

Abstract
RATIONALE
N-Alkylation of sulfonylbenzamides was reported recently to cause a dramatic and surprising change in electron ionization mass spectrometry (EIMS), leading to a closed-shell base peak. Only an incomplete, speculative mechanism was available at that time. The fragmentation mechanism is determined in the present work and set in the context of related compounds.
METHODS
Candidate reaction mechanisms were evaluated theoretically using modest density-functional calculations. The fragmentation mechanism with the lowest barriers was identified and one of its implications tested successfully by experimental 18O-isotopic substitution.
RESULTS
The amide oxygen atom attacks the arylsulfonyl group at the ipso position (Smiles-type rearrangement), displacing a molecule of SO2. The resulting carboximidate radical cation has a weak C–O bond that breaks easily. The incipient aryloxyl radical abstracts a proton from the amide nitrogen to form the dominant product ion, but if the molecule is N-alkylated this cannot occur. Instead, the neutral aryloxyl radical is lost and a closed-shell, N-alkyl nitrilium ion is the major product.
CONCLUSIONS
The Smiles-type ion fragmentation mechanism is facile for the title compounds, despite the necessity for carbonyl oxygen to serve as a nucleophile. This rearrangement probably occurs in many of the mass spectra reported for structurally similar compounds, in which the nucleophile may be a thione, arylthio, imine, methylene, or methine moiety.
Electron ionization mass spectrometry (EIMS), often in combination with gas chromatography (GC), remains the most popular method for qualitative and quantitative analysis of a wide range of organic compounds. As part of the augmentation of the NIST/NIH/EPA library of mass spectra and GC retention indices,[1] GC/EIMS data for new chemical compounds are measured routinely. In many cases, important analytes are not amenable to GC analysis because of poor volatility or thermal instability. In such cases, chemical derivatization is often effective for improving the thermophysical properties and achieving successful analyses.[2] During the technical review of such series of related spectra, new chemistry is sometimes encountered.
One example was reported recently, in which alkylation gave an unexpected and puzzling result.[3] When the amidic nitrogen is not alkylated, the dominant base peak in the mass spectra of simple sulfabenzamide derivatives is the corresponding phenol radical cation,[3] as expected from established fragmentation rules for sulfones. In particular, Meyerson and co-workers[4] described a sulfone-sulfinate rearrangement giving rise to arylsulfoxyl and aryloxyl ions. When followed by a hydrogen migration, this corresponds to the mechanism shown in Scheme 1 for R1 = H. However, amidic N-alkylation (R1 = alkyl) suppresses the phenol radical cation completely. Instead, the base peak in the mass spectrum is a closed-shell alkyl benzonitrilium ion.[3] This indicates that the amidic oxygen atom is migrating.
Scheme 1.
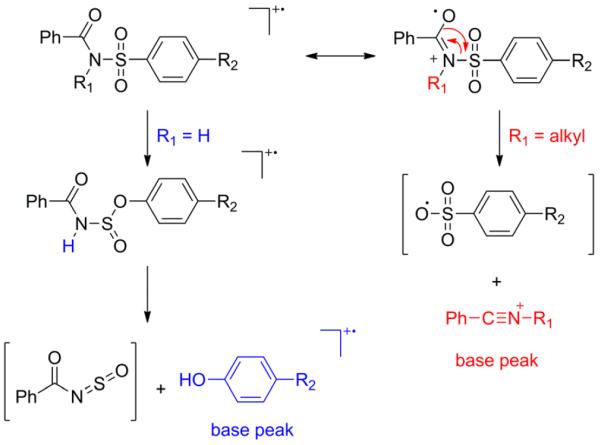
Dominant fragment ions and originally suggested mechanism.
Although migration of hydrogen atoms,[5,6] such as the McLafferty rearrangement,[7] is well known in mass spectrometry, migration of a carbon-bound oxygen atom is rare. Clark and co-workers[8] reported an example in which closed-shell N-methylnitrilium ions are formed from perfluoroalkanoylated methamphetamines. The four-centered mechanism shown in Scheme 1 for R1 = alkyl is based upon the mechanism suggested by Clark et al. for their amides.[8,9]
Both branches of Scheme 1 are consistent with published mechanisms, but it is not clear why the substituent (R1) should affect the outcome so strongly. A different mechanism, presented below in detail, provides a satisfactory explanation. The initial step is a Smiles-type rearrangement.[10] Smiles rearrangements typically require a good nucleophile and electron-withdrawing groups.[11] In contrast, a carbonyl oxygen serves here as the nucleophile, and the p-amino group is electron-donating.
TECHNICAL DETAILS
Materialsa
N-(4-Aminophenylsulfonyl) benzamide (sulfabenzamide), 4-methylphenylsulfonamide, 4-acetylaminophenylsulfonamide and benzoyl chloride were obtained commercially (Sigma-Aldrich, St. Louis, MO, USA) in stated purity >99%. 18O-Benzoyl chloride (70% isotopic enrichment) was also obtained commercially (Icon Isotopes, Summit, NJ, USA).
Synthesis
Benzoyl chloride (or 18O-benzoyl chloride) (0.03 mL) was added to a solution of 10 mg of 4-methylphenylsulfonamide in 0.25 mL pyridine and 1 mL triethylamine and the mixture held for 20 min at 25 °C. Reaction products were verified by GC/EIMS analysis. The same procedure was used for the reaction of 4-acetylaminophenylsulfonamide with benzoyl chloride (or 18O-benzoyl chloride).
Instrumentation
EI mass spectra were recorded on GC/MS systems with quadrupole (Agilent 6890/5973: Agilent Technologies, Santa Clara, CA, USA; ionization energy 70 eV and ion source temperature 230 °C) and magnetic sector analyzers (Finnigan MAT95XL: Thermo Scientific, Bremen, Germany; ionization energy 70 eV and temperature of ionization chamber 220 °C). Samples were introduced via GC inlet (injection temperature 270 °C). Separation was achieved on a fused quartz capillary column (30 m, 0.25 mm i.d.; non-polar stationary liquid phase: polydimethylsiloxane + 5% phenyl groups) with a temperature ramp from 60 °C to 270 °C at 5 °C/min.
Theoretical methods
Four plausible reaction mechanisms were conjectured for the N-methylated derivative, based upon chemical intuition or by using an isopotential searching algorithm[12] on a semiempirical PM6[13] potential energy surface. The corresponding thermochemistry and reaction barriers were estimated using the B3LYP hybrid density functional[14] with standard 6-31G (d) basis sets. Vibrational zero-point energy was scaled by an empirical factor of 0.9757.[15] Hessian analysis characterized each optimized structure as either a minimum or a transition structure. In the latter case, intrinsic reaction coordinate (IRC) calculations[16] verified that the transition structure mediates the reaction of interest. In the most important part of the potential energy diagram, higher-level fc-CCSD(T)/6-311G(d,p)//fc-MP2/6-311G(d,p) calculations were used to verify the results. All calculations were spin-unrestricted and were performed using the Gaussian 03 software package.[17]
RESULTS AND DISCUSSION
Computations
Quantum chemistry calculations were performed for four candidate mechanisms for the rearrangement of the N-methyl derivative (R1 = CH3, R2 = NH2): (1) attack of amide oxygen at the ortho position of the aryl ring; (2) attack of amide oxygen at the ipso position; (3) attack of amide nitrogen at the ipso position; (4) migration of the sulfonyl group to the amide oxygen. Routes (3) and (4) both appear to require passing through an arylsufonate radical intermediate, which is about 169 kJ/mol less stable than the initial radical cation. The highest-energy intermediate for route 1 is only ~85 kJ/mol above the precursor ion, but the barrier for internal H-atom migration is high, lying 187 kJ/mol above that intermediate. Route (2) is clearly the best; all intermediates are more stable than the precursor ion and the products, and the highest barrier lies only ~40 kJ/mol above the precursor ion. This mechanism is illustrated in Fig. 1(b).
Figure 1.

Quantum chemistry calculations of (a) N-[(4-aminophenyl)sulfonyl] benzamide (i.e., sulfabenzamide; R1 = H, R2 = NH2) and (b) its methylated derivative (R1 = CH3). The observed pathways are marked in red.
In more detail, ipso attack of the amide oxygen upon the aryl sulfonyl moiety is favored by an unusually small energy barrier (about 57 kJ/mol when R1 = H and 40 kJ/mol when R1 = CH3). Sulfur dioxide is then expelled, which is entropically favorable, producing a carboximidate intermediate (3a and 3b in Fig. 1). In the N-methyl case (Fig. 1(b)), the carboximidate crosses a barrier to C–O bond cleavage forming a non-bonded complex between a phenoxyl radical and N-methyl benzonitrilium cation (the observed base peak; see Scheme 1). A larger barrier, for methyl-group transfer, must be crossed subsequently to reach the thermodynamically favored products (an anisole radical cation and neutral benzonitrile), which are not observed experimentally. Although this barrier is only slightly higher than the observed products, it is entropically unfavorable and will have a much smaller kinetic prefactor than simple dissociation of a non-bonded complex. Higher-level CCSD(T) calculations, described above, confirm these important relative energies with only small differences: the barrier between ‘complex’ and ‘complex_alt’ is larger by 1 kJ/mol and the dissociation energy for ‘complex’ is smaller by 4 kJ/mol. Because of the barrier, only the kinetic product is observed.
In the N-H case (Fig. 1(a)), the carboximidate intermediate is followed by a barrier for trans-cis isomerization (i.e., rotation of the N–H bond) and another barrier for C–O bond cleavage accompanied by H-atom transfer. This produces a non-bonded complex that dissociates to the thermodynamically favored products: a phenol radical cation (the observed base peak; see Scheme 1) and neutral benzonitrile.
Thus, the same mechanism explains the base peak in all situations. Following initial ipso attack, when R1 = H, the thermodynamically favored products are formed. When R1 = alkyl, there is a barrier to alkyl transfer, leading to the kinetically favored products. This is summarized in Scheme 2.
Scheme 2.
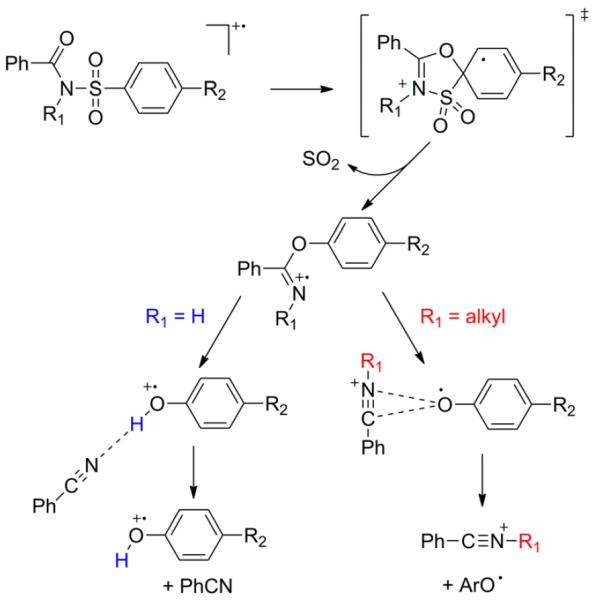
Currently supported mechanism.
Isotope labeling
The original mechanism (Scheme 1) predicts that the oxygen atom in the phenol radical cation (when R1 = H) derives from the sulfonyl group, while the new mechanism (Scheme 2) predicts that it derives from the carbonyl group. To distinguish these alternatives, we prepared two compounds (R1 = H with 18 R2 = NHAc; R1 = H with R2 = CH3) with 18O-labeled carbonyl groups and compared their mass spectra with those of the unlabeled analogs. The spectra are shown in Fig. 2 for the second pair of isotopologs (R1 = H with R2 = CH3). For the unlabeled compound, the base peak, m/z 108, corresponds to the cresol radical cation as indicated in Scheme 1. (The major peak at m/z 105 corresponds to the benzoyl cation, a common fragment whenever a benzoyl moiety is present.) In the 18O-labeled compound, the base peak is shifted to m/z 110. A smaller peak at m/z 108 remains because the 18O enrichment is only 70%. Spectra for R2 = NHAc are analogous and are shown in the Supporting Information. These results clearly refute the original mechanism and support the new mechanism.
Figure 2.

EI mass spectra of (a) N-benzoyl-p-toluenesulfonamide and (b) its 18O analog.
Precedent
After identifying the first step as a Smiles-type rearrangement, we were able to locate earlier mass-spectrometric examples of rearrangements analogous to that shown in Fig. 1(a). The earliest examples were reported in 1963 by Spiteller and Kaschnitz for a variety of sulfonamides.[18] No mechanism was proposed at that time. Later, in a careful study of tolbutamide (N-butyl-N’-tosylurea), Grostic et al. discovered that the urea oxygen and, to a smaller extent, the proximal nitrogen can attack the ipso position to displace SO2.[19] The former, dominant route is the Smiles-type rearrangement.[10]
Brown and Djerassi speculated that this rearrangement is not restricted to α-carbonyl sulfonamides.[20] This was soon confirmed by the discovery of the Smiles-type rearrangement in the mass spectra of several thiourea derivatives.[21] In these compounds, the sulfur atom of the thiocarbonyl group acts as the nucleophile.
An interesting extension to N-arylsulfonyl carbamates was reported by Daly and Heurtevant.[22] Phenol radical cations were abundant, as expected from Scheme 1. However, they were suppressed completely when a tert-butyl group was present on the carbamate oxygen; elimination of CO2 became easier. N-Methylation restored the Smiles-type chemistry, indicating that its barrier was reduced. This is consistent with the present calculations (Fig. 1), which predict that the barrier becomes about 17 kJ/mol smaller upon N-methylation.
Despite N-methylation, the tert-butyl carbamates do not follow the variation shown in Fig. 1(b), in which a closed-shell, N-alkylated ion is produced.[22] This variation was first reported by Sabih as a minor peak in the mass spectrum of N-methyl tolbutamide.[23] It was a major peak in the subsequent, extensive study by Braselton et al., who examined several methylated sulfonylureas. Closed-shell methyl cyanamide ions, analogous to the N-methyl nitrilium ions in the present study, were observed in all cases.[24]
In solution-phase chemistry, a close analogy to the right branch of Scheme 2 was reported by Huber and Bartsch, who found that acyl chlorides can be converted into nitriles by an anionic Smiles rearrangement of corresponding deprotonated N-acyl arylsulfonamides.[25] As in the present study, the acyl oxygen atom serves as the nucleophile. Besides the desired nitrile, the co-products are SO2 and a substituted phenoxide anion.
The present compounds are radical cations, in contrast to the closed-shell anions typical in solution.[26] Uncharged radicals are also known to undergo Smiles-type rearrangements. For example, primary alkyl[27] and ketomethyl[28] radicals may serve as the nucleophile.
Substituent effects
The conventional Smiles rearrangement is facilitated by electron-withdrawing substituents on the aryl ring.[29] Reaction barrier heights were computed for a variety of N-methyl arylsulfonamides, to learn whether gas-phase radical cations behave unconventionally. The resulting Hammett-type plot is shown in Fig. 3, where we have used σp+ parameters from the review by Hansch et al.[30] The pattern is conventional; electron-donating substituents increase the barrier, with the highest barrier for R2 = NH2 (sulfabenzamide itself). There is little variation among the electron-withdrawing substituents, except that the barrier is anomalously high for R2 = NO2. Numerical results are tabulated in the Supporting Information.
Figure 3.

Computed Smiles barrier heights (ΔH0‡) for radical cations of N-methyl arylsulfonamides bearing various para substituents (R2).
Non-oxygen analogs
For comparison among analogs, Smiles-type energy barriers were computed for several compounds and are collected in Table 1. (For the notation for the analogs of N-acyl sulfonamides, as used in Table 1, see Scheme 3.) As mentioned above, sulfur can serve instead of oxygen as the nucleophile in Scheme 2,[21] although it has a slightly higher barrier (compare rows 5 and 6 of Table 1). Additional analogs were discovered by Thyagarajan and coworkers, particularly when the nucleophilic atom is the sulfur of an aryl thioether.[31,32] Earlier examples,[33,34] in which the attacking group is ≡CH or = CH2, were believed to proceed through a [2 + 2]-cycloaddition reaction, but the barriers (Table 1) are low enough for a Smiles-type mechanism to be plausible. Nitrogen can apparently serve as the nucleophile in the elimination of SO2 from the radical cation of sulfaguanidine.[18,35] However, the nitrile analog (row 12 of Table 1) of phenylpropargylsulfone (row 11) does not appear to undergo the Smiles-type rearrangement.
Table 1.
Smiles-type barrier heights (kJ/mol) for arylsulfonamides and analogs
| Row | R2 | X | Nu | R3 | Δ H 0 ‡ | EIMS ref. |
|---|---|---|---|---|---|---|
| 1 | NH2 | NH | O | Ph | 57 | 3 |
| 2 | NH2 | NMe | O | Ph | 40 | 3 |
| 3 | NH2 | NH | O | Me | 67 | 18,35 |
| 4 | Me | NH | O | OEt | 30 | 22 |
| 5 | Me | NH | O | NHBu | 17 | 19 |
| 6 | H | NH | S | NHMe | 37 | 21 |
| 7 | NH2 | NH | NH | NH2 | 11 | 18,35 |
| 8 | Me | CH2 | O | Me | 61 | 35 |
| 9 | H | CH2 | CH2 | H | 51 | 34 |
| 10 | NH2 | CH2 | CH2 | H | 61 | 34 |
| 11 | H | CH2 | ≡CH | None | 40 | 33 |
| 12 | H | CH2 | ≡N | None | 86 | 35 |
| 13 | Me | CH+ | S-p-tolyl | Me | 109 | 31 |
| 14 | H | CH2 | CH2 | SPh | 75 | 32 |
| 15 | H | CH2 | -SPh | =CH2 | 124 | 32 |
Scheme 3.
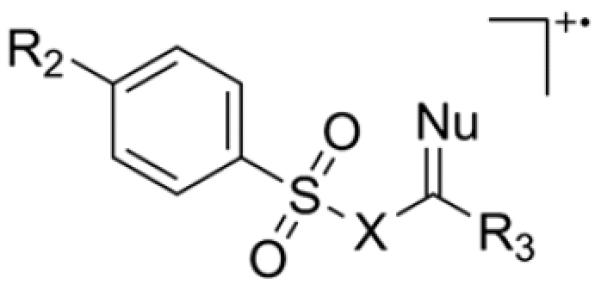
Notation for analogs of N-acyl sulfonamides, as used in Table 1.
Rows 14 and 15 in Table 1 are for the same compound, 3-phenylsulfonyl-2-phenylthiopropene, but considering either the terminal = CH2 group or the thioether sulfur atom as the nucleophile. The mass spectrum shows a copious diphenyl sulfide ion, corresponding to loss of SO2 + C3H4.[32] This suggests that phenylthio attack (row 15 in Table 1) is more important than carbon attack (row 14), which is contrary to the Smiles barrier heights. Further mechanistic study would be necessary to reconcile this conflict.
CONCLUSIONS
Smiles-type rearrangements appear to be common in the cation radicals of typical sulfa drugs. Moreover, a variety of functional groups can serve as the nucleophile. The rearrangement is facilitated by electron-withdrawing substituents on the aryl ring, as it is in conventional solution-phase chemistry. The final products can be remarkably sensitive to structural details, such as N-methylation, because of reactions subsequent to the Smiles rearrangement.
Supplementary Material
Acknowledgements
We are grateful to Dr. Anzor I. Mikaia for guidance and encouragement during these investigations.
Footnotes
Additional supporting information may be found in the online version of this article at the publisher’s website.
Certain commercial materials and equipment are identified in this paper in order to specify procedures completely. In no case does such identification imply recommendation or endorsement by the National Institute of Standards and Technology, nor does it imply that the material or equipment identified is necessarily the best available for the purpose.
REFERENCES
- [1].Ausloos P, Clifton CL, Lias SG, Mikaya AI, Stein SE, Tchekhovskoi DV, Sparkman OD, Zaikin V, Zhu DJ. NIST Standard Reference Database Number 11. National Institute of Standards and Technology; Gaithersburg, MD: The critical evaluation of a comprehensive mass spectral library. [DOI] [PubMed] [Google Scholar]; J. Am. Soc. Mass Spectrom. 1999;10:287. [Google Scholar]
- [2].Zaikin V, Halket J. A Handbook of Derivatives for Mass Spectrometry. IM Publications; Chichester: 2009. [Google Scholar]
- [3].Todua NG, Tretyakov KV, Borisov RS, Zhilyaev DI, Zaikin VG, Stein SE, Mikaia AI. Electron ionization mass spectra of alkylated sulfabenzamides. Rapid Commun. Mass Spectrom. 2011;25:750. doi: 10.1002/rcm.4918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Meyerson S, Drews H, Fields EK. Mass spectra of diaryl sulfones. Anal. Chem. 1964;36:1294. [Google Scholar]
- [5].Happ GP, Stewart DW. Rearrangement peaks in the mass spectra of certain aliphatic acids. J. Am. Chem. Soc. 1952;74:4404. [Google Scholar]
- [6].Nicholson AJC. The protochemical decomposition of the aliphatic methyl ketones. Trans. Faraday Soc. 1954;50:1067. [Google Scholar]
- [7].McLafferty FW. Mass spectrometric analysis. Molecular rearrangements. Anal. Chem. 1959;31:82. [Google Scholar]
- [8].Clark CR, DeRuiter J, Valaer AK, Noggle FT. GC-MS analysis of acylated derivatives of methamphetamine and regioisomeric phenethylamines. J. Chromatogr. Sci. 1995;33:485. [Google Scholar]
- [9].Awad T, Maher HM, DeRuiter J, Clark CR. Studies on the formation of N-methylperfluoroalkylnitrile cations from perfluoroacylphenethylamines in electron ionization mass spectrometry: unique marker ion fragments in methamphetamine analysis. Eur. J. Mass Spectrom. 2012;18:287. doi: 10.1255/ejms.1185. [DOI] [PubMed] [Google Scholar]
- [10].Warren LA, Smiles S. Dehydro-2-naphtholsulphone. J. Chem. Soc. 1930:1327. [Google Scholar]
- [11].Eichinger PCH, Bowie JH, Hayes RN. The gas-phase Smiles rearrangement: a heavy atom labeling study. J. Am. Chem. Soc. 1989;111:4224. [Google Scholar]
- [12].Irikura KK, Johnson RD., III Predicting unexpected chemical reactions by isopotential searching. J. Phys. Chem. A. 2000;104:2191. [Google Scholar]
- [13].Stewart JJP. Optimization of parameters for semiempirical methods V: Modification of NDDO approximations and application to 70 elements. J. Mol. Model. 2007;13:1173. doi: 10.1007/s00894-007-0233-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Lee C, Yang W, Parr RG. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B. 1988;37:785. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]; Becke AD. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993;98:5648. [Google Scholar]; Stephens PJ, Devlin FJ, Chabalowski CF, Frisch MJ. Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. J. Phys. Chem. 1994;98:11623. [Google Scholar]
- [15].Irikura KK, Johnson RD, III, Kacker RN, Kessel RJ. Uncertainties in scaling factors for ab initio vibrational zero-point energies. Chem. Phys. 2009;130:1. doi: 10.1063/1.3086931. [DOI] [PubMed] [Google Scholar]; Irikura KK, Johnson RD, III, Kacker RN, Kessel RJ. Erratum. Chem. Phys. 2009;131:1. doi: 10.1063/1.3086931. [DOI] [PubMed] [Google Scholar]
- [16].Gonzalez C, Schlegel HB. Reaction-path following in mass-weighted internal coordinates. J. Phys. Chem. 1990;94:5523. [Google Scholar]
- [17].Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA, Jr, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA. Gaussian 03. version E.01 Gaussian, Inc.; Wallingford, CT: 2004. [Google Scholar]
- [18].Spiteller G, Kaschnitz R. Anwendung der Massenspektrometrie zur Untersuchung von Arzneimitteln, 1. Mitt.: Sulfonamide. Monatsh. Chem. 1963;94:964. [Google Scholar]
- [19].Grostic MF, Wnuk RJ, MacKellar FA. The mass spectrometry of sulfonylureas. I. Mechanisms for the loss of sulfur dioxide. J. Am. Chem. Soc. 1966;88:4664. doi: 10.1021/ja00972a026. [DOI] [PubMed] [Google Scholar]
- [20].Brown P, Djerassi C. Electron-impact induced rearrangement reactions of organic molecules. Angew. Chem. Int. Ed. 1967;6:477. [Google Scholar]
- [21].Duffield AM, Djerassi C, Neidlein R, Heukelbach E. Mass spectrometry in structureal and stereochemical problems – CLXXIV: Electron-impact induced fragmentation of some aryl- and alkylsulfonylthioureas. Org. Mass Spectrom. 1969;2:641. [Google Scholar]
- [22].Daly WH, Heurtevant CW. The mass spectrometry of O-alkyl-N-arylsulfonyl carbamates. Org. Mass Spectrom. 1970;4:165. [Google Scholar]
- [23].Sabih K. High resolution mass spectrometric analysis of methylated tolbutomide derivatives. Biomed. Mass Spectrom. 1974;1:163. doi: 10.1002/bms.1200010303. [DOI] [PubMed] [Google Scholar]
- [24].Braselton WE, Jr., Bransome ED, Jr., Ashline HC, Stewart JT, Honigberg IL. Gas chromatographic and mass spectral properties of sulfonylurea N-methyl-N’-perfluoroacyl derivatives. Anal. Chem. 1976;48:1386. doi: 10.1021/ac50003a030. [DOI] [PubMed] [Google Scholar]
- [25].Huber VJ, Bartsch RA. Preparation of nitriles from carboxylic acids: A new, synthetically useful example of the Smiles rearrangement. Tetrahedron. 1998;54:9281. [Google Scholar]
- [26].Bowden K, Williams PR. Intramolecular catalysis. Part 7. The Smiles rearrangement of substituted 2-hydroxy-2′-nitro- and -2′,4′-dinitro-diphenyl sulphones, as well as 2-amino-2′,4′-dinitrodiphenyl sulphide, 2-[(2-aminophenyl) thio]-3-nitropyridine and 2-hydroxy-2′,4′-dinitrodiphenyl ether. J. Chem. Soc.-Perkin Trans. 1991;2:215. [Google Scholar]
- [27].Tada M, Shijima H, Nakamura M. Smiles-type free radical rearrangement of aromatic sulfonates and sulfonamides: syntheses of arylethanols and arylethylamines. Org. Biomol. Chem. 2003;1:2499. doi: 10.1039/b303728b. [DOI] [PubMed] [Google Scholar]
- [28].Bacqué E, El Qacemi M, Zard SZ. An Unusual Radical Smiles Rearrangement. Org. Lett. 2005;7:3817. doi: 10.1021/ol051568l. [DOI] [PubMed] [Google Scholar]
- [29].Wang TF, Nibbering NMM, Bowie JH. The gas phase Smiles rearrangement of anions PhO(CH2)nO− (n = 2–4). A joint theoretical and experimental approach. Org. Biomol. Chem. 2010;8:4080. doi: 10.1039/c0ob00064g. [DOI] [PubMed] [Google Scholar]
- [30].Hansch C, Leo A, Taft RW. A survey of Hammett substituent constants and resonance and field parameters. Chem. Rev. 1991;91:165. [Google Scholar]
- [31].Hill CJ, Thyagarajan BS, Bates DK, Spangler RJ. Mass spectral rearrangements of aryl propenyl sulfones - electron-impact induced smiles type rearragement. Org. Mass Spectrom. 1977;12:379. [Google Scholar]
- [32].Glaspy PE, Hancock RA, Thyagarajan BS. Mass spectral rearrangements of 3-arylsulphonyl-2-arylthiopropenes and N-(4′-arylsulphonyl-2′-butynyl)-N-(4′′-arylthio-2′′-butynyl) anilines. Org. Mass Spectrom. 1985;20:281. [Google Scholar]
- [33].Bates DK, Thyagarajan BS. A novel elimination of sulfur dioxide from aryl propargyl sulfones under electron impact. Int. J. Sulfur Chem. 1973;8:57. [Google Scholar]
- [34].Thyagarajan BS, Majumdar KC, Bates DK. Electron-impact induced eliminations. 3. Elimination of SO2 vs remote group-interactions in aryl allyl sulfones, 1,4-bis-(arylsulfonyl)-2-butenes, 1-aryloxy-4-arylsulfonyl-2-butenes, and 4-(phenylsulfonyl)methyl-2H-1-benzopyrans. Phosphorus Sulfur Relat. Elem. 1976;1:67. [Google Scholar]
- [35].Linstrom PJ, Mallard WG. NIST Standard Reference Database Number 69. National Institute of Standards and Technology; Gaithersburg, Maryland: 2003. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.