

Abstract
Utilizing a mouse model of ‘active’ developmental cigarette smoke exposure (CSE) [gestational day (GD) 1 through postnatal day (PD) 21] characterized by offspring low birth weight, the impact of developmental CSE on liver proteome profiles of adult offspring at 6 months of age was determined. Liver tissue was collected from Sham- and CSE-offspring for 2D-SDS-PAGE based proteome analysis with Partial Least Squares-Discriminant Analysis (PLS-DA). A similar study conducted at the cessation of exposure to cigarette smoke documented decreased gluconeogenesis coupled to oxidative stress in weanling offspring. In the current study, exposure throughout development to cigarette smoke resulted in impaired hepatic carbohydrate metabolism, decreased serum glucose levels, and increased gluconeogenic regulatory enzyme abundances during the fed-state coupled to decreased expression of SIRT1 as well as increased PEPCK and PGC1α expression. Together these findings indicate inappropriately timed gluconeogenesis that may reflect impaired insulin signaling in mature offspring exposed to ‘active’ developmental CSE.
Keywords: Cigarette smoke, liver, proteome, murine, developmental, tobacco
1 INTRODUCTION
Maternal environmental exposures, nutrition, and lifestyle play critical roles in fetal growth and development [1–4]. According to the Center for Disease Control and Prevention, approximately 20% of women smoke cigarettes at some time during pregnancy [5] though both epidemiological evidence and animal studies suggest that consumption of tobacco products during pregnancy is hazardous to the fetus [5–15]. Maternal cigarette smoke exposure during gestation is associated with a host of adverse reproductive outcomes, congenital anomalies, and a high incidence of intrauterine growth restriction and resulting infant low birth weight [5, 11, 12, 14–16]. Moreover, children manifest adverse outcomes of fetal exposure to tobacco smoke that persist into childhood such as an increased incidence of respiratory infections and asthma as well as a propensity towards infant/childhood behavioral and cognitive deficits [5, 11, 12, 14–16]. Interestingly, more recent studies have established compelling linkages between pre-/perinatal exposures to cigarette smoke and an increased risk of offspring obesity and metabolic disease [17–25]. While the ability of prenatal and early-life environmental exposures such as cigarette smoke to elicit developmental programming and long-lived alterations in adult health/disease risk has been well documented [1, 4, 26–31], the precise biological mechanisms by which such exposures dysregulate the development, function, and adaptability of the organism’s key metabolic regulatory tissues/organs are still largely unknown.
Cigarette smoke contains a multitude of combustion gases, heavy metals, flavorings and other additives including addictive substances such as nicotine [32–35]; The toxins in cigarette smoke are generally transferred across the placenta to the embryo/fetus [36], exposing rapidly proliferating and differentiating cells/tissues to a mixture of over 8000 toxic substances [37]. Although a primary site of exogenous compound detoxification in adult humans [38, 39], the liver has limited capacity to detoxify such compounds in the embryo/fetus [40–42] and, as such, the developing organ is functionally challenged, in a premature state, by this myriad of toxins. The diversity of potentially toxic substances in cigarette smoke, along with multiplicity of metabolic organs that are targeted by exposure, makes it difficult to elucidate the mechanisms through which developmental exposure to cigarette smoke elicits long-lived metabolic consequences in exposed offspring. Key insights may be found by examining alterations in genomic/epigenomic transcriptomic, proteomic, and metabolomic signatures of the primary metabolic tissues between exposed and non-exposed offspring. Such strategies have been utilized successfully to delineate the mechanistic underpinnings of ‘developmental reprogramming’ and long-lived metabolic consequences of in utero and early-life xenobiotic exposures such as cigarette smoke [43–52].
In order to elucidate the cellular and molecular mechanisms underlying the linkage between developmental exposure to cigarette smoke and an increased risk of offspring metabolic disease, we have utilized a well-characterized mouse inhalation exposure model which simulates ‘active’ smoke exposure spanning both fetal and early neonatal developmental periods (GD1-PD21) and examined the impact of such exposure on the proteome of liver, kidney and hippocampus at varied times during the offspring’s lifespan. The present report details the impact of developmental cigarette smoke exposure on hepatic proteome profiles of offspring at 6 months of age – 5 months past cessation of their exposure – from littermates of pups from the same litters utilized in our prior study of developmental (GD1-PD21) cigarette smoke exposure. Parallel studies from our laboratory concerning the impact of such developmental CSE on liver, kidney and hippocampus proteome profiles at weaning (PD21) [53–55] and at adulthood [56, 57] documented an impact of exposure on tissue metabolic activity. We previously reported that weanling (PD21) offspring who were developmentally exposed to cigarette smoke exhibited hepatic oxidative stress, impaired gluconeogenesis, altered lipid metabolism, impaired small molecule and amino acid metabolism, and impaired cellular morphology networks [54]. In the present report, we document sustained deficits in offspring growth, suppressed serum blood glucose levels, as well as disruption of basal gluconeogenesis in the fed state demonstrating a continued impact of developmental CSE on metabolic pathway function in adult offspring aged 6 months well past the cessation of exposure.
2. MATERIALS AND METHODS
2.1. Animal Exposures
Adult C57BL/6J mice were purchased from Jackson Labs (Bar Harbor, ME). Animals were housed and maintained in the University of Louisville Research Resources Center, an Association for Assessment and Accreditation of Laboratory Animal Care accredited facility. Animals were maintained in a controlled temperature/humidity environment with a 12 hour light/dark cycle and free access to Purina LabDiet 5015 and water throughout the duration of the experiment (both prenatal and postnatal exposure periods as well as following cessation of exposure to cigarette smoke. Female mice were age-matched at the outset of the study and timed pregnancies were obtained by overnight mating of a single mature male with two nulliparous females. The presence of a vaginal plug was considered evidence of mating and the time designated as gestational day 1 (GD 1). Pregnant mice were weighed and randomly assigned to either the Sham exposure (Sham, n=9) or Cigarette Smoke Exposure (CSE; n=9) groups. Animals were exposed from GD1, throughout the entirety of gestation; following parturition maternal animals were exposed with offspring until postnatal day 21 (PD21).
Animals were exposed to ambient air or cigarette smoke for 6 hours per day, 7 days per week, from GD1-PD21. A Teague TE-10C whole body smoke inhalation exposure system (Teague Enterprises; Davis, CA)[58] was utilized to generate and deliver mixed mainstream/sidestream cigarette smoke at the rate of 40 cigarettes smoked per 6 hour period. Cigarette smoke was generated from Philip Morris Marlboro Red brand cigarettes™ (Philip Morris; Richmond, VA; 15mg of tar/cigarette; 1.1mg nicotine/cigarette; additives), selected since it represents the most popular brand of cigarettes consumed among 18–25 year olds - the age group containing the majority of maternal smokers [59–62]. Cigarettes were smoked using the standard Federal Trade Commission method: a two second, 35 cm3 puff, once a minute for a total of 9 min [58]. For the duration of the exposure period, the dam/litter combinations were individually housed in the cages utilized for exposures. For quality control purposes, dual exposure chambers (one receiving cigarette smoke and one receiving ambient air) were characterized twice during each daily exposure session for: total suspended particulates (TSP), temperature, carbon monoxide levels, and humidity (Table 1) [54].
Table 1. Animal exposure conditions and outcomes.
Mean Carbon Monoxide and Total Suspended Particulates (TSP) were calculated from measures taken twice daily in the inhalation exposure chambers from GD1-PD21. Plasma cotinine levels were determined by tail vein blood sampling of the dam and offspring within 1 hour of cessation of exposure on PD21 (*p < 0.05).
| Outcome/Condition | SHAM | CSE |
|---|---|---|
| Carbon Monoxide (ppm) | 0 | 138 ± 19.8* |
| TSP (mg/m3) | 0 | 25.4 ± 6.5* |
| Dam Cotinine (ng/mL) | <4 | 89.7 ± 37.3* |
| Pup Cotinine (ng/mL) | <4 | 244.3 ± 132.4* |
Tail blood was collected from representative animals immediately following the 6 hour exposure session at various time points throughout the 6 week exposure regimen for determination of plasma cotinine levels. Cotinine, the principal metabolite of nicotine, is a well-documented marker of ‘active’ tobacco smoking and passive/environmental tobacco smoke exposure [63–66]. Cotinine levels were assayed by electrospray tandem mass spectrometry (ESI-MS/MS) utilizing a direct inject platform (Nanomate) coupled to a 7T LTQ-FT-ICR-MS.
Following discontinuation of cigarette smoke exposure on PD21, offspring were maintained at normal temperature and humidity without exposure to any agent. At 6 months of age, following behavioral and cognitive assessments [67], offspring were euthanized by asphyxiation with carbon dioxide followed by thoracotomy and cardiac puncture. Tissues were harvested and stored at −80°C until analysis. Proteome profiling of the liver and other tissues was conducted on identically exposed littermates at the time of weaning (PD21) [53–55]. The current report of liver proteome profiles from 6 month old animals that were previously developmental exposed to cigarette smoke serves as the lead manuscript for the coordinated three part evaluation of tissue specific proteome profiling studies, and includes the companion studies on kidney and hippocampus proteome profiles [56, 57].
2.2.1. 2D-SDS-PAGE
The liver samples (Sham n=4; CSE n=6) were homogenized in sample preparation buffer [7M urea, 2M thiourea, 40mM dithiothreitol (DTT)]. Protein concentration for each of the samples was determined using the Bradford Assay [68]. Four hundred micrograms of protein in rehydration buffer (8M urea, 2% CHAPS, 2 μl IPG buffer pH 3–10, 2.5 mg/ml DTT, 0.002% bromophenol blue) was applied to IPGphor Drystrips (Nonlinear, 3–11, 180 mm × 3 mm × 0.5 mm, GE Healthcare, Piscataway, NJ). First dimension separation by isoelectric focusing at 22,000 Volt hours (Vhrs) was performed with a hold at 100 Volts until further processing. The IEF strips were stored at −80°C for 1 hr followed by: 1) equilibration for 60 minutes in reducing buffer (6M urea, 75 mM Tris-HCl pH 8.8, 29.3% glycerol, 2% SDS, 0.002% bromophenol blue with 3.5 mg/ml DTT) and 2) equilibration in alkylating buffer (same buffer with 45 mg/ml iodoacetamide instead of DTT) for an additional 30 minutes. Second dimension SDS-PAGE separation (25cm × 20.5cm 15% polyacrylamide gels) was performed overnight (18 hrs; 100V). Protein spots were visualized by Colloidal Coomassie Blue G-250.
2.2.2. Image Analysis
Gels were scanned using an Epson Expression 10000 XL scanner with transparency attachment. Densitometric analysis of gel images was performed with Progenesis SameSpots software (Nonlinear Dynamics; New Castle-on-Tyne, UK). Protein spots were detected automatically and manually adjusted where necessary for accuracy. For each protein spot, the intensity was measured, background was subtracted, and individual spot density was normalized by total pixel density of each gel. Spots with average normalized pixel depth of ≤1000 relative abundance units and non-normalized areas with pixel depth below 100 were removed as noise. The averaged normalized spot abundance was compared between groups to determine fold differences in abundance.
2.2.3. Mass Spectrometry Based Protein Identification
Protein spots were excised and destained with 50% ethanol in 50mM ammonium bicarbonate for a minimum of 5 washes. Excised gel spots were then dehydrated in acetonitrile (ACN), dried, and rehydrated with 10 ng/μl trypsin and 40mM ammonium bicarbonate. Proteins were digested at room temperature for approximately 18 hrs. Peptides were eluted in acidified acetonitrile and stored at −20C until analysis. The mass to charge ratio of peptides was determined by direct inject LTQ/FT-ICR-MS/MS (or HPLC interface on occasion) with collision induced dissociation for structural feature identification. Peptide identification was performed with the Mascot (Matrix Sciences v 2.2.2) search algorithm utilizing the NCBInr (with decoy) database (updated June 1, 2010). Search parameters included: mammalian class, 2 missed cleavages, carbamidomethyl C variable modification, enzyme trypsin/P, and an allowed peptide charge of 1+, 2+, or 3+. Positive protein identification required a total MOWSE absolute probability entire protein score of ≥100 composed of a minimum of two peptides with individual scores MOWSE absolute probability scores ≥50 [53–55].
2.2.4. Statistical Analysis
Analysis of Variance (ANOVA, two way, p<0.05 as significant) and a series of Partial Least Squares-Discriminant Analysis (PLS-DA) models were utilized to determine the protein spots which differed in intensity and described the differences between the groups. Multiple PLS-DA models were constructed utilizing Variable Importance in Projection (VIP)-ranked protein spots of interest with recursive feature elimination identifying protein spots with VIP≥1.75 as important in defining the separation between groups [69]. Sequential removal of top ranked protein spots was performed until the variance between groups was eliminated. All protein spots included in further analysis were significantly different between groups based on ANOVA (p<0.05).
2.3. Ingenuity® Pathway Analysis
Ingenuity® Pathway Analysis was used to determine families of proteins and metabolic pathways of the proteins that were identified (Ingenuity Systems, 2010). Once the proteins were identified, the GI numbers of the proteins were entered into the IPA algorithm. Networks of interactions between the proteins and their respective genes were generated by the program. Networks of interactions between the proteins and biological pathways were generated with sub-categorization by increased or decreased abundance. For the canonical pathways analysis, the following settings were employed: Benjamin-Hochberg p-value greater than or equal to 1.5, a threshold of 0.5 z-score, and a Benjamin-Hochberg Multiple Testing Correction p-value utilized for scoring. In the associated figure (Figure 5), solid lines indicated a direct interaction while dotted lines indicate an indirect interaction. Geometric shapes identify classes of proteins: phosphatases (triangle), kinases (inverted triangle), enzymes (vertical diamond), transcription regulators (horizontal ellipse), transporters (trapezoid), and other important molecules (circles).
Figure 5. Serum glucose levels in 6 month old offspring that were sham exposed or exposed to cigarette smoke from GD1 through PD21.
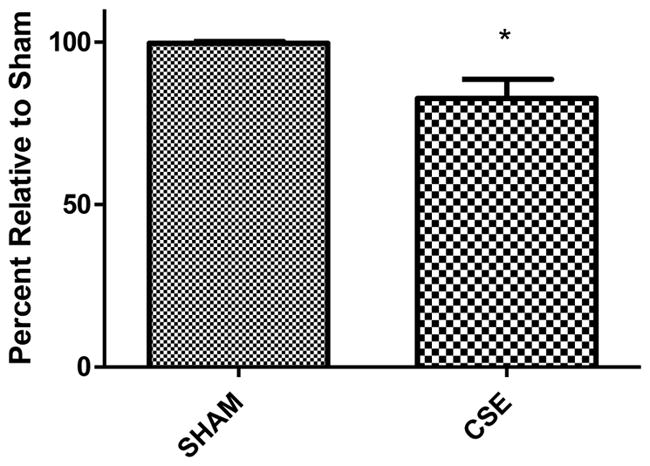
Fed state serum glucose levels, collected at the time of euthanization, are suppressed by 17% in CSE offspring at 6 months of age (*p<0.0001).
2.4. Serum Glucose
Serum glucose levels in offspring in the fed-state were analyzed by a handheld glucometer (OneTouch Ultra, LifeScan Inc.) in representative offspring from 5 litters per group.
2.5. Glutathione (GSH and GSSG) Assay
The ratio of reduced/oxidized glutathione (GSH/GSSG) was utilized as an indicator of oxidative stress. To measure total glutathione (GSH + 2GSSG), GSH standards (or sample homogenate) was combined with 50mM phosphate buffer (pH=7.2) containing 2mM EDTA with the addition of 2μUnits of glutathione reductase, and incubated in the presence of DNTB for 30 minutes in the dark followed by spectral acquisition at 405nm [70, 71]. A separate GSSG standard curve was constructed for GSSG measurement with the modification of a pre-incubation of samples or standards with 100mM 2-vinylpyridine for 60 minutes followed by addition of glutathione reductase and subsequent incubation in the presence of DNTB as described for total glutathione measurements. All glutathione measurements were normalized to total protein as measured by the Bradford assay [68]. The amount of free GSH was calculated from the total glutathione minus the GSSG levels in each sample. The ratio of GSH/GSSG was then calculated.
2.6. Glutathione-S-Transferase (GST) and Glutathione Reductase (GR) Assay
Liver GST activity was measured as an indicator of detoxification activity [72]. The total GST activity of the liver was measured using the enzyme driven conjugation of 1-chloro-2, 4-dinitrobenzene (CDNB) to reduced glutathione (absorbance read at 340 nm each minute for 15 minutes; Cayman Chemical Company). The absorbance per minute was divided by amount of protein (mg) to determine the specific activity for each sample [73]. Glutathione reductase activity was measured spectrophotometrically by mixing an aliquot of tissue homogenate with GSSG and measuring the GST enzyme driven conjugation to 1-chloro-2,4-dinitrobenzene (CDNB) to reduced glutathione (absorbance read at 340 nm each minute for 15 minutes; Cayman Chemical Company) [74].
2.7 Western Blot
Liver protein homogenates from the 2D-SDS-PAGE preparations were mixed 1:1 with Laemmli buffer (0.25M Tris pH 6.8, glycerol, 10% SDS, bromophenol blue trace) then heated at 70° C for 10 minutes. Twenty-five μg total protein was separated by 10% PAGE for 2 hours at a 100V in Tris-glycine run buffer (0.025 M Tris Base, 0.192 M glycine, 0.1% SDS) followed by electrophoretic transfer to PVDF membrane at 90V for 1 hour in transfer buffer (0.025M Tris base, 0.192M glycine, 10% ethanol). Following blocking in 4% non-fat dry milk, the blots were incubated overnight at 4°C with primary antibody diluted 1:500 in non-fat dry milk (SIRT1, sc-19857; PEPCK, sc-271029; PGC1α, sc-13067; Santa Cruz Biotechnology, Dallas, TX). After three washes of 15 minutes in cold PBS-Tween, blots were incubated with secondary antibody complexed to horse radish peroxidase (1:1000, Santa Cruz Biotechnology, Dallas, TX) diluted in non-fat dry milk at room temperature for 1 hour. After three washes in cold PBS-Tween, blots were developed with chemiluminescence substrate and visualized with an ImageQuant LAS 4000 (G.E. Healthcare Life Sciences, Pittsburgh, PA). Following visualization, blots were washed three times with PBS-Tween, incubated in 2M glycine for 30 minutes followed by another three washes in PBS-Tween and subsequent incubation with β-actin primary antibody (1:1000 dilution; sc-81173, Santa Cruz Biotechnology, Dallas TX) and secondary antibody and visualization as described above [75].
3. RESULTS
3.1. Exposure Conditions and Offspring Weights
Mean CO and TSP levels in the cigarette smoke exposure chamber were 138 ± 19.8 ppm and 25.4 ± 6.5 mg/m3, respectively with measures in the Sham group found to be less than the limit of detection for each assay. Cotinine, a metabolite of nicotine, was used as an indicator of cigarette smoke exposure. Cotinine levels were greater than 50ng/mL in the CSE group indicating an ‘active’ exposure model with Sham exposure group cotinine levels below the detection limit of 4 ng/mL (Table 1). Low birth weight was evident in the CSE offspring (~15% decrease relative to Sham) [54] with persistence of decrements in weight throughout weaning and into maturity (maintenance on Purina 5015 diet) [67]. Six month old offspring from the CSE group remained reduced in weight at the time of cognitive and behavioral assessment [67] and at the time of tissue collections for the current study (data not shown).
3.2. Liver Proteome Profiles
As shown in Figure 1, 2D-SDS-PAGE gels of liver proteins from Sham and CSE mice were similar at 6 months of age (5 months since cessation of exposure to cigarette smoke). The proteins on the gels spanned an isoelectric focusing range of 3 to 10, with the acidic proteins on the left and the basic proteins on the right of the gel image and descending molecular weights ranging from ~80 kDa to ~11 kDa. The predominant variance between groups was in the intensity of protein spot abundances rather than the appearance/disappearance of protein spots. The average ± standard deviation of the total spot density of the Sham group equaled 4,814,147,401 ± 277,980,203 while the CSE group equaled 4,827,210,729 ± 282,064,550.
Figure 1. Liver proteome profiles of 6 month old offspring previously developmentally exposed to cigarette smoke.

The proteins with altered abundance that contributed to the separation of the groups within the PLS-DA model and possessed the highest VIP values (≥1.75) are numbered. Numbers in blue represent increased abundance and the numbers in red represent decreased abundance. The gel image is highly contrasted to enable visualization of low abundant protein spots. Refer to Table 2 for protein identifications.
3.3. Partial Least Squares- Discriminant Analysis
Iterative PLS-DA models were generated encompassing spot abundances of all proteins not determined to be noise; sequential removal of top VIP ranked protein spots contributing to the separation of groups followed by re-plotting of group separation until the loss of separation of groups resulted in identification of a set of features that were essential to describing the differences between groups. Sixty protein spots (VIP≥ 1.75, p<0.05) were found to contribute to the separation in proteome profiles between the Sham and CSE groups. When abundances of all protein spots were included (noise excluded), the first latent factor of the PLS-DA model accounted for 87% of the variance between the Sham and CSE groups and the second latent factor accounted for an additional 12% of the variance. As shown in Figure 2, the proteome profiles of the Sham and CSE groups are distinct. The separation between the groups is depicted by graphing latent factors 1 and 2.
Figure 2. The PLS-DA model effectively describes the differences between the liver proteome profiles of Sham and developmental CSE offspring at 6 months of age.

Plotting the first two vectors within the PLS-DA model visualizes the variance in liver proteome profiles between the Sham and CSE groups. Each circle represents a single sample subjected to 2D-SDS-PAGE.
3.4. Proteins Impacted by CSE
Forty-two proteins of interest were identified by proteolytic digestion and ESI-MS/MS. These proteins represented all spots of sufficient intensity for clarity of boundaries and all spots that included p-values within 0.10. Identification of the remaining protein spots was not attempted although though they did not represent background noise. Of the 42 protein spots whose identification was attempted, 36 were identified unambiguously (see Table 2). Proteins that represent the predominant contribution to the spot intensity, as determined by a minimum of 200% of the MOWSE score of the next ranked protein identified, are listed. Proteins with altered abundance in liver of CSE mice when compared to that of Sham-exposed mice were grouped by membership in metabolic networks via IPA analysis. As shown in Figure 3, proteins with altered abundance in the liver of 6 month old mice previously developmentally exposed to cigarette smoke belonged to the Sucrose Degradation, NRF2-mediated Oxidative Stress Response, Glycolysis, Gluconeogenesis, Methionine Degradation, Methylglyoxal Degradation, and Aldosterone signaling pathways.
Table 2. Protein spots with altered abundances that were ranked of greatest import within the PLS-DA model, VIP values (≥1.75; p<0.05).
Numbers in red represent decreased abundance and the numbers in blue represent increased abundance of proteins in 6 month old offspring previously developmentally exposed to cigarette smoke (GD1-PD21), as compared to the Sham group. Corresponds to Figure 1.
| Spot Number | GI Number | Abbreviation1 | Protein Identification | VIP | Percent Change |
|---|---|---|---|---|---|
| 1 | 24691 | PRSS2 | Anionic trypsin-1 | 1.92 | −26 |
| 3 | 1174637 | VCP | Transitional endoplasmic reticulum ATPase | 2.00 | −21 |
| 4 | 341941239 | P4HB | Protein disulfide-isomerase | 2.25 | −19 |
| 5 | 51702252 | HSPD1 | 60 kDa heat shock protein, mitochondrial | 2.13 | −35 |
| 7 | 225913 | Bifunctional ATP-dependent dihydroxyacetone kinase/FAD-AMP lyase (cyclizing) | 1.78 | −37 | |
| 11 | 11669 | ALDH2 | Aldehyde dehydrogenase, mitochondrial | 1.87 | −29 |
| 17 | 15516 | HSP90AB1 | Heat shock protein HSP 90-beta | 2.02 | −32 |
| 24 | 14085 | FAH | Fumarylacetoacetase | 1.92 | −36 |
| 2 | 341942170 | RDX | Radixin | 2.07 | 95 |
| 6 | 16691 | KRT8 | Keratin, type II cytoskeletal 8 | 1.77 | |
| 8 | 14661 | GLUD1 | Glutamate dehydrogenase 1, mitochondrial | 1.98 | 61 |
| 9 | Q03265 | ATP5A1 | ATP synthase subunit alpha, mitochondrial | 1.80 | 21 |
| 10 | 12116 | BHMT | Betaine-homocysteine S-methyltransferase 1 | 1.89 | 47 |
| 12 | 20342 | SELENBP1 | Selenium-binding protein 2 | 1.97 | 80 |
| 13 | 11657 | ALB | Serum albumin | 1.96 | 46 |
| 14 | 29443 | AHCY | Adenosylhomocysteinase | 2.18 | 80 |
| 15 | 52815 | Probable D-lactate dehydrogenase, mitochondrial | 1.88 | 53 | |
| 16 | 11465 | F-actin | Actin, cytoplasmic 2 | 1.84 | 34 |
| 18 | 19733 | RGN | Regucalcin | 1.89 | 34 |
| 19 | 25659 | KHK | Ketohexokinase | 1.99 | 39 |
| 20 | 14121 | FBP1 | Fructose-1,6-bisphosphatase 1 | 2.20 | 43 |
| 21 | 56357 | Isovaleryl-CoA dehydrogenase, mitochondrial | 1.93 | 86 | |
| 22 | 13637776 | ENO1 | Alpha-enolase | 1.78 | 47 |
| 23 | 11846 | Arginase-1 | 1.82 | 51 | |
| 25 | 14555 | Glycerol-3-phosphate dehydrogenase (NAD+), cytoplasmic | 2.01 | 69 | |
| 26 | 107766 | 3-hydroxyanhranilate 3,4-dioxygenase | 1.99 | 41 | |
| 27 | 63938 | 3-hydroxyisobutyrate dehydrogenase, mitochondrial | 2.25 | 39 | |
| 28 | Not identified | 1.81 | 23 | ||
| 29 | 3122065 | Delta(3,5)-Delta(2,4)-dienoyl-CoA isomerase, mitochondrial | 1.95 | 55 | |
| 30 | 107766 | 3-hydroxyanhranilate 3,4-dioxygenase | 1.86 | 42 | |
| 31 | 66302 | Regulator of microtubule dynamics protein 1 | 1.85 | 120 | |
| 32 | 3219774 | PRDX6 | Peroxiredoxin-6 | 1.98 | 30 |
| 33 | 14862 | GSTM5 | Glutathione S-transferase Mu 1 | 2.27 | 15 |
| 34 | 18477 | PRDX1 | Peroxiredoxin-1 | 2.14 | 37 |
| 35 | 11430 | ACOX1 | Peroxisomal acyl-coenzyme A oxidase 1 | 2.26 | 95 |
| 36 | 20042 | 40S ribosomal protein S12 | 1.75 | 49 |
Denotes abbreviation used in Figure 4. VIP (Variable Importance in Projection); GI Number (NCBI protein sequence GenInfo Identifier).
Figure 3. The top seven ranked protein interaction networks and pathways impacted within the liver of 6 month old offspring who were previously developmentally exposed to cigarette smoke.

The distance from the threshold value (vertical orange line) depicts the intensity of change between Sham exposure and CSE groups.
3.4.1. Carbohydrate Metabolism Impacted by CSE
The abundance of two proteins belonging to the Sucrose Degradation pathway (total of nine protein members for a 22% pathway coverage) was altered at in the liver of 6 month old offspring who were developmental exposed to cigarette smoke; dihydroxyacetone kinase 2 (Spot 7; decreased 37%) and ketohexokinase (Spot 19; increased 39%). The conversion of dihyroxyacetone to the phosphorylated form utilizes ATP as a cofactor, while the conversion of D-fructose to D-fructose-1-phosphate generates AMP. The opposing impact may signal a cellular prioritization of ATP utilization for energetic processes. Enolase 1 (Spot 22; increased 47%) is a member of both the Glycolysis/Gluconeogenesis pathways while fructose-1, 6-bisphosphatase 1 (Spot 20; increased 43%) is a key regulatory step of the gluconeogenesis pathway; both of these enzymes were increased in the liver of adult offspring aged six months who were developmentally exposed to cigarette smoke. The Gluconeogenesis pathway has a total of 9 direct and 16 support members (2/9 and 22% or 2/25 and 8% coverage respectively. As shown in Figure 4, the hepatic carbohydrate metabolism pathway was impacted in offspring aged 6 months who had undergone developmental exposure to cigarette smoke, with insulin signaling likely impacted. Serum glucose levels in these same CSE offspring were decreased (~15%; Figure 5). As validation of the purported impact of developmental CSE on hepatic gluconeogenesis at a time 5 months past cessation of exposure, we measured the expression of three metabolic regulatory proteins that influence gluconeogenesis protein expression (Figure 6A–6C). SIRT1 expression was inversely correlated to PEPCK in the current study, similar to the established literature, though the current study did not directly measure SIRT1 enzymatic activity [76]. PGC1α, which enhances gluconeogenic gene expression and is a known a target of SIRT1 deacetylase activity, exhibited increased expression in the current study.
Figure 4. The hepatic carbohydrate metabolism pathway in 6 month old offspring is impacted by previous developmental CSE.

As shown below, insulin signaling is a central node that regulates an abundance of metabolic proteins found to be altered in the liver of offspring developmentally exposed to cigarette smoke. In the associated figure, solid lines indicated a direct interaction while dotted lines indicate an indirect interaction. Geometric shapes identify classes of proteins: phosphatases (triangle), kinases (inverted triangle), enzymes (vertical diamond), transcription regulators (horizontal ellipse), transporters (trapezoid), and other important molecules (circles).
Figure 6.



Figure 6A: Western blot analysis of SIRT1 expression in whole liver homogenates from 6 month old offspring that were sham exposed or exposed to cigarette smoke from GD1 through PD21. Liver homogenates from CSE offspring at age 6 months exhibited a decrease of approximately 10% in expression of the metabolic regulatory protein SIRT1 (*p<0.05; n=3 per group).
Figure 6B: Western blot analysis of PEPCK expression in whole liver homogenates from 6 month old offspring that were sham exposed or exposed to cigarette smoke from GD1 through PD21. Liver homogenates from CSE offspring at age 6 months exhibited an increase of approximately 44% in PEPCK expression in the fed state (*p<0.05; n=4 per group).
Figure 6C: Western blot analysis of PGC1α expression in whole liver homogenates from 6 month old offspring exposed to cigarette smoke from GD1 through PD21. Liver homogenates from CSE offspring at age 6 months exhibited an increase of approximately 86% in PGC1α expression in the fed state (*p<0.05; n=4 per group).
3.4.2. NRF2-mediated Oxidative Stress Response Impacted by CSE
The abundance of four proteins belonging to the NRF2-mediated Oxidative Stress Response pathway (180 members of pathway with total coverage of 2.2%) was altered in the liver of 6 month old adult offspring by developmental CSE: γ-actin (Spot 16; increased 34%), glutathione S-transferase μ (Spot 33; increased 15%; GST), peroxiredoxin 1 (Spot 34; increased 37%; PXR1), and transitional endoplasmic reticulum ATPase (Spot 3; decreased 21%; ER ATPase). The pool of glutathione (reduced, GSH; oxidized, GSSG,; and total, GSH+2GSSG; Figure 7) was decreased by 14% in the liver of adult offspring who were developmentally exposed to cigarette smoke indicating possible suppression of glutathione synthesis though a key marker of oxidative stress, the GSH/GSSG ratio remained unaltered and no impact on GR or GST activity was noted (data not shown)..
Figure 7. Glutathione levels in liver of 6 month old offspring that were sham exposed or exposed to cigarette smoke from GD1 through PD21.

Developmental CSE reduces hepatic glutathione levels at maturity. Hepatic total glutathione (GSH+2GSSG), oxidized glutathione (GSSG), and reduced glutathione (GSH) were reduced at maturity in offspring developmentally exposed to cigarette smoke (n=7 per group, *p<0.05) without an impact on the overall reduced/oxidized glutathione ratio (GSH/GSSG).
3.4.3. Amino Acid Degradation Impacted by CSE
The abundance of betaine-homocysteine S-methyltransferase (Spot 10; increased 47%) and adenosylhomocysteinase (Spot 14; increased 80%), two enzymes involved in methionine degradation (2/32 members, 6.2 % pathway coverage), was increased within the liver of 6 month old adult offspring following developmental CSE. The abundance of 3-hydroxyisobutyrate dehydrogenase (Spot 27; increased 39%; valine degradation), isovaleryl-CoA dehydrogenase (Spot 21; increased 86%; leucine degradation); and arginase (Spot 23; increased 51%; arginine metabolism to urea) also was increased in the liver of the adult offspring who were developmentally exposed to cigarette smoke. These five enzymes participate in amino acid metabolism, including the modulation of branched chain amino acid catabolism in support of gluconeogenesis.
3.4.4. Aldosterone Signaling Impacted by CSE
The abundance of adenosylhomocysteinase (Spot 14; increased 80%), HSP 90 (Spot 17; decreased 32%) and HSP 60 (Spot 5; decreased 35%) were altered in the liver of adult offspring aged 6 months who had undergone developmental exposure to cigarette smoke. Two members of this pathway are involved in the chaperoning of other proteins during cellular stress. Similarly, three proteins with increased abundance from the NRF-2 mediated oxidative stress response pathway were identified as cellular stress response proteins (GST, PXR1, and ER ATPase) with peroxiredoxin 6 (Spot 32, increased 30%) also acting as an antioxidant and cellular stress response protein. Coupled with the state of oxidative stress, heightened hepatic stress likely exists in the 6 month old adult offspring, who were developmentally exposed to cigarette smoke, that would lead to an impaired response to a secondary stressor.
3.4.5. Cytoskeletal Structural Proteins Impacted by CSE
The abundance of radixin (Spot 2, increased 95%), actin 2 (Spot 16, increased 34%), and regulator of microtubule dynamics protein 1 (Spot 31, increased 120%) was increased in the liver of 6 month old adult offspring developmentally exposed to cigarette smoke. Radixin 1 regulates head-to-tail association and plasma membrane binding of actin filaments while regulator of microtubule dynamics protein 1 modulates microtubule assembly. An increase in the levels of these proteins indicates an increased cellular requirement for cytoskeletal stabilization protein expression likely in response to increased oxidative stress.
3.4.6. Lipid Metabolism Proteins Impacted by CSE
The abundance of delta(3,5)-delta(2,4)-dienoyl-CoA isomerase (Spot 29; increased 55%; fatty acid isomerization) and glycerol-3-phosphate dehydrogenase (Spot 25; increased 69%; links carbohydrate and lipid metabolism) was increased in the liver of adult offspring aged 6 months who had undergone developmental exposure to cigarette smoke, possibly signaling an increase in fatty acid oxidation and mitochondrial activity. A trend toward increased serum triglycerides was found (p=0.09; data not shown) in these same CSE offspring.
4. DISCUSSION
The mouse model of developmental CSE (GD1-PD21) that was employed in the present study exhibited low birth weight and sustained suppression of weight at both weaning (PD21; time of cessation of exposure) and at PD180 - the time of tissue collection for the present study [54]. Serum cotinine levels in excess of 50 ng/mL – as utilized in the current study – are typically classified as an ‘active’ smoking exposure paradigm [77]. The sustained decrements in weight of 6 month old adult offspring who were developmentally exposed to ‘active’ cigarette smoke are a feature of the current study and the parallel studies on the hippocampus and kidney proteomes of these animals [56, 57]. As such, the exposure system models low birth weight outcomes in infants whose mothers smoked throughout gestation [54, 67, 78, 79] and mirrors the persistence of weight decrements seen in such exposed individuals through childhood and into adulthood [80–84]. The impact of developmental cigarette smoke exposure likely reflects systemic toxicity that is coordinately manifested within individual tissues as reflected within the proteomics studies reported within this model system and in conjunction with the prior report describing behavioral alterations within these same offspring [53–57, 67].
In a study examining the impact of gestational ‘passive’ cigarette smoke exposure on mouse fetal liver, genotoxic damage (including DNA adduct formation and oxidative nucleotide damage) was observed, along with transcriptomic alterations in xenobiotic metabolic pathways; stress response proteins; protein repair mediators, protein removal and folding effectors; cell cycle regulators; growth factors; and cytoskeletal proteins [38]. We previously reported that juvenile offspring (i.e. at PD21 at weaning) who were developmentally exposed to cigarette smoke in an identical manner to the current study, exhibited alterations in proteomic abundances including antioxidant proteins (mixed impact), glucose metabolic enzymes (suppressed gluconeogenesis), and protein members of metabolic networks including lipid metabolism (mixed impact), small molecule and amino acid metabolism (mixed impact), and cellular morphology networks (inhibited) [54]. In the present report, we document a continued impact of developmental CSE on several of these same metabolic pathways in 6 month old adult offspring, 5 months post-cessation of exposure.
The carbohydrate metabolic pathway was impacted in the liver of 6 month old offspring who previously were developmentally exposed to cigarette smoke with insulin signaling likely impacted. In the current study, proteins enolase 1 (gluconeogenesis/glycolysis pathway) and fructose-1, 6-bisphosphatase (gluconeogenesis only) were increased which points to a dysregulation of glucose metabolic activity within the fed-state. A mixed impact on dihydroxyacetone kinase 2 and ketohexokinase was also observed. The conversion of dihyroxyacetone to the phosphorylated form utilizes ATP as a cofactor while the conversion of D-fructose to D-fructose-1-phosphate generates AMP. This opposing impact may signal a cellular prioritization of ATP utilization for energetic processes. Serum glucose levels in these same CSE offspring were decreased and point to the limited systemic availability of energetic substrates. SIRT1 expression, an NAD+-dependent protein deacetylase, coordinately regulates hepatic gluconeogenesis through the post-translational modification of PGC1α [85] and leads to the deacetylation and degradation of the cAMP response element binding protein (CREB) regulated transcription coactivator 2 (CRTC2) with attenuation of gluconeogenesis [86]. In the current study, we report a suppression of SIRT1 protein expression which was inversely associated with PEPCK and PGC1α protein expression. We believe this is the first report of an impaired “switch” from gluconeogenesis to glycolysis in the fed state in 6 month old adult animals that were previously developmentally exposed to cigarette smoke. We propose that the failure to equivalently limit hepatic gluconeogenesis in the fed state acts as a survival mechanism to provide additional systemic glucose and may indicate a failure to adequately respond to insulin signaling with the attendant implication that mitochondrial insufficiency and leakage may lead to diet-associated metabolic syndrome.
Systemic oxidative stress is a characteristic feature of ‘active’ CSE in similar animal model systems. At weaning (PD21) in our model system, hepatic oxidative stress was evident at the time of cessation of developmental CSE [54]. In the present study we report a sustained decrement in the availability of gluthatione pools (total, free, and oxidized) though the ratio of GSH/GSSG remained unchanged in 6 month old adult offspring who were exposed throughout developmental to cigarette smoke. This lack of reducing equivalents, coupled with the increase in antioxidant enzymes, indicates a systemic stress response.
DNA methylation is dependent on one-carbon metabolism and homocysteine availability. A recent report demonstrated that maternal smoking during pregnancy resulted in sex-specific impairment of one-carbon metabolism in human fetal liver [87]. We report that betaine-homocysteine S-methyltransferase and adenosylhomocysteinase, two enzymes involved in methionine degradation and specifically within the one-carbon metabolic pathway, were increased in abundance in the liver of 6 month old adult offspring who had undergone prior developmental cigarette smoke exposure. These alterations were coupled to a more generalized increase in the abundance of amino acid catabolic enzymes and indicate increased requirements for DNA methylation substrates as well as gluconeogenic precursors.
In summary, ‘active’ CSE from GD1-PD21 results in growth retardation that persists into adulthood well past the cessation of offspring exposure. Liver metabolic networks including glucose metabolism that were suppressed in PD21 weanling animals at the time of cessation of exposure continued to be impacted in adults, though gluconeogenic activity in the fed state appears to be elevated, with an accompanying suppression of fed-state serum glucose availability. We propose that a continued suppression of energetic precursor availability not only impacts hepatic metabolic pathways but influences systemic metabolic function. The accompanying hepatic oxidative stress that is present both in PD21 animals at the time of cessation of exposure as well as in 6 month old adult animals likely influences both cellular/tissue structure and function. The developmental basis of adult disease states that an adverse environment during development yields a propensity to diet-induced obesity, metabolic syndrome, diabetes, and a host of associated deleterious health outcomes. We propose that developmental exposure to ‘active’ maternal cigarette smoking falls within this paradigm. The observed hepatic metabolic dysregulation noted in exposed offspring may influence the development of metabolic syndrome at maturity within a high fat diet challenge paradigm. In future studies, we will address the impact of developmental ‘passive’ and ‘active’ cigarette smoke exposure on offspring sex-specific hepatic function within the context of diet-induced metabolic syndrome.
Highlights.
Developmental exposure to cigarette smoke results in persistant alterations in hepatic proteome.
Sustained impairment of carbohydrate metabolism in 6 month old CSE offspring
Weight gain may reflect impaired carbohydrate metabolism.
Acknowledgments
Research described in this article was supported in part by PHS grants NIH P20 RR/DE-17702, NIH R21 DA027466, NIH P30 ES014443 and by the University of Louisville CREAM Center from NSF EPSCoR grant EPS-0447479 (MS).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Langley-Evans SC. Nutrition in early life and the programming of adult disease: a review. J Hum Nutr Diet. 2015;28(Suppl 1):1–14. doi: 10.1111/jhn.12212. [DOI] [PubMed] [Google Scholar]
- 2.Lassi ZS, Imam AM, Dean SV, Bhutta ZA. Preconception care: caffeine, smoking, alcohol, drugs and other environmental chemical/radiation exposure. Reprod Health. 2014;11(Suppl 3):S6. doi: 10.1186/1742-4755-11-S3-S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Triunfo S, Lanzone A. Impact of maternal under nutrition on obstetric outcomes. J Endocrinol Invest. 2014 doi: 10.1007/s40618-014-0168-4. [DOI] [PubMed] [Google Scholar]
- 4.Wigle DT, Arbuckle TE, Turner MC, Berube A, Yang Q, Liu S, et al. Epidemiologic evidence of relationships between reproductive and child health outcomes and environmental chemical contaminants. J Toxicol Environ Health B Crit Rev. 2008;11:373–517. doi: 10.1080/10937400801921320. [DOI] [PubMed] [Google Scholar]
- 5.Surgeon General’s report highlights the health impact of smoking among women. Clin J Oncol Nurs. 2001;5:189. [PubMed] [Google Scholar]
- 6.State-specific prevalence of current cigarette smoking among adults, and policies and attitudes about secondhand smoke--United States, 2000. MMWR Morb Mortal Wkly Rep. 2001;50:1101–6. [PubMed] [Google Scholar]
- 7.From the Centers for Disease Control and Prevention. Cigarette smoking among adults--United States, 1999. JAMA. 2001;286:2802–4. [PubMed] [Google Scholar]
- 8.Smoking and women. How to overcome barriers and quit. Mayo Clin Womens Healthsource. 2001;5:1–2. [PubMed] [Google Scholar]
- 9.From the Centers for Disease Control and Prevention. State-specific prevalence of current cigarette smoking among adults, and policies and attitudes about secondhand smoke--United States, 2000. JAMA. 2002;287:309–10. [PubMed] [Google Scholar]
- 10.Cigarette smoking among adults--United States, 2006. MMWR Morb Mortal Wkly Rep. 2007;56:1157–61. [PubMed] [Google Scholar]
- 11.State-specific prevalence of cigarette smoking among adults and quitting among persons aged 18–35 years--United States, 2006. MMWR Morb Mortal Wkly Rep. 2007;56:993–6. [PubMed] [Google Scholar]
- 12.Current cigarette smoking prevalence among working adults--United States, 2004–2010. MMWR Morb Mortal Wkly Rep. 2011;60:1305–9. [PubMed] [Google Scholar]
- 13.Current cigarette smoking among adults - United States, 2011. MMWR Morb Mortal Wkly Rep. 2012;61:889–94. [PubMed] [Google Scholar]
- 14.Abbott LC, Winzer-Serhan UH. Smoking during pregnancy: lessons learned from epidemiological studies and experimental studies using animal models. Crit Rev Toxicol. 2012;42:279–303. doi: 10.3109/10408444.2012.658506. [DOI] [PubMed] [Google Scholar]
- 15.Abel EL. Smoking during pregnancy: a review of effects on growth and development of offspring. Hum Biol. 1980;52:593–625. [PubMed] [Google Scholar]
- 16.Current tobacco use and secondhand smoke exposure among women of reproductive age--14 countries, 2008–2010. MMWR Morb Mortal Wkly Rep. 2012;61:877–82. [PubMed] [Google Scholar]
- 17.Mattsson K, Jonsson I, Malmqvist E, Larsson HE, Rylander L. Maternal smoking during pregnancy and offspring type 1 diabetes mellitus risk: accounting for HLA haplotype. Eur J Epidemiol. 2015;30:231–8. doi: 10.1007/s10654-014-9985-1. [DOI] [PubMed] [Google Scholar]
- 18.La Merrill MA, Cirillo PM, Krigbaum NY, Cohn BA. The impact of prenatal parental tobacco smoking on risk of diabetes mellitus in middle-aged women. J Dev Orig Health Dis. 2015;6:242–9. doi: 10.1017/S2040174415000045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.van Dijk SJ, Molloy PL, Varinli H, Morrison JL, Muhlhausler BS. Epigenetics and human obesity. Int J Obes (Lond) 2015;39:85–97. doi: 10.1038/ijo.2014.34. [DOI] [PubMed] [Google Scholar]
- 20.Cupul-Uicab LA, Skjaerven R, Haug K, Travlos GS, Wilson RE, Eggesbo M, et al. Exposure to tobacco smoke in utero and subsequent plasma lipids, ApoB, and CRP among adult women in the MoBa cohort. Environ Health Perspect. 2012;120:1532–7. doi: 10.1289/ehp.1104563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cupul-Uicab LA, Skjaerven R, Haug K, Melve KK, Engel SM, Longnecker MP. In utero exposure to maternal tobacco smoke and subsequent obesity, hypertension, and gestational diabetes among women in the MoBa cohort. Environ Health Perspect. 2012;120:355–60. doi: 10.1289/ehp.1103789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Behl M, Rao D, Aagaard K, Davidson TL, Levin ED, Slotkin TA, et al. Evaluation of the association between maternal smoking, childhood obesity, and metabolic disorders: a national toxicology program workshop review. Environ Health Perspect. 2013;121:170–80. doi: 10.1289/ehp.1205404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mamun AA, O’Callaghan MJ, Williams GM, Najman JM. Maternal smoking during pregnancy predicts adult offspring cardiovascular risk factors - evidence from a community-based large birth cohort study. PLoS One. 2012;7:e41106. doi: 10.1371/journal.pone.0041106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bakker H, Jaddoe VW. Cardiovascular and metabolic influences of fetal smoke exposure. Eur J Epidemiol. 2011;26:763–70. doi: 10.1007/s10654-011-9621-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Oken E, Levitan EB, Gillman MW. Maternal smoking during pregnancy and child overweight: systematic review and meta-analysis. Int J Obes (Lond) 2008;32:201–10. doi: 10.1038/sj.ijo.0803760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alexander BT, Dasinger JH, Intapad S. Fetal programming and cardiovascular pathology. Compr Physiol. 2015;5:997–1025. doi: 10.1002/cphy.c140036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Heindel JJ, Vandenberg LN. Developmental origins of health and disease: a paradigm for understanding disease cause and prevention. Curr Opin Pediatr. 2015;27:248–53. doi: 10.1097/MOP.0000000000000191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nye MD, Fry RC, Hoyo C, Murphy SK. Investigating Epigenetic Effects of Prenatal Exposure to Toxic Metals in Newborns: Challenges and Benefits. Med Epigenet. 2014;2:53–9. doi: 10.1159/000362336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vickers MH. Early life nutrition, epigenetics and programming of later life disease. Nutrients. 2014;6:2165–78. doi: 10.3390/nu6062165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schug TT, Barouki R, Gluckman PD, Grandjean P, Hanson M, Heindel JJ. PPTOX III: environmental stressors in the developmental origins of disease--evidence and mechanisms. Toxicol Sci. 2013;131:343–50. doi: 10.1093/toxsci/kfs267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koletzko B, Brands B, Poston L, Godfrey K, Demmelmair H. Early nutrition programming of long-term health. Proc Nutr Soc. 2012;71:371–8. doi: 10.1017/S0029665112000596. [DOI] [PubMed] [Google Scholar]
- 32.Borgerding MF, Hicks RD, Bodnar JE, Riggs DM, Nanni EJ, Fulp GW, Jr, et al. Cigarette smoke composition. Part 1. Limitations of FTC method when applied to cigarettes that heat instead of burn tobacco. J Assoc Off Anal Chem. 1990;73:605–9. [PubMed] [Google Scholar]
- 33.Counts ME, Morton MJ, Laffoon SW, Cox RH, Lipowicz PJ. Smoke composition and predicting relationships for international commercial cigarettes smoked with three machine-smoking conditions. Regul Toxicol Pharmacol. 2005;41:185–227. doi: 10.1016/j.yrtph.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 34.Haussmann HJ. Use of hazard indices for a theoretical evaluation of cigarette smoke composition. Chem Res Toxicol. 2012;25:794–810. doi: 10.1021/tx200536w. [DOI] [PubMed] [Google Scholar]
- 35.Ingebrethsen BJ. Numerical simulation of the effects of dilution level, depth of inhalation, and smoke composition on nicotine vapor deposition during cigarette smoking. Inhal Toxicol. 2006;18:1071–6. doi: 10.1080/08958370600945457. [DOI] [PubMed] [Google Scholar]
- 36.Zdravkovic T, Genbacev O, McMaster MT, Fisher SJ. The adverse effects of maternal smoking on the human placenta: a review. Placenta. 2005;26(Suppl A):S81–6. doi: 10.1016/j.placenta.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 37.Rodgman A, Perfetti TA. The chemical components of tobacco and tobacco smoke. 2. Boca Raton, Fla: CRC Press; 2013. [Google Scholar]
- 38.Izzotti A, Balansky RM, Cartiglia C, Camoirano A, Longobardi M, De Flora S. Genomic and transcriptional alterations in mouse fetus liver after transplacental exposure to cigarette smoke. FASEB J. 2003;17:1127–9. doi: 10.1096/fj.02-0967fje. [DOI] [PubMed] [Google Scholar]
- 39.De Flora S, D’Agostini F, Balansky R, Camoirano A, Bennicelli C, Bagnasco M, et al. Modulation of cigarette smoke-related end-points in mutagenesis and carcinogenesis. Mutat Res. 2003;523–524:237–52. doi: 10.1016/s0027-5107(02)00340-8. [DOI] [PubMed] [Google Scholar]
- 40.Stanley EL, Hume R, Coughtrie MW. Expression profiling of human fetal cytosolic sulfotransferases involved in steroid and thyroid hormone metabolism and in detoxification. Mol Cell Endocrinol. 2005;240:32–42. doi: 10.1016/j.mce.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 41.Shenefelt PD. Fetal detoxification of maternal protoporphyria. J Am Acad Dermatol. 1998;38:129–30. doi: 10.1016/s0190-9622(98)70558-4. [DOI] [PubMed] [Google Scholar]
- 42.Corbel T, Perdu E, Gayrard V, Puel S, Lacroix MZ, Viguie C, et al. Conjugation and deconjugation reactions within the fetoplacental compartment in a sheep model: a key factor determining bisphenol A fetal exposure. Drug Metab Dispos. 2015;43:467–76. doi: 10.1124/dmd.114.061291. [DOI] [PubMed] [Google Scholar]
- 43.Hivert MF, Perng W, Watkins SM, Newgard CS, Kenny LC, Kristal BS, et al. Metabolomics in the developmental origins of obesity and its cardiometabolic consequences. J Dev Orig Health Dis. 2015;6:65–78. doi: 10.1017/S204017441500001X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nielsen CH, Larsen A, Nielsen AL. DNA methylation alterations in response to prenatal exposure of maternal cigarette smoking: A persistent epigenetic impact on health from maternal lifestyle? Arch Toxicol. 2014 doi: 10.1007/s00204-014-1426-0. [DOI] [PubMed] [Google Scholar]
- 45.Maccani JZ, Koestler DC, Houseman EA, Marsit CJ, Kelsey KT. Placental DNA methylation alterations associated with maternal tobacco smoking at the RUNX3 gene are also associated with gestational age. Epigenomics. 2013;5:619–30. doi: 10.2217/epi.13.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Menni C, Kastenmuller G, Petersen AK, Bell JT, Psatha M, Tsai PC, et al. Metabolomic markers reveal novel pathways of ageing and early development in human populations. Int J Epidemiol. 2013;42:1111–9. doi: 10.1093/ije/dyt094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Joubert BR, Haberg SE, Nilsen RM, Wang X, Vollset SE, Murphy SK, et al. 450K epigenome-wide scan identifies differential DNA methylation in newborns related to maternal smoking during pregnancy. Environ Health Perspect. 2012;120:1425–31. doi: 10.1289/ehp.1205412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xiao R, Perveen Z, Paulsen D, Rouse R, Ambalavanan N, Kearney M, et al. In utero exposure to second-hand smoke aggravates adult responses to irritants: adult second-hand smoke. Am J Respir Cell Mol Biol. 2012;47:843–51. doi: 10.1165/rcmb.2012-0241OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Suter M, Ma J, Harris A, Patterson L, Brown KA, Shope C, et al. Maternal tobacco use modestly alters correlated epigenome-wide placental DNA methylation and gene expression. Epigenetics. 2011;6:1284–94. doi: 10.4161/epi.6.11.17819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mukhopadhyay P, Horn KH, Greene RM, Michele Pisano M. Prenatal exposure to environmental tobacco smoke alters gene expression in the developing murine hippocampus. Reprod Toxicol. 2010;29:164–75. doi: 10.1016/j.reprotox.2009.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huuskonen P, Storvik M, Reinisalo M, Honkakoski P, Rysa J, Hakkola J, et al. Microarray analysis of the global alterations in the gene expression in the placentas from cigarette-smoking mothers. Clin Pharmacol Ther. 2008;83:542–50. doi: 10.1038/sj.clpt.6100376. [DOI] [PubMed] [Google Scholar]
- 52.Rouse RL, Boudreaux MJ, Penn AL. In utero environmental tobacco smoke exposure alters gene expression in lungs of adult BALB/c mice. Environ Health Perspect. 2007;115:1757–66. doi: 10.1289/ehp.10358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Neal RE, Chen J, Jagadapillai R, Jang H, Abomoelak B, Brock G, et al. Developmental cigarette smoke exposure: hippocampus proteome and metabolome profiles in low birth weight pups. Toxicology. 2014;317:40–9. doi: 10.1016/j.tox.2014.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Canales L, Chen J, Kelty E, Musah S, Webb C, Pisano MM, et al. Developmental cigarette smoke exposure: liver proteome profile alterations in low birth weight pups. Toxicology. 2012;300:1–11. doi: 10.1016/j.tox.2012.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jagadapillai R, Chen J, Canales L, Birtles T, Pisano MM, Neal RE. Developmental cigarette smoke exposure: kidney proteome profile alterations in low birth weight pups. Toxicology. 2012;299:80–9. doi: 10.1016/j.tox.2012.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Neal R, Jagadapillai R, Chen J, Stocke K, Gambrell C, Greene RM, Pisano MM. Developmental Cigarette Smoke Exposure II: Kidney Proteome Profile Alterations in Adult Offspring. Reprod Toxicol. 2016;X:X. doi: 10.1016/j.reprotox.2016.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Neal RE, Jagadapillai R, Chen J, Stocke K, Webb C, Greene RM, Pisano MM. Developmental Cigarette Smoke Exposure II: Hippocampus Proteome and Metabolome Profiles in Adult Offspring. Reprod Toxicol. 2016;x:x. doi: 10.1016/j.reprotox.2016.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Teague SV, K, Pinkerton E, Goldsmith M, Gebremichael A, Chang S, Jenkins RA, Moneyhun JH. Sidestream cigarette smoke generation and exposure system for environmental tobacco smoke studies. Inhalation Toxicology. 1994;6:79–93. [Google Scholar]
- 59.CDC. Incidence of initiation of cigarette smoking--United States, 1965–1996. MMWR Morb Mortal Wkly Rep. 1998;47:837–40. [PubMed] [Google Scholar]
- 60.Husten CG, Chrismon JH, Reddy MN. Trends and effects of cigarette smoking among girls and women in the United States, 1965–1993. J Am Med Womens Assoc. 1996;51:11–8. [PubMed] [Google Scholar]
- 61.Nelson DE, Giovino GA, Shopland DR, Mowery PD, Mills SL, Eriksen MP. Trends in cigarette smoking among US adolescents, 1974 through 1991. Am J Public Health. 1995;85:34–40. doi: 10.2105/ajph.85.1.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.CDC. Comparison of the cigarette brand preferences of adult and teenaged smokers--United States, 1989, and 10 U.S. communities, 1988 and 1990. MMWR Morb Mortal Wkly Rep. 1992;41:169–73. 79–81. [PubMed] [Google Scholar]
- 63.Seccareccia F, Zuccaro P, Pacifici R, Meli P, Pannozzo F, Freeman KM, et al. Serum cotinine as a marker of environmental tobacco smoke exposure in epidemiological studies: the experience of the MATISS project. Eur J Epidemiol. 2003;18:487–92. doi: 10.1023/a:1024672522802. [DOI] [PubMed] [Google Scholar]
- 64.Pichini S, Basagana XB, Pacifici R, Garcia O, Puig C, Vall O, et al. Cord serum cotinine as a biomarker of fetal exposure to cigarette smoke at the end of pregnancy. Environ Health Perspect. 2000;108:1079–83. doi: 10.1289/ehp.001081079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Scherer G, Richter E. Biomonitoring exposure to environmental tobacco smoke (ETS): a critical reappraisal. Hum Exp Toxicol. 1997;16:449–59. doi: 10.1177/096032719701600806. [DOI] [PubMed] [Google Scholar]
- 66.Granella M, Priante E, Nardini B, Bono R, Clonfero E. Excretion of mutagens, nicotine and its metabolites in urine of cigarette smokers. Mutagenesis. 1996;11:207–11. doi: 10.1093/mutage/11.2.207. [DOI] [PubMed] [Google Scholar]
- 67.Amos-Kroohs RM, Williams MT, Braun AA, Graham DL, Webb CL, Birtles TS, et al. Neurobehavioral phenotype of C57BL/6J mice prenatally and neonatally exposed to cigarette smoke. Neurotoxicol Teratol. 2013;35:34–45. doi: 10.1016/j.ntt.2013.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–54. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 69.Karp NA, Griffin JL, Lilley KS. Application of partial least squares discriminant analysis to two-dimensional difference gel studies in expression proteomics. Proteomics. 2005;5:81–90. doi: 10.1002/pmic.200400881. [DOI] [PubMed] [Google Scholar]
- 70.Baker MA, Cerniglia GJ, Zaman A. Microtiter plate assay for the measurement of glutathione and glutathione disulfide in large numbers of biological samples. Anal Biochem. 1990;190:360–5. doi: 10.1016/0003-2697(90)90208-q. [DOI] [PubMed] [Google Scholar]
- 71.Tietze F. Enzymic method for quantitative determination of nanogram amounts of total and oxidized glutathione: applications to mammalian blood and other tissues. Anal Biochem. 1969;27:502–22. doi: 10.1016/0003-2697(69)90064-5. [DOI] [PubMed] [Google Scholar]
- 72.Boyland E, Chasseaud LF. Glutathione S-aralkyltransferase. Biochem J. 1969;115:985–91. doi: 10.1042/bj1150985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jakoby WB. The glutathione S-transferases: a group of multifunctional detoxification proteins. Adv Enzymol Relat Areas Mol Biol. 1978;46:383–414. doi: 10.1002/9780470122914.ch6. [DOI] [PubMed] [Google Scholar]
- 74.Staal GE, Visser J, Veeger C. Purification and properties of glutathione reductase of human erythrocytes. Biochim Biophys Acta. 1969;185:39–48. doi: 10.1016/0005-2744(69)90280-0. [DOI] [PubMed] [Google Scholar]
- 75.Gershoni JM. Protein blotting: a manual. Methods Biochem Anal. 1988;33:1–58. doi: 10.1002/9780470110546.ch1. [DOI] [PubMed] [Google Scholar]
- 76.Boutant M, Canto C. SIRT1 metabolic actions: Integrating recent advances from mouse models. Mol Metab. 2014;3:5–18. doi: 10.1016/j.molmet.2013.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Koren G, Klein J, Forman R, Graham K, Phan MK. Biological markers of intrauterine exposure to cocaine and cigarette smoking. Dev Pharmacol Ther. 1992;18:228–36. [PubMed] [Google Scholar]
- 78.Horn KH, Esposito ER, Greene RM, Pisano MM. The effect of cigarette smoke exposure on developing folate binding protein-2 null mice. Reprod Toxicol. 2008;26:203–9. doi: 10.1016/j.reprotox.2008.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Esposito ER, Horn KH, Greene RM, Pisano MM. An animal model of cigarette smoke-induced in utero growth retardation. Toxicology. 2008;246:193–202. doi: 10.1016/j.tox.2008.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gray TR, Eiden RD, Leonard KE, Connors G, Shisler S, Huestis MA. Nicotine and metabolites in meconium as evidence of maternal cigarette smoking during pregnancy and predictors of neonatal growth deficits. Nicotine Tob Res. 2010;12:658–64. doi: 10.1093/ntr/ntq068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lindsay CA, Thomas AJ, Catalano PM. The effect of smoking tobacco on neonatal body composition. Am J Obstet Gynecol. 1997;177:1124–8. doi: 10.1016/s0002-9378(97)70027-9. [DOI] [PubMed] [Google Scholar]
- 82.Mansi G, Raimondi F, Pichini S, Capasso L, Sarno M, Zuccaro P, et al. Neonatal urinary cotinine correlates with behavioral alterations in newborns prenatally exposed to tobacco smoke. Pediatr Res. 2007;61:257–61. doi: 10.1203/pdr.0b013e31802d89eb. [DOI] [PubMed] [Google Scholar]
- 83.Reeves S, Bernstein I. Effects of maternal tobacco-smoke exposure on fetal growth and neonatal size. Expert Rev Obstet Gynecol. 2008;3:719–30. doi: 10.1586/17474108.3.6.719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Espy KA, Fang H, Johnson C, Stopp C, Wiebe SA. Prenatal tobacco exposure: developmental outcomes in the neonatal period. Dev Psychol. 2011;47:153–6. doi: 10.1037/a0020724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature. 2005;434:113–8. doi: 10.1038/nature03354. [DOI] [PubMed] [Google Scholar]
- 86.Liu Y, Dentin R, Chen D, Hedrick S, Ravnskjaer K, Schenk S, et al. A fasting inducible switch modulates gluconeogenesis via activator/coactivator exchange. Nature. 2008;456:269–73. doi: 10.1038/nature07349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Drake AJ, O’Shaughnessy PJ, Bhattacharya S, Monteiro A, Kerrigan D, Goetz S, et al. In utero exposure to cigarette chemicals induces sex-specific disruption of one-carbon metabolism and DNA methylation in the human fetal liver. BMC Med. 2015;13:18. doi: 10.1186/s12916-014-0251-x. [DOI] [PMC free article] [PubMed] [Google Scholar]