

Abstract
Background
The coffee species Coffea canephora is commercially identified as “Conilon” when produced in Brazil, or “Robusta” when produced elsewhere in the world. It represents approximately 40 % of coffee production worldwide. While the genetic diversity of wild C. canephora has been well studied in the past, only few studies have addressed the genetic diversity of currently cultivated varieties around the globe. Vietnam is the largest Robusta producer in the world, while Mexico is the only Latin American country, besides Brazil, that has a significant Robusta production. Knowledge of the genetic origin of Robusta cultivated varieties in countries as important as Vietnam and Mexico is therefore of high interest.
Results
Through the use of Sequencing-based diversity array technology-DArTseq method-on a collection of C. canephora composed of known accessions and accessions cultivated in Vietnam and Mexico, 4,021 polymorphic SNPs were identified. We used a multivariate analysis using SNP data from reference accessions in order to confirm and further fine-tune the genetic diversity of C. canephora. Also, by interpolating the data obtained for the varieties from Vietnam and Mexico, we determined that they are closely related to each other, and identified that their genetic origin is the Robusta Congo – Uganda group.
Conclusions
The genetic characterization based on SNP markers of the varieties grown throughout the world, increased our knowledge on the genetic diversity of C. canephora, and contributed to the understanding of the genetic background of varieties from very important coffee producers. Given the common genetic origin of the Robusta varieties cultivated in Vietnam, Mexico and Uganda, and the similar characteristics of climatic areas and relatively high altitude where they are grown, we can state that the Vietnamese and the Mexican Robusta have the same genetic potential to produce good cup quality.
Electronic supplementary material
The online version of this article (doi:10.1186/s12870-016-0933-y) contains supplementary material, which is available to authorized users.
Keywords: Genetic diversity, DArTseq, Coffea canephora, Mexico, Vietnam
Background
Canephora coffee produced by the coffee species Coffea canephora is named either “Conilon” when produced in Brazil, or “Robusta” when produced elsewhere in the world. In 2014, Canephora (hence Conilon and Robusta) coffee represented around 40 % of coffee production worldwide, while the remaining part corresponded to (http://www.ico.org/).
C. canephora is a rubiaceous plant originated from the sub-equatorial plains of Africa. It belongs to the Coffea genus, which comprises 124 species, originating from Africa, Madagascar, the Mascarene Islands, Asia and Oceania [1]. C. canephora and Coffea species are lowland, generally allogamous and diploids (2n = 2x = 22), with the notable exception of the highland, self-fertilizing allotetraploid (2n = 4x = 44) C. arabica [2]. Wild C. canephora plants are naturally distributed within intertropical Africa, stretching from Guinea to Uganda and from Central African Republic to Angola. Natural populations are composed of few individuals, subjected to gene flows from neighboring populations up to a few kilometers away [3, 4].
Based on former genetic studies [5–7], five main regions of wild genetically distant populations can be recognized: (i) West Africa (Guinea and Ivory Coast); (ii) Central Africa, Cameroon and Congo; (iii) the Atlantic frontage from Gabon to Angola; (iv) the Congo central basin; and (v) Uganda. The genetic diversity of C. canephora has been analyzed using isozyme markers [8, 9], microsatellites [10–12] and RFLPs [7, 13]. While these former analyses gave consistent results regarding the number and geographic origin of genetic groups, each independent work gave different names ending up with some confusion for the coffee community, suggesting the importance of precisely defining a general nomenclature. In this paper, we have therefore chosen, for clarity’s sake, the use of a new unified nomenclature for the five previously referenced genetic groups of C. canephora, which will be explained in detail in the plant materials section.
Whereas C. arabica was cultivated early (since the XIVth century) in Ethiopia and Yemen, C. canephora cultivation dates back to the end of the XIXth century, based on the use of local landraces populations. C. canephora was introduced to the main current producers of Robusta coffee by colonists during the 19th century [14]. Until recently, it was thought that most of the cultivated C. canephora trees were derived from common sources reported to belong to the Congo basin [15, 16]. While former genetic diversity studies of C. canephora have focused on wild accessions from Africa and several Brazilian cultivated varieties [17], nothing is known about the genetic origin of coffee cultivated in Robusta-producing countries as important as Vietnam and Mexico. Vietnam is the first C. canephora producer (http://faostat3.fao.org), yet the genetic origin of the coffee plants grown by more than 400 000 cultivators in over 600 000 ha, within relatively high altitudes for Robusta coffee (>600 m.a.s.l), remains unknown. From 2012 to 2015, Vietnam produced 23 to 27 million 60 kg-bags of coffee, while Brazil produced 43 to 51 million of Arabica and Robusta taken together. In Latin America, apart from Brazil, only Mexico has a significant C. canephora production, producing 3.5 to 4.3 million (http://www.ico.org/). The qualities of the beans from Mexico and Vietnam have limited their marketability. Notably, Vietnamese beans are typically used in cheap soluble Western coffee. As a consequence of climate change, C. arabica growing will be affected in hotter lower (600–800 m.a.s.l.) production zones [18]. C. canephora could thus represent a good alternative for millions of small coffee farmers. In the near future, Mexican C. canephora varieties will probably become the sources of varieties for Central America, where C. canephora cultivation is rapidly expanding due to its resistance to several diseases. Knowing the genetic origin of the accessions cultivated in Vietnam and Mexico is therefore of the greatest interest.
As mentioned before, C. canephora genetic diversity has been analyzed using a limited number of isozyme, SSR and RFLP markers, representing only a restricted fraction of the C. canephora genome. In contrast to classical molecular markers, SNPs (Single nucleotide polymorphisms) are the most abundant markers, particularly in the non-coding regions of the genome [19]. New sequencing technologies (so called Next generation sequencing or NGS) used jointly with different complexity reduction methods, like the ones used in RADseq (Restriction site associated DNA sequencing) [20], GBS (Genotyping by sequencing) [21] and DArTseq (Sequencing-based diversity array technology) [22] methods, enable a large-scale discovery of SNPs in a wide variety of non-model organisms. When such techniques are applied to hundreds of genotypes, they provide measures of genetic divergence and genetic diversity within the major genetic clusters that comprise crop germplasm [23]. Indeed, the recently sequenced and assembled C. canephora genome, representing 64 % of the 710 Mb genome [24], facilitates the use of such marker technology and further analyses of the obtained data.
For this new extended study of the genetic diversity of C. canephora, we report the use of SNPs markers. In this study, DArTseq [22], a technique based on complexity reduction by the use of restriction enzymes targeting gene-rich regions and NGS sequencing, was used to study the genetic diversity of C. canephora. The specific objectives of the present study are (i) to test the performance of DArTseq method-derived markers in coffee: repeatability, error rates and genome wide representation of the markers; (ii) to assess consistency of C. canephora genetic diversity structures as compared to previous studies with ancient markers; and (iii) to identify the genetic origin of the coffee plants cultivated in Vietnam and Mexico, and to discuss possible consequences for coffee quality and breeding. By evaluating DArTseq-derived SNP markers from a set of well-known and unknown C. canephora accessions, it was possible to confirm and further fine-tune the genetic diversity of C. canephora, and to identify the genetic origin of accessions cultivated in two climate change susceptible zones, Vietnam and Mexico.
Methods
Plant material
Since each previous independent study has given different names to the genetic groups found, in this paper we have therefore chosen the use of the following nomenclature for the five previously referenced genetic groups of C. canephora: (i) “Guinean” Group (sometimes called D group), it is the genetic group originating from the Ivory Coast-Guinea area in West Africa; (ii) “Nana” group (sometimes called C group), stands for the coffee originating from the fringes of South-East Cameroon, South-West Central Africa and Northern Congo; (iii) “Conilon” group (sometimes called SG1 or A) represented by the Luki, Niaouli and Kouilou domesticated populations, originating from the south of Gabon; (iv) “Robusta Congo-Central Africa” group (sometimes called B), constituted by the wild coffees from the north of the Congo central basin and the south of Central Africa; and (v) “Robusta Congo-Uganda” group (sometimes called SG2) corresponding to the wild populations or cultivated varieties native to Uganda and the Congo basin.
A collection of 105 individuals from 87 accessions of C. canephora was analyzed in this study, from which 81 were used to analyze the diversity structure present in C. canephora. Known accessions, provided by the IRD (Institut de recherche pour le développement), were used as biological and technical replicates, to structure C. canephora diversity; while lyophilized leaves of plants cultivated in Mexico and Vietnam were supplied by AMSA (Agroindustrias unidas de México). Details on the accessions are given in Table 1 and Additional file 1: Table S1. C. canephora accessions are coded using the following rules: The first letter depicts their agronomical interest: wild (W) or cultivated (C). The following two letters represent their country of origin: Central African Republic (Ca), Congo (Cg), Ivory Coast (Ci), Cameroon (Cm), Uganda (Ug), Mexico (Mx), and Vietnam (Vn). The remaining numbers correspond to the plant number. Full siblings are named with “_” followed by the corresponding number. Biological replicates are named with “-” followed by the corresponding number. Accessions with technical replicates are marked as “-a” or “-b”.
Table 1.
List of C. canephora accessions evaluated with DArTseq SNP markers
| Wild/cultivated | Origin (prospection or cultivated) | No. of individuals | Provider | Putative genetic group | Reference | Markers | |
|---|---|---|---|---|---|---|---|
| Active individuals | Wild | South - East Cameroon | 9 | IRD | Nana | [7] | RFLP |
| Wild | South - West Central African Republic | 5 | IRD | Nana | [7] | RFLP | |
| Wild | South Central African Republic | 9 | IRD | Robusta Congo-Central Africa | [7] | RFLP | |
| Wild/Cultivated | Ivory Coast | 3 | IRD | Guinean | [7] | RFLP | |
| Cultivated | Ivory Coast | 2 | CIRAD | Conilon | [35] | Isozymes | |
| Cultivated | Central America | 2 | Catie | Robusta Congo-Uganda | [10] | SSR | |
| Cultivated | Uganda | 4 | Cori, Uganda | Robusta Congo-Uganda | [10] | SSR | |
| Subtotal | 34 | ||||||
| Supplemental Individuals | Cultivated | Vietnam | 6 | AMSA | Unknown | ||
| Cultivated | Mexico, Chiapas | 41 | AMSA | Unknown | |||
| Subtotal | 47 | ||||||
| Biological replicates | Wild/Cultivated | Various | 20 | IRD/ CIRAD | Various | ||
| Additional technical replicates | Wild/Cultivated | Various | 4 | IRD/CIRAD | Various | ||
| Total | 105 |
Active individuals in multivariate analysis are those whose putative genetic group could be deduced from past studies. Other individuals, whose genetic group was unknown, were considered as supplementary individuals. Biological replicates are plants representing four different accessions
DNA extraction and genotyping
Genomic DNA was extracted from leafs using the ADNid method (http://www.adnid.fr/index-2-4A.html). Technical replicates from two independent DNA extractions were used for some accessions and several accessions were represented by more than one tree, as biological replicates (Additional file 1: Table S1). Genotyping was carried out at DArT P/L in Canberra-Australia, using a combination of HiSeq 2000 (Illumina) next-generation sequencing with DArT technology, as previously described [22]. The SNP markers obtained were used for data analysis after discarding markers with more than 10 % of missing data and a minor allele frequency (MAF) below 1 %.
Data analysis
In order to obtain the genotyping error rates of the DArTseq method when applied to coffee, the identical allele call rates in technical and biological replicates were evaluated with the “Similarity of Individuals” function from the Joinmap 4.1 software [25], based on SNPs with no missing data within the entire panel of replicates. Then, the error rates were calculated as the number of allelic differences between replicates, divided by the total number of markers analyzed [26].
All the genetic statistical analyses were carried using R, version 3.2.3 [27]. The polymorphic information content (PIC) for each SNP marker was calculated using the equation with p2 i representing the squared frequency of allele i at each locus. Statistics such as the mean observed heterozygosity (Ho), and mean expected heterozygosity (He) were calculated with the “adegenet” 2.0.2 package [28]. The Fixation index (FST) was calculated with the “fstat” function of the “hierfstat” 0.04–22 pakage [29]. The percentage of missing data and MAF were calculated using the “SRPRelate” 1.4.2 package [30]. Diversity structure present in the C. canephora collection was analyzed using a Discriminant Analysis of Principal Components (DAPC) multivariate analysis implemented in “adegenet” [31], as follows: First, 34 known individuals (Table 1) corresponding to the previously described diversity groups [10, 32] were used to model the diversity present in the panel, after centering the data. The most probable number of groups that define the diversity evaluated were inferred using the “find.cluster” function, running successive K-means with an increasing number of clusters (k) from one to ten, and with the Bayesian Information Criterion (BIC) as the statistical measure of goodness of fit. The number of retained Principal Components (PC) to be used in the discriminant analysis was determined using the “xvalDapc” function with the default parameters. Second, individuals with a probability of membership over 80 % to each genetic group were subjected to another round of DAPC analysis in order to find possible subgroups, following the same procedure. Using a threshold calculated with the median hierarchical clustering method implemented in the “snpzip” function from “adegenet”, a set of alleles with the highest contribution to the between-population structure was identified. Additionally, we used the outlier test based on the joint distributions of expected heterozygosity and FST under an island model of migration, implemented in LOSITAN [33], in order to identify the SNP loci under selection and to compare them to the ones discriminating the genetic groups identified. A first run consisting of 100,000 simulations was used to remove outlier candidate SNPs outside the 99 % confidence interval. A neutral FST value was then recalculated, and with it, outlier SNPs were identified after 100,000 simulations, as the ones outside the 1 to 99 % confidence interval, with a false discovery rate smaller than 0.05.
Finally, individuals of unknown groups were projected onto the discriminant functions found with DAPC, using the “predict” function from the package.
To illustrate the genetic relationships between individuals, unrooted NJ trees were constructed with the package “poppr” 2.1.0 [34], based on a Nei’s genetic distance matrix, modified to measure distances between individuals. Bootstrap analyses were also computed with “poppr”, using 100 iterations.
Sequence comparisons
The sequences obtained by the DArTseq method, containing the filtered SNPs markers, were mapped against C. canephora pseudo-molecules [24] and predicted C. canephora genes (available at http://coffee-genome.org), using the Bowtie2 algorithm [35] with the very sensitive, end-to-end alignment option. Markers with the highest contribution to the between-population structure were similarly mapped on the C. canephora pseudo-molecules and genes. Graphical representations of the hits were drawn with the “Circos” program [36].
Results
Marker descriptions and distribution
After sequencing 105 individuals from C. canephora, we obtained 10,806 DArTseq-derived SNP markers. The average missing data and MAF percentages were 16.3 % and 12.8 %, respectively. After removing markers with more than 10 % of missing data and MAF below 1 %, 4,021 polymorphic SNPs remained for the analysis, with an average missing data of 3.1 %, a MAF percentage of 12.6 %, and an average PIC of 0.159 for the whole sample panel. The mean Ho and mean He calculated for the 4,021 markers were 0.124 and 0.162, respectively, estimated based on a panel of depurated biological and technical replicates (81 unique accessions) in order to avoid any bias on the measure.
The 4,021 DArTseq-derived SNP markers were obtained from 3,388 unique sequences (Additional file 1: Table S2). These sequences showed a tendency towards gene-rich regions when mapped on the recently sequenced C. canephora genome (Fig. 1), with 90.8 % of sequences aligned on the pseudo-molecules, and 35.7 % within annotated gene sequences. The average density in the genome was one marker per 178 kb.
Fig. 1.
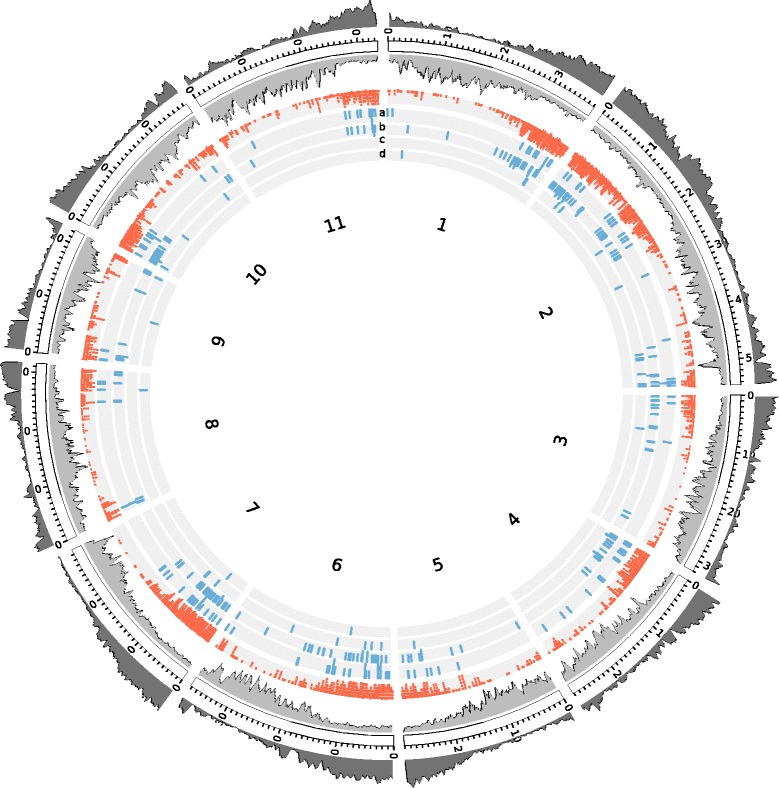
Distribution of DArTseq-derived SNP markers in the C. canephora genome. Graphical representation of the eleven pseudo-molecules of C. canephora showing the density of genes (dark gray) and transposable elements (light gray), along with the location of the 4,021 DArTseq SNP markers used for the analysis (red). Markers with the highest contribution (blue) to the first (a), second (b), third (c) and fourth (d) discriminant axes deciphering the genetic structure of C. canephora are also shown
Technical and biological replicates allowed us to assess the reliability of the DArTseq method in coffee. Genotyping error rates in technical and biological replicates for the 2,616 SNPs with no missing data within the entire panel of replicates were 4.0 % (s = 1.0) and 4.3 % (s = 0.8), respectively Additional file 2: Figure S1. The difference between the two types of replicates was not significant (p-value = 0.2887). Taken together, these results suggest that the overall error rate in allele calls for the DArTseq method in C. canephora would be near 4 %.
Genetic structure of the C. canephora collection
The observed and expected heterozygosities calculated with 4,021 SNPs for the 34 analyzed accessions were 0.1405 and 0.1933, respectively (Table 2).
Table 2.
Observed and expected heterozygosities found for the five C. canephora genetic groups
| Group1 | Group 2 | Group 3 | Group 4 | Group 5 | Total | |
|---|---|---|---|---|---|---|
| Ho | 0.1360 | 0.1641 | 0.1215 | 0.0530 | 0.1347 | 0.1405 |
| He | 0.1199 | 0.1642 | 0.1007 | 0.0456 | 0.1283 | 0.1933 |
In order to interpret C. canephora diversity in a whole genome context, the DArTseq SNP data obtained from a collection of 34 C. canephora members of previously known diversity groups was analyzed using a DAPC multivariate analysis.
The first four principal components of the principal component analysis (PCA), which explained 25.4 %, 10.3 %, 9.5 % and 7.0 % of the variance, respectively, were retained for the discriminant analysis with the DAPC function. Genetic diversity, as revealed by the DArTseq-derived SNP markers, confirms the genetic diversity previously revealed by RFLPs and SSRs, as five genetic clusters were identified (Fig. 2a). A detailed observation on the accessions belonging to the obtained groups allowed us to find equivalences, as follows: (i) Group 1 encloses cultivated individuals from Congo and Uganda, known to belong to the Robusta Congo-Uganda group; (ii) Group 2 represents the accessions previously described in the Nana group, from Cameroon and the Central African Republic; (iii) Group 3 is equivalent to the Conilon group, with cultivated individuals from the Ivory Coast; (iv) Group 4 is made up of only wild and cultivated Guinean accessions collected in the Ivory Coast; and finally, (v) Group 5 is composed of wild individuals from the Central African Republic belonging to the Robusta Congo-Central Africa group. The first discriminant axis of the DAPC clearly separates the Guinean and Conilon groups from the three others, while the second axis opposes the Conilon group against the rest of the groups. The third axis discriminates the Robusta Congo-Central Africa group from the Nana group; and the fourth axe separates the Robusta Congo-Uganda group from the others. The observed and expected heterozygosities estimated for the groups ranged from 0.0530 to 0.1641, and from 0.0456 to 0.1642, respectively (Table 2).
Fig. 2.
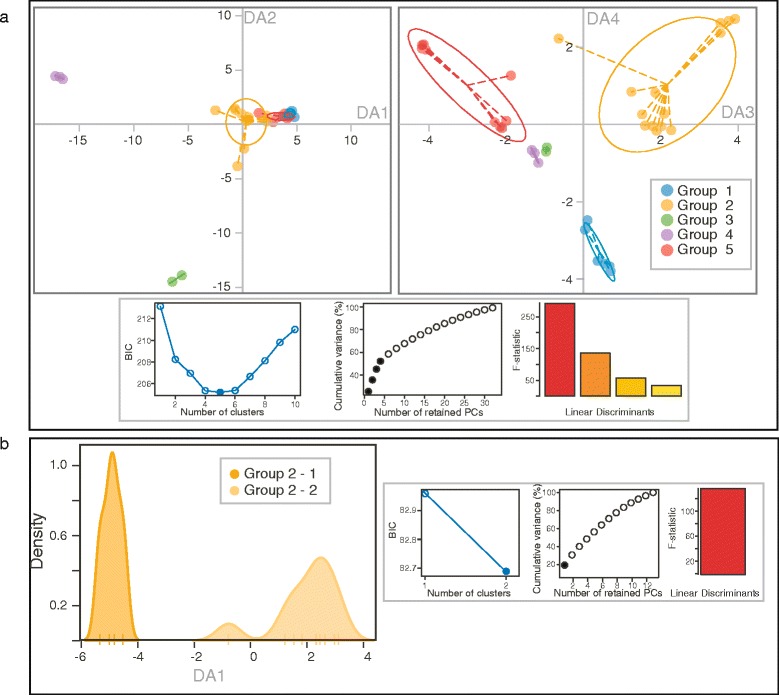
Genetic structure of C. canephora individuals evaluated with 4,021 DArTseq SNP markers. Scatter plots from the DAPC analysis carried out with 34 C. canephora accessions. a Discriminant axes 1 and 2 (left) and 3 and 4 (right) representing the five groups (inertia ellipses) determined by the DAPC. Group 1 encloses cultivated individuals from Congo and Uganda, known to belong to the Robusta Congo-Uganda group; Group 2 represents the accessions previously described into the Nana group, from Cameroon and the Central African Republic; Group 3 is equivalent to the Conilon group, with cultivated individuals from the Ivory Coast; Group 4 is made up of only wild and cultivated Guinean accessions collected in the Ivory Coast; and finally, Group 5 is composed of wild individuals from the Central African Republic belonging to the Robusta Congo-Central Africa group. b First discriminant axis deciphering the genetic relationships between individuals from the two sub-groups of group 2. For each DAPC analysis (a and b), the Bayesian information criterion (BIC) used to determine the optimal k number of clusters (blue dot), the percentage of cumulative variance for the retained PCA eigenvectors (black dots), and the F-statistic of the between/within group variance ratio for the discriminant functions (colored bars) are also exposed below each DAPC plot
In order to identify the genomic regions contributing to the population structure found in C. canephora, the identity and genome location of the SNPs discriminating the five groups were determined, taking advantage of the recently available C. canephora genome [24]. Out of 149, 240, 33 and 8 structural alleles contributing to the four discriminating axes (Additional file 1: Table S3), respectively, 125, 205, 26, and 5 were mapped only once to the C. canephora genome; while 15, 17, 5 and 2 mapped more than once, and 54, 99, 12, and 2 fell into an annotated gene. Their putative functions and gene ontologies show a large range of putative functions (Additional file 1: Table S3), with a high representation of genes involved in signal transduction, and a higher distribution on gene-rich regions on the C. canephora pseudo-molecules (Fig. 1).
In order to identify SNP loci under selection and to compare them to the ones discriminating the genetic groups identified, an outlier test based on the joint distributions of expected heterozygosity and FST was used. An initial FST of 0.3307 was calculated based on the 4,021 markers. After candidates for outliers were removed, a simulated FST of 0.4815 was found. From the 4,021 SNPs, 793 were found to be under balancing selection, 107 under positive selection, while the rest was found to be under neutral selection (Additional file 1: Table S4, and Additional file 3: Figure S2). When comparing the discriminant markers identified by the DAPC analysis to the ones found by the outlier test, we found that 12.9 % (55 SNPs) are subject to positive selection, while the rest are under neutral selection (Additional file 1: Table S3).
In order to establish a more detailed structure of the species, a second DAPC analysis was carried out with groups containing a sufficient number of individuals. In this manner, a more profound genetic structure was found only for Group 2, with two subgroups (Fig. 2b). Group 2–1 includes all but one individuals from the south-western Central African Republic from the Nana group, and Group 2–2 consists of all the South-Eastern Cameroon individuals evaluated in the study.
Taken together, the present analysis corroborates the previous structure of the C. canephora diversity, and adds a higher level of resolution to the observed structure.
Genetic structure of cultivated overseas accessions
With the aim of assessing group membership of cultivated accessions in Vietnam and Mexico and to identify their putative origin, the DArTseq SNP data obtained from the evaluation of 47 additional C. canephora accessions were interpolated into the DAPC analysis (Fig. 3a). All newly incorporated accessions collocated closely with individuals of the Robusta Congo-Uganda group. Membership probabilities for each accession were close to 100 % (Fig. 3b).
Fig. 3.
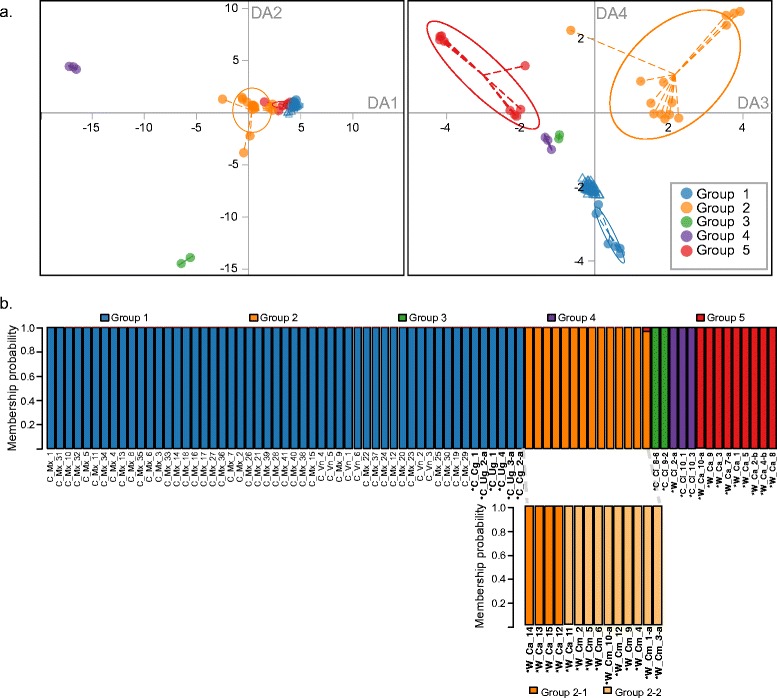
Genetic origin of C. canephora cultivated overseas accessions. Scatter plots from the DAPC analysis, showing the 81 C. canephora accessions analyzed. a Discriminant axes 1 and 2 (left) and 3 and 4 (right) representing the five groups (inertia ellipses) determined by the DAPC, as explained in Fig. 2. Empty circles represent reference accessions used to identify genetic groups, as in Fig. 2, while empty triangles represent interpolated individuals from Vietnam and Mexico. b Bar plots of the posterior membership probabilities obtained with the DAPC analysis. The top barplot represents the five groups found, while the bottom shows the sub-groups derived from group 2. The names of the 34 accessions used to identify the genetic groups are highlighted with an (*), and written in bold characters
In order to obtain a more complete picture of the genetic relationships linking the C. canephora accessions evaluated in the present study, a NJ tree was constructed using the 4,021 SNP markers (Fig. 4). The tree comprises at least eight well-defined branches, all in agreement with the DAPC results. Two branches encompass the Vietnamese and Mexican accessions from the Robusta Congo-Uganda group, as well as one Congolese accession; another branch includes the Ugandan and one Congolese individuals from the same genetic group; at least one branch encompasses the Robusta Congo-Central Africa group; at least two correspond to the Nana group; and there is one branch for each of the Guinean and the Conilon groups.
Fig. 4.
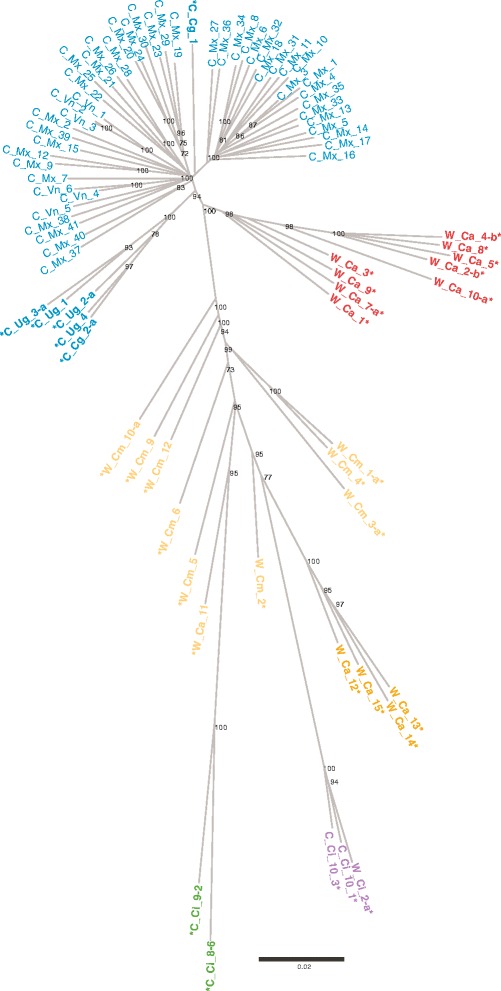
Neighbor Joining tree based on SNP marker evaluations. Unrooted tree using the Neighbor-joining algorithm based on Nei’s genetic distances between 81 individuals of C. canephora. Accessions marked with an (*) are active individuals used in the DAPC analysis to determine the genetic groups. The color patterns are equivalent to the barplots in Figs. 2 and 3, where blue represents cultivated individuals from Congo, Uganda, Vietnam and Mexico, known to belong to the Robusta Congo – Uganda group; Orange and yellow represent the accessions previously described into the Nana group, from Cameroon and the Central African Republic; Green is equivalent to the Conilon group; purple represents wild and cultivated Guinean accessions collected in the Ivory Coast; and finally, red represents wild individuals from the Central African Republic belonging to the Robusta Congo-Central Africa group. For clarity’s sake only bootstrap values over 70 are exposed
Discussion
In the present study, we have employed a DArTseq method on a C. canephora collection. After evaluation, we found an overall genotyping error for the obtained SNP markers close to 4 %, which is similar to what has been previously reported for NGS derived data [37]. The number of exploitable SNPs, repeatability and missing data is similar to what has been obtained using the same technique with other crops [22, 38, 39]. The obtained SNP markers seem to be located mostly in gene-rich parts of the genome, making them an excellent resource for traditional gene mapping or even association mapping assays in coffee trees. The DArTseq method is therefore particularly reliable and easy to use as part of genetic diversity studies. Also, the implementation of these markers in germplasm collections represents an appreciable tool for the curation and optimization of such resources, as it enables a simple means for eliminating redundant or mistagged accessions. From our analysis, we found Ho and He not very distant from the ones calculated previously with microsatellites [5] when evaluated for the complete reference panel, while the observed and expected heterozygosity estimates for the groups were almost half of what has been observed in the past in C. canephora groups using microsatellites [5]. In addition, the data obtained in the present study has allowed us to decipher the diversity of C. canephora in a genome-wide context, and to identify the possible origin of several cultivated accessions from countries where C. canephora has a crucial economic importance. Our C. canephora genetic diversity analysis soundly supports previous studies based on a restricted number of molecular markers [7–13], with all groups unambiguously identified using the DArTseq-derived SNP markers. Compared to former analyses, our study provides a better characterization of the Nana group, through subgroups: one composed of accessions from Southeastern Cameroon and the other from Southwestern Central African Republic. It is clear that a more complete collection evaluated with SNPs derived from one of the NGS technologies would give a better look of the species diversity, especially for groups that were under-represented in our analysis.
By comparing the 427 unique discriminant SNPs identified by the DAPC analysis with the outliers found based on the joint distributions of expected heterozygosity and FST, we were able to infer that nearly 87 % of the differential alleles found with the DAPC analysis seem to have been fixed randomly within the populations. The remaining discriminant alleles found to be under positive selection may have been differentially fixed in the populations as an adaptation to local environmental conditions encountered at the sites of origin of each group.
Although it is not possible to ensure whether all the identified differential alleles are actively or directly involved in the evolutionary differentiation between the groups, or whether they are simply highly linked to the actual causal factor, it is still interesting to seek out the putative molecular function of the genes in which they reside. Most of the markers are located in annotated genes coding for proteins involved in signal transduction, while others reside in proteins constituting cellular organelles, and even DNA-interacting proteins.
In contrast with the C. canephora cultivated trees from Brazil, which originated mainly from the Conilon group [17], here we revealed for the first time that Mexican and Vietnamese C. canephora cultivars form a cluster with the “Robusta Congo-Uganda group”. The genetic origin of populations grown in Mexico and Vietnam appears to be the same as that of Ugandan cultivars, for which Cubry and coworkers [10] showed that they were not distinguishable from wild Ugandan C. canephora individuals. Therefore, the genetic basis introduced in Vietnam, Mexico, and Brazil reflects the wild African genetic groups from where they are originated, indicating that the two main producers of Robusta coffee in the world (i.e., Vietnam and Brazil) produce beans from two very different genetic origins.
In Vietnam as well as in Mexico and Uganda, cultivated C. canephora trees are grown at relatively high altitudes (>600 m.a.s.l.), as compared to the usual 0–400 m.a.s.l range [40] used elsewhere. It is interesting to note that in Mexico and Vietnam coffee trees are distributed over the same latitude range (Latitude: 12.00° N to 20.00° N). In both countries, the optimum coffee-producing zone is at an altitude between 300 and 900 m.a.s.l. In Uganda, the same coffee group is grown near the equator between 300 and 1,100 m. This data suggests that there is a wide adaptability of the “Robusta Congo-Uganda group”, since it is able to adapt in mountainous areas with rather cool climates and fairly high latitude areas, as well as in low-lying areas and low latitudes. This is also observed in Indonesia (the third biggest Robusta producer) that grows coffee from the same genetic group at latitudes ranging between 5 and 11° latitude to 300 to 1,200 m.a.s.l. Since Robusta coffee produced in Uganda has a very good reputation in terms of quality, we can deduce that the relatively bad reputation of Robusta produced in Vietnam (in intensive and full-sun systems), and in a lesser extent in Mexico and Indonesia (in extensive and agroforestry systems), is probably mainly due to poor quality of post-harvest treatments.
In the long term, climate changes-particularly, global warming-will affect not only the three biggest producing countries (i.e., Vietnam, Indonesia and Brazil), but also several producing countries like Mexico. Is the “Conilon” genetic group present in Brazil more adapted to climate change than the “Robusta Congo-Uganda group” present in Asia or Mexico? This issue needs to be addressed by researchers to predict supply scenarios for the industry and growers. We strongly recommend comparing the performance of Robusta to Conilon cultivars under abiotic stresses. We also suggest comparing those origins with hybrids produced between genetics groups.
In the majority of Robusta-producing countries, the current genetic diversity available for breeding programs is very low [41]. The introduction of a core collection representing the genetic diversity of the species is a priority for breeding programs in a climate change context. Thus, a similar initiative to that implemented by the World Coffee Research (http://www.ico.org/) for Arabica should be undertaken urgently for C. canephora, in order to cope with future challenges brought about by the evolving climate conditions.
Conclusions
In the present study, we established that markers obtained from NGS approaches are easily exploitable in coffee, with an error rate similar to what has been observed for other crops. The genetic characterization based on SNP markers of the varieties grown throughout the world increased our knowledge on the genetic diversity of C. canephora, and contributed to the understanding of the genetic background of varieties from very important coffee producers. Also, the discriminant SNP markers identified in our work represent a valuable tool that could be used by breeders to discriminate between C. canephora genetic groups in Robusta germplasm.
The quality of Mexico and Vietnamese coffee are traded at a price lower than Uganda. Given the similar characteristics of climatic areas and relatively high altitude where Robusta is grown in the three countries, and given the common genetic origin of the varieties cultivated, we can state that the Vietnamese and Mexican Robusta accessions have the genetic potential to increase the quality of Robusta they produce.
Acknowledgements
We thank Diana Lopez-Alvarez at BIOS for advice on the methodology for the identification of SNP loci under selection, and to Andrea Gonzales at BIOS for improving the use of English in the manuscript.
Funding
The work was supported by the French agricultural research and international cooperation organization-CIRAD.
Availability of data and materials
The complete dataset obtained during the current study is available from the corresponding author on reasonable request. Sequences corresponding to the analyzed SNPs are included in the Additional files 1, 2 and 3 of this published article.
Authors’ contributions
CM and BB participated in the design of the study. AG performed the data analysis. RG preformed the sequence analysis. AG, CM and BB wrote the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Abbreviations
- AMSA
Agroindustrias Unidas de México
- BIC
Bayesian Information Criterion
- CIRAD
Centre de coopération internationale en recherche agronomique pour le développement / French agricultural research and international cooperation organization
- DA
Discriminant axis
- DAPC
Discriminant analysis of principal components
- DArTseq
Sequencing-based diversity array technology
- GBS
Genotyping by sequencing
- He
Mean expected heterozygosity
- HL
Homozygocity by loci
- Ho
Mean observed heterozygosity
- INEAC
Institut national d’études agronomiques du Congo Belge
- IRD
Institut de recherche pour le développement
- m.a.s.l
Meters above sea level
- MAF
Minor allele frequencies
- Na
Mean allele number
- NGS
Next generation sequencing
- NJ
Neighbor-joining
- PC
Principal component
- PCA
Principal component analysis
- PCR
Polymerase chain reaction
- PIC
Polymorphic information content
- RADseq
Restriction site associated DNA sequencing
- RFLPs
Restriction fragment length polymorphism
- SNPs
Single nucleotide polymorphism
- WCR
World coffee research
Additional files
List of plants evaluated with DarTseq markers. Table S2. List of DArTseq markers and used in the analysis. Table S3. Structural SNPs contributing to the population structure in C. canephora. Table S4. Putative outlier marker loci identified by the Fst outlier method implemented in Lositan. (XLSX 457 kb)
Genotyping error rates in technical and biological replicates. Genotyping error rates for 15 and 4 accessions with technical (a) and biological (b) replicates evaluated with SNPs. Ratios were obtained using all 4,021 markers (blue) or by selectively ignoring markers having differential missing data (red). (EPS 683 kb)
Selection test for each of the 4021 DArTseq SNP markers in C. canephora Plotted distribution of the empirical FST values versus the expected heterozygosity. The red and blue lines indicate 99 % and 1 % confidence limits, respectively, while the gray line corresponds to the median value. (PDF 192 kb)
Contributor Information
Andrea Garavito, Email: andrea.garavito@bios.co.
Christophe Montagnon, Email: christophe.montagnon@rd2vision.com.
Romain Guyot, Email: romain.guyot@ird.fr.
Benoît Bertrand, Phone: (33) 467 41 62 73, Email: benoit.bertrand@cirad.fr.
References
- 1.Davis AP, Tosh J, Ruch N, Fay MF. Growing coffee: Psilanthus (Rubiaceae) subsumed on the basis of molecular and morphological data; implications for the size, morphology, distribution and evolutionary history of Coffea. Bot J Linn Soc. 2011;167(4):357–377. doi: 10.1111/j.1095-8339.2011.01177.x. [DOI] [Google Scholar]
- 2.Yu Q, Guyot R, de Kochko A, Byers A, Navajas-Perez R, Langston BJ, Dubreuil-Tranchant C, Paterson AH, Poncet V, Nagai C, et al. Micro-collinearity and genome evolution in the vicinity of an ethylene receptor gene of cultivated diploid and allotetraploid coffee species (Coffea) Plant J. 2011;67(2):305–317. doi: 10.1111/j.1365-313X.2011.04590.x. [DOI] [PubMed] [Google Scholar]
- 3.Berthaud J. Gene flow and population structure in Coffea canephora coffee populations in Africa. In: Jacquart P, Heim G, Antonovics J, editors. Genetic differentiation and dispersal in plants. Berling: Springer Verlag; 1985.
- 4.Montagnon C, Leroy T, Yapo A. Caractérisation et évaluation de caféiers Coffea canephora prospectés dans des plantations de Côte-d’Ivoire. Café, Cacao, Thé. 1993;37(2):115–119. [Google Scholar]
- 5.Cubry P, de Bellis F, Avia K, Bouchet S, Pot D, Dufour M, Legnate H, Leroy T. An initial assessment of linkage disequilibrium (LD) in coffee trees: LD patterns in groups of Coffea canephora Pierre using microsatellite analysis. BMC Genomics. 2013;14:10. doi: 10.1186/1471-2164-14-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Montagnon C, Leroy T, Yapo A. Diversité génotypique et phénotypique de quelques groupes de caféiers (Coffea canephora Pierre) en collection. Conséquences sur leur utilisation en sélection. Café, Cacao, Thé. 1992;36(3):187–198. [Google Scholar]
- 7.Dussert S, Lashermes P, Anthony F, Montagnon C, Trouslot P, Combes MC, Berthaud J, Noirot M, Hamon S. Le caféier, Coffea canephora. In: Diversité génétique des plantes tropicales cultivées. Edited by P H, M S, X P, C. GJ. Montpellier: CIRAD; 1999:175–194.
- 8.Montagnon C, Guyot B, Cilas C, Leroy T. Genetic parameters of several biochemical compounds from green coffee, Coffea canephora. Plant Breed. 1998;117(6):576–8.
- 9.Berthaud J. Les ressources génétiques pour l’amélioration des caféiers africains diploïdes : évaluation de la richesse génétique des populations sylvestres et de ses mécanismes organisateurs. Conséquences pour l’application. Paris: ORSTOM; 1986. [Google Scholar]
- 10.Cubry P, De Bellis F, Pot D, Musoli P, Leroy T. Global analysis of Coffea canephora Pierre ex Froehner (Rubiaceae) from the Guineo-Congolese region reveals impacts from climatic refuges and migration effects. Genet Resour Crop Evol. 2013;60(2):483–501. doi: 10.1007/s10722-012-9851-5. [DOI] [Google Scholar]
- 11.Cubry P, Musoli P, Legnaté H, Pot D, de Bellis F, Poncet V, Anthony F, Dufour M, Leroy T. Diversity in coffee assessed with SSR markers: structure of the genus Coffea and perspectives for breeding. Genome. 2008;51(1):50–63. doi: 10.1139/G07-096. [DOI] [PubMed] [Google Scholar]
- 12.Musoli P, Cubry P, Aluka P, Billot C, Dufour M, De Bellis F, Pot D, Bieysse D, Charrier A, Leroy T. Genetic differentiation of wild and cultivated populations: diversity of Coffea canephora Pierre in Uganda. Genome. 2009;52(7):634–646. doi: 10.1139/G09-037. [DOI] [PubMed] [Google Scholar]
- 13.Gomez C, Dussert S, Hamon P, Hamon S, de Kochko A, Poncet V. Current genetic differentiation of Coffea canephora Pierre ex A. Froehn in the Guineo-Congolian African zone: cumulative impact of ancient climatic changes and recent human activities. BMC Evolutionary Biology. 2009;9(1):1–19. C7 - 167. [DOI] [PMC free article] [PubMed]
- 14.Portères R. Etude sur les caféiers spontanés de la section Eucoffea, Leur répartition, leur habitat, leur mise en culture et leur sélection en Côte d’Ivoire. Annales de l’Afrique Occidental Française et Etrangère. 1937;1:68–91. [Google Scholar]
- 15.Portères R. Valeur agronomique des caféiers des types Kouilou et Robusta cultivés en Côte d’ivoire. Café Cacao Thé. 1959;3:3–13. [Google Scholar]
- 16.Thomas A. Types of Robusta coffee and their selection in Uganda. East Afr Agric For J. 1935;1:193–198. [Google Scholar]
- 17.Souza FF, Caixeta ET, Ferrão LFV, Pena GF, Sakiyama NS, Zambolim EM, Zambolim L, Cruz CD. Molecular diversity in Coffea canephora germplasm conserved and cultivated in Brazil. Crop Breed Appl Biotechnol. 2013;13:221–227. doi: 10.1590/S1984-70332013000400001. [DOI] [Google Scholar]
- 18.Bunn C, Läderach P, Ovalle Rivera O, Kirschke D. A bitter cup: climate change profile of global production of Arabica and Robusta coffee. Clim Change. 2014;129(1):89–101. [Google Scholar]
- 19.Mondini L, Noorani A, Pagnotta MA. Assessing Plant Genetic Diversity by Molecular Tools. Diversity. 2009;1(1):19–35. doi: 10.3390/d1010019. [DOI] [Google Scholar]
- 20.Baird NA, Etter PD, Atwood TS, Currey MC, Shiver AL, Lewis ZA, Selker EU, Cresko WA, Johnson EA. Rapid SNP Discovery and Genetic Mapping Using Sequenced RAD Markers. PLoS ONE. 2008;3(10):e3376. doi: 10.1371/journal.pone.0003376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Elshire RJ, Glaubitz JC, Sun Q, Poland JA, Kawamoto K, Buckler ES, Mitchell SE. A Robust, Simple Genotyping-by-Sequencing (GBS) Approach for High Diversity Species. PLoS ONE. 2011;6(5):e19379. doi: 10.1371/journal.pone.0019379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cruz VMV, Kilian A, Dierig DA. Development of DArT Marker Platforms and Genetic Diversity Assessment of the U.S. Collection of the New Oilseed Crop Lesquerella and Related Species. PLoS ONE. 2013;8(5):e64062. doi: 10.1371/journal.pone.0064062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kumar S, Banks TW, Cloutier S. SNP Discovery through Next-Generation Sequencing and Its Applications. Int J Plant Genomics. 2012;2012:15. doi: 10.1155/2012/831460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Denoeud F, Carretero-Paulet L, Dereeper A, Droc G, Guyot R, Pietrella M, Zheng C, Alberti A, Anthony F, Aprea G, et al. The coffee genome provides insight into the convergent evolution of caffeine biosynthesis. Science. 2014;345(6201):1181–1184. doi: 10.1126/science.1255274. [DOI] [PubMed] [Google Scholar]
- 25.Stam P. Construction of integrated genetic linkage maps by means of a new computer package: Join Map. Plant J. 1993;3(5):739–744. doi: 10.1111/j.1365-313X.1993.00739.x. [DOI] [Google Scholar]
- 26.Hoffman JI, Tucker R, Bridgett SJ, Clark MS, Forcada J, Slate J. Rates of assay success and genotyping error when single nucleotide polymorphism genotyping in non-model organisms: a case study in the Antarctic fur seal. Mol Ecol Resour. 2012;12(5):861–872. doi: 10.1111/j.1755-0998.2012.03158.x. [DOI] [PubMed] [Google Scholar]
- 27.R Core Team . R: A Llanguage and environment for statistical computing. Vienna: R Core Team; 2014. [Google Scholar]
- 28.Jombart T. adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics. 2008;24(11):1403–1405. doi: 10.1093/bioinformatics/btn129. [DOI] [PubMed] [Google Scholar]
- 29.Goudet J. hierfstat, a package for r to compute and test hierarchical F-statistics. Mol Ecol Notes. 2005;5(1):184–186. doi: 10.1111/j.1471-8286.2004.00828.x. [DOI] [Google Scholar]
- 30.Zheng X, Levine D, Shen J, Gogarten SM, Laurie C, Weir BS. A high-performance computing toolset for relatedness and principal component analysis of SNP data. Bioinformatics. 2012;28(24):3326–3328. doi: 10.1093/bioinformatics/bts606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jombart T, Devillard S, Balloux F. Discriminant analysis of principal components: a new method for the analysis of genetically structured populations. BMC Genetics. 2010;11(1):1–15. doi: 10.1186/1471-2156-11-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Montagnon C, Cubry P, Leroy T. Amélioration génétique du caféier Coffea canephora Pierre : connaissances acquises, stratégies et perspectives. Cah Agricultures. 2012;21(2–3):143–153. [Google Scholar]
- 33.Antao T, Lopes A, Lopes RJ, Beja-Pereira A, Luikart G. LOSITAN: A workbench to detect molecular adaptation based on a F st -outlier method. BMC Bioinformatics. 2008;9(1):1–5. doi: 10.1186/1471-2105-9-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kamvar ZN, Tabima JF, Grünwald NJ. An R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. Peer J. 2014;2(3):e281. doi: 10.7717/peerj.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9(4):357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA. Circos: an information aesthetic for comparative genomics. Genome Res. 2009;19(9):1639–1645. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wall JD, Tang LF, Zerbe B, Kvale MN, Kwok P-Y, Schaefer C, Risch N. Estimating genotype error rates from high-coverage next-generation sequence data. Genome Res. 2014;24(11):1734–9. doi: 10.1101/gr.168393.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ren R, Ray R, Li P, Xu J, Zhang M, Liu G, Yao X, Kilian A, Yang X. Construction of a high-density DArTseq SNP-based genetic map and identification of genomic regions with segregation distortion in a genetic population derived from a cross between feral and cultivated-type watermelon. Mol Genet Genomics. 2015;290:1–14. doi: 10.1007/s00438-015-0997-7. [DOI] [PubMed] [Google Scholar]
- 39.Hahn V, Würschum T. Molecular genetic characterization of Central European soybean breeding germplasm. Plant Breed. 2014;133(6):748–755. doi: 10.1111/pbr.12212. [DOI] [Google Scholar]
- 40.Wintgens JN. The Coffee Plant. In: Coffee: Growing, Processing, Sustainable Production. Wiley-VCH Verlag GmbH. 2008. pp. 1–24. [Google Scholar]
- 41.Leroy T, De Bellis F, Legnate H, Musoli P, Kalonji A, Loor Solórzano R, Cubry P. Developing core collections to optimize the management and the exploitation of diversity of the coffee Coffea canephora. Genetica. 2014;142(3):185–199. doi: 10.1007/s10709-014-9766-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The complete dataset obtained during the current study is available from the corresponding author on reasonable request. Sequences corresponding to the analyzed SNPs are included in the Additional files 1, 2 and 3 of this published article.