

Abstract
Total syntheses of oridamycin A, triptoquinones B and C and isoiresin are accomplished from a common intermediate prepared via iridium catalyzed alcohol C-H tert-(hydroxy)-prenylation; a byproduct-free process that forms an all-carbon quaternary stereocenter with excellent control of diastereo- and enantioselectivity.
Terpenoid natural products are a large class of secondary metabolites that have found broad use in human medicine, agriculture and the flavor/fragrance industry.1 The biosynthesis2 and de novo chemical synthesis3 of terpenoid natural products is largely reliant on the cascade polycyclization of polyolefins.2,3 While polycyclization strategies enable rapid increases in molecular complexity, their use in de novo chemical synthesis is often accompanied by a lack of convergency, impeding the design of more concise routes.3c Recently, in connection with the development of methods for direct alcohol C-H functionalization via C-C bond forming transfer hydrogenation,4 we devised a diastereo- and enantioselective protocol for primary alcohol C-H tert-(hydroxy)-prenylation.5 This process creates a structural motif bearing an all-carbon quaternary stereocenter that is found in over 2000 terpenoid natural products (eq. 1).1
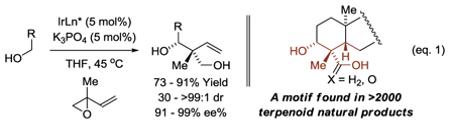 |
Here, our first efforts to utilize alcohol C-H tert-(hydroxy)-prenylation vis-à-vis terpenoid construction are described. This effort has resulted in the total synthesis of oridamycin A,6,7 triptoquinones B and C,8,9 and isoiresin10,11 from a common intermediate (Figure 1). Notably, each natural product is prepared in fewer steps than in any prior approach7,9,11 and, with the exception of isoiresin, protecting groups are not required. Thus, alcohol C-H tert-(hydroxy)-prenylation offers a powerful, modular means of preparing diverse terpenoid natural products beyond cascade polycyclization.
Figure 1.

Oridamycin A, triptoquinone B and C and isoiresin and summary of prior total syntheses.a
aFor graphical summaries of prior total syntheses, see Supporting Information. Longest Linear Sequence (LLS); Total Steps (TS).
Retrosynthetically, a modular strategy was envisioned (Scheme 1). The union of Fragment A with Fragments B-I or B-II via Suzuki cross-coupling followed by Friedel-Crafts cyclization serves as a conduit to oridamycin A and triptoquinones B and C, respectively. For isoiresin, Diels-Alder cycloaddition with iso-Fragment A and dimethyl acetylene dicarboxylate represents an alternative to Friedel-Crafts cyclization. Fragment A, the common intermediate en route to each natural product, is prepared in 4 steps through anti-diastereo- and enantioselective tert-(hydroxy)prenylation5 of commercially available alcohol 1 followed by ring-closing metathesis12 and intramolecular Sakurai allylation.13 Sakurai allylation, when conducted under more forcing conditions, converts Fragment A to iso-Fragment A. Fragments B-I, B-II or B-III symbolize the diversity of structural components that may be applied in this strategy, which serves as a convergent conduit to numerous members of the terpenoid class.
Scheme 1.

Modular retrosynthetic analysis of oridamycin A, triptoquinones B and C and isoiresin.
The synthesis of Fragment A, the common intermediate in the total synthesis of oridamycin A, triptoquinones B and C, and isoiresin is accomplished in only 4 steps (Scheme 2). The commercially available alcohol 1 is exposed to isoprene oxide in the presence of the π-allyliridium C,O-benzoate complex derived from 4-CN-3-NO2-benzoic acid and (R)-Tol-BINAP in THF at 60 °C. The desired product of anti-diastereo- and enantioselective tert-(hydroxy)prenylation 2 is formed in 90% yield with remarkable anti-diastereoselectivity (34:1) and enantioselectivity (98% ee). Compound 2 exists in equilibrium with the 5-membered lactol and upon exposure to acid forms a cyclic ketal (not shown). Hence, chromatographic isolation required pretreatment of the silica gel with triethylamine. Exposure of 2 to allyldimethylsilyl chloride results in chemoselective functionalization of the primary neopentyl alcohol to provide silyl ether 3 in 73% yield, which upon ring-closing metathesis delivers the cyclic allylsilane 4.12 Compounds 3 and 4 exist in equilibrium with their 5-membered lactols, suggesting the [2.2.1]oxabicycle Fragment A may be formed upon intramolecular Sakurai allylation by way of an endocyclic oxacarbenium ion.13 Indeed, exposure of allylsilane 4 to ZnCl2 provides Fragment A as a single diastereomer in 92% yield. Using a stronger Lewis acid, BF3•OEt2, Fragment A eliminates in situ to iso-Fragment A in 63% yield.
Scheme 2.
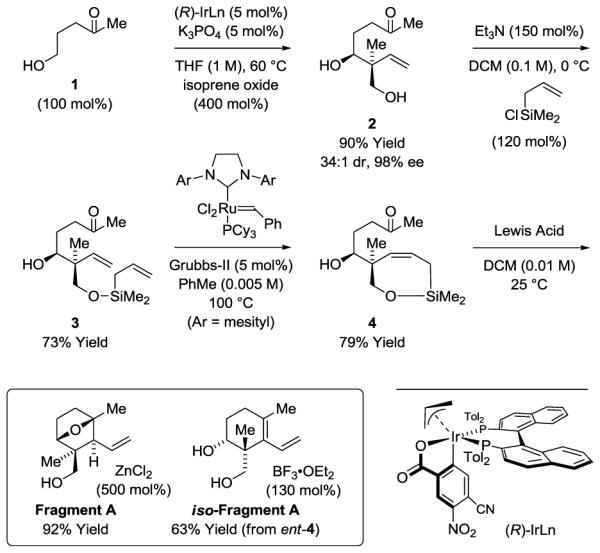
Synthesis of Fragment A via anti-diastereo- and enantioselective alcohol C-H tert-(hydroxy)prenylation.a
aYields are of material isolated by silica gel chromatography. Enantioselectivity was determined by chiral stationary phase HPLC analysis. See Supporting Information for further experimental details.
Acquisition of Fragment A sets the stage for the synthesis of oridamycin A and tryptoquinones B and C. Construction of oridamycin A was readily accomplished as follows (Scheme 3). Suzuki cross-coupling14 of Fragment A with 2-bromocarbazole Fragment B-I was conducted by way of the alkyl 9-BBN derivative using KF as base.15 The desired product of cross-coupling 5 was obtained in 66% isolated yield. Recognizing that the oxidation of primary alcohols to aldehydes using IBX16 and the Pinnick oxidation17 aldehydes to carboxylic acids can both be conducted in DMSO solvent,18 a direct one-pot conversion of the primary alcohol 5 to the carboxylic acid 6 was developed. Friedel-Crafts cyclization19 of carboxylic acid 6 would complete the synthesis of oridamycin A. Here, numerous Lewis acids and Brønsted acids were evaluated. In most cases, cyclization occurred in good yield, however, regioselectivity for the carbazole 1- vs 3-position was problematic. Optimal results were obtained using TiCl4, which gave the product of Friedel-Crafts cyclization in 81% yield as a 3:1 mixture of regioisomers favoring oridamycin A. The present route to oridamycin A, which is protecting group-free,20 is the most concise total synthesis and the first asymmetric total synthesis of this natural product.
Scheme 3.
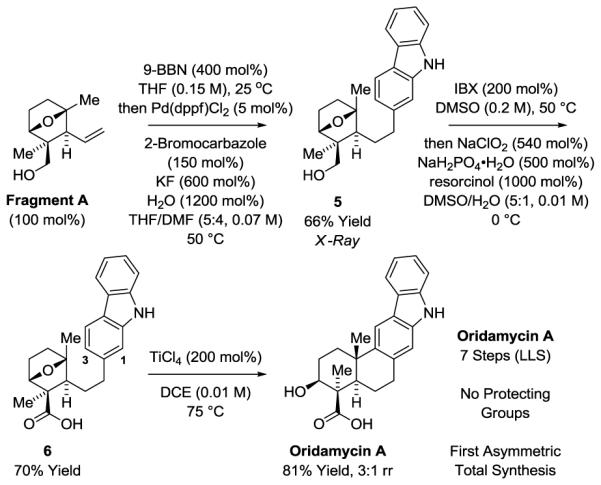
Total synthesis of oridamycin A.a
aYields are of material isolated by silica gel chromatography. See Supporting Information for further experimental details.
The conversion of Fragment A to triptoquinones B and C illustrates the modularity and generality of this convergent approach to terpenoid construction (Scheme 4). Suzuki cross-coupling14,15 of Fragment A with Fragment B-II was accomplished in 53% yield using the palladium catalyst modified by tri-tert-butylphosphine.21 Exposure of the Suzuki coupling product 7 to ZrCl4 resulted in Friedel-Crafts cyclization to furnish the tricyclic compound 8 in 57% yield, which upon treatment with 4-iodophenoxyacetic acid in the presence of Oxone® delivered triptoquinone C,22 representing a formal synthesis of triptoquinone B.9
Scheme 4.
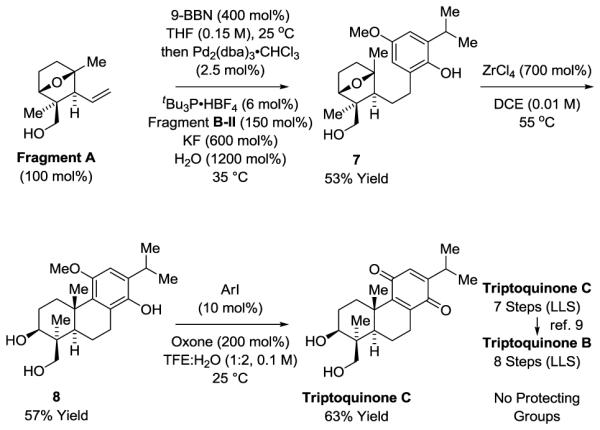
Total synthesis of triptoquinones B and C.a
aYields are of material isolated by silica gel chromatography. See Supporting Information for further experimental details. ArI = 4-I-PhOCH2CO2H.
To further demonstrate the flexibility of the present approach to terpenoid construction, the total synthesis of isoiresin was undertaken using a complementary strategy (Scheme 5). iso-Fragment A was heated in the presence of dimethyl acetylene dicarboxylate, Fragment B-III, however, the desired product of [4+2] cycloaddition 9 was not formed.23 For the corresponding bis-acetate and bis-isobutyrate derivatives, the desired cycloadducts were generated in good yield with diastereoselectivities of 2.5:1 and 4.5:1, respectively. These data led us to investigate the cycloaddition of the bis-pivalate 10, which delivered the product of cycloaddition 11 in 74% yield as a 10:1 mixture of diastereomers. Exposure of the tetraester 11 to LiAlH4 provided the tetraol 12 in 78% yield. Finally, oxidative lactonization of the 1,4-ene-diol moiety11b in the presence of the saturated 1,3-diol followed by chemoselective Shenvi reduction of the less substituted olefin converted tetraol 12 to isoiresin.24
Scheme 5.
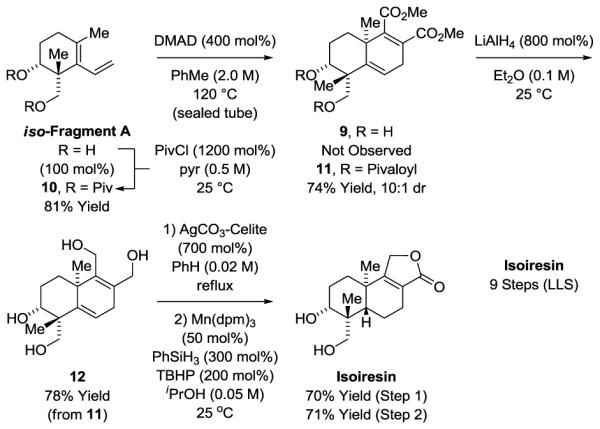
Total synthesis of isoiresin.a
aYields are of material isolated by silica gel chromatography. See Supporting Information for further experimental details.
In summary, a modular, convergent and step-economic strategy for terpenoid construction beyond cascade polycyclizations has been defined, as illustrated by the total synthesis of oridamycin A, triptoquinones B and C and isoiresin from a common precursor. Our strategy is uniquely enabled by a catalytic method for primary alcohol C-H tert-(hydroxy)-prenylation that forms an all-carbon quaternary center with excellent control of diastereo- and enantioselectivity. In particular, alcohol C-H tert-(hydroxy)-prenylation facilitates axial placement of the oxygenated A-ring methyl group (CH2OH or CO2R), which is difficult to achieve through classical cascade polycyclization.25 The effectiveness of this protecting-group free strategy for convergent terpenoid construction is borne out by the fact that each natural product is now prepared in significantly fewer steps than in any prior approach. Future studies will focus on the development and application of related catalytic methods for C-C bond formation that transcend use of stoichiometric organometallic reagents.
Supplementary Material
Acknowledgments
The Robert A. Welch Foundation (F-0038) and the NIH-NIGMS (RO1-GM093905) are acknowledged for partial support of this research. Yi-An Guo is acknowledged for preliminary studies on the synthesis of the diene iso-Fragment A.
Footnotes
Supporting Information Available: Experimental procedures and spectral data for all new compounds. Single crystal X-ray diffraction data corresponding to compound 5. This material is available free of charge via the internet at http://pubs.acs.org.
REFERENCES
- (1).(a) Connolly JD, Hill RA. Dictionary of Terpenoids. Springer; Dordrecht: 1991. For selected reviews on terpenoid natural products, see. [Google Scholar]; (b) Breitmaier E. Terpenes: Flavors, Fragrances, Pharmaca, Pheromones. Wiley-VCH; Weinheim, Germany: 2006. [Google Scholar]; (c) Harrewijn P, van Oosten AM, Piron PGM. Natural Terpenoids as Messengers. Springer; Dordrecht: 2000. [Google Scholar]
- (2).(a) Davis EM, Croteau R. Top. Curr. Chem. 2000;209:53. For selected reviews on terpenoid biosynthesis, see. [Google Scholar]; (b) Christianson DW. Chem. Rev. 2006;106:3412. doi: 10.1021/cr050286w. [DOI] [PubMed] [Google Scholar]; (c) Dewick PM. Medicinal Natural Products: A Biosynthetic Approach. 3rd. Wiley & Sons; Chichester, U. K.: 2009. pp. 187–306. “The mevalonate and methylerythritol phosphate pathways: terpenoids and steroids,”. Chapter 5. [Google Scholar]; (d) Tantillo DJ. Nat. Prod. Rep. 2011;28:1035. doi: 10.1039/c1np00006c. [DOI] [PubMed] [Google Scholar]
- (3).(a) Yoder RA, Johnston JN. Chem. Rev. 2005;105:4730. doi: 10.1021/cr040623l. For selected reviews on de novo terpenoid synthesis, see. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Maimone TJ, Baran PS. Nature Chem. Biol. 2007;3:396. doi: 10.1038/nchembio.2007.1. [DOI] [PubMed] [Google Scholar]; (c) Urabe D, Asaba T, Inoue M. Chem. Rev. 2015;115:9207. doi: 10.1021/cr500716f. [DOI] [PubMed] [Google Scholar]
- (4).(a) Hassan A, Krische MJ. Org. Proc. Res. Devel. 2011;15:1236. doi: 10.1021/op200195m. For selected reviews on hydrogenative and transfer hydrogenative C-C coupling, see. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ketcham JM, Shin I, Montgomery TP, Krische MJ. Angew. Chem. Int. Ed. 2014;53:9142. doi: 10.1002/anie.201403873. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Bower JF, Krische MJ. Top. Organomet. Chem. 2011;34:107. doi: 10.1007/978-3-642-15334-1_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Feng J, Garza VJ, Krische MJ. J. Am. Chem. Soc. 2014;136:8911. doi: 10.1021/ja504625m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).(a) Takada K, Kajiwara H, Imamura N. J. Nat. Prod. 2010;73:698. doi: 10.1021/np1000522. For the isolation and structure determination of oridamycin A and B, and xiamycin, see. [DOI] [PubMed] [Google Scholar]; (b) Ding L, Münch J, Goerls H, Maier A, Fiebig HH, Lin WH, Hertweck C. Bioorg. Med. Chem. Lett. 2010;20:6685. doi: 10.1016/j.bmcl.2010.09.010. [DOI] [PubMed] [Google Scholar]; (c) Ding L, Maier A, Fiebig HH, Lin WH, Hertweck C. Org. Biomol. Chem. 2011;9:4029. doi: 10.1039/c1ob05283g. [DOI] [PubMed] [Google Scholar]; (d) Zhang Q, Mandi A, Li SM, Chen Y, Zhang W, Tian XP, Zhang H, Li HX, Zhang W, Zhang S, Ju JH, Kurtan T, Zhang C. Eur. J. Org. Chem. 2012:5256. [Google Scholar]; (e) Kim S-H, Ha T-K-Q, Oh WK, Shin J, Oh D-C. J. Nat. Prod. 2016;79:51. doi: 10.1021/acs.jnatprod.5b00634. [DOI] [PubMed] [Google Scholar]
- (7).(a) Rosen BR, Werner EW, O'Brien AG, Baran PS. J. Am. Chem. Soc. 2014;136:5571. doi: 10.1021/ja5013323. For total syntheses of oridamycin A and B, and xiamycin, see. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Meng Z, Yu H, Li L, Tao W, Chen H, Wan M, Yang P, Edmonds DJ, Zhong J, Li A. Nat. Commun. 2015;6:6096. doi: 10.1038/ncomms7096. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Trotta AH. Org. Lett. 2015;17:3358. doi: 10.1021/acs.orglett.5b01629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).(a) Takaishi Y, Shishido K, Wariishi N, Shibuya M, Goto K, Kido M, Ono Y. Tetrahedron Lett. 1992;33:7177. For the isolation and structure determination of triptoquinones B and C, see. [Google Scholar]; (b) Shishido K, Nakano K, Wariishi N, Tateishi H, Omodani T, Shibuya M, Goto K, Ono Y, Takaishi Y. Phytochemistry. 1994;35:731. [Google Scholar]; (c) Morota T, Qin W-Z, Takagi K, Xu L-H, Maruno M, Yang B-H. Phytochemistry. 1995;40:865. [Google Scholar]; (d) Duan HQ, Takaishi Y, Momota H, Ohmoto Y, Taki T, Jia YF, Li D. J. Nat. Prod. 1999;62:1522. doi: 10.1021/np9902183. [DOI] [PubMed] [Google Scholar]; (e) Tanaka N, Ooba N, Duan H, Takaishi Y, Nakanishi Y, Bastow K, Lee K-H. Phytochemistry. 2004;65:2071. doi: 10.1016/j.phytochem.2004.04.032. [DOI] [PubMed] [Google Scholar]; (f) Wang X-D, Jia W, Gao W-Y, Zhang R, Zhang Y-W, Zhang J, Takaishi Y, Duan H-Q. J. Asian Nat. Prod. Res. 2005;7:755. doi: 10.1080/1028602042000325618. [DOI] [PubMed] [Google Scholar]; (g) Zhang Y, Fan Y, Wang X, Gao W, Duan H. Zhong Cao Yao. 2007;38:493. [Google Scholar]; (h) Wang Y, Huang R, Yang J. Zhong Cao Yao. 2010;41:1252. [Google Scholar]
- (9).(a) Shishido K, Goto K, Tsuda A, Takaishi Y, Shibuya M. J. Chem. Soc., Chem. Commun. 1993:793. For total syntheses of triptoquinones B and C, see. [Google Scholar]; (b) Yamamura I, Fujiwara Y, Yamato T, Irie O, Shishido K. Tetrahedron Lett. 1997;38:4121. [Google Scholar]
- (10).(a) Djerassi C, Sengupta P, Herran J, Walls F. J. Am. Chem. Soc. 1954;76:2966. For the isolation and structure determination of iresin and isoiresin, see. [Google Scholar]; (b) Crabbé P.; Burstein S, Djerassi C. Bull. Soc. Chim. Belg. 1958;67:632. [Google Scholar]; (c) Bratoeff E, Perez-Amador MC, Ramirez E. Phyton. 1996;58:119. [Google Scholar]; (d) Djerassi C, Rittel W, Nussbaum AL, Donovan FW, Herran J. J. Am. Chem. Soc. 1954;76:6410. Structure Elucidation. [Google Scholar]; (e) Djerassi C, Rittel W. J. Am. Chem. Soc. 1957;79:3528. [Google Scholar]; (f) Djerassi C, Donovan FW, Burstein S, Mauli R. J. Am. Chem. Soc. 1958;80:1972. [Google Scholar]; (g) Rossmann MG, Lipscomb WN. J. Am. Chem. Soc. 1958;80:2592. [Google Scholar]; (h) Djerassi C, Burstein S. J. Am. Chem. Soc. 1958;80:2593. [Google Scholar]; (i) Djerassi C, Burstein S. Tetrahedron. 1959;7:37. [Google Scholar]; (j) Rossmann MG, Lipscomb WN. Tetrahedron. 1958;4:275. X-ray structure. [Google Scholar]; (k) Rios MY, Berber LA. Magn. Reson. Chem. 2005;43:339. doi: 10.1002/mrc.1550. NMR Assignments. [DOI] [PubMed] [Google Scholar]
- (11).(a) Pelletier SW, Prabhakar S. J. Am. Chem. Soc. 1968;90:5318. For total syntheses of iresin and isoiresin, see. [Google Scholar]; (b) Wang B-L, Gao H-T, Li W-DZ. J. Org. Chem. 2015;80:5296. doi: 10.1021/acs.joc.5b00365. [DOI] [PubMed] [Google Scholar]
- (12).Taylor RE, Engelhardt FC, Schmitt MJ, Yuan H. J. Am. Chem. Soc. 2001;123:2964. For a related cross-metathesis, see. [Google Scholar]
- (13).Noguchi N, Nakada M. Org. Lett. 2006;8:2039. doi: 10.1021/ol060437x. For intramolecular Sakurai allylation of a 5-membered lactol, see. [DOI] [PubMed] [Google Scholar]
- (14).Chemler SR, Trauner D, Danishefsky SJ. Angew. Chem. Int. Ed. 2001;40:4544. doi: 10.1002/1521-3773(20011217)40:24<4544::aid-anie4544>3.0.co;2-n. For a review on the use of B-alkyl Suzuki cross-couplings in natural product synthesis, see. [DOI] [PubMed] [Google Scholar]
- (15).(a) Wright SW, Hageman DL, McClure LD. J. Org. Chem. 1994;59:6095. For use of KF as a base in Suzuki coupling, see. [Google Scholar]; (b) Wolfe JP, Singer RA, Yang BH, Buchwald SL. J. Am. Chem. Soc. 1999;121:9550. [Google Scholar]
- (16).(a) Hartman C, Meyer V. Chem. Ber. 1893;26:1727. [Google Scholar]; (b) Frigerio M, Santagostino M. Tetrahedron Lett. 1994;35:8019. [Google Scholar]
- (17).(a) Lindgren BO, Nilsson T. Acta Chem. Scand. 1973;27:888. [Google Scholar]; (b) Kraus GA, Taschner MJ. J. Org. Chem. 1980;45:1175. [Google Scholar]; (c) Bal BS, Childers WE, Jr., Pinnick HW. Tetrahedron. 1981;37:2091. [Google Scholar]
- (18).(a) Granger BA, Jewett IT, Butler JD, Martin SF. Tetrahedron. 2014;70:4094. doi: 10.1016/j.tet.2014.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Boger DL, Kim SH, Mori Y, Weng J-H, Rogel O, Castle SL, McAtee JJ. J. Am. Chem. Soc. 2001;123:1862. doi: 10.1021/ja003835i. [DOI] [PubMed] [Google Scholar]
- (19).(a) Harrowven DC, Dainty RF. Tetrahedron Lett. 1997;38:7123. For intramolecular Friedel-Crafts reactions of tethered tetrahydrofurans, see. [Google Scholar]; (b) Harrowven DC, Sibley GEM. Tetrahedron Lett. 1999;40:8299. [Google Scholar]
- (20).Saicic RN, Saicic RN. Tetrahedron. Tetrahedron. 2014;2015;7071:8183, 2777. For a recent review on protecting group-free total synthesis, see. Addition-correction. [Google Scholar]
- (21).(a) Netherton MR, Fu GC. Org. Lett. 2001;3:4295. doi: 10.1021/ol016971g. [DOI] [PubMed] [Google Scholar]; (b) Lou S, Fu GC. Adv. Synth. Catal. 2010;352:2081. doi: 10.1002/adsc.201000267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Yakura T, Omoto M, Yamauchi Y, Tian Y, Ozono A. Tetrahedron. 2010;66:5833. [Google Scholar]
- (23).Hollinshead DM, Howell SC, Ley SV, Mahon M, Ratcliffe NM. J. Chem. Soc. Perkin Trans. I. 1983:1579. For Diels-Alder reaction of the corresponding non-hydroxylated compound, see. [Google Scholar]
- (24).Iwasaki K, Wan KK, Oppedisano A, Crossley SWM, Shenvi RA. J. Am. Chem. Soc. 2014;136:1300. doi: 10.1021/ja412342g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Cherney EC, Green JC, Baran PS. Angew. Chem. Int. Ed. 2013;52:9019. doi: 10.1002/anie.201304609. An elegant solution to this problem is found in Baran’s approach to the ent-kaurane and beyerane diterpenoids. Also, see reference 7b. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.