

Abstract
RNAs fold into intricate and precise secondary structures. These structural patterns regulate multiple steps of the RNA lifecycle, while also conferring catalytic and scaffolding functions to certain transcripts. Therefore, a full understanding of RNA posttranscriptional regulation requires a comprehensive picture of secondary structure. Here, we review several high throughput sequencing-based methods to globally survey plant RNA secondary structure. These methods are more accurate than computational prediction, and more scalable than physical techniques such as crystallography. We note hurdles to reliably measuring secondary structure, including RNA-binding proteins, RNA base modifications, and intramolecular duplexes. Finally, we survey the functional knowledge that has been gleaned from each of these methods, and identify some unanswered questions that remain.
Graphical Abstract
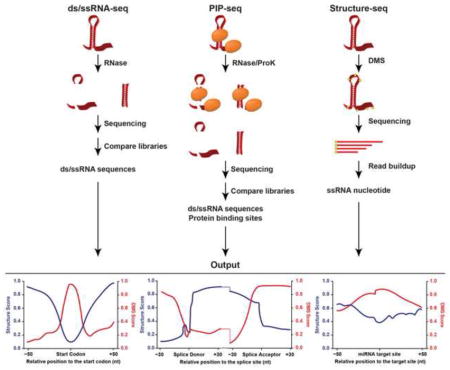
INTRODUCTION
Both coding and noncoding RNAs fold into intricate secondary structures via intramolecular base-pairing. These secondary structures, often in conjunction with RNA-binding proteins (RBPs), form the basis for higher-order tertiary structures that can direct catalysis, form scaffolds, and regulate RNA posttranscriptionally [1]. Such regulation affects multiple steps of the RNA lifecycle, including transcription [2], addition of the 5′ cap [3], splicing [4–8], polyadenylation [9,10], nuclear export [11], subcellular localization [12,13], translation [14–16], and turnover [17]. Notably, specific classes of RNAs, such as microRNAs (miRNAs) and transfer RNAs (tRNAs) require secondary structure for correct processing and subsequent functionality [18–20]. Furthermore, structural scaffolds include many long noncoding RNAs (lncRNAs) [21], ribosomal RNAs (rRNAs) [22], and tRNAs. Thus, determining the patterns of RNA folding across the transcriptome is crucial to fully understanding RNA function and regulation.
Moreover, RNA secondary structure may be an important sensor and signal integrator. Specifically, RNA folding is a dynamic process in which double-and single-stranded RNA (ds- and ssRNA, respectively) can alter their conformations in response to fluctuations in temperature, cellular osmolarity, covalently modified nucleotides, or other signals. For instance, certain RNA structures inhibit translation [23], but are destabilized at higher temperatures, thus forming “RNA thermometers” that link translation regulation to temperature [24,25]. While best characterized in prokaryotes, such RNA thermometers are appealing candidates for RNA regulation in plants [26], which experience wide temperature fluctuations due to their sessile nature. The strong effect of osmolarity on RNA secondary structure [27–29] is likewise of particular interest in plant biology, given the host of osmotic stresses, such as flooding, drought, soil salinity, or nutrient content, that can translate to large-scale changes in intracellular osmolite concentrations [30–32]. Additionally, there are over 150 naturally occurring covalent RNA modifications [33] that modulate RNA secondary structure, alter RNA-protein interactions, and influence posttranscriptional processing [34]. Like structure, these modifications are reversible, demonstrating dynamic patterns during the cell cycle [35,36] and cellular differentiation [37]. Thus, RNA secondary structure is uniquely suited to rapidly sense changing environmental stimuli. Nonetheless, the landscape and functions of plant RNA secondary structure are still largely uncharacterized, presenting a broad opportunity for future study.
The fundamental importance of RNA secondary structure to biological systems has spurred the development of numerous methods to map this feature. While the first and highest fidelity models of secondary structure come from physical methods such as crystallography and NMR, these techniques are labor intensive, can only be performed on single transcripts, and have been rarely applied to plant RNAs. In contrast, the more recently developed high throughput sequencing-based structure probing can be rapidly applied in parallel across the entire plant transcriptome [38–41]. These techniques fall into two broad categories based on the reagents used for structural analysis, and either probe with dsRNA and ssRNA-specific ribonucleases (dsRNases and ssRNases, respectively) or with small chemicals that preferentially modify unpaired RNA. The resulting data from these approaches can be used to constrain folding algorithms (e.g. RNAfold [42]), producing more accurate secondary structure predictions when compared to free energy minimization alone [39–41,43–45]. In total, these scalable genome-wide approaches are uncovering the patterns and functionality of RNA secondary structure on a transcriptome-wide scale, transforming our understanding of this fundamental biological feature.
Here, we review a variety of high-throughput techniques for empirically measuring plant RNA secondary structure on a global scale. Studies using these techniques observe specific structural patterns over splice sites, RBP binding sites, miRNA target sites, and translation start and stop codons, including those in upstream open reading frames (uORFs). Moreover, there are correlations between structure and ribosome association, RNA cleavage, and smRNA production that would not be visible without such transcriptome-wide measurements.
Interrogating RNA secondary structure in plants
Nuclease-based techniques
The first studies to probe RNA folding in plants on a genome-wide scale were the nuclease-based dsRNA-seq and ssRNA-seq techniques performed on total RNA from Arabidopsis thaliana (hereafter Arabidopsis) unopened flower buds [40,41]. These studies first denatured and reannealed the purified RNA prior to digestion. To construct a dsRNA-seq library, this in vitro refolded RNA was then treated with RNase I, an ssRNase that cleaves any unpaired nucleotide, allowing full digestion of all ssRNA. To construct a complementary ssRNA-seq library, aliquots of RNA from the same sample are treated with RNase V1, a dsRNase that cleaves any paired nucleotide. Subsequently, high throughput sequencing libraries were made from these digested RNA samples to map RNA secondary structure in the plant transcriptome [41]. These complementary libraries reveal that RNA folding is a dynamic process, as few sequences are present solely in the ds- or ssRNA library. Thus, the ratio of coverage in each library is used to calculate a structure score representing the degree of pairing or unpairing for each nucleotide. The transcriptome-wide distribution of structure scores was then used to identify regions that are significantly structured (hotspots) or unstructured (coldspots). These structural hotspots and coldspots were found to be significantly more conserved than flanking sequences, suggesting that they have some functional relevance within the parent transcripts [40]. These data were further validated by RT-PCR of structure hotspots using RNA digested with ss- or dsRNases, demonstrating these regions to be in the predicted paired confirmation [41].
These datasets were also used to probe for global structural patterns, resulting in several interesting observations. For instance, a significant structural dip was observed over the start and stop codons of Arabidopsis mRNAs [39–41], as was also observed in Saccharomyces cerevisiae, Drosophila melanogaster, Caenorhabditis elegans, and humans [44–47]. This structural dip likely makes the start codon more accessible than flanking sequences to the scanning ribosome. Additionally, these studies demonstrated that the 5′ and 3′ untranslated regions (UTRs) are on average both less structured than the coding sequence (CDS) [40,41]. This decreased structure in the 5′UTR and at the start codon is of particular interest as previous reports have shown that high secondary structure can inhibit translation initiation [48]. Secondary structure was also probed at known miRNA target sites. These sites were significantly less structured than flanking sequences, indicating that they are more accessible to miRNA-incorporated RNA-induced silencing complex (miRISC) binding [40]. Similar studies in C. elegans [45] yielded the same results.
Furthermore, these nuclease-based studies tested for relationships between transcriptome-wide structure scores and posttranscriptional regulatory processes. Overall, structure score was observed to anti-correlate with transcript abundance, consistent with the observations that euchromatic histone modifications were enriched in genes containing structure coldspots. Furthermore, high structure scores correlated with the abundance of both endogenous fragmented transcripts and small RNAs (smRNAs), indicating that many highly structured transcripts are processed into smRNAs prior to, or during, their turnover. Together, these data revealed that highly abundant transcripts are generally more unstructured [26,40]. Interestingly, highly structured RNAs were also found to be more heavily ribosome-bound, likely due to ribosome stalling over their numerous structural elements [40]. In total, these findings suggest that RNA secondary structure regulates multiple levels of the RNA lifecycle by promoting or inhibiting ribosome and RBP binding. However, these early studies identified structural features using in vitro deproteinated RNA, and measuring native RNA required the development of more sophisticated techniques.
Protein interaction profile sequencing (PIP-seq) is a modified version of ds/ssRNA-seq that identifies both native, deproteinated RNA secondary structure, as well as all protein-bound sequences in a single experimental approach [49,50] (Figure 1A). This technique was first developed and used on two different human cell lines [49,50]. However, it was most recently used to probe RNA secondary structure and RNA-RBP interaction sites throughout the Arabidopsis nuclear transcriptome [39]. This powerful approach identified more than 40,000 distinct protein-bound nuclear sequences with an average size of 68 nucleotides. Furthermore, it obtained a more in vivo glimpse at the RNA secondary structure landscape of the Arabidopsis nucleus.
Figure 1. Confounding factors in PIP-seq.
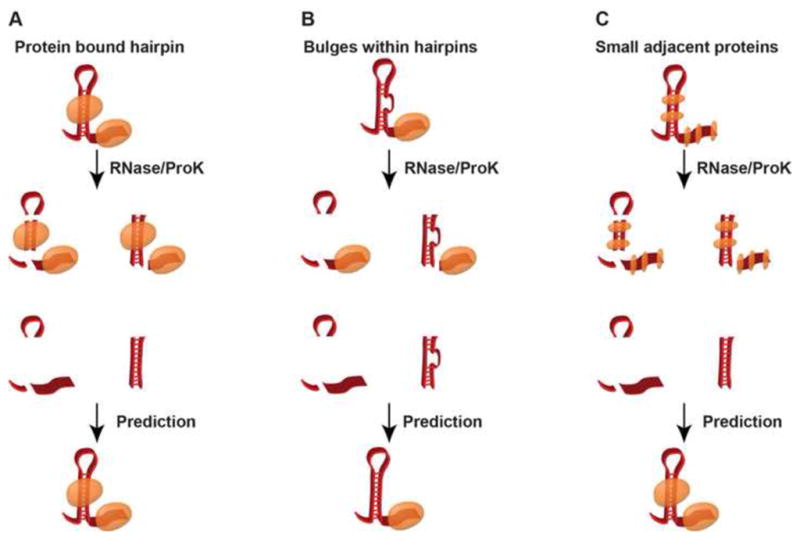
Diagrammatic representation of PIP-seq limitations in RNA secondary structure and RNA-protein interaction site probing. (A) A schematic of the ideal case in which a transcript has a large structural feature and distinct protein binding sites. (B) An example of a small bulge within a highly structured region, which may be undetectable by PIP-seq. (C) Multiple adjacent proteins, will likely undergo crosslinking during formaldehyde treatment and become represented as one large protein interaction site.
When applied to Arabidopsis nuclear RNA, these PIP-seq data both recapitulated and expanded upon the findings from in vitro ds/ssRNA-seq studies. For instance, when examining structural patterns along the CDS, a structural dip was again observed at the start codon, as well as at uORF starts, but was not observed at stop codons. Interestingly, this nuclear-focused study also revealed that both UTRs are more structured than the CDS, which was the opposite pattern observed when structure for the whole (mostly cytoplasmic) transcriptome was interrogated (see above) [38–40]. Thus, there are likely distinct structural patterns of mRNAs in the nucleus as compared to the cytoplasm, which is a hypothesis that will require further testing.
Additionally, constitutive and alternative splice sites were examined for differential structural features, revealing a significant dip in secondary structure at the splice donor site. This is analogous to the dip at the start codon, which likely also makes this region more accessible to binding by the U1 small nuclear RNA (snRNA) to initiate splicing [51]. Interestingly, alternatively spliced cassette exons and retained introns also showed distinct patterns of RNA folding and protein binding when compared to constitutive introns. These data indicate that the secondary structure of alternative splice sites is fundamentally different from constitutive splice sites, likely functioning in their regulation. A close and specific examination of these differential structure patterns and their effects on alternative splicing will significantly increase our understanding of how this process is regulated in plants.
Chemical-based structure probing
Dimethyl sulphate (DMS) is a tissue-permeable chemical adduct that specifically modifies single-stranded adenines and cytosines [52,53]. These DMS modifications inhibit reverse transcriptase extension during first strand complementary DNA (cDNA) synthesis, resulting in a buildup of molecules terminating at the same nucleotide. This nucleotide is then inferred to be in a single-stranded conformation [54,55]. Two groups have recently developed high throughput sequencing techniques in which tissues are treated with DMS prior to RNA fragmentation, reverse transcription, and high throughput sequencing. The RT stops are then measured in each library and normalized to stops from an untreated library [38,46], allowing structural information for unpaired bases to be obtained and used to constrain folding algorithms for a genome-wide RNA folding analysis.
This approach is termed Structure-seq, and has been performed on the total (mostly cytoplasmic) transcriptome of Arabidopsis seedlings, and recapitulated many of the findings from nuclease-based techniques [38]. For instance, Structure-seq also revealed a structural dip over the start and stop codons and globally increased structure in the CDS as compared to the UTRs, similar to the in vitro ds/ssRNA-seq studies of the total transcriptome from unopened flower buds [39–41]. The authors also observed significantly increased structure at alternative splice donor sites compared to control sequences with similar nucleotide composition. This is consistent with results from the nuclear PIP-seq study [39], and indicate that alternatively spliced exons and introns have inherent structural differences compared to those that are constitutively spliced.
However, nuclease and chemical probing yielded different observed structural signatures at alternative polyadenylation sites. In Structure-seq, the authors find a decrease in DMS signal upstream of these sites and an increase in signal directly downstream [38], while no such signature was observed in the nuclear PIP-seq study [39]. This discrepancy could be due to structural differences between nuclear and mostly cytoplasmic whole transcriptome profiles. Alternatively, the high level of protein binding found at alternative polyadenylation sites [39] could hinder DMS modification leading to aberrant structure predictions, as described below. Regardless, future studies will need to more closely examine the secondary structure near both constitutive and alternative polyadenylation sites for the nuclear and cytoplasmic transcriptomes.
Hurdles to measuring RNA secondary structure
Although extremely powerful, there are also specific confounding factors that need to be addressed when measuring secondary structure through these high throughput sequencing-based approaches. For instance, the nuclease-based methodologies have limited resolution of small nucleotide bulges and loops, since RNases used for this approach are somewhat bulky (Figure 1B). Additionally, the use of formaldehyde as the cross-linking agent in the PIP-seq version of this approach can induce protein-protein crosslinks as well as protein-RNA bonds, so small adjacent protein binding events are likely to be represented by one long protein-bound sequence in data analysis (Figure 1C). Finally, unlike certain chemical probes such as DMS, nucleases cannot readily diffuse into plant cells. Thus, these RNase-based approaches must be performed after cell lysis, which may affect the native secondary structures. Despite these drawbacks, the nuclease-based techniques have reproducibly generated transcriptome-wide structural and RBP interaction patterns that provide new functional insights into these features of the plant transcriptome [39,49].
In chemical probing, a significant cause for concern is the presence of naturally occurring covalent RNA modifications, many of which can induce reverse transcriptase (RT) drop off (Figure 2A). Those most likely to cause RT inhibition are the modifications affecting the Watson-Crick base pairing edge of RNA nucleotides [56]. As Structure-seq relies upon RT stops to determine the location of DMS adduct addition (Figure 2B), RNA bases that are modified with an RT-interfering chemical addition will likely be misinterpreted as unpaired, regardless of actual pairing state. Thus, an untreated control library must be used to differentiate between DMS adduct-induced RT stalling, and natural modifications that can result in the same molecular phenotype (Figure 2C). However, even with the proper control libraries the structure at modified nucleotides cannot be reliably measured with DMS treatment. Given the multitude of modified nucleotides in mRNAs [57–63], this will require careful controls be developed and performed when these chemical-dependent structure probing methodologies are used in future experiments.
Figure 2. Covalent RNA modifications and limitations to Structure-seq.

(A) The stopping power for each covalently modified nucleotide, defined as the percentage of reads terminating via reverse transcriptase stalling normalized to total read coveraqe. * p < 0.05, ** p < 1×10−10. (B) An example of the ideal case for Structure-seq in which a transcript is unbound and has no covalently modified nucleotides. Yellow hexagons represent the DMS adduct in all figures. (C) An example of covalent modifications leading to increased RT stops, resulting in a predicted ssRNA sequence in a paired region. Green hexagons represent covalent modifcations to specific nucleotides. (D) A schematic showing proteins occluding DMS addition, leading to a predicted stretch of dsRNA in what is actually a single-stranded region.
An additional concern for chemical probing approaches is that RBP binding will occlude chemical adduct addition in protein interacting regions, leading to stretches of nucleotides where data is lacking [64]. The nuclease-based PIP-seq methodology overcomes this problem by probing both proteinated and deproteinated RNAs [39,49,50], which is how this approach is able to specifically identify protein interaction sites. In contrast, Structure-seq is highly sensitive to protein binding [64] as adduct addition will be occluded in these regions. This is a significant problem given that this approach does not have a direct measurement of paired bases, and high structure is simply inferred from the absence of chemical labeling in specific RNA regions. Therefore, RBP-bound sites, which merely lack adduct addition, will be called as double-stranded regardless of their actual pairing state, leading to incorrect structural models (Figure 2D). Thus, these methodologies would be significantly improved through an additional library preparation that directly assesses paired bases, as is currently part of the nuclease-based approaches. This shortcoming will need to be addressed for future methodologies dependent on chemical probing of RNA secondary structure in plants.
Finally, no currently available sequencing-based structure probing method is able to precisely identify the exact nucleotides that are directly based paired in dsRNA regions. While constrained folding algorithms can predict specific partners, these methods cannot directly resolve true intramolecular secondary structure. To do so, will require future methodologies to adopt a step where intramolecularly paired regions are ligated to each other followed by high-throughput sequencing library preparation. Similar approaches have been previously developed [65,66], and can be modified for the purpose outlined here.
Conclusions and future directions
In summary, high throughput sequencing-based approaches for measuring RNA secondary structure have already provided new functional insights for a wide array of RNAs (especially mRNAs) in plants, most of which have not been previously studied using traditional physical approaches. In fact, these approaches have revealed relationships between mRNA secondary structure and stability, translation, smRNA production, transcript cleavage [26,40], and alternative splicing [38,39] in the model organism Arabidopsis (Figure 3). Future studies will need to focus on the mechanism by which RNA secondary structure directs posttranscriptional regulation. This will require improved base pair predictions and systematic mutation of specific structural elements. Fully understanding this fundamental transcriptome feature will require use of these structure probing techniques over a broader range of plant species and specific cell types. In total, transcriptome-wide probing of RNA secondary structure has and will continue to offer incredible new insights into the physiologically important regulatory functions of plant RNA secondary structure.
Figure 3. Examples of RNA secondary structure data identified by PIP-seq and Structure-seq.

(A) An example of a deproteinated, folded mRNA molecule. (B–D) Example structure score and DMS score profiles at (B) the mRNA start codon, (C) splice donor and acceptor sites, and (D) a miRNA target site.
Both nuclease and chemical-based structure probing methods have been developed
Sequencing-based structure probing is more accurate than folding algorithms alone
Distinct structural profiles have been observed across numerous mRNA regions
There are technical limitations to all experimental RNA structure probing methods
Acknowledgments
We thank members of the Gregory lab for their helpful discussions and comments on the manuscript. This work was funded by the NSF (Career Award MCB-1053846 and MCB-1243947 to BDG) and the National Institute of General Medical Sciences (5T32GM007229-37 to LEV). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Abbreviations
- RBP
RNA binding protein
- miRNA
microRNA
- tRNA
transfer RNA
- lncRNA
long noncoding RNA
- rRNA
ribosomal RNA
- dsRNA
double-stranded RNA
- ssRNA
single-stranded RNA
- dsRNase
double-stranded ribonuclease
- ssRNase
single-stranded ribonuclease
- uORF
upstream open reading frame
- UTR
untranslated region
- CDS
coding sequence
- PIP-seq
protein interaction profile sequencing
- snRNA
small nuclear RNA
- smRNA
small RNA
- DMS
dimethyl-sulfide
- cDNA
complimentary DNA
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Cruz JA, Westhof E. The Dynamic Landscapes of RNA Architecture. Cell. 2009;136:604–609. doi: 10.1016/j.cell.2009.02.003. [DOI] [PubMed] [Google Scholar]
- 2.Wanrooij PH, Uhler JP, Simonsson T, Falkenberg M, Gustafsson CM. G-quadruplex structures in RNA stimulate mitochondrial transcription termination and primer formation. Proceedings of the National Academy of Sciences. 2010;107:16072–16077. doi: 10.1073/pnas.1006026107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dong H, Ray D, Ren S, Zhang B, Puig-Basagoiti F, Takagi Y, Ho CK, Li H, Shi P-Y. Distinct RNA Elements Confer Specificity to Flavivirus RNA Cap Methylation Events. Journal of Virology. 2007;81:4412–4421. doi: 10.1128/JVI.02455-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Buratti E, Baralle FE. Influence of RNA Secondary Structure on the Pre-mRNA Splicing Process. Molecular and Cellular Biology. 2004;24:10505–10514. doi: 10.1128/MCB.24.24.10505-10514.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jin Y, Yang Y, Zhang P. New insights into RNA secondary structure in the alternative splicing of pre-mRNAs. RNA Biology. 2011;8:450–457. doi: 10.4161/rna.8.3.15388. [DOI] [PubMed] [Google Scholar]
- *6.Liu HX, Goodall GJ, Kole R, Filipowicz W. Effects of secondary structure on pre-mRNA splicing: hairpins sequestering the 5′ but not the 3′ splice site inhibit intron processing in Nicotiana plumbaginifolia. The EMBO Journal. 1995;14:377–388. doi: 10.1002/j.1460-2075.1995.tb07012.x. An early study demonstrating that artificially introduced secondary structure can modulate splicing in plants. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Raker VA, Mironov AA, Gelfand MS, Pervouchine DD. Modulation of alternative splicing by long-range RNA structures in Drosophila. Nucleic Acid Research. 2009;37:4533–4544. doi: 10.1093/nar/gkp407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Warf MB, Berglund JA. Role of RNA structure in regulating pre-mRNA splicing. Trends in Biochemical Sciences. 2010;35:169–178. doi: 10.1016/j.tibs.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Klasens BI, Das AT, Berkhout B. Inhibition of polyadenylation by stable RNA secondary structure. Nucleic Acids Research. 1998;26:1870–1876. doi: 10.1093/nar/26.8.1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Oikawa D, Tokuda M, Hosoda A, Iwawaki T. Identification of a consensus element recognized and cleaved by IRE1 alpha. Nucleic Acids Research. 2010;38:6265–6273. doi: 10.1093/nar/gkq452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grüter P, Tabernero C, von Kobbe C, Schmitt C, Saavedra C, Bachi A, Wilm M, Felber BK, Izaurralde E. TAP, the human homolog of Mex67p, mediates CTE-dependent RNA export from the nucleus. Molecular Cell. 1998;1:649–659. doi: 10.1016/s1097-2765(00)80065-9. [DOI] [PubMed] [Google Scholar]
- 12.Bullock SL, Ringel I, Ish-Horowicz D, Lukavsky PJ. A′-form RNA helices are required for cytoplasmic mRNA transport in Drosophila. Nature Structural & Molecular Biology. 2010;17:703–709. doi: 10.1038/nsmb.1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Subramanian M, Rage F, Tabet R, Flatter E, Mandel J-L, Moine H. G-quadruplex RNA structure as a signal for neurite mRNA targeting. EMBO reports. 2011;12:697–704. doi: 10.1038/embor.2011.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kozak M. Leader length and secondary structure modulate mRNA function under conditions of stress. Molecular and Cellular Biology. 1988;8:2737–2744. doi: 10.1128/mcb.8.7.2737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Svitkin YV, Pause A, Haghighat A, Pyronnet S, Witherell G, Belsham GJ, Sonenberg N. The requirement for eukaryotic initiation factor 4A (elF4A) in translation is in direct proportion to the degree of mRNA 5′ secondary structure. RNA (New York, NY) 2001;7:382–394. doi: 10.1017/s135583820100108x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wen J-D, Lancaster L, Hodges C, Zeri A-C, Yoshimura SH, Noller HF, Bustamante C, Tinoco I. Following translation by single ribosomes one codon at a time. Nature. 2008;452:598–603. doi: 10.1038/nature06716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goodarzi H, Najafabadi HS, Oikonomou P, Greco TM, Fish L, Salavati R, Cristea IM, Tavazoie S. Systematic discovery of structural elements governing stability of mammalian messenger RNAs. Nature. 2012;485:264–268. doi: 10.1038/nature11013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bhaskaran H, Rodriguez-Hernandez A, Perona JJ. Kinetics of tRNA folding monitored by aminoacylation. RNA. 2012;18:569–580. doi: 10.1261/rna.030080.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Francklyn CS, Minajigi A. tRNA as an active chemical scaffold for diverse chemical transformations. FEBS letters. 2010;584:366–375. doi: 10.1016/j.febslet.2009.11.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carthew RW, Sontheimer EJ. Origins and Mechanisms of miRNAs and siRNAs. Cell. 2009;136:642–655. doi: 10.1016/j.cell.2009.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rinn JL, Chang HY. Genome regulation by long noncoding RNAs. Annu Rev Biochem. 2012;81 doi: 10.1146/annurev-biochem-051410-092902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tsai M-C, Manor O, Wan Y, Mosammaparast N, Wang JK, Lan F, Shi Y, Segal E, Chang HY. Long noncoding RNA as modular scaffold of histone modification complexes. Science (New York, NY) 2010;329:689–693. doi: 10.1126/science.1192002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang H, Chung PJ, Liu J, Jang IC, Kean MJ, Xu J, Chua NH. Genome-wide identification of long noncoding natural antisense transcripts and their responses to light in Arabidopsis. Genome Research. 2014;24:444–453. doi: 10.1101/gr.165555.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kortmann J, Narberhaus F. Bacterial RNA thermometers: molecular zippers and switches. Nature Reviews Microbiology. 2012;10:255–265. doi: 10.1038/nrmicro2730. [DOI] [PubMed] [Google Scholar]
- 25.Narberhaus F. Translational control of bacterial heat shock and virulence genes by temperature-sensing mRNAs. RNA Biology. 2010;7:84–89. doi: 10.4161/rna.7.1.10501. [DOI] [PubMed] [Google Scholar]
- *26.Vandivier L, Li F, Zheng Q, Willmann M, Chen Y, Gregory B. Arabidopsis mRNA secondary structure correlates with protein function and domains. Plant Signaling & Behavior. 2013;8:e24301. doi: 10.4161/psb.24301. This study further analyzes ds/ssRNA-seq data from Arabidopsis and compares these data to in silico structure predictions. The authors find that the constrained structure data correlates much more strongly with gene expression, smRNA processing, and ribosome association data than in silico predicted scores. Additionally, the authors find specific gene ontology terms to be enriched in the most highly and lowly structured transcripts. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Draper DE. RNA Folding: Thermodynamic and Molecular Descriptions of the Roles of Ions. Biophysical Journal. 2008;95:5489–5495. doi: 10.1529/biophysj.108.131813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kilburn D, Roh JH, Guo L, Briber RM, Woodson SA. Molecular Crowding Stabilizes Folded RNA Structure by the Excluded Volume Effect. Journal of the American Chemical Society. 2010;132:8690–8696. doi: 10.1021/ja101500g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lambert D, Draper DE. Effects of Osmolytes on RNA Secondary and Tertiary Structure Stabilities and RNA-Mg2+ Interactions. Journal of Molecular Biology. 2007;370:993–1005. doi: 10.1016/j.jmb.2007.03.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bartels D, Sunkar R. Drought and Salt Tolerance in Plants. Critical Reviews in Plant Sciences. 2005;24:23–58. [Google Scholar]
- 31.Seki M, Umezawa T, Urano K, Shinozaki K. Regulatory metabolic networks in drought stress responses. Current Opinion in Plant Biology. 2007;10:296–302. doi: 10.1016/j.pbi.2007.04.014. [DOI] [PubMed] [Google Scholar]
- 32.Taji T, Ohsumi C, Iuchi S, Seki M, Kasuga M, Kobayashi M, Yamaguchi-Shinozaki K, Shinozaki K. Important roles of drought- and cold-inducible genes for galactinol synthase in stress tolerance in Arabidopsis thaliana. The Plant Journal. 2002;29:417–426. doi: 10.1046/j.0960-7412.2001.01227.x. [DOI] [PubMed] [Google Scholar]
- 33.Dunin-Horkawicz S, Czerwoniec A, Gajda MJ, Feder M, Grosjean H, Bujnicki JM. MODOMICS: a database of RNA modification pathways. Nucleic Acids Research. 2006;34:D145–D149. doi: 10.1093/nar/gkj084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu N, Dai Q, Zheng G, He C, Parisien M, Pan T. N6-methyladenosine-dependent RNA structural switches regulate RNA–protein interactions. Nature. 2015;518:560–564. doi: 10.1038/nature14234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schwartz S, Mumbach MR, Jovanovic M, Wang T, Maciag K, Bushkin GG, Mertins P, Ter-Ovanesyan D, Habib N, Cacchiarelli D, et al. Perturbation of m6A Writers Reveals Two Distinct Classes of mRNA Methylation at Internal and 5′ Sites. Cell Reports. 2014;8:284–296. doi: 10.1016/j.celrep.2014.05.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Horiuchi K, Kawamura T, Iwanari H, Ohashi R, Naito M, Kodama T, Hamakubo T. Identification of Wilms’ tumor 1-associating protein complex and its role in alternative splicing and the cell cycle. J Biol Chem. 2013;288:33292–33302. doi: 10.1074/jbc.M113.500397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Batista PJ, Molinie B, Wang J, Qu K, Zhang J, Li L, Bouley DM, Lujan E, Haddad B, Daneshvar K, et al. m6A RNA Modification Controls Cell Fate Transition in Mammalian Embryonic Stem Cells. Cell Stem Cell. 2014;15:707–719. doi: 10.1016/j.stem.2014.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **38.Ding Y, Tang Y, Kwok CK, Zhang Y, Bevilacqua PC, Assmann SM. In vivo genome-wide profiling of RNA secondary structure reveals novel regulatory features. Nature. 2014;505:696–700. doi: 10.1038/nature12756. This is the first chemical-based structure probing study in Arabidopsis. The authors reported structural dips at translation start and stop codons, as well as distinct structural signatures at alternatve polyadenylation and alternative splice sites. [DOI] [PubMed] [Google Scholar]
- **39.Gosai SJ, Foley SW, Wang D, Silverman IM, Selamoglu N, Nelson ADL, Beilstein MA, Daldal F, Deal RB, Gregory BD. Global Analysis of the RNA-Protein Interaction and RNA Secondary Structure Landscapes of the Arabidopsis Nucleus. Mol Cell. 2015;57:376–388. doi: 10.1016/j.molcel.2014.12.004. The authors performed PIP-seq on nuclei from Arabidopsis seedlings, simultaneously probing secondary structure and protein binding sites. They reported structural dips at both canonical and noncanonical start codons, and observed distinct structural profiles at cassette exon and intron retention sites. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **40.Li F, Zheng Q, Vandivier LE, Willmann MR, Chen Y, Gregory BD. Regulatory Impact of RNA Secondary Structure across the Arabidopsis Transcriptome. The Plant Cell. 2012;24:4346–4359. doi: 10.1105/tpc.112.104232. This study analyzes ds/ssRNA-seq data from unopened Arabidopsis flower buds. The authors describe structural dips at miRNA binding sites, as well as translation start and stop codons. Additionally, the study reports correlations between transcriptome-wide secondary structure and expression, degradation, smRNA production, and ribosome association. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **41.Zheng Q, Ryvkin P, Li F, Dragomir I, Valladares O, Yang J, Cao K, Wang LS, Gregory BD. Genome-Wide Double-Stranded RNA Sequencing Reveals the Functional Significance of Base-Paired RNAs in Arabidopsis. PLoS Genetics. 2010;6:e1001141. doi: 10.1371/journal.pgen.1001141. This study describes the first genome-wide structure probing analysis in Arabidopsis unopened flower buds. The authors perform dsRNA-seq and compare their constrained RNA folding predictions to in silico free energy minimization algorithms. This study validates dsRNA-seq as a higher fidelity alternative to in silico techniques, while also showing increased conservation at structure hotspots relative to flanking regions. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gruber AR, Lorenz R, Bernhart SH, Neuböck R, Hofacker IL. The Vienna RNA Websuite. Nucleic Acids Research. 2008;36:W70–W74. doi: 10.1093/nar/gkn188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *43.Mathews DH, Disney MD, Childs JL, Schroeder SJ, Zuker M, Turner DH. Incorporating chemical modification constraints into a dynamic programming algorithm for prediction of RNA secondary structure. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:7287–7292. doi: 10.1073/pnas.0401799101. One of the first studies proposing constraint of RNA folding algorithms with empirical RNA secondary structure data. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **44.Kertesz M, Wan Y, Mazor E, Rinn JL, Nutter RC, Chang HY, Segal E. Genome-wide measurement of RNA secondary structure in yeast. Nature. 2010;467:103–107. doi: 10.1038/nature09322. One of the first studies to empirically measure RNA secondary structure across a eukaryotic transcriptome. The authors focus on translation regulation, and observe structural features across the start and stop codon, correlation between secondary structure and ribosome density, and a three nucleotide periodicity in structure across the coding sequence. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li F, Zheng Q, Ryvkin P, Dragomir I, Desai Y, Aiyer S, Valladares O, Yang J, Bambina S, Sabin LR, et al. Global Analysis of RNA Secondary Structure in Two Metazoans. Cell Reports. 2012;1:69–82. doi: 10.1016/j.celrep.2011.10.002. [DOI] [PubMed] [Google Scholar]
- 46.Rouskin S, Zubradt M, Washietl S, Kellis M, Weissman JS. Genome-wide probing of RNA structure reveals active unfolding of mRNA structures in vivo. Nature. 2014;505:701–705. doi: 10.1038/nature12894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wan Y, Kun Qu K, Zhang QC, Flynn RA, Manor O, Ouyang Z, Zhang JS, RC, Snyder MP, Segal E, Chang HY. Landscape and variation of RNA secondary structure across the human transcriptome. Nature. 2014;505:706–709. doi: 10.1038/nature12946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *48.Wang LS, Wessler SR. Role of mRNA Secondary Structure in Translational Repression of the Maize Transcriptional Activator Lc1,2. Plant Physiology. 2001;125:1380–1387. doi: 10.1104/pp.125.3.1380. Showed that previously characterized effects of secondary structure on mRNA translation also occur in maize. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **49.Silverman IM, Li F, Alexander A, Goff L, Trapnell C, Rinn JL, Gregory BD. RNase-mediated protein footprint sequencing reveals protein-binding sites throughout the human transcriptome. Genome Biology. 2014;15:R3. doi: 10.1186/gb-2014-15-1-r3. One of the first studies to measure RBP binding across a eukaryotic transcriptome. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Silverman IM, Gregory BD. Transcriptome-wide ribonuclease-mediated protein footprinting to identify RNA–protein interaction sites. Methods. 2015;72:76–85. doi: 10.1016/j.ymeth.2014.10.021. [DOI] [PubMed] [Google Scholar]
- 51.Wahl MC, Will CL, Luhrmann R. The Spliceosome: Design Principles of a Dynamic RNP Machine. Cell. 2009;136:701–718. doi: 10.1016/j.cell.2009.02.009. [DOI] [PubMed] [Google Scholar]
- 52.Peattie DA, Gilbert W. Chemical probes for higher-order structure in RNA. Proc Natl Acad Sci. 1980;77:4679–4682. doi: 10.1073/pnas.77.8.4679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Peattie DA. Direct chemical method for sequencing RNA. Proc Natl Acad Sci. 1979;76:1760–1764. doi: 10.1073/pnas.76.4.1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *54.Lempereurl L, Nicolosol M, Riehl N, Ehresmann C, Ehresmann B, Bachelleriel J-P. Conformation of yeast 18S rRNA. Direct chemical probing of the 5′ domain in ribosomal subunits and in deproteinized RNA by reverse transcriptase mapping of dimethylsulfate accessible sites. Nucleic Acid Research. 1985;13:8339–8357. doi: 10.1093/nar/13.23.8339. An early example of DMS probing for secondary structure analyses. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Inoue T, Cech TR. Secondary structure of the circular form of the Tetrahymena rRNA intervening sequence: A technique for RNA structure analysis using chemical probes and reverse transcriptase. Proc Natl Acad Sci. 1985;82:648–652. doi: 10.1073/pnas.82.3.648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *56.Ryvkin P, Leung YY, Silverman IM, Childress M, Valladares O, Dragomir I, Gregory BD, Wang LS. HAMR: high-throughput annotation of modified ribonucleotides. RNA. 2013;19:1684–1692. doi: 10.1261/rna.036806.112. The first method to empirically predict multiple types of modification sites across a whole transcriptome using RNA-seq data. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dominissini D, Moshitch-Moshkovitz S, Salmon-Divon M, Amariglio N, Rechavi G. Transcriptome-wide mapping of N6-methyladenosine by m6A-seq based on immunocapturing and massively parallel sequencing. Nature Protocols. 2013;8:176–189. doi: 10.1038/nprot.2012.148. [DOI] [PubMed] [Google Scholar]
- 58.Schwartz S, Bernstein Douglas A, Mumbach Maxwell R, Jovanovic M, Herbst Rebecca H, León-Ricardo Brian X, Engreitz Jesse M, Guttman M, Satija R, Lander Eric S, et al. Transcriptome-wide Mapping Reveals Widespread Dynamic-Regulated Pseudouridylation of ncRNA and mRNA. Cell. doi: 10.1016/j.cell.2014.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell. 2012;149:1635–1646. doi: 10.1016/j.cell.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Squires JE, Patel HR, Nousch M, Sibbritt T, Humphreys DT, Parker BJ, Suter CM, Preiss T. Widespread occurrence of 5-methylcytosine in human coding and non-coding RNA. Nucleic Acid Research. 2012;40:5023–5033. doi: 10.1093/nar/gks144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li JB, Levanon EY, Yoon JK, Aach J, Xie B, LeProust EM, Zhang K, Gao Y, Church GM. Genome-wide identification of human RNA editing sites by parallel DNA capturing and sequencing. Science. 2009;324:1210–1213. doi: 10.1126/science.1170995. [DOI] [PubMed] [Google Scholar]
- 62.Wulff BE, Sakurai M, Nishikura K. Elucidating the inosinome: global approaches to adenosine-to-inosine RNA editing. Nat Rev Genetics. 2011;12:81–85. doi: 10.1038/nrg2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Carlile TM, Rojas-Duran MF, Zinshteyn B, Shin H, Bartoli KM, Gilbert WV. Pseudouridine profiling reveals regulated mRNA pseudouridylation in yeast and human cells. Nature. 2014;6:134–136. doi: 10.1038/nature13802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Talkish J, May G, Lin Y, Woolford JL, Jr, McManus CJ. Mod-seq: high-throughput sequencing for chemical probing of RNA structure. RNA. 2014;20:1–8. doi: 10.1261/rna.042218.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Helwak A, Tollervey D. Mapping the miRNA interactome by cross-linking ligation and sequencing of hybrinds (CLASH) Nature Protocols. 2014;9:711–728. doi: 10.1038/nprot.2014.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kudla G, Granneman S, Hahn D, Beggs JD, Tollervey D. Cross-linking, ligation, and sequencing of hybrids reveals RNA–RNA interactions in yeast. Proc Natl Acad Sci. 2011;108:10010–10015. doi: 10.1073/pnas.1017386108. [DOI] [PMC free article] [PubMed] [Google Scholar]