1016/j.addr.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 8.Vasconcelos T, Sarmento B, Costa P. Solid dispersions as strategy to improve oral bioavailability of poor water soluble drugs. Drug Discov Today. 2007;12:1068–75. doi: 10.1016/j.drudis.2007.09.005. [DOI] [PubMed] [Google Scholar]
- 9.Yan HM, Sun E, Cui L, Jia XB, Jin X. Improvement in oral bioavailability and dissolution of tanshinone IIA by preparation of solid dispersions with porous silica. J Pharm Pharmacol. 2015 doi: 10.1111/jphp.12423. doi: 10.1111/jphp.12423. [DOI] [PubMed] [Google Scholar]
- 10.Guo Z, Lu M, Li Y, Pang H, Lin L, Liu X, et al. The utilization of drug-polymer interactions for improving the chemical stability of hot-melt extruded solid dispersions. J Pharm Pharmacol. 2014;66:285–96. doi: 10.1111/jphp.12145. [DOI] [PubMed] [Google Scholar]
- 11.Gu B, Linehan B, Tseng YC. Optimization of the Büchi B-90 spray drying process using central composite design for preparation of solid dispersions. Int J Pharm. 2015;491:208–17. doi: 10.1016/j.ijpharm.2015.06.006. [DOI] [PubMed] [Google Scholar]
- 12.Kim SA, Kim SW, Choi HK, Han HK. Enhanced systemic exposure of saquinavir via the concomitant use of curcumin-loaded solid dispersion in rats. Eur J Pharm Sci. 2013;49:800–4. doi: 10.1016/j.ejps.2013.05.029. [DOI] [PubMed] [Google Scholar]
- 13.Cho Y, Ha ES, Baek IH, Kim MS, Cho CW, Hwang SJ, et al. Enhanced supersaturation and oral absorption of sirolimus using an amorphous solid dispersion based on eudragit® e. Molecules. 2015;20:9496–509. doi: 10.3390/molecules20069496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ghebremeskel AN, Vemavarapu C, Lodaya M. Use of surfactants as plasticizers in preparing solid dispersions of poorly soluble API: selection of polymer-surfactant combinations using solubility parameters and testing the processability. Int J Pharm. 2007;328:119–29. doi: 10.1016/j.ijpharm.2006.08.010. [DOI] [PubMed] [Google Scholar]
- 15.Van den Mooter G, Weuts I, De Ridder T, Blaton N. Evaluation of Inutec SP1 as a new carrier in the formulation of solid dispersions for poorly soluble drugs. Int J Pharm. 2006;316:1–6. doi: 10.1016/j.ijpharm.2006.02.025. [DOI] [PubMed] [Google Scholar]
- 16.Wang Y, Sun L, Jiang T, Zhang J, Zhang C, Sun C, et al. The investigation of MCM-48-type and MCM-41-type mesoporous silica as oral solid dispersion carriers for water insoluble cilostazol. Drug Dev Ind Pharm. 2014;40:819–28. doi: 10.3109/03639045.2013.788013. [DOI] [PubMed] [Google Scholar]
- 17.Zhang Y, Wang H, Li C, Sun B, Wang Y, Wang S, et al. A novel three-dimensional large-pore mesoporous carbon matrix as a potential nanovehicle for the fast release of the poorly water-soluble drug, celecoxib. Pharm Res. 2014;31:1059–70. doi: 10.1007/s11095-013-1227-9. [DOI] [PubMed] [Google Scholar]
- 18.Niu X, Wan L, Hou Z, Wang T, Sun C, Sun J, et al. Mesoporous carbon as a novel drug carrier of fenofibrate for enhancement of the dissolution and oral bioavailability. Int J Pharm. 2013;452:382–9. doi: 10.1016/j.ijpharm.2013.05.016. [DOI] [PubMed] [Google Scholar]
- 19.Charnay C, Bégu S, Tourné-Péteilh C, Nicole L, Lerner DA, Devoisselle JM, et al. Inclusion of ibuprofen in mesoporous templated silica: drug loading and release property. Eur J Pharm Biopharm. 2004;57:533–40. doi: 10.1016/j.ejpb.2003.12.007. [DOI] [PubMed] [Google Scholar]
- 20.Thomas MJ, Slipper I, Walunj A, Jain A, Favretto ME, Kallinteri P, et al. Inclusion of poorly soluble drugs in highly ordered mesoporous silica nanoparticles. Int J Pharm. 2010;387:272–7. doi: 10.1016/j.ijpharm.2009.12.023. [DOI] [PubMed] [Google Scholar]
- 21.Vinu A, Miyahara M, Ariga K. Biomaterial immobilization in nanoporous carbon molecular sieves: influence of solution pH, pore volume, and pore diameter. J Phys Chem B. 2005;109:6436–41. doi: 10.1021/jp050454o. [DOI] [PubMed] [Google Scholar]
- 22.Zhang Y, Wang H, Gao C, Li X, Li L. Highly ordered mesoporous carbon nanomatrix as a new approach to improve the oral absorption of the water-insoluble drug, simvastatin. Eur J Pharm Sci. 2013;49:864–72. doi: 10.1016/j.ejps.2013.05.031. [DOI] [PubMed] [Google Scholar]
- 23.Liang C, Li Z, Dai S. Mesoporous carbon materials: synthesis and modification. Angew Chem Int Ed Engl. 2008;47:3696–717. doi: 10.1002/anie.200702046. [DOI] [PubMed] [Google Scholar]
- 24.Zhao P, Wang L, Sun C, Jiang T, Zhang J, Zhang Q, et al. Uniform mesoporous carbon as a carrier for poorly water soluble drug and its cytotoxicity study. Eur J Pharm Biopharm. 2012;80:535–43. doi: 10.1016/j.ejpb.2011.12.002. [DOI] [PubMed] [Google Scholar]
- 25.Lai MY, Hsiu SL, Tsai SY, Hou YC, Chao PD, et al. Comparison of metabolic pharmacokinetics of baicalin and baicalein in rats. J Pharm and Pharmacol. 2003;55:205–9. doi: 10.1211/002235702522. [DOI] [PubMed] [Google Scholar]
- 26.Lloyd GR, Craig DQ, Smith A. A calorimetric investigation into the interaction between paracetamol and polyethlene glycol 4000 in physical mixes and solid dispersions. Eur J Pharm Biopharm. 1999;48:59–65. doi: 10.1016/s0939-6411(99)00022-3. [DOI] [PubMed] [Google Scholar]
- 27.Qian KK, Bogner RH. Application of mesoporous silicon dioxide and silicate in oral amorphous drug delivery systems. J Pharm Sci. 2012;101:444–63. doi: 10.1002/jps.22779. [DOI] [PubMed] [Google Scholar]
- 28.Sanganwar GP, Gupta RP. Dissolution-rate enhancement of fenofibrate by adsorption onto silica using supercritical carbon dioxide. Int J Pharm. 2008;360:213–8. doi: 10.1016/j.ijpharm.2008.04.041. [DOI] [PubMed] [Google Scholar]
- 29.Yan HM, Zhang ZH, Jiang YR, Ding DM, Sun E, Jia XB, et al. Preparation of baicalin colon-specific solid dispersion and evaluation on its in vitro release. Zhongguo Zhong Yao Za Zhi. 2014;39:71–4. [PubMed] [Google Scholar]
- 30.Marasini N, Tran TH, Poudel BK, Cho HJ, Choi YK, Chi SC, et al. Fabrication and evaluation of pH-modulated solid dispersion for telmisartan by spray-drying technique. Int J Pharm. 2013;441:424–32. doi: 10.1016/j.ijpharm.2012.11.012. [DOI] [PubMed] [Google Scholar]
- 31.Emara LH, Badr RM, Elbary AA. Improving the dissolution and bioavailability of nifedipine using solid dispersions and solubilizers. Drug Dev Ind Pharm. 2002;28:795–07. doi: 10.1081/ddc-120005625. [DOI] [PubMed] [Google Scholar]
- 32.Li X, Gu L, Xu Y, Wang Y. Preparation of fenofibrate nanosuspension and study of its pharmacokinetic behavior in rats. Drug Dev Ind Pharm. 2009;35:827–33. doi: 10.1080/03639040802623941. [DOI] [PubMed] [Google Scholar]