

Abstract
Our previous study showed that the therapeutic effects of mesenchymal stem cells (MSCs) transplantation were improved by enhancing migration. MicroRNA-211 (miR-211) can modulate the migratory properties of some cell types by mechanisms that are not fully understood. This study was designed to investigate a possible role for miR-211 in MSC migration, and whether genetic manipulation of miR-211 in MSCs could be used to enhance its beneficial effects of cell transplantation. Transwell assays confirmed that MSCs migration of was significantly impaired by miR-211 knockdown but enhanced by miR-211 overexpression. MiR-211 overexpressing MSCs also exhibited significantly increased cell engraftment in the peri-infarct areas of female rat hearts 2 days after intravenous transplantation of male MSCs as shown by GFP tracking and SYR gene quantification. This conferred a significant decrease in infarct size and improved cardiac performance. By using a loss or gain of gene function approach, we demonstrated that miR-211 targeted STAT5A to modulate MSCs migration, possibly by interacting with MAPK signaling. Furthermore, the beneficial effects of miR-211 overexpression in MSCs were abolished by simultaneous overexpression of STAT5A whereas the negative effects of miR-211 silencing on MSC migration were rescued by simultaneous downregulation of STAT5A. Finally, using ChIP-PCR and luciferase assays, we provide novel evidence that STAT3 can directly bind to promoter elements that activate miR-211 expression. STAT3/miR-211/STAT5A signaling plays a key role in MSCs migration. Intravenous infusion of genetically modified miR-211 overexpressing MSCs conveys enhanced protection from adverse post-MI remodeling compared with unmodified MSCs.
Keywords: Mesenchymal stem cells, MicroRNA-211, STAT5A, Migration, Retention, Myocardial infarction
Introduction
Stem cell transplantation is a promising therapeutic strategy for ischemic heart diseases that involves paracrine actions and enhanced regenerative capacity of the myocardium that convey protection against adverse remodeling post-myocardial infarction (MI). Bone marrow-derived mesenchymal stem cells (MSCs) have unique properties that make them ideally suited for off-the-shelf clinical cell transplantation; they are immune privileged with multilineage potential and easy to isolate and amplify [1]. However, the poor survival, retention, and engraftment of MSCs following transplantation [2, 3] compromises the benefits of cell therapy [4]. Thus, there is a need to find safe and effective strategies to enhance survival and engraftment of MSCs and optimize their therapeutic effects. We hypothesized that enhancement of the migratory properties of MSCs may provide such a strategy to improve retention and therapeutic effects after transplantation.
MicroRNAs are small, noncoding RNAs that regulate gene expression at the post-transcriptional level by degrading mRNA or inhibiting translation [5]. An increasing amount of data suggests that microRNAs play important roles in regulating stem cell self-renewal, differentiation, and pluripotency [6, 7]. In addition, microRNAs can regulate proliferation, senescence, migration, and survival of MSCs [8]. Previous studies have shown that microRNAs-211 (miR-211) plays key roles in cancer cell migratory; however, it can either promote [9–11] or suppress tumor cell migration [12], depending on the cell types and pathological conditions. Roles for miR-211 in regulating stem cell migration including MSCs have not been described.
Previous studies [13, 14], including our own [15, 16] have shown that MSCs pre-exposed to hypoxic preconditioning (HP) exhibited enhanced migration and therapeutic capacities. We also found that HP of MSCs was associated with significantly enhanced miR-211 expression (Hu et al., unpublished observation). Therefore, in this study we used a gain and loss of function approach to investigate the cell signaling functions of miR-211 in MSCs and the possible contribution(s) of miR-211 to MSC transplantation therapy in a rat MI model. We show that activated STAT3 activity during HP confers enhanced expression of miR-211 that in turn targets STAT5A thereby modulating the migratory properties of MSCs. Intravenous delivery of miR-211 overexpressing MSCs conferred enhanced protection against adverse post-MI remodeling and improved cardiac function. These beneficial effects were lost when miR-211 expression was silenced or STAT5A was simultaneously overexpressed in MSCs.
Materials and Methods
The animal research study protocols were in compliance with National Institutes of Health (NIH) Guide for the Care of Use of Laboratory Animals (NIH Pub. No. 85-23, Revised 1996) and the Chinese Guidelines for the Care and Use of Laboratory Animals and were approved by the Zhejiang University Animal Care Committee. The human research protocol was carried out according to the principles of the Declaration of Helsinki and approved by the Human Ethics Committee of the Second Affiliated Hospital of Zhejiang University. All the donors enrolled in the study provided a written informed consent for the use of their samples for research purposes.
Rat and Human MSCs Isolation, Culture, and Characterization
The methodology of isolation, culture, and characterization of rat MSCs were the same as in our previous study [16, 17]. The human MSCs (hMSCs) were also obtained from three old (age over 65) and three young donor patients who underwent orthopedic surgery. The method is described in detail in Supporting Information data.
Plasmids and Vectors Construction
For miR-211 overexpression in MSCs, we constructed lentivirus that contained the miR-211 expressing plasmid (miR-211-over), while an empty plasmid served as control (miR-211-control). Two fragments encompassing precusor miR-211 sequence were synthesized chemically and then cloned into the EcoRI and NotI sites in plvx-IRES-ZsGreen1 (#632187; Clontech Laboratories, Mountain View, CA, http://www.clontech.com). To downregulate miR-211 expression in MSCs, lentivirus-based miR-211 silencing sponge plasmid (miR-211-shRNA) and the control sponge plasmid (miR-211-scramble) were constructed, respectively (GenePharma, Shanghai, People’s Republic of China, http://www.genepharma.com/).
To overexpress STAT5A and STAT3 in MSCs, the cDNA for STAT5A and STAT3 was amplified by polymerase chain reaction (PCR) and cloned into pLentivirus-CMV-IRES-GFP and pCNDA3.0, respectively (Newgene, Shanghai, People’s Republic of China) to generate STAT5A (STAT5A-over) and STAT3 (STST3-over) overexpression lentivirus-based plasmids. Empty plasmids were used as controls for STAT5A (STAT5A-control) and STAT3 (STAT3-control). To downregulate STAT5A expression in MSCs, lentiviral plasmids encoding a short hairpin ribonucleic acid (shRNA) targeting STAT5A mRNA (STAT5A-shRNA) and a nonspecific shRNA (STAT5A-scramble) were generated based on GV115 (Genechem, Shanghai, People’s Republic of China, www.genechem.com). Similarly, lentiviral plasmids encoding a shRNA targeting STAT3 mRNA (STAT3-shRNA) and a nonspecific shRNA (STAT3-scramble) were also generated to knockdown STAT3 in MSCs.
To test the specific binding of miR-211 to 3′-UTR (untranslated region) region of the STAT5A mRNA in MSCs, total RNA was extracted from rat MSCs, and a PCR was performed using specific primers for both wild-type 3′-UTRs of STAT5A and mutant 3′-UTRs of STAT5A containing the predicted binding sites for miR-211. GV306-STAT5A-3′-UTR and GV306-STAT5A-3′UTRs-mut vector were generated (Genechem).
To test the specific binding of STAT3 to promoter of miR-211, genomic DNA was extracted from MSCs, and different promoter regions of miR-211 were amplified, isolated, and cloned into pGL3B (Newgene, Xiamen, People’s Republic of China). The sequences of primers for plasmid construction for different promoter regions are listed in Supporting Information Table.
Lentivirus Infection of MSCs
Rat and hMSCs were initially plated at a density of 1.5 × 105 cells per well in a six-well plate or at a density of 1 × 106 in a 10-cm dish for 16 hours before infection. MSCs were infected with lentiviral particles (multiplicity of infection, MOI=75) with polybrene (final concentration 8 μg/ml) (H9268, Sigma, St. Louis, MI). Lentivirus containing miR-211-over plasmids as described above were used for infection to overexpress miR-211 in MSCs (MSCsmiR-211-over), MSCs infection done with miR-211-control served as controls (MSCsmiR-211-control); likewise, infection with lectivirus containing miR-211-shRNA plasmid was performed to knockdown miR-211 in MSCs (MSCsmiR-211-shRNA) and MSCs infected with miR-211-scramble plasmid as controls (MSCsmiR-211-scramble). To overexpress STAT5A and STAT3 in MSCs, lentivirus containing STAT5A and STAT3 plasmid were used, while lentivirus with empty vectors as their respective controls. The lentivirus containing specific shRNA targeting STAT5A or STAT3 was used to knockdown STAT5A or STAT3 in MSCs, with their nonspecific scrambles used as controls. The related plasmids were described as above. All infections were carried out for 24 hours with incubation at 37 °C, the medium was then replaced with fresh medium to exclude polybrene. Seventy-two hours after infection, the virus infection efficiency was confirmed by detecting green fluorescent protein (GFP) in MSCs, quantifying the expression levels of miR-211 in MSCs by real-time PCR (RT-PCR), or measuring the STAT3 or STAT5a protein expression by Western blot.
Hypoxic Preconditioning
Using the same method as described in our previous report [16], MSCs were plated at 1 × 105 cells per cm2 in complete culture medium and incubated under normoxia (21% O2, 5% CO2) or hypoxia (0.5% O2, 5% CO2) using finely controlled ProOx-C-chamber system (Biospherix, Redfield, NY) for 24 hours.
Chormatin Immunoprecipitation (ChIP)-Quantitative PCR Assay
A total of 1 × 106 MSCs were treated with 1% formaldehyde for crosslinking by incubation at room temperature for 10 minutes, 10 × glycine buffer was added to terminate the crosslinking. The cells were then washed and lysed in 1% SDS lysis buffer. Sonication was carried out at 320 W for 15 cycles of 30 seconds followed by 30 seconds rest interval. Part of the DNA obtained from the cells lysates was used as input (as positive control), the remaining DNA was subjected to immunoprecipitation using a specific antibodies against STAT3 (sc-482x; Santa Cruz Biotechnology, Santa Cruz, CA, http://www.scbt.com) or IgG (as negative control) by incubation for 3 hours at 4 °C. The precipitated STAT3-DNA complexes were subjected to RNaseA and proteinase K treatment, recovered with phenol/chloroform extraction and ethanol precipitation. The pellets were washed with 70% ethanol, air-dried, and resuspended. PCRs with miR-211 promoter specific primers were carried out. PCR products were separated on a 1% ethidium bromide agarose gel for densitometry analysis. Primer sequences are listed in Supporting Information Table.
Dual Luciferase Report Assays
Luciferase assay was performed using the Dual-Luciferase Reporter Assay System (Promega, Madison, WI, http://www.promega.com) according to the manufacturer’s instructions. To test the binding of miR-211 to STAT5A, Hela cells were transfected with GV306-STAT5A or GV306-mutant-STAT5A vector in the presence of pLVX-IRES-ZsGreen1-miR-211 or empty vector using Lipofectamine 2000 (Life Technologies, Grand Island, NY, http://www.lifetech.com). To test the binding of STAT3 to the promoter region of miR-211, HEK 293T cells were cotransfected with a pGL3B vector either containing the different promoter region of miR-211 or an empty pGL3B vector. Vector pCDNA3.0 containing cDNA STAT3 was transfected using Lipofectamine. The luciferase activity was measured 48 hours after transfection using GloMax TM 20/20n Luminometry System (Promega).
RNA Isolation and Quantitative RT-PCR
RNA was isolated using standard procedure using the Trizol reagent (Invitrogen, Carlsbad, CA, http://www.invitrogen.com) and quantitative RT–PCR (qRT-PCR) was performed using SYBR green fluorescence for quantification. The detailed procedure and the primer sequences are described in Supporting Information Data.
Western Blot Analysis
The protein expression levels of STAT5A, MMP9, STAT3, and phosphorylation levels of STAT3, ERK1/2, p38, JNK and their total expression levels were quantified by Western blot using the same method as in our previous report [16, 17]. The detailed method is described in Supporting Information Data.
Transwell Assay
The transwell assay as described in our previous report [17] was used to measure the migratory ability of MSCs. The detailed methodology is described in Supporting Information data.
In Vivo Rat MI Model and Cell Transplantation
Female Sprague-Dawley rats (8 weeks old, 150—200 g) were anesthetized with 4% chloral hydrate (300 mg/kg, administered intraperitoneally) and then intubated. Rat MI model was induced by ligation of left anterior descending branch of coronary artery, using the same method as described previously [16, 18]. A total of 5×106 MSCs from male rats were intravenously delivered 24 hours after the surgical procedure via a tail vein. A detailed procedure is available in Supporting Information Data.
MSCs Cell Retention Assay by qRT-PCR of Y-Chromosomal DNA
The retention of transplanted MSCs in heart tissue was evaluated by quantifying the copies of SRY gene of male donor MSCs, using qRT-PCR. A detailed procedure description is available Supporting Information Data.
Cardiac Echocardiographic and Left Ventricle Catheterization Examination
Twenty-eight days post-MI, echocardiography was performed to assess left ventricular (LV) morphology and performance, LV intracardiac pressure was obtained by catheterization to evaluate the hemodynamics [18]. A detailed procedure is described in Supporting Information Data.
Immunofluorescence and Masson Trichrome Staining
After completion of the examination, rat hearts were OCT embedded, and frozen tissue slices were prepared for immunofluorescence staining, using antibody against GFP and CD31, respectively. Masson trichrome staining was done using a commercially available kit to quantify the infarct size. The detailed methodology is described in Supporting Information Data.
Statistical Analysis
All data are presented as means±SE. Statistical analyses were performed by one-way ANOVA using the SPSS 17.0 software. A p value of less than .05 was considered to be statistically significant.
Results
MiR-211 Modulates MSC Migration
Firstly we observed that HP, a treatment that has been shown to enhance the therapeutic effects of MSCs, increased the expression of miR-211 (Supporting Information Fig. 1). As a strategy to investigate possible biological consequences of increased miR-211 expression, MSCs were infected with lentiviral vectors to overexpress (miR-211-over) or decrease (miR-211-shRNA) miR-211 expression with respective control as described in Materials and Methods section. Following infection, the respective effects on miR-211 expression were confirmed by RT-PCR (Supporting Information Fig. 2). In vitro transwell assays (Fig. 1) showed that MSCsmiR-211-shRNA exhibited a significant decrease in migration compared with MSCsmiR-211-scramble (p<.01. Fig. 1A, 1B), whereas MSCsmiR-211-over had significantly enhanced migration compared with MSCsmiR-211-control (p<.01. Fig. 1A, 1B), suggesting that miR-211 levels may contribute to the migratory properties of MSCs in vitro culture.
Figure 1.
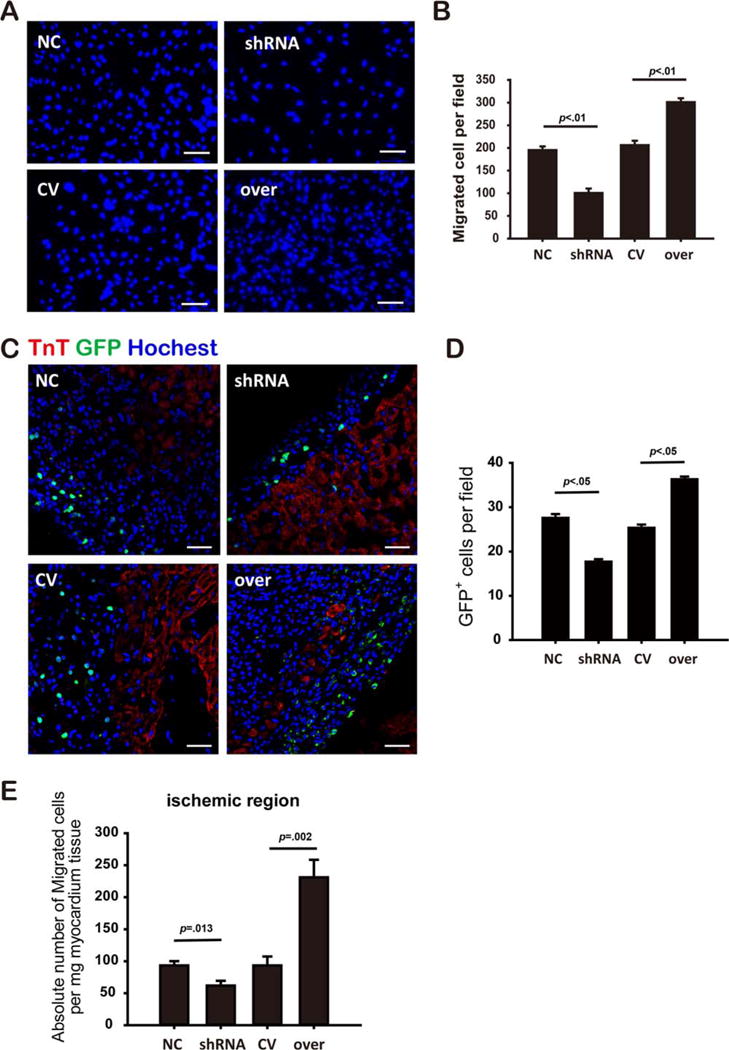
MiR-211 modulated mesenchymal stem cell (MSC) migration. Migrated MSCs assessed by transwell assay (A, scale bar=100 μm), and cell number was quantified for each group (B, n>3 per group). Fluorescent image showed green fluorescent protein (GFP) positive MSCs in the peri-infarct area (C, scale bar=50 μm), and number of GFP positive cells was quantified (D, n≥3 heart samples per group). TnT and Hoechst costaining were performed as shown in red and purple, respectively. SRY gene copies were quantified by real time polymerase chain reaction, the migrated MSCs cells were determined as the absolute number of SRY gene copies per mg myocardium tissue (E, n=4 per group). MSCsmiR-211-over was abbreviated as over, MSCsmiR-211-shRNA as shRNA, and their controls, MSCsmiR-211-control and MSCsmiR-211-scramble, are abbreviated as CV and NC, respectively. Abbreviations: GFP, green fluorescent protein; TnT, Troponin T.
To determine whether the in vitro effects translated in vivo, we used an in vivo rat MI cell transplantation model to test the effects of miR-211 on MSCs therapy. Twenty-four hours after coronary ligation, male GFP-tagged MSCs were delivered intravenously as a bolus to infarcted female rats as described in Materials and Methods section. Recipient rats were sacrificed 2 days after cell transplantation, and hearts were collected to evaluate the retention of MSCs. GFP positive MSCs were quantified under fluorescent microscopy to track cell retention and migration. Rats that received MSCsmiR-211-shRNA had markedly fewer GFP positive cells compared with MSCsmiR-211-scramble, effects were reversed when MI rats were treated with MSCsmiR-211-over, indicating that miR-211 expression correlated closely with the migration ability of MSCs (Fig. 1C, 1D). In parallel, by quantifying the SRY gene expression using quantitative real time PCR, we showed that there were less MSCsmiR-211-shRNA (61.92±7.43) compared with MSCsmiR-211-scramble (93.30±6.70, p=.013. Fig. 1E) in the peri-infarct area; in contrast, more MSCs migrated to the ischemic region in rats receiving MSCsmiR-211-over (231.16±27.34) compared with MSCsmiR-211-control (93.32±14.08, p=.002. Fig. 1E). These results suggest roles for miR-211 in regulating MSCs migration both in vitro and in vivo.
Intravenous Delivery of miR-211 Overexpressing MSCs Improved Post-mi Remodeling
Echocardiography was performed 28 days after coronary ligation (Fig. 2A) to evaluate the therapeutic effects of MSCs on post-MI remodeling. Intravenous delivery of MSCsmiR-211-over significantly improved cardiac function as evidenced by increased fractional shortening (24.41%±2.15%) compared with phosphate buffer saline (PBS) group (17.54%±2.42%, p<.05) and miR-211-control group (CV group, 19.32%±0.57%, p>.05. Fig. 2B). LV chamber pressure recording also showed that the MSCsmiR-211-over group had lower left ventricular end-diastolic pressure (LVEDP) compared with either PBS or CV group (Fig. 2C). Higher +dP/dtmax and −dP/dtmax were also observed compared with PBS treated but not CV group rats (Fig. 2D, 2E). This improved cardiac function was associated with decreased infarct size in the MSCsmiR-211-over group as shown by Masson trichrome staining of heart sections at 28 days after MI (Fig. 2F, 2G). On the other hand, the beneficial effects on post-MI remodeling process were absent when rats received MSCsmiR-211-shRNA infusion compared with MSCsmiR-211-over, however, no significant difference was observed when compared with rats that were treated with MSCsmiR-211-scramble (Supporting Information Fig. 3).
Figure 2.
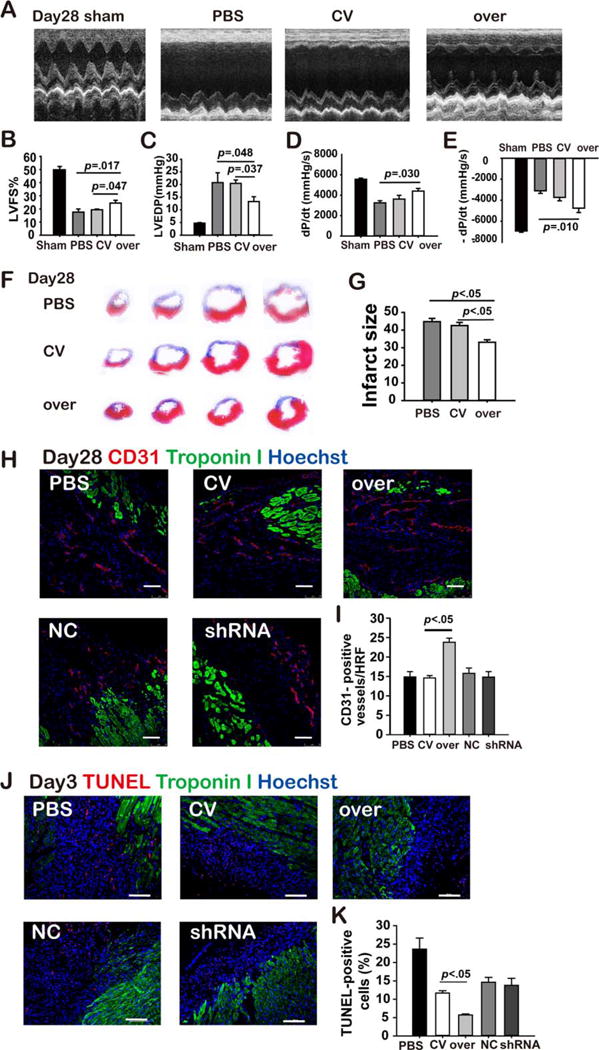
MiR-211 overexpressing mesenchymal stem cells (MSCs) improve post-myocardial infarction (MI) remodeling. Echocardiography examination was performed for each group rats at 28 days after MI (A), and fractional shortening was quantified (B). Left ventricle (LV) pressure recording was also obtained, and LV-end-diastolic pressure (C), and ±dP/dtmax (D, E) then measured. Masson trichrome staining was performed (F) to evaluate the infarct size (G). Immunofluorescence microscopy with antibody targeting CD31 was performed, and CD31 positive structure was quantified in the peri-infarct area using the heart tissue obtained at day 28 after infarction, where troponin I and Hoechst were costained for identifying myocardium and nuclei (H, I, scale bar=75 μm). Terminal Deoxynucleotidyl Transferase dUTP Nick End Labeling (TUNEL) staining was also done and positive cells quantified (K) in the peri-infarct area using the heart tissue obtained at day 3 after infarction (J, scale bar=100 μm). All the abbreviations are the same as in Figure 1. Abbreviations: HRF, high resolution field; LVFS, left ventricular fractional shortening; PBS, phosphate buffer solution; shRNA, short hairpin ribonucleic acid; TUNEL, terminal deoxynucleotidyl transferase dUTP nick end labeling.
To explore the underlying mechanisms by which infarct size was decreased in hearts of the MSCsmiR-211-over group, we quantified apoptosis in the peri-infarct area using heart tissue slices obtained at 3 days after infarction while angiogenesis was also evaluated by counting CD31 staining positive neovessels at the peri-infarct area using the heart tissue obtained at 28 days after infarction. These assays suggested that the beneficial effects conferred by MSCsmiR-211-over correlated with enhanced angiogenesis, as evidenced by immunofluorescence staining for CD31 in the peri-infarct area (Fig. 2H, 2I). Interestingly, decreased infarct size in MSCsmiR-211-over group rats was associated with less terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining compared with MSCsmiR-211-control (Fig. 2J, 2K). No significant differences in either CD31 or TUNEL staining positive cells were seen in the MSCsmiR-211-shRNA group relative to control (Fig. 2H–2K).
STAT5A is a Direct Target of miR-211
To investigate possible miR-211 target genes, we searched the Targetscan database. Based on the Targetscan database, the 3′-UTR mRNA of rat STAT5A is complementary to miR-211 (Fig. 3A). We then infected HeLa cells with both GV306-STAT5A-3′-UTR and miR-211-over and found that these cells exhibited decreased luciferase activity compared with those infected with GV306-STAT5A-3′-UTR and miR-211-control (Fig. 3B). This indicated an inhibitory effect of miR-211 on STAT5A expression. This inhibitive effect by miR-211 was absent in HeLa cells that were infected with GV306-STAT5A-3′-UTR-mut, further suggesting a specific interaction and possibly binding of miR-211 to the 3′-UTR of STAT5A mRNA (Fig. 3B).
Figure 3.
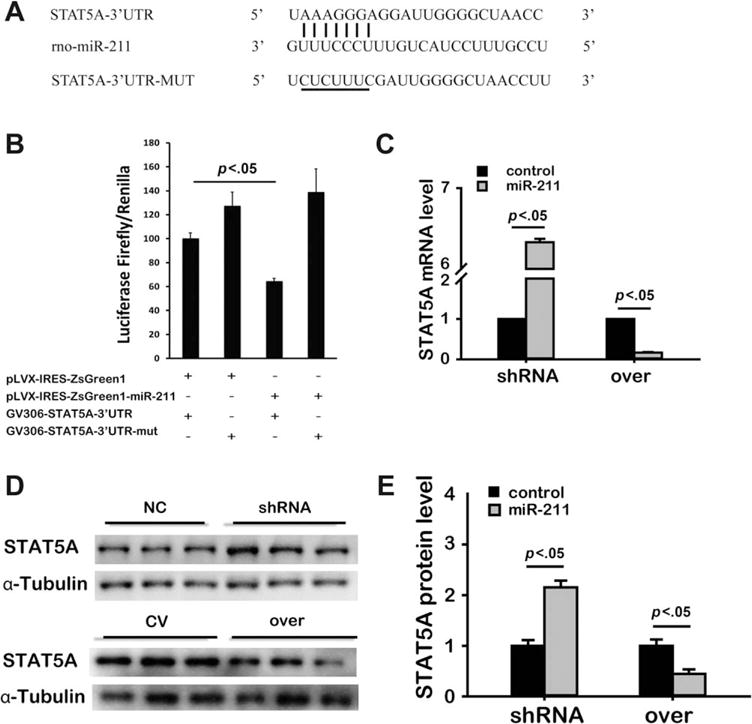
Identification of STAT5A as a target of miR-211. Schematic of the miR-211 putative binding site alignment with rat 3′-UTR mRNA of STAT5A, the mutated STAT5A 3′-UTR binding site for miR-211 are illustrated with the mutated nucleotides underlined (A). Using Hela cells, dual luciferase report assay was performed. Renilla and Firefly Luciferase activities were measured 48 hours after transfection with Renilla used as baseline control (B, n=3). Using real time polymerase chain reaction, the effect of miR-211 on mRNA level of STAT5A was tested in mesenchymal stem cells after infected with miR-211-shRNA or miR-211-over (in grey bars and their respective controls shown in black bars, respectively. C). Western blot was also done (representative bands in D) to quantify the effect of miR-211 on STAT5A protein expression in MSCs (E, the layout is the same as in (C), n=3). All the abbreviations are the same as above. Abbreviations: shRNA, short hairpin ribonucleic acid; UTR, untranslated region; WT, wild type.
Consistent with the data shown above, we showed that the MSCmiR-211-shRNA group had a significant increase in both mRNA (Fig. 3C) and protein levels (Fig. 3D, 3E) of STAT5A, whereas MSCsmiR-211-over showed markedly decreased STAT5A mRNA (Fig. 3C) and protein (Fig. 3D, 3E) expression. Collectively, these results support the possibility that miR-211 targets the 3′-UTR of STAT5A mRNA and inhibits STAT5A expression in MSCs.
Role of STAT5A in miR-211-Mediated MSC Migration
To investigate a possible role for STAT5 in miR-211 mediated MSC migration, MSCs were transduced with STAT5A-shRNA or STAT5A-over lentiviral vectors with appropriate control groups as described in Materials and Methods section. The effects of infection were confirmed by Western blotting to quantify the STAT5A expression levels in MSCs (Supporting Information Fig. 4A, 4B). Using an in vitro transwell assay, we first showed that MSCs infected with STAT5A-shRNA had increased numbers of migrated MSCs, whereas MSCs with STAT5A-over showed markedly lower numbers of migrated MSCs (Supporting Information Fig. 4C, 4D), indicating negative correlations of STAT5A with migration ability of MSCs.
We then coinfected MSCsmiR-211-over with STAT5A-over lentiviral vectors and the STAT5A expression of MSCs that received both miR-211-over and STAT5A over were measured by Western blot compared with miR-211-over or STAT5A-over alone (Supporting Information Fig. 5). The enhanced migration of the MSCsmiR-211-over group was significantly inhibited by simultaneous STAT5A overexpression (Fig. 4A, 4B). Note that STAT5A-over alone significantly suppressed MSC migration. Similarly, the suppressed migration of MSCs with miR-211-shRNA was rescued when coinfected with STAT5A-shRNA (Fig. 4C, 4D). The STAT5A expression of MSCs that received both miR-211-shRNA and STAT5A-shRNA were also quantified by Western blot (Supporting Information Fig. 5). Again, STAT5A-shRNA alone significantly enhanced the MSCs migration. The effects of miR-211/STAT5A signaling on MSCs migration were also evaluated in our in vivo rat MI model. MSCs infected with different viruses as described above were delivered intravenously as described in Materials and Methods section. Quantification of GFP positive cells demonstrated that the increased number of migrated MSCs mediated by miR-211-over was significantly suppressed when MSCs were coinfected with STAT5A-over (p<.001. Fig. 4E, 4F). In contrast, the inhibited migration ability by miR-211-shRNA was partially rescued by coinfection with STAT5A-shRNA (p<.05. Fig. 4E, 4F). PCR quantification of SRY gene copies showed the same pattern of changes as GFP positive cells quantification (Fig. 4G). Thus, our data suggest the regulation of STAT5A expression by miR-211 contributes significantly to MSCs migration.
Figure 4.
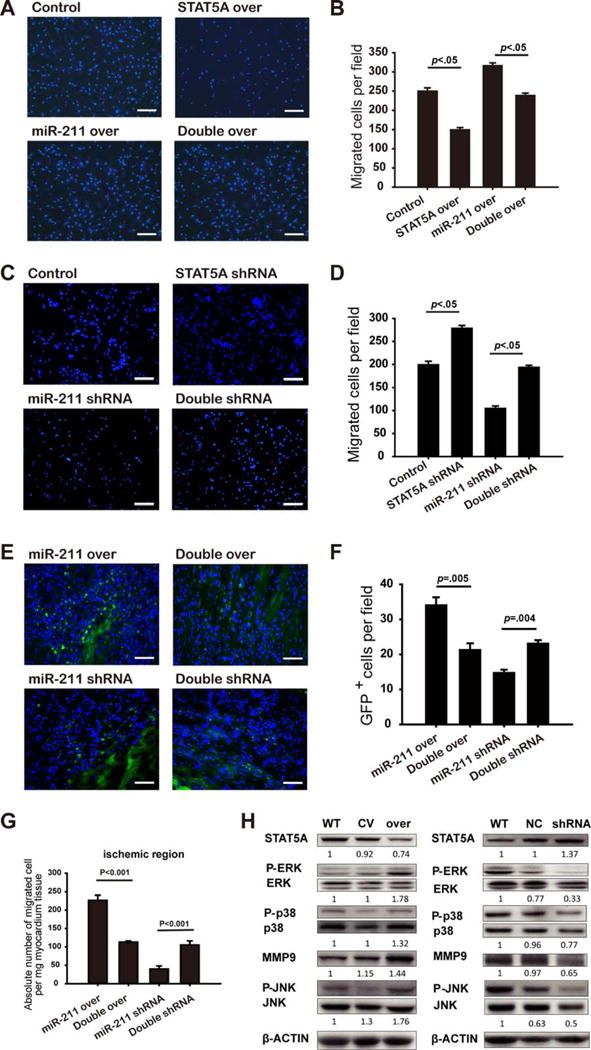
Role of STAT5A in miR-211-mediated mesenchymal stem cell (MSC) migration. Transwell assay was performed for MSCsmiR-211 over, MSCs infected with STAT5 over alone or MSCs simultaneously infected with miR-211-over and STAT5A-over (double over), representative image was shown in (A) (scale bar=200 μm) and cell number quantified in (B) (n=3). MSCsmiR-211-shRNA, MSCs infected with STAT5 shRNA, or MSCs simultaneously infected with miR-211-shRNA and STAT5A-shRNA (double shRNA) were used for transwell assay, with representative image shown in (C) (scale bar=200 μm) and quantified in (D) (n=3). MSCs obtained from male rats that were infected with miR-211-over alone or Double over, miR-211-shRNA alone or double shRNA were delivered in female MI rats, the number of engrafted MSCs was measured by either counting GFP positive cells in the peri-infarct area (representative image in (E) (scale bar=50 μm) and quantified in (F), n≥3 per group) or quantifying the copy of SRY gene per mg heart tissue (G, n≥3 per group). Western blot showed phosphorylation levels of ERK1/2, p38MAPK and JNK, and MMP9 protein expression respectively for MSCs when infected with miR-211-over (abbreviated as over, CV for empty vector, and WT as control) or miR-211 shRNA (denoted as shRNA, NC for miR-211 scramble and WT as control) with representative blots shown in (H) where β-Actin was used as loading controls (data repeated for three times). Abbreviations: GFP, green fluorescent protein; shRNA, short hairpin ribonucleic acid; WT, wild type.
To further explore possible downstream targets of STAT5A that may signal cellular responses to miR-211, we quantified the phosphorylation levels of the three members of mitogen activated protein kinase, including ERK, p-38 MAPK, and JNK. Interestingly, downregulation of STAT5A by miR-211 overexpression was accompanied by increases in their phosphorylation levels of ERK, p38 MAPK, and JNK, whereas these phosphorylation levels were decreased when STAT5A was upregulated with silenced miR-211 expression (Fig. 4H). Similar patterns of changes in MMP9 protein expression were also observed (Fig. 4H).
Role of STAT5A in Improved Post-MI Remodeling by miR-211 Overexpressing MSCs
We used the same rat MI model to explore a role for STAT5A in the enhanced protection conferred by miR-211 overexpression in transplanted MSCs. Cardiac functions were evaluated by echocardiography and LV catheterization examination 28 days after MSCs transplantation as described in Materials and Methods. We found that the improved cardiac performance conferred by transplantation of MSCsmiR-211-over was abolished by coinfection with STAT5A-over. These effects were evidenced by impacts on fractional shortening measured by echocardiography (Fig. 5A–5E). However, these effects were not observed in LVEDP and maximal changes in LV pressure (±−dP/dtmax) obtained by LV pressure recording. In contrast, loss of beneficial effects on post-MI remodeling process with transplantation of MSCsmiR-211-shRNA manifested by improved fractional shortening were rescued by coinfection of STAT5A-shRNA(Fig. 5A–5E). These in vivo experiments further support our hypothesis that STAT5 is an essential intermediate in the protective effects of MSCs by miR-211 overexpression.
Figure 5.
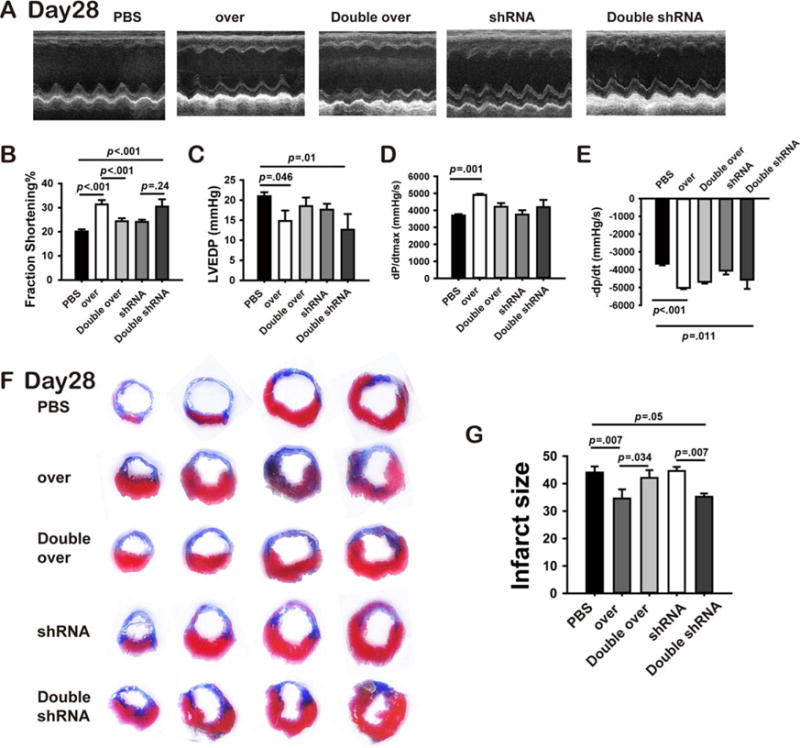
MiR-211 overexpressing mesenchymal stem cells (MSCs) improved post-myocardial infarction (MI) remodeling via STAT5A. Male MSCs infected with different lentivirus as described in Figure 4E were delivered for female MI rats, another group MI rats received PBS served as controls. At 28 days after MI, before the rat were sacrificed, echocardiography was performed for each group rats (A, n≥5) and fractional shortening quantified (B). With LV pressure recording, LVEDP (C),+dP/dtmax (D) and −dP/dtmax (E) were analyzed for each group rats. After rats were sacrificed, heart tissue slices were obtained for Masson’s trichrome staining was then performed (F) and the infarct size was quantified (G). Abbreviations: LVEDP, left ventricular end-diastolic pressure; PBS, phosphate buffer solution; shRNA, short hairpin ribonucleic acid.
Consistent with the changes in cardiac morphology and performance, quantification of infarct size by Masson trichrome staining demonstrated the same pattern of cardioprotection (Fig. 5F, 5G). We found that the decrease in infarct size with MSCsmiR-211-over therapy was reversed when cells were coinfected with STAT5A-over. Again, loss of decrease in infarct size by MSCsmiR-211-shRNA was rescued by coinfection with sh-STAT5A (Fig. 5F, 5G).
STAT3 as a Transcription Factor for miR-211 Induced by HP
Consistent with our previous study, we found that HP increased STAT3 phosphorylation (Supporting Information Fig. 6A), that paralleled the increased expression of miR-211. Therefore, we hypothesized that activated STAT3 is responsible for upregulation of miR-211 expression in MSCs by HP. To test this, MSCs were infected with either STAT3-over or STAT3-shRNA and appropriate controls as described in Materials and Methods section. The effects of different groups were confirmed by Western blotting (Supporting Information Fig. 7A, 7B). MSCs cultured under normoxia (N-MSC) and infected with STAT3-over exhibited increased miR-211 expression compared with STAT3-control (1.47±0.16-fold changes, p=.045), and this was further enhanced in HP-treated MSCs (9.28±0.61-fold increase, p<.01; Fig. 6A). In contrast, the increase in miR-211 expression induced by HP was abolished in MSCs infected with STAT3-shRNA (0.27±0.07-fold decrease, p<.001) and significantly reduced in normoxia culture (0.15±0.02-fold decrease, p<.001; Fig. 6B). The results provide strong evidence that STAT3 regulates miR-211 expression.
Figure 6.
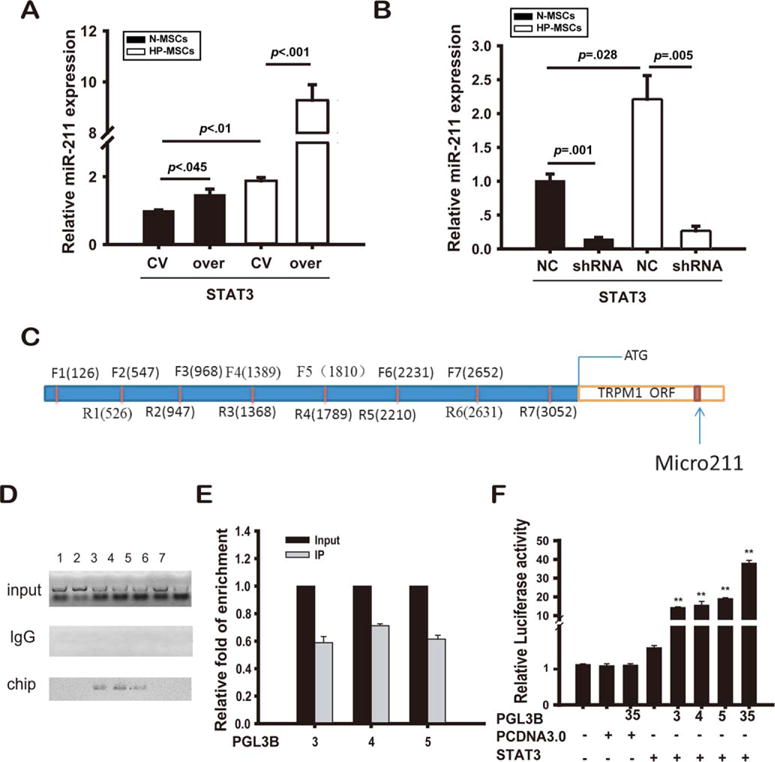
STAT3 directly mediated hypoxia-induced miR-211 expression. MiR-211 expression levels were analyzed by polymerase chain reaction (PCR) for both normoxia-treated MSCs (N-MSCs, in black bars) and HP-treated MSCs (HP-MSCs, in white bars) when they were infected with STAT3-over (over, CV as control, shown in A) or STAT3-shRNA (shRNA, NC as control, shown in B), respectively (three separate experiments). The promoter region of miR-211 was divided into seven consecutive segments (C), and ChIP with STAT3 specific antibody was carried out and the pull-down precipitates were analyzed with quantitative PCR, PCR products were run in the ethidium bromide agarose gel, and the image was captured with the grey-scale inverted for better illustration (D), the relative fold of enrichment was normalized to the input (E). Luciferase assay showed that STAT3 can positively regulate the transcription activities specific for the different promoter regions 3, 4, and 5 of miR-211 (F). The experiments were repeated for three times. **Denotes p less than .01 vs. groups that were not infected with vectors containing both STAT3 and specific promoter regions of 3–5 of miR-211. Abbreviations: HP, hypoxic preconditioning; MSCs, mesenchymal stem cells; shRNA, short hairpin ribonucleic acid.
To further confirm the regulation of STAT3 on miR-211 expression, ChIP-qRT-PCR and luciferase assay were performed. The promoter region of miR-211 was divided into seven consecutive segments, and specific primer sets were then designed (Fig. 6C; Supporting Information Table). These primers which were used for ChIP-qRT-PCR assay to detect possible binding sites for STAT3. We showed that STAT3 could bind to three binding sites (segment 3, 4, and 5) in the miR-211 promoter (Fig. 6D, 6E). Using HEK 293T cells that were cotransfected with STAT3-over combined with a pGL3B vector that contained each of the three different promoter regions of miR-211 alone or in combination (primer sequences for constructing plasmids that contain different promoter regions were listed in Supporting Information Table), luciferase assays clearly showed that STAT3 upregulated the transcriptional activities of miR-211 (Fig. 6F).
Downregulated miR-211 Expression in Aged hMSCs is Associated with Impaired
Migration To provide possible physiological relevance for these studies, we examined miR-211 activity in hMSC as a function of age. We found that the miR-211 expression level was markedly lower in hMSCs obtained from aged individuals (0.41±0.05-fold) compared with young donors (p<.05. Fig. 7A), and this correlated with a decrease in migration capability of aged hMSCs (93.57±2.19 cells per field) compared with young hMSCs (142.90±6.13 cells per field, p<.05; Fig. 7B, 7C). Interestingly, hMSCs infected with miR-211-over resulted in significantly increased migratory capability of both young (253.13±12.44 cells per field, p<.05) and aged hMSCs (212±12.10 cells per field, p<.05) (Fig. 7B, 7C). These findings suggest that miR-211 may be a novel target to improve cell migration and possibly counter the negative effects of aging in this respect.
Figure 7.
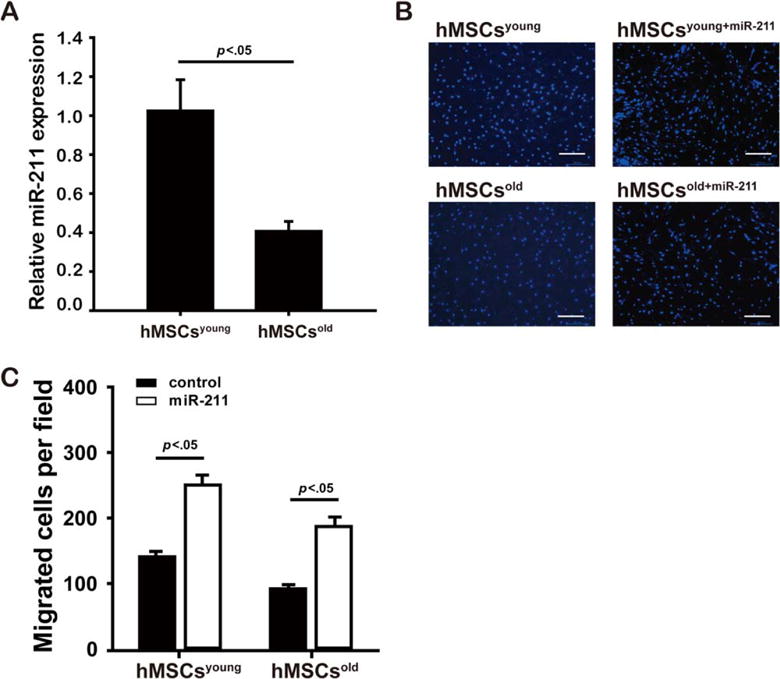
Low miR-211 expression impairs migration of aged hMSCs ability. Downregulated miR-211 expression levels were detected by quantitative real time polymerase chain reaction in aged hMSCs (hMSCsold, n=3) compared with young hMSCs (hMSCsyoung, n=3. A). Migration ability of hMSCs was assessed by transwell assay when both young and old hMSCs were infected with miR-211-over (denoted as hMSCsyoung+miR-211 and hMSCsold+miR-211, respectively) compared with hMSCsyoung and hMSCsold controls, respectively, with representative images shown in (B) (scale bar=200 μm) and the fold changes in number of migrated cells (mean value from three wells and total number of five fields counted for each well, C). Scale bar=200 μm. Abbreviation: hMSCs, human mesenchymal stem cells.
Discussion
In this study, we show that miR-211 modulates migration of MSCs at least in part by regulating STAT5A and associated MAPK signaling. Intravenous transplantation of MSCs that overexpress miR-211 significantly increased cell retention and reduced infarct size in a Rat MI model, leading to improved post-MI cardiac performance. Consistent with our previous study, we found that STAT3 was activated by HP and plays a role in regulating miR-211. Thus our results suggest a novel STAT3/miR-211/STAT5A signaling pathway that regulates MSC migration. Our results implicate genetically modified miR-211 overexpressing MSCs as novel therapeutic agents with improved therapeutic potential over unmodified MSCs. The enhanced migration conferred by miR-211 may be extrapolated to other stem cell therapies.
Optimal cell homing and migration are essential ingredients of cell-based therapy, and better understanding of the underlying mechanisms of these processes following transplantation will lead to improved therapy. It is increasingly recognized that microRNAs play important roles in the regulation of stem cell activities including cell survival, differentiation, and migration. Biological roles of miR-211 have been reported, however, there are controversial findings with reports that miR-211 promotes [9, 10] or inhibits cancer cell growth [12] by targeting different proteins. Using gain and loss of function, our study demonstrates that MSCs migration maybe a prime target for miR-211 function. Furthermore, our findings demonstrate that transcription factor STAT3 positively regulates miR-211 perhaps by binding to specific promoter elements on the host gene, and the effects on migration are conferred by miR-211 regulation of STAT5A. Thus, our study provides a novel mechanism and pathway for regulating MSCs migration. Of note, while our data supports previous reports that activated STAT5A can inhibit cell migration [19], contradictory data are also available demonstrating that STAT5 can promote EPC or stem cell survival and enhance endothelial function [20–22].
A wealth of information are available showing that MAPKs signals govern cell migration with p38MAPK, JNK, and ERK1/2 all being involved, exhibiting their differential roles in mediating downstream targets [23]. For example, with MSCs therapy alveolar epithelial cells could exhibit enhanced migration via phosphorylation of both JNK and p38MAPK [24]. In another recent study, MAPKs were studied for their roles in mediating the skin wound healing process, and p38MAP and JNK were differentially activated by Src [25]. Our data showed that when miR-211 was overexpressed in MSCs, three members of MAPKs were all activated and STAT5A expression was downregulated, which were associated with enhanced MSCs migration, indicating that these proteins could interact with STAT5A in miR-211 mediated MSCs migration. Moreover, miR-211 knockdown in MSCs was accompanied by a reversal of MAPKs phosphorylation levels and STAT5A expression, thus further supporting the important roles for MAPKs in miR-211/STAT5A signaling pathway. In fact, previous data have shown that STAT5A can interact with ERK1/2 and respond to various cell stimuli [26–31]. The STAT5A-ERK1/2 pathway can take actions either in a possible synergistic way [29–31] or in a counter-inhibitory way [27, 28]. In this study we found that downregulation of STAT5A due to miR-211 overexpression in MSCs was associated with increased phosphorylation of ERK1/2, whereas increased STAT5A expression by silenced miR-211 resulted in a lower activity of ERK1/2. We reason that STAT5A can bind to ERK1/2 and interfere with its activation [26], and this may account for ERK1/2 activation by downregulated STAT5A and consequent promotion of MSC migration capability [32]. However, in this study we failed to further show how each component of MAPK signal contributed to the enhanced angiogenesis process. Further study is certainly needed to elucidate the underlying mechanisms.
It is generally acknowledged that the retention and engraftment of cells after stem cell transplantation is low [3], and new approaches are needed to deliver cells in ways that improve engraftment [33]. A finely designed catheter with a helical tip for better cell injection [34] has been successfully used in a clinical trial [35]. In addition, a combination of different microRNAs to synergistically improve the survival and engraftment of stem cells has also been investigated with promising results [36]. Our study shows that miR-211 overexpressing MSCs delivered intravenously have stronger migration capacity and appear to be recruited more efficiently to the injured heart tissue. This certainly offers enhanced therapeutic effects compared to intravenous delivery of regular MSCs without any pretreatment, that is, either HP or miR-211 overexpression. In contrast, a low miR-211 expression level was detected in hMSCs obtained from aged human donors that may contribute to the impaired migration capacity of these cells. This also explains why intravenous delivery of MSCsmiR-211-shRNA did not offer any beneficial effects, as we expected a loss of migratory function due to increases in the expression of STAT5A and MAPK components, which were responsible for impaired migration capability and hence much less MSCs recruited into the infracted heart tissue as evidence by retention assay on day 3 after MI. Importantly we found that miR-211 engineering reversed the compromised migration ability of aged human cells. Thus, we reason that an approach to genetically modify miR-211 expression may be developed to treat aged patients and recover circulating MSCs functions. Most importantly, our results clearly show that transplantation of genetically modified MSCs with miR-211 overexpression are therapeutically superior to unmodified MSCs in a rat MI model.
We have shown that STAT3 plays a key role in HP-induced protection of MSCs during ischemia and enhanced survival and engraftment after transplantation in a mouse MI model [16]. Regulatory roles for STAT3 signaling in cell migration have been reported previously by others [37, 38]. Previous work has also linked microRNAs to these pathways. Haider et al. [39], using a rat MI model, showed that activated STAT3 in myoblasts regulated angiogenesis by inducing miR-21 expression. In tumor cells roles for miR-34a and activated STAT3 were shown to regulate IL6 signaling [40]. In this study, we identified miR-211 as another novel target gene of STAT3. HP-activated STAT3 was found to bind to the promoter region of miR-211 and upregulate its expression as validated by both ChIP-qPCR and luciferase assays. Our results support a unique pathway wherein miR-211 is activated by STAT3 in MSCs and the migration signal is delivered by STAT5A. The work may have important implications both for cell therapy and bone marrow hematopoietic cell aging.
Conclusion
We have demonstrated that miR-211 can enhance MSCs migration via regulating STAT5A, and genetically modified miR-211 overexpressing MSCs delivered intravenously confer enhanced protect against adverse post-MI remodeling. This is closely related to reduced infarct size and enhanced angiogenesis after the MI insult. Thus our experiments provide a novel approach to markedly enhance stem cell therapies.
Supplementary Material
Significance Statement.
This study showed that: (a) MiR-211 overexpression augmented MSCs migration which resulted in a significantly increased retention and enhanced therapeutic effects in an in vivo rat MI model. In contrast, silencing of miR-211 impaired MSCs migration capability and abolished its therapeutic effects; (b) Impaired migration associated with downregulated miR-211 in aging human MSCs can be rescued by miR-211 overexpression; (c) MiR-211 targeted STAT5A to modulate MSCs migration. STAT5A overexpression can attenuate the promigratory effects of miR-211 on MSCs and vice versa; (d) Finally, at the transcriptional levels, miR-211 expression was positively regulated by STAT3 which can be activated by hypoxia preconditioning.
Acknowledgments
This work was supported by grants from the National Key Basic Research Development Plan (973 plan, No. 2014CB965103), the National High-tech R&D Program 863 Program (No. 2013AA020101 for X.Y.H., No. 2015AA020922 for X.B.l.), the National Natural Science Foundation of China (No. 81320108003, 31371498 for J.W., No. 81370247 for X.Y.H., No. 81570233 for X.B.l., No. 81573641 for L.Z., 31271585 for H.Y., and 81370346 for W.Z.), grants from the Education Department of Zhejiang Province (Nos. Y201329862, 20130525, 20130668) and Science and Technology Department of Zhejiang province public welfare projects (No. 2014C33190 for R.W.), Zhejiang Provincial Natural Science Foundation (No. LY16H280003 for L.Z.), and the Fundamental Research Funds for the Central Universities(2016XZZX002-03).
Footnotes
Author Contributions
X.H.: conception and design, data analysis and interpretation, manuscript writing, and final approval of manuscript; P.C.: conception and design, collection and/or assembly of data, and manuscript writing; Y.W.: collection and/or assembly of data; K.W.: collection and interpretation of the data; Y.X., data analysis and interpretation; H.C.: data analysis and interpretation;, L.Z. and R.W. collection and/or assembly of data; K.A.W.: data analysis and interpretation and manuscript preparation; H.Y.: data interpretation and manuscript preparation; W.Z.: conception and design, data analysis and interpretation, manuscript writing, and final approval of manuscript; J.W.: conception and design, financial support, provision of study material or patients, and final approval of manuscript. X.H. and P.C. contributed equally to this article.
See www.StemCells.com for supporting information available online.
Disclosure of Potential Conflicts of Interest
The authors indicate no potential conflicts of interest.
References
- 1.Williams AR, Hare JM. Mesenchymal stem cells: Biology, pathophysiology, translational findings, and therapeutic implications for cardiac disease. Circ Res. 2011;109:923–940. doi: 10.1161/CIRCRESAHA.111.243147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dixon JA, Gorman RC, Stroud RE, et al. Mesenchymal cell transplantation and myocardial remodeling after myocardial infarction. Circulation. 2009;120:S220–S229. doi: 10.1161/CIRCULATIONAHA.108.842302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hou D, Youssef EA, Brinton TJ, et al. Radiolabeled cell distribution after intramyocardial, intracoronary, and interstitial retrograde coronary venous delivery: Implications for current clinical trials. Circulation. 2005;112:I150–I156. doi: 10.1161/CIRCULATIONAHA.104.526749. [DOI] [PubMed] [Google Scholar]
- 4.Bartunek J, Behfar A, Dolatabadi D, et al. Cardiopoietic stem cell therapy in heart failure: The C-CURE (Cardiopoietic stem Cell therapy in heart failURE) multicenter randomized trial with lineage-specified biologics. J Am Coll Cardiol. 2013;61:2329–2338. doi: 10.1016/j.jacc.2013.02.071. [DOI] [PubMed] [Google Scholar]
- 5.Small EM, Olson EN. Pervasive roles of microRNAs in cardiovascular biology. Nature. 2011;469:336–342. doi: 10.1038/nature09783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Heinrich EM, Dimmeler S. MicroRNAs and stem cells: Control of pluripotency, reprogramming, and lineage commitment. Circ Res. 2012;110:1014–1022. doi: 10.1161/CIRCRESAHA.111.243394. [DOI] [PubMed] [Google Scholar]
- 7.Jakob P, Landmesser U. Role of micro-RNAs in stem/progenitor cells and cardiovascular repair. Cardiovasc Res. 2012;93:614–622. doi: 10.1093/cvr/cvr311. [DOI] [PubMed] [Google Scholar]
- 8.Clark EA, Kalomoiris S, Nolta JA, et al. Concise review: MicroRNA function in multipotent mesenchymal stromal cells. STEM CELLS. 2014;32:1074–1082. doi: 10.1002/stem.1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cai C, Ashktorab H, Pang X, et al. Micro-RNA-211 expression promotes colorectal cancer cell growth in vitro and in vivo by targeting tumor suppressor CHD5. PLoS One. 2012;7:e29750. doi: 10.1371/journal.pone.0029750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chu TH, Yang CC, Liu CJ, et al. miR-211 promotes the progression of head and neck carcinomas by targeting TGFbetaRII. Cancer Lett. 2013;337:115–124. doi: 10.1016/j.canlet.2013.05.032. [DOI] [PubMed] [Google Scholar]
- 11.Liu Q, Huang J, Zhou N, et al. LncRNA loc285194 is a p53-regulated tumor suppressor. Nucleic Acids Res. 2013;41:4976–4987. doi: 10.1093/nar/gkt182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Levy C, Khaled M, Iliopoulos D, et al. Intronic miR-211 assumes the tumor suppressive function of its host gene in melanoma. Mol Cell. 2010;40:841–849. doi: 10.1016/j.molcel.2010.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tang YL, Zhu W, Cheng M, et al. Hypoxic preconditioning enhances the benefit of cardiac progenitor cell therapy for treatment of myocardial infarction by inducing CXCR4 expression. Circ Res. 2009;104:1209–1216. doi: 10.1161/CIRCRESAHA.109.197723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pasha Z, Ashraf M. Preconditioning in regenerative medicine: Authors’ retrospective. Cardiovasc Res. 2012;96:210–213. [Google Scholar]
- 15.Hu X, Yu SP, Fraser JL, et al. Transplantation of hypoxia-preconditioned mesenchymal stem cells improves infarcted heart function via enhanced survival of implanted cells and angiogenesis. J Thorac Cardiovasc Surg. 2008;135:799–808. doi: 10.1016/j.jtcvs.2007.07.071. [DOI] [PubMed] [Google Scholar]
- 16.Hu X, Wu R, Jiang Z, et al. Leptin signaling is required for augmented therapeutic properties of mesenchymal stem cells conferred by hypoxia preconditioning. STEM CELLS. 2014;32:2702–2713. doi: 10.1002/stem.1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hu X, Zhang L, Jin J, et al. Heparanase released from mesenchymal stem cells activates integrin beta1/HIF-2alpha/Flk-1 signaling and promotes endothelial cell migration and angiogenesis. STEM CELLS. 2015;33:1850–1862. doi: 10.1002/stem.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wei M, Xin P, Li S, et al. Repeated remote ischemic postconditioning protects against adverse left ventricular remodeling and improves survival in a rat model of myocardial infarction. Circ Res. 2011;108:1220–1225. doi: 10.1161/CIRCRESAHA.110.236190. [DOI] [PubMed] [Google Scholar]
- 19.Sultan AS, Xie J, LeBaron MJ, et al. Stat5 promotes homotypic adhesion and inhibits invasive characteristics of human breast cancer cells. Oncogene. 2005;24:746–760. doi: 10.1038/sj.onc.1208203. [DOI] [PubMed] [Google Scholar]
- 20.Schroder K, Kohnen A, Aicher A, et al. NADPH oxidase Nox2 is required for hypoxia-induced mobilization of endothelial progenitor cells. Circ Res. 2009;105:537–544. doi: 10.1161/CIRCRESAHA.109.205138. [DOI] [PubMed] [Google Scholar]
- 21.Trombetta A, Togliatto G, Rosso A, et al. Increase of palmitic acid concentration impairs endothelial progenitor cell and bone marrow-derived progenitor cell bioavailability: Role of the STAT5/PPARgamma transcriptional complex. Diabetes. 2013;62:1245–1257. doi: 10.2337/db12-0646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang X, Meyer K, Friedl A. STAT5 and prolactin participate in a positive autocrine feedback loop that promotes angiogenesis. J Biol Chem. 2013;288:21184–21196. doi: 10.1074/jbc.M113.481119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang C, Jacobson K, Schaller MD. MAP kinases and cell migration. J Cell Sci. 2004;117:4619–4628. doi: 10.1242/jcs.01481. [DOI] [PubMed] [Google Scholar]
- 24.Chen J, Li Y, Hao H, et al. Mesenchymal stem cell conditioned medium promotes proliferation and migration of alveolar epithelial cells under septic conditions in vitro via the JNK-P38 signaling pathway. Cell Physiol Biochem. 2015;37:1830–1846. doi: 10.1159/000438545. [DOI] [PubMed] [Google Scholar]
- 25.Su L, Fu L, Li X, et al. Loss of CAR promotes migration and proliferation of HaCaT cells, and accelerates wound healing in rats via Src-p38 MAPK pathway. Sci Rep. 2016;6:19735. doi: 10.1038/srep19735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pircher TJ, Petersen H, Gustafsson JA, et al. Extracellular signal-regulated kinase (ERK) interacts with signal transducer and activator of transcription (STAT) 5a. Mol Endocrinol. 1999;13:555–565. doi: 10.1210/mend.13.4.0263. [DOI] [PubMed] [Google Scholar]
- 27.Arnaud M, Crouin C, Deon C, et al. Phosphorylation of Grb2-associated binder 2 on serine 623 by ERK MAPK regulates its association with the phosphatase SHP-2 and decreases STAT5 activation. J Immunol. 2004;173:3962–3971. doi: 10.4049/jimmunol.173.6.3962. [DOI] [PubMed] [Google Scholar]
- 28.Chen AY, Kleiboeker S, Qiu J. Productive parvovirus B19 infection of primary human erythroid progenitor cells at hypoxia is regulated by STAT5A and MEK signaling but not HIFalpha. PLoS Pathog. 2011;7:e1002088. doi: 10.1371/journal.ppat.1002088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lin SK, Kok SH, Yeh FT, et al. MEK/ERK and signal transducer and activator of transcription signaling pathways modulate oncostatin M-stimulated CCL2 expression in human osteoblasts through a common transcription factor. Arthritis Rheum. 2004;50:785–793. doi: 10.1002/art.20058. [DOI] [PubMed] [Google Scholar]
- 30.Neilson LM, Zhu J, Xie J, et al. Coactivation of janus tyrosine kinase (Jak)1 positively modulates prolactin-Jak2 signaling in breast cancer: Recruitment of ERK and signal transducer and activator of transcription (Stat)3 and enhancement of Akt and Stat5a/b pathways. Mol Endocrinol. 2007;21:2218–2232. doi: 10.1210/me.2007-0173. [DOI] [PubMed] [Google Scholar]
- 31.Nishioka C, Ikezoe T, Yang J, et al. Long-term exposure of leukemia cells to multi-targeted tyrosine kinase inhibitor induces activations of AKT, ERK and STAT5 signaling via epigenetic silencing of the PTEN gene. Leukemia. 2010;24:1631–1640. doi: 10.1038/leu.2010.145. [DOI] [PubMed] [Google Scholar]
- 32.Meriane M, Duhamel S, Lejeune L, et al. Cooperation of matrix metalloproteinases with the RhoA/Rho kinase and mitogen-activated protein kinase kinase-1/extracellular signal-regulated kinase signaling pathways is required for the sphingosine-1-phosphate-induced mobilization of marrow-derived stromal cells. STEM CELLS. 2006;24:2557–2565. doi: 10.1634/stemcells.2006-0209. [DOI] [PubMed] [Google Scholar]
- 33.Barbash IM, Chouraqui P, Baron J, et al. Systemic delivery of bone marrow-derived mesenchymal stem cells to the infarcted myocardium: Feasibility, cell migration, and body distribution. Circulation. 2003;108:863–868. doi: 10.1161/01.CIR.0000084828.50310.6A. [DOI] [PubMed] [Google Scholar]
- 34.Trachtenberg B, Velazquez DL, Williams AR, et al. Rationale and design of the transendocardial injection of autologous human cells (bone marrow or mesenchymal) in chronic ischemic left ventricular dysfunction and heart failure secondary to myocardial infarction (TAC-HFT) trial: A randomized, double-blind, placebo-controlled study of safety and efficacy. Am Heart J. 2011;161:487–493. doi: 10.1016/j.ahj.2010.11.024. [DOI] [PubMed] [Google Scholar]
- 35.Heldman AW, DiFede DL, Fishman JE, et al. Transendocardial mesenchymal stem cells and mononuclear bone marrow cells for ischemic cardiomyopathy: The TAC-HFT randomized trial. JAMA. 2014;311:62–73. doi: 10.1001/jama.2013.282909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hu S, Huang M, Nguyen PK, et al. Novel microRNA prosurvival cocktail for improving engraftment and function of cardiac progenitor cell transplantation. Circulation. 2011;124:S27–S34. doi: 10.1161/CIRCULATIONAHA.111.017954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Orgaz JL, Pandya P, Dalmeida R, et al. Diverse matrix metalloproteinase functions regulate cancer amoeboid migration. Nat Commun. 2014;5:4255. doi: 10.1038/ncomms5255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhou C, Tong Y, Wawrowsky K, et al. PTTG acts as a STAT3 target gene for colorectal cancer cell growth and motility. Oncogene. 2014;33:851–861. doi: 10.1038/onc.2013.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Haider KH, Idris NM, Kim HW, et al. MicroRNA-21 is a key determinant in IL-11/ Stat3 anti-apoptotic signalling pathway in preconditioning of skeletal myoblasts. Cardiovasc Res. 2010;88:168–178. doi: 10.1093/cvr/cvq151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rokavec M, Oner MG, Li H, et al. IL-6R/ STAT3/miR-34a feedback loop promotes EMT-mediated colorectal cancer invasion and metastasis. J Clin Invest. 2014;124:1853–1867. doi: 10.1172/JCI73531. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.