

Abstract
The objective was to examine the influence of Pluronic block-copolymers on the interaction between the drug efflux transporter, P-glycoprotein and HIV-1 protease inhibitors (PIs). The ATPase assay determined the effect of various Pluronics on PI-stimulated P-gp ATPase activity. Cellular accumulation studies were conducted using MDCKII and LLC-PK1 cells transfected with human MDR1 to assess Pluronic modulation of PI efflux. Pluronic P85 inhibited both basal and nelfinavir-stimulated P-gp ATPase activity, while Pluronic F127 had no effect. In cell accumulation studies, Pluronic P85 restored the accumulation of nelfinavir in MDCKII-MDR1 cells while Pluronic F127 and F88 had no effect. Pluronic P85 increased saquinavir accumulation in wild-type and MDR1-transfected cells in both the MDCKII and LLC-PK1 cell models, suggesting inhibition of multiple transporters, including MRPs. In conclusion, this study provides evidence that a block-copolymer, Pluronic P85, effectively inhibits the interaction of P-gp with nelfinavir and saquinavir. These data indicate that effective inhibition of HIV-1 PI efflux by Pluronic P85 may influence the distribution of antiretroviral agents to sites protected by efflux mechanisms, such as the blood–brain barrier, and possibly increase the brain exposure of these drugs resulting in suppression of viral replication and reduction in the incidence of drug resistant mutants.
Keywords: efflux pumps, HIV-1 protease inhibitors, pluronics, polymers, ATPase P-glycoprotein, blood–brain barrier
INTRODUCTION
Protease inhibitors (PIs) are drugs used in the treatment of HIV-1 infection and are an integral part of active retroviral therapy (ART), which involves the use of a combination of different classes of antiretroviral drugs. PIs prevent the formation of mature HIV-1 particles by inhibition of the viral protease. By decreasing the morbidity and mortality related to HIV-1 infection and AIDS, PIs have become the mainstay of anti-HIV-1 ART therapy.
Given the frequent mutations the virus undergoes due to its retro-viral nature, development of resistance to PIs has been reported1 and these mutants may persist even after PI therapy is stopped.2 This phenomenon is more pronounced in tissues such as the brain, which has been implicated as a sanctuary site for HIV-1. Proliferation of the virus in brain tissue can lead to such conditions as the AIDS dementia complex and HIV-1 encephalopathy.3 Many PIs have been shown to have much lower concentrations in CNS compared to plasma,4,5 resulting in drug levels that provide an ideal environment for the evolution of more virulent forms of the virus.6
The low concentration of PIs in brain tissue has been shown to be a result of active efflux of these drugs out of the brain.7–9 One of the main transport proteins responsible for this active efflux is P-glycoprotein. P-glycoprotein (P-gp, ABCB1) is a 170 kD trans-membrane protein belonging to the ATP binding cassette (ABC) super-family that is expressed in a number of tissues important for the ADME of drugs, such as the gut wall, liver canaliculi and proximal tubules in kidney.10 In brain, P-gp is expressed at the luminal surface of the blood–brain barrier11 and uses the energy generated by ATP hydrolysis to efflux drug substrates out of the brain against a concentration gradient, thereby decreasing brain levels. P-gp has been shown to efflux PIs both in vitro and in vivo,7,12–15 in addition to a number of structurally diverse compounds. In vitro biochemical assays such as the ATPase assay have shown the ability of a number of PIs to stimulate P-gp ATPase activity.16,17 Also, PIs have been shown to compete with substrates for the P-gp binding site.9 In vitro cell accumulation studies have shown the ability of P-gp to limit cellular accumulation of PIs both in P-gp transfected cell models,18 as well as primary cultures of brain capillary endothelial cells.16 Directional flux studies across P-gp transfected cell monolayers have shown the preferential transport of PIs in the direction of P-gp mediated efflux.8 There is some evidence regarding the ability of PIs to up-regulate P-gp expression.19 In vivo studies in different animal models have shown that the brain-to-plasma ratio for PIs is much lower than unity following intravenous, as well as oral dosing.8,14,15 Inhibition of P-gp using small molecule inhibitors has been shown to increase cellular accumulation and directional transport of PIs in P-gp over-expressing cells.7,8 The brain-to-plasma ratio for PIs was significantly increased on treatment with P-gp inhibitors or in transgenic P-gp knockout mouse models.18,20
Small molecules have been traditionally employed as inhibitors for P-gp both in vitro and in vivo. Recent reports have provided evidence that surfactant and polymeric molecules, used primarily as excipients, have biologic activity against P-gp function.21 Surfactants such as TPGS,22–24 Cremophor EL,25 Tween 8026 and polymers such as Pluronics27,28 and amphiphilic diblock copolymers29 have been shown to inhibit P-gp function both in vitro and to enhance drug exposure in vivo. The effect of excipients on passive drug permeation across the plasma membrane has also been investigated.30
Pluronics consist of a combination of polyethylene oxide and polypropylene oxide chains arranged as a tri-block copolymer. Pluronic P85, an amphiphilic block copolymer with two chains of hydrophilic polyethylene oxide groups flanking a hydrophobic polypropylene oxide group, has been shown to inhibit P-gp efflux activity.28
The objective of the study was to evaluate the ability of Pluronic P85 to modulate the interaction of HIV-1 PIs with P-glycoprotein using the biochemical ATPase assay and in vitro cell accumulation studies.
MATERIALS AND METHODS
Chemicals
[3H]-Vinblastine sulfate, [3H]-saquinavir, and [14C]-nelfinavir were obtained from Moravek Biochemicals (Brea, CA). Verapamil was obtained from the Sigma Chemical Co. (St. Louis, MO). Pluronics P85, F127, and F88 were a gift from BASF (Florham Park, NJ). Human P-gp expressing membranes were purchased from BD Biosciences (San Jose, CA). All other chemicals used were HPLC or reagent grade.
Cell Lines
Wild-type (WT) and MDR1-transfected epithelial Madin-Darby canine kidney (MDCKII) cells, as well as wild-type and MDR1-transfected porcine kidney epithelial cells (LLC-PK1 WT and LLC-PK1 MDR1) were obtained from Dr. Piet Borst (The Netherlands Cancer Institute, Amsterdam, The Netherlands). The MDCKII cells were maintained in Dulbecco’s modified eagle medium (Mediatech, Inc., Herndon, VA) fortified with 10% heat deactivated fetal bovine serum (Sera-Care Life Sciences, Inc., Oceanside, CA) and the LLC-PK1 cells were maintained in Medium 199 (Mediatech, Inc.) and fortified with 3% fetal bovine serum. Both the canine and porcine cell lines were treated with 100 U/mL of penicillin and 100 μg/mL of streptomycin (Sigma–Aldrich, St. Louis, MO) and incubated at 37°C under humidity and 5% CO2 tension. The MDCKII-MDR1 cell growth media additionally contained 80 ng/mL of colchicine to maintain positive selection pressure for P-gp expression. Cells between passages 5 and 15 were used in all experiments.
P-gp ATPase Assay
P-gp ATPase activity was evaluated using a suspension of human P-gp over-expressing membranes obtained from baculovirus-infected insect cells (High Five, BTI-TN5B1-4) in TMEP buffer. Care was taken to ensure that all the membranes had the same lot number to minimize lot to lot variability. Upon arrival, the membranes from different vials (0.5 mL/vial at 5 mg/mL of protein) were pooled together. Two hundred microliter aliquots in individual tubes were stored at −80°C for future use. This was done in order to minimize repeated freeze-thaw denaturation of the membranes.
Briefly, a suspension of P-gp over-expressing membranes was incubated at 37°C for 20 min in presence of 4 mM ATP with and without different concentrations of the test compound. An identical reaction mixture containing 100 μM sodium orthovanadate was assayed in parallel. Orthovanadate inhibits P-gp by trapping Mg2+ ADP in the nucleotide-binding site. Thus, the ATPase activity measured in the presence of orthovanadate represents non-P-gp ATPase activity and was subtracted from the total activity measured in the nonorthovanadate treated case to yield the P-gp mediated ATPase activity. The reaction was stopped by the addition of 10% SDS and the liberated inorganic phosphate was detected by a colorimetric reaction of inorganic phosphate with a solution of ascorbic acid and ammonium molybdate at 650 nm. The nanomoles of phosphate released were determined from the absorbance values, using a standard curve generated from inorganic potassium phosphate standards (0–60 nM range). The same procedure was followed for control membranes (also obtained from BD Biosciences) that do not express P-gp.
Verapamil (20 μM) was used as a positive control for P-gp ATPase stimulatory activity. Initially, the Pluronic P85 was evaluated in the verapamil stimulated P-gp ATPase system for its ability to inhibit P-gp ATPase activity. Subsequently, the kinetics of P-gp ATPase activity was determined in isolated membranes using up to 50 μM concentrations of the model P-gp substrate verapamil with and without 0.01% (w/v) P85. Finally, nelfinavir was used in the P-gp ATPase assay at a concentration of 20 μM and Pluronics used at 0.01% (w/w) P85 (22 μM), 0.01% (w/w) F127 (7 μM), and 0.01% (w/w) F88 (9 μM), were evaluated for their ability to inhibit nelfinavir stimulated P-gp ATPase activity.
Cellular Accumulation Studies in MDCKII and LLC-PK1 Cells
For the accumulation experiments, cells were seeded in clear polyester 12-well plates (TPP© cell culture plate) at a seeding density of 2 × 105 cells/well. The media were changed every other day and the cells formed confluent monolayers in 4 days. On the day of the experiment, the media were aspirated and the confluent monolayer was washed twice with prewarmed (37°C) assay buffer. The cells were preincubated for 30 min with 1 mL of assay buffer, after which the buffer was aspirated and the cells were exposed to a tracer solution of radiolabeled drug in 1 mL assay buffer per well. The plates were continuously agitated at 60 rpm in an orbital shaker and maintained at 37°C for the duration of the experiment. Following a 3-h accumulation period the supernatant was aspirated and the cells were first washed three times with 2 mL of ice cold phosphate buffered saline (PBS) and then solubilized using 1 mL of mammalian protein extraction reagent (MPER; Pierce Biotechnology, Inc., Rockford, IL). A 200 μL sample was drawn from each well in triplicate and 4 mL of scintillation fluid (ScintiSafe Econo1 cocktail; Fisher Scientific Co., Fair Lawn, NJ) was added to each sample and counted using liquid scintillation counting (LS-6500, Beckman Coulter, Inc., Fullerton, CA) to determine the radioactivity associated with the cells. Accumulated drug was normalized to cellular protein using the BCA protein assay (Pierce Biotechnology, Inc.).
In the case of inhibition studies, the cells were pretreated with 0.01% (w/w) Pluronic for 30 min followed by incubation with both test drug and inhibitor for the duration of the accumulation. Drug accumulation was expressed as a percentage of the accumulation in WT cells with the WT cells being designated as 100. All the stocks were diluted to working concentrations using assay buffer (NaCl, 122 mM; NaHCO3, 25 mM; Glucose, 10 mM; HEPES, 10 mM; KCl, 3 mM; MgSO4·7 H20, 1.2 mM; CaCl2·H20, 1.4 mM; K2HPO4, 0.4 mM; pH of 7.4).
Model Validation
Confluent monolayers of MDCKII WT and MDR1 cells were trypsinized and the cells washed with cold PBS to remove trypsin. Four to five million cells were used for total RNA extraction using the RNeasy Mini Kit (Qiagen, Valencia, CA). The concentration, as well as purity of the extracted RNA, was determined by the use of UV spectrophotometry (Beckman Coulter, DU 530 UV/Visible spectrophotometer). Equal amounts of total RNA for each cell line was immediately reverse transcribed to obtain stable cDNA using the Omni script RT kit (Qiagen). Quantitative RT-PCR was performed for each cell line and the reaction tube contained QPCR master mix (Brilliant SYBR green qPCR master mix, Stratagene, La Jolla, CA), template (cDNA), primers synthesized for the gene of interest, and SYBR green dye. Expression of the gene of interest was always normalized to the housekeeping gene β-actin. Quantitative real time RT-PCR was conducted using a Stratagene Mx3000P qRT-PCR system. The machine was set to run for 40 cycles with a thermal profile made up of heating at 95°C for 30 s followed by 1 min at 55°C and 30 s at 72°C. The results were evaluated using the Mx3000P software to obtain the threshold cycle (Ct) values for each gene and the relative expression of the MDR1 gene signal in the P-gp over expressing MDCKII cells compared to the MDCKII WT cells was computed. The nontemplate controls lacking the cDNA template required for signal amplification were also used in the experiment.
For evaluation of P-gp function [3H]-vinblastine was used as a model P-gp substrate and its cellular accumulation was used to validate increased P-gp function in the MDCKII-MDR1 cell model compared to MDCKII WT cells.
Cellular Accumulation Studies With Protease Inhibitors
[14C]-Nelfinavir cellular accumulation was studied in MDCKII WT and MDR1-transfected cell lines to determine the interaction between nelfinavir and P-gp. Subsequently, the ability of three different Pluronics (P85, F127, and F88) to modulate the interaction between nelfinavir and P-gp was evaluated. [3H]-Saquinavir cellular accumulation was studied in both MDCKII and LLC-PK1 cell models and the effect of Pluronic P85 on the interaction between saquinavir and P-gp was evaluated.
Statistical and Data Analysis
The Michaelis–Menten model for saturable enzyme kinetics was fit to the data for P-gp ATPase activity with increasing concentrations of the substrate verapamil. The enzyme kinetics module in Sigma Plot (version 9.0.1, SYSTAT software) with nonlinear regression was used to fit the Michaelis–Menten model to the velocity data and estimates of the kinetic parameters, Vmax and Km were obtained. For the rate versus substrate concentration plot in presence of the inhibitor P85, the data were fit by linear regression to the equation of a straight line with intercept and the ratio of Vmax/Km was determined as the slope of the line.
Statistical comparisons between two groups were made using the two sample t-test at a p < 0.05 level of significance. Multiple groups were compared using ANOVA with Holm–Sidak post-hoc test at a p < 0.05 level of significance.
RESULTS
P-gp ATPase Assay
Verapamil P-gp ATPase Assay
The P-gp ATPase assay was initially validated using verapamil in P-gp over-expressing membranes. Treatment with 20 μM verapamil stimulated the P-gp ATPase activity and resulted in a 10-fold increase in the amount of inorganic phosphate generated compared to the control case (Fig. 1). Treatment with 0.01% (w/w) of Pluronics P85 and F127 resulted in significant inhibition of verapamil-stimulated P-gp ATPase activity (p < 0.05; Fig. 1). The basal P-gp ATPase activity was completely abolished by Pluronic P85 but not by F127 (p < 0.05; Fig. 2).
Figure 1.
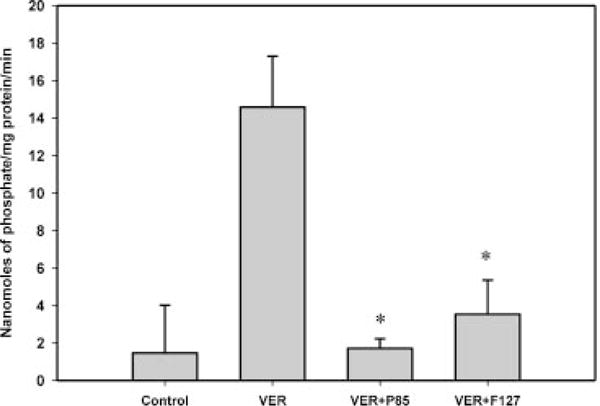
P-gp ATPase activity expressed as nanomoles of phosphate generated/mg protein/min. The positive control verapamil (20 μM) significantly stimulated P-gp ATPase activity. Pluronic P85 and F127 significantly inhibited the verapamil-stimulated P-gp ATPase activity (*p < 0.05); n = 9.
Figure 2.

Effect of Pluronics on basal P-gp ATPase activity. Pluronic P85, but not Pluronic F127, significantly inhibited basal P-gp ATPase activity in human P-gp expressing membranes (*p < 0.05); n = 9.
The interaction of P-gp ATPase with verapamil showed concentration dependence and was saturable (Fig. 3). The Vmax and Km (mean ± SE) were estimated to be 40.6 ± 1.7 nmol phosphate/mg protein/min and 3.4 ± 0.7 μM, respectively. The R2 value for the fit of the model to the data was 0.91. For the experiments with 0.01% (w/w) P85, there was significant inhibition of ATPase activity at all concentrations of verapamil and the maximal rate obtainable was no more than 6–8 nmol phosphate/mg protein/min at 50 μM verapamil (Fig. 3). The Vmax/Km was determined to be 0.072 ± 0.02 mL/min/mg protein from the fit of the equation for a straight line to the data. The linear regression was significant with a p-value of 0.0113.
Figure 3.
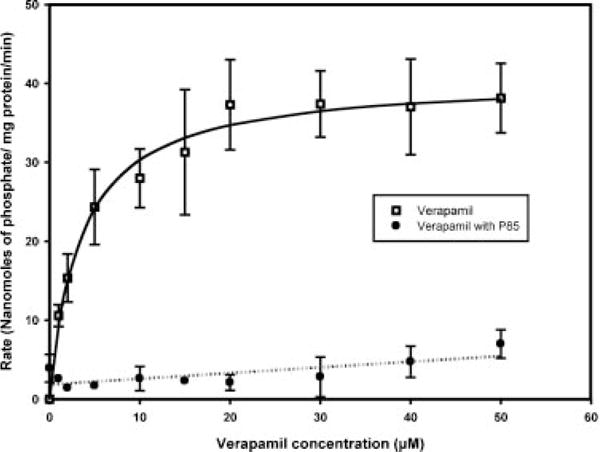
Plot of the P-gp ATPase activity with increasing concentrations of verapamil and the effect of Pluronic P85 on this interaction. The interaction of P-gp ATPase with verapamil was saturable (open squares) and was modeled using the Michealis–Menten enzyme kinetics model (solid line) to obtain estimates of Vmax and Km. Pluronic P85 inhibited the verapamil-stimulated P-gp ATPase activity (filled circles) and the equation of a straight line was fit to the data (dotted line) to obtain the estimate of Vmax/Km. Results are expressed as mean ± SD with three replicates. SD not shown for data points measured in duplicate.
Nelfinavir P-gp ATPase Assay
Nelfinavir, used at a concentration of 20 μM, significantly stimulated the P-gp ATPase activity and Pluronic P85 completely abolished the nelfinavir mediated P-gp ATPase stimulation (p < 0.05). As seen in Figure 4, Pluronic F127 and F88 did not significantly inhibit nelfinavir stimulated P-gp ATPase activity (p < 0.05).
Figure 4.
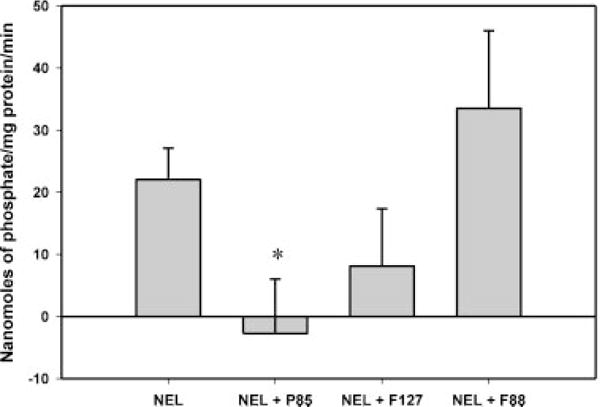
Effect of different Pluronics on nelfinavir-stimulated P-gp ATPase activity. Pluronic P85 significantly inhibited nelfinavir-stimulated P-gp ATPase activity (*p < 0.05) while Pluronic F127 and F88 did not show inhibition; n = 9.
Cell Accumulation Studies in MDCKII Cells
MDCKII Model Validation
P-gp Gene Expression
Figure 5a shows that the Ct value for P-gp expression in the MDR1 transfects was much lower than the WT (17.16 vs. 23.79) indicating a much higher level of expression of the MDR1 gene in the human P-gp over-expressing cells. No difference was observed in the Ct values for the housekeeping gene in both the WT and MDR1-transfected MDCKII cells (14.14 and 14.73). The nontemplate controls for MDR1 and β-actin did not undergo amplification.
Figure 5.
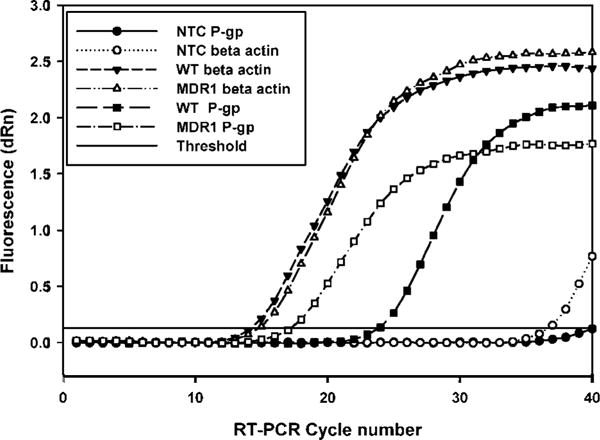
Amplification plots for MDR1 and GAPDH genes using quantitative Real-time Polymerase Chain Reaction (qRT-PCR). The threshold cycle number (Ct) for the MDR1 gene was lower for the MDR1-transfected MDCKII cells (open squares) when compared to MDCK Wild-type cells (filled squares). The Ct values for the housekeeping gene, GAPDH were similar between the WT (filled inverted triangles) and the MDR1-transfected cell lines (open triangles). The non-template controls for MDR1 (filled circles) and GAPDH (open circles) did not contain primers in the amplification PCR mix and were used as negative controls; n = 3.
P-gp Function
The accumulation of vinblastine in the P-gp over-expressing MDCKII-MDR1 cells was significantly (p < 0.05) lower than in the wild-type cells. Vinblastine accumulation in the MDR1 cells was only ~20% of that observed in the wild-type MDCKII cells (data not shown).
Cellular Accumulation Studies With Protease Inhibitors
Nelfinavir Accumulation in Wild Type (WT) and P-gp Transfected MDCKII-MDR1 Cells
Nelfinavir accumulation in MDCKII-MDR1 cells was significantly lower than in the WT cells (p < 0.05; Fig. 6). There was ~85% lower accumulation of nelfinavir in the MDR1 cells compared to the WT cells indicating that P-gp efflux is responsible for lower accumulation in the MDR1 cells. Pluronic P85 at a concentration of 0.01% (w/w) significantly enhanced the accumulation of nelfinavir in MDR1 cells such that it was comparable to the level in WT cells (p < 0.05). F88 and F127 did not show any increase in accumulation in the MDR1 cells compared to the control case. Treatment of WT cells with Pluronic P85 resulted in a slight but not significant increase in nelfinavir accumulation (p < 0.05). Pluronics F127 and F88 did not show any effect on nelfinavir accumulation in the WT cells.
Figure 6.

Effect of different Pluronics on the cellular accumulation of nelfinavir in MDCKII WT and MDR1-transfected cells. Nelfinavir accumulation in the MDR1 cells (gray bars) was significantly lower than in the WT cells (black bars). Pluronic P85 significantly increased the accumulation of nelfinavir in the MDR1 cells (*p < 0.05). Pluronic F127 and F88 did not have any significant effect on nelfinavir accumulation in the MDR1 cells. Nelfinavir accumulation in the WT cells was not affected by treatment with Pluronics. Results are expressed as mean ± SD (as percentage of wild-type control); n = 9.
Saquinavir Accumulation in Wild Type (WT) and P-gp Transfected MDCKII-MDR1 Cells
Saquinavir accumulation in the MDCKII-MDR1 cells was ~40% of that seen in the WT cells (p < 0.05; Fig. 7). Treatment with 0.01% (w/w) P85 resulted in a twofold increase in saquinavir accumulation in MDR1 cells. Interestingly, the WT cells showed a greater increase (approximately sixfold) in accumulation on treatment with 0.01% (w/w) P85. The positive control used in this study, vinblastine, also showed a fourfold increase in accumulation in the WT cells but a greater (sevenfold) increase in accumulation in the MDR1 cells on treatment with P85.
Figure 7.
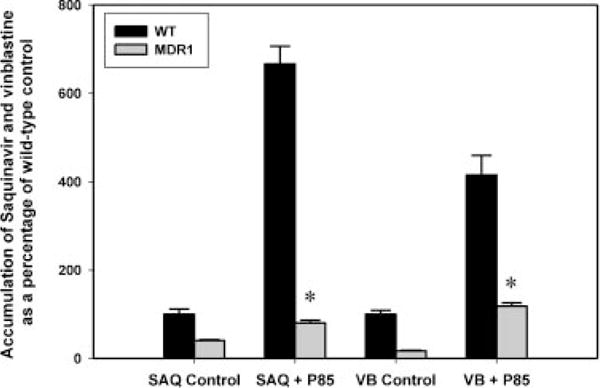
Effect of Pluronic P85 on the cellular accumulation of saquinavir in MDCKII wild-type and MDR1-transfected cells. Saquinavir accumulation in the MDR1 cells (gray bars) was significantly lower than in the WT cells (black bars). Pluronic P85 significantly increased the accumulation of saquinavir in both WT and MDR1 cells (*p < 0.05). Vinblastine was used as the positive control and Pluronic P85 significantly increased vinblastine accumulation in both WT and MDR1 cells. Results are expressed as mean ± SD (as percentage of wild-type control); n = 9.
Saquinavir Accumulation in Wild Type (WT) and P-gp Transfected LLC-PK1 MDR1 Cells
In the LLC-PK1 cell model, saquinavir accumulation in the MDR1 cells was only ~20% of that seen in LLC-PK1 WT cells (Fig. 8). Treatment with 0.01% (w/w) P85 significantly increased saquinavir accumulation by about fivefold in the MDR1 cells (p < 0.05) while in the WT cells the increase was about 3.5-fold.
Figure 8.

Saquinavir accumulation in the LLC-PK1 cell model. Saquinavir accumulation in the LLC-PK1 MDR1 cells (gray bars) that over-express human P-gp was significantly lower than in the LLC-PK1 WT cells (black bars). Pluronic P85 significantly enhanced the cellular accumulation of saquinavir in the human P-gp over-expressing LLC-PK1 MDR1 cells (*p < 0.05). Results are expressed as mean ± SD (as percentage of wild-type control); n = 9.
DISCUSSION
P-glycoprotein is an efflux drug transporter that utilizes energy released from the hydrolysis of ATP (ATPase) for its efflux action.9 Drugs that interact with P-gp are known to affect its ATPase activity. One of the objectives of this study was to evaluate the inhibitory effect of Pluronics on the interaction between a HIV-1 PI and human P-gp ATPase. The ability of Pluronic P85 to inhibit drug-stimulated P-gp ATPase activity has previously been reported.31 Initially, verapamil, a classic substrate of P-gp32 was used as a model P-gp substrate. Pluronic P85 showed significant inhibition of P-gp ATPase activity in verapamil-stimulated membranes (Fig. 1), as well as basal P-gp ATPase activity (Fig. 2). This is in agreement with previous reports describing the ability of P85 to significantly inhibit basal, as well as drug-stimulated P-gp ATPase activity in the same membrane system.31,33
An interesting observation in this study was the effect of Pluronic F127 on verapamil-stimulated ATPase activity. In a previous study, Batrakova et al.,33 reported that F127 did not inhibit basal P-gp ATPase activity. A similar observation was made in this study where there was no difference between basal ATPase activity and activity in unstimulated membranes treated with Pluronic F127 (Fig. 2). However, treatment with Pluronic F127 in the presence of verapamil resulted in significant inhibition of the verapamil-stimulated P-gp ATPase activity (Fig. 1), suggesting that P-gp interaction with a ligand may be required for Pluronic F127 to show its inhibitory effect.
The interaction of P-gp ATPase with verapamil in human P-gp expressing membranes followed Michealis–Menten kinetics allowing for the estimation of Vmax and Km (Fig. 3). For the hydrolysis of ATP the Vmax and Km values (Vmax—40.6 nmol/mg/min and Km—3.4 μM), were comparable to the values previously observed for verapamil in Sf9 insect cell membranes (Vmax—45 nmol/mg/min, Km—2.5 μM) by Sharom et al.34 The efficiency of P-gp ATPase in the presence of verapamil was calculated as Vmax/Km and was ~11 mL/min/mg protein. A similar value for the efficiency of P-gp ATPase has been reported for the interaction between P-gp membranes and vinblastine.35 Batrakova et al., reported that the vinblastine-stimulated P-gp ATPase activity followed saturable kinetics with an efficiency of 11 mL/min/mg which was decreased to 0.46 mL/min/mg upon treatment with 0.01% (w/w) Pluronic P85.
Upon treatment with 0.01% (w/w) P85, the rate of formation of inorganic phosphate by P-gp ATPase was much lower when compared to the verapamil only case (Fig. 3). The Michealis–Menten model was deemed unsuitable for analyzing these data since the velocity of the reaction did not show saturation over the range of verapamil concentrations used and was essentially linear. For this reason the equation of a straight line with intercept was used which would provide a Vmax/Km ratio from the slope of the line. The Vmax/Km ratio as given by the slope was 0.072 mL/min/mg protein. The ability of Pluronic P85 to dramatically decrease the efficiency of P-gp ATPase activity, stimulated by an excellent substrate of P-gp, shows its ability to strongly inhibit the interaction between drug and P-gp in the ATPase assay possibly by altering both the Vmax and/or Km.
HIV-1 PIs have been shown to interact with isolated P-gp membranes and stimulate ATPase activity.9 This activity has been shown to be inhibited by small molecule inhibitors like PSC833.9 In contrast, the ability of relatively large polymers such as Pluronics to modulate nelfinavir-stimulated P-gp ATPase activity was evaluated in this study. Nelfinavir stimulation of P-gp ATPase activity (Fig. 4) resulted in a rate of hydrolysis similar to what has been previously reported in the same membrane system.16 Bachmeier et al., reported a Vmax of 20.8 ± 6.2 nmol/mg/min (20 μM nelfinavir), which was similar to the rate of inorganic phosphate generation measured in this study (22 ± 5.1 nmol/mg/min) with 20 μM nelfinavir. The complete inhibition of nelfinavir-mediated P-gp activity by Pluronic P85 provides evidence toward its ability to effectively inhibit the interaction between P-gp and nelfinavir.
Pluronics F88 and F127 did not exhibit significant inhibition of P-gp ATPase activity and these treatments did not result in ATPase activity significantly different from control (Fig. 4). Interestingly, when verapamil was used as the stimulating agent in place of nelfinavir, Pluronic F127 did show significant inhibition (p < 0.05) of verapamil-stimulated P-gp ATPase activity (Fig. 1). Pluronic F127-mediated inhibition of verapamil-stimulated but not nelfinavir-stimulated ATPase activity or basal P-gp ATPase activity, suggests that inhibition of P-gp by Pluronic F127 may depend not only on the interaction of P-gp with the ligand, but also possibly on the site of interaction of the ligand with P-gp. Nelfinavir and verapamil could be interacting with P-gp at different sites leading to the differential inhibitory effect of F127. P-gp possesses multiple binding sites36 and has been shown to interact with different drugs at different binding sites. Orlowski et al.,37 have reported that the inhibition of basal, as well as verapamil-stimulated, P-gp ATPase activity by various detergents at concentrations below their CMC does not correlate with the amount of detergents incorporated in the membrane, suggesting the existence of specific interactions between P-gp and detergents. These observations support the possibility of specific interaction of Pluronics with the different binding sites on P-gp. It is possible that nelfinavir and verapamil could be interacting with P-gp at different sites on the protein and Pluronic F127 may be inhibiting the interaction at the site of verapamil interaction but not at the site of nelfinavir interaction with P-gp. This specificity in binding sites may be more apparent in the in vitro ATPase assay than in a cell based system where Pluronics may also show inhibitory effect by depletion of ATP and limited access to the cytoplasm could result in lack of inhibitory effect.31 This is true for Pluronic F127 which did not show any inhibitory effect on P-gp mediated efflux of drugs in cellular accumulation studies, as seen in this and previous works.33
Pluronic P85 on the other hand was able to inhibit P-gp ATPase activity under both unstimulated and stimulated conditions31 (see Figs. 1 and 2). This has been attributed to presence of an intermediate hydrophobic chain length for Pluronic P85 that may allow it to be incorporated into the bi-lipid layer resulting in a decrease in the membrane micro-viscosity. A decrease in micro-viscosity would unfavorably affect the conformation of P-gp resulting in loss of its function.33
The P-gp ATPase assay allowed the study of the interaction between the PI, nelfinavir and P-gp, as well as the effect of Pluronic P85 on this interaction in isolated human P-gp membranes without the interfering effects of other transporters that may confound any conclusive results. On the other hand, it must be noted that the ATPase assay can be subject to substantial variability and may therefore provide results that are subject to question. Therefore, the P85 inhibitory effect was evaluated in an intact cell based system which also provides the advantage of a more complete system. Pluronic P85 was evaluated for its ability to enhance cellular accumulation of PIs in the human P-gp over-expressing MDCKII cell model. This model has been routinely employed for evaluation of substrate status and to measure the ability of a molecule to inhibit P-gp-mediated efflux.38,39
Quantitative real-time RT-PCR40 was used to examine the higher expression of the human MDR1 gene in the MDCKII-MDR1 transfects compared to the MDCKII-WT cells (Fig. 5). Higher expression of the MDR1 gene translated to lower accumulation of the model probe [3H]-vinblastine in the MDCKII-MDR1 cells compared to WT cells, indicating greater efflux function of P-gp in the transfects.
The sixfold lower accumulation of nelfinavir in the P-gp over-expressing cells (Fig. 6) demonstrates P-gp mediated efflux of nelfinavir. Treatment with Pluronic P85 significantly (p < 0.05) increased the nelfinavir cellular accumulation in the MDCKII-MDR1 cells by about fivefold while there was a 1.2-fold increase in nelfinavir accumulation in the P85 treated WT cells. The increase in accumulation could be attributed to inhibition of endogenous P-gp in the MDCKII WT cells. Neither Pluronic F88 nor F127 enhanced nelfinavir accumulation in MDR1 or WT cells (Fig. 6). This result is in agreement with the lack of effect observed for these Pluronics in the nelfinavir P-gp ATPase assay.
In a recent study, Chandler et al., reported the enhanced accumulation of nelfinavir in wild-type HIV-1 infected MT4 cells, treated with the P-gp inhibitor verapamil, compared to untreated cells. The incidence of drug resistant mutations was significantly reduced in MT4 cells treated with verapamil possibly as a result of higher intracellular exposure to nelfinavir.41 Similarly, effective inhibition of P-gp at the blood–brain barrier could increase the exposure of target cells in the brain to nelfinavir resulting in suppression of viral replication and reduction in the incidence of drug resistant mutants. Adequate antiretroviral drug levels in the brain could compromise its status as a sanctuary site for the HIV-1 virus.
Saquinavir accumulation in the MDCKII cell system appears to be a more complex phenomenon than for nelfinavir (Fig. 7). The significant enhancement in saquinavir (sevenfold) and vinblastine (fourfold) accumulation in the WT cells on treatment with Pluronic P85 may be a result of its inhibitory activity against multiple drug transporters. Saquinavir has been shown to be directionally transported both in MDCKII-MRP2 and -MRP1 over-expressing cell lines. Indeed, the MDCKII WT cells were also shown to directionally transport saquinavir.42,43 Western blot analysis data for MDCKII WT cells indicated the expression of both MRP1 and MRP2 along with P-gp in these cells.44 In vivo, saquinavir brain penetration is limited by P-gp, as well as MRPs in the mouse.45 Vinblastine has also been shown to be a MRP substrate.43,44 Moreover, Pluronic P85 can inhibit MRP1 and MRP2 ATPase activity in isolated membranes35 and MRP1 and MRP2 mediated efflux in MDCKII-MRP1 and MRP2 transfected cells.44,46 Taken together, all the above studies provide evidence for both P-gp and MRP being responsible for the transport of both saquinavir and vinblastine in the MDCKII cell model.
The LLC-PK1 cell line has been shown, using western blot and functional studies, to have much lower endogenous P-gp compared to the MDCKII cells.47–49 Consequently, saquinavir cellular accumulation was also evaluated in the porcine kidney cell model (LLC-PK1) using LLC-PK1 WT and LLC-PK1-MDR1 cell lines. The LLC-PK1 cell model has also been used to study the effect of P-gp on cellular accumulation of drugs.50,51 The cellular accumulation of saquinavir in the LLC-PK1 WT cells was higher than in the MDCKII-WT cells, indicating lower expression of P-gp in these cells and transport of saquinavir by P-gp. In the LLC-PK1 cell system the accumulation of saquinavir in the P-gp over-expressing LLC-PK1-MDR1 cells was sixfold lower than in the WT cells indicating that the LLC-PK1 cell model may be a better system to test the interaction between saquinavir and P-gp (Fig. 8). Pluronic P85 treatment significantly increased saquinavir accumulation in both WT and MDR1 cells, similar to the result seen in the MDCKII cell model. Nonetheless, the fold increase in accumulation of saquinavir in the P85-treated MDR1 cells was greater than in the WT cells (3.5-fold in WT vs. fivefold in MDR1), indicating that the MRPs may be having less of a confounding effect on the results possibly due to lower endogenous expression.47 Taken together, all the above data for saquinavir demonstrated the ability of Pluronic P85 to effectively inhibit both P-gp and MRPs to enhance the cellular accumulation of saquinavir. Inhibition of multiple efflux transporters may considerably enhance the therapeutic concentrations in hard to reach sites such as the brain.
Given that Pluronic P85 significantly increased the cellular accumulation of PIs, one question that arises is whether or not the P85 treatment itself changed the expression of the efflux proteins. If so, the change in substrate accumulation may be in part a response to differing expression of the transporters. Other lipid excipients have been shown to decrease the P-glycoprotein expression upon very long-term exposure.52 It is important to note that Pluronic P85 has been implicated in the transcriptional activation of gene expression.53,54 A recent study evaluated the effect of Pluronic P85, used alone or in conjunction with the cytotoxic anticancer agent doxorubicin, on expression of the MDR1 gene in the human breast cancer MCF7 cell line.55 It was shown that a number of MDR1-related genes were up-regulated in cells cultured in medium containing doxorubicin alone, but were not up-regulated in cells cultured in medium containing a mixture of doxorubicin and Pluronic P85. However, cells cultured in medium containing Pluronic P85 alone over 305 days did not show any change in expression of the MDR1 gene compared to drug sensitive parental MCF7 cells.55 Therefore, the increase in cellular accumulation of drug with the P85 treatment in the current study is unlikely to be due to an effect of Pluronic P85 on MDR1 gene expression.
CONCLUSION
In summary, this study used biochemical and cellular accumulation assays to determine the inhibitory effect of Pluronics on the interaction between HIV-1 PIs and P-gp. The polymeric inhibitor Pluronic P85 inhibited the interaction between PIs and P-gp both in the ATPase assay, as well as in cell accumulation studies. Pluronic P85 inhibited both basal and drug-stimulated P-gp ATPase activity. This study also exhibited the ability of Pluronic P85 to reduce the efficiency of verapamil-stimulated P-gp ATPase activity. Pluronic F127 on the other hand, did not inhibit basal ATPase activity. F127 required the presence of a ligand to inhibit P-gp ATPase activity and inhibited verapamil-stimulated but not nelfinavir-stimulated, ATPase activity. These data suggested that F127 inhibitory effect may be contingent on P-gp binding a ligand at a specific binding site. Cellular accumulation studies with nelfinavir provided conclusive evidence that P85, but not F127 and F88, inhibits P-gp mediated efflux of nelfinavir demonstrating that P85 inhibitory effect is a specific phenomenon rather than a general membrane effect. Cellular accumulation studies with the PI saquinavir in two different cell lines exhibited the ability of Pluronic P85 to inhibit multiple transporters. In conclusion, this is the first study to provide evidence of the ability of a block copolymer, Pluronic P85, to effectively inhibit the interaction of P-gp and the HIV-1 PIs nelfinavir and saquinavir.
Acknowledgments
This work was supported by National Institutes of Health Grant NS42549. We thank Dr. Piet Borst from the Netherlands Cancer Institute for generously providing MDCKII and LLC-PK1 cells. We would like to thank BASF for generously providing us with the Pluronics.
References
- 1.Shafer RW, Rhee SY, Pillay D, Miller V, Sandstrom P, Schapiro JM, Kuritzkes DR, Bennett D. HIV-1 protease and reverse transcriptase mutations for drug resistance surveillance. Aids. 2007;21:215–223. doi: 10.1097/QAD.0b013e328011e691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.van Maarseveen NM, Wensing AM, de Jong D, Taconis M, Borleffs JC, Boucher CA, Nijhuis M. Persistence of HIV-1 variants with multiple protease inhibitor (PI)-resistance mutations in the absence of PI therapy can be explained by compensatory fixation. J Infect Dis. 2007;195:399–409. doi: 10.1086/510533. [DOI] [PubMed] [Google Scholar]
- 3.Ances BM, Ellis RJ. Dementia and neurocognitive disorders due to HIV-1 infection. Semin Neurol. 2007;27:86–92. doi: 10.1055/s-2006-956759. [DOI] [PubMed] [Google Scholar]
- 4.Lafeuillade A, Solas C, Halfon P, Chadapaud S, Hittinger G, Lacarelle B. Differences in the detection of three HIV-1 protease inhibitors in non-blood compartments: Clinical correlations. HIV Clin Trials. 2002;3:27–35. doi: 10.1310/WMWL-6W9Y-PXV2-X148. [DOI] [PubMed] [Google Scholar]
- 5.Kravcik S, Gallicano K, Roth V, Cassol S, Hawley-Foss N, Badley A, Cameron DW. Cerebrospinal fluid HIV RNA and drug levels with combination ritonavir and saquinavir. J Acquir Immune Defic Syndr. 1999;21:371–375. [PubMed] [Google Scholar]
- 6.Smit TK, Brew BJ, Tourtellotte W, Morgello S, Gelman BB, Saksena NK. Independent evolution of human immunodeficiency virus (HIV) drug resistance mutations in diverse areas of the brain in HIV-infected patients, with and without dementia, on antiretroviral treatment. J Virol. 2004;78:10133–10148. doi: 10.1128/JVI.78.18.10133-10148.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Choo EF, Leake B, Wandel C, Imamura H, Wood AJ, Wilkinson GR, Kim RB. Pharmacological inhibition of P-glycoprotein transport enhances the distribution of HIV-1 protease inhibitors into brain and testes. Drug Metab Dispos. 2000;28:655–660. [PubMed] [Google Scholar]
- 8.Kim RB, Fromm MF, Wandel C, Leake B, Wood AJ, Roden DM, Wilkinson GR. The drug transporter P-glycoprotein limits oral absorption and brain entry of HIV-1 protease inhibitors. J Clin Invest. 1998;101:289–294. doi: 10.1172/JCI1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee CG, Gottesman MM, Cardarelli CO, Ramachandra M, Jeang KT, Ambudkar SV, Pastan I, Dey S. HIV-1 protease inhibitors are substrates for the MDR1 multidrug transporter. Biochemistry. 1998;37:3594–3601. doi: 10.1021/bi972709x. [DOI] [PubMed] [Google Scholar]
- 10.Schinkel AH, Jonker JW. Mammalian drug efflux transporters of the ATP binding cassette (ABC) family: An overview. Adv Drug Deliv Rev. 2003;55:3–29. doi: 10.1016/s0169-409x(02)00169-2. [DOI] [PubMed] [Google Scholar]
- 11.Cordon-Cardo C, O’Brien JP, Casals D, Rittman-Grauer L, Biedler JL, Melamed MR, Bertino JR. Multidrug-resistance gene (P-glycoprotein) is expressed by endothelial cells at blood–brain barrier sites. Proc Natl Acad Sci USA. 1989;86:695–698. doi: 10.1073/pnas.86.2.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huisman MT, Smit JW, Wiltshire HR, Hoetelmans RM, Beijnen JH, Schinkel AH. P-glycoprotein limits oral availability, brain, and fetal penetration of saquinavir even with high doses of ritonavir. Mol Pharmacol. 2001;59:806–813. doi: 10.1124/mol.59.4.806. [DOI] [PubMed] [Google Scholar]
- 13.Ronaldson PT, Lee G, Dallas S, Bendayan R. Involvement of P-glycoprotein in the transport of saquinavir and indinavir in rat brain microvessel endothelial and microglia cell lines. Pharm Res. 2004;21:811–818. doi: 10.1023/b:pham.0000026433.27773.47. [DOI] [PubMed] [Google Scholar]
- 14.Polli JW, Jarrett JL, Studenberg SD, Humphreys JE, Dennis SW, Brouwer KR, Woolley JL. Role of P-glycoprotein on the CNS disposition of amprenavir (141W94), an HIV protease inhibitor. Pharm Res. 1999;16:1206–1212. doi: 10.1023/a:1018941328702. [DOI] [PubMed] [Google Scholar]
- 15.Kaddoumi A, Choi SU, Kinman L, Whittington D, Tsai CC, Ho RJ, Anderson BD, Unadkat JD. Inhibition of P-glycoprotein activity at the primate blood–brain barrier increases the distribution of nelfinavir into the brain but not into the cerebrospinal fluid. Drug Metab Dispos. 2007;35:1459–1462. doi: 10.1124/dmd.107.016220. [DOI] [PubMed] [Google Scholar]
- 16.Bachmeier CJ, Spitzenberger TJ, Elmquist WF, Miller DW. Quantitative assessment of HIV-1 protease inhibitor interactions with drug efflux transporters in the blood–brain barrier. Pharm Res. 2005;22:1259–1268. doi: 10.1007/s11095-005-5271-y. [DOI] [PubMed] [Google Scholar]
- 17.Jorajuria S, Dereuddre-Bosquet N, Becher F, Martin S, Porcheray F, Garrigues A, Mabondzo A, Benech H, Grassi J, Orlowski S, Dormont D, Clayette P. ATP binding cassette multidrug transporters limit the anti-HIV activity of zidovudine and indinavir in infected human macrophages. Antivir Ther. 2004;9:519–528. [PubMed] [Google Scholar]
- 18.Washington CB, Wiltshire HR, Man M, Moy T, Harris SR, Worth E, Weigl P, Liang Z, Hall D, Marriott L, Blaschke TF. The disposition of saquinavir in normal and P-glycoprotein deficient mice, rats, and in cultured cells. Drug Metab Dispos. 2000;28:1058–1062. [PubMed] [Google Scholar]
- 19.Perloff MD, von Moltke LL, Fahey JM, Greenblatt DJ. Induction of P-glycoprotein expression and activity by ritonavir in bovine brain microvessel endothelial cells. J Pharm Pharmacol. 2007;59:947–953. doi: 10.1211/jpp.59.7.0006. [DOI] [PubMed] [Google Scholar]
- 20.Salama NN, Kelly EJ, Bui T, Ho RJ. The impact of pharmacologic and genetic knockout of P-glycoprotein on nelfinavir levels in the brain and other tissues in mice. J Pharm Sci. 2005;94:1216–1225. doi: 10.1002/jps.20344. [DOI] [PubMed] [Google Scholar]
- 21.Constantinides PP, Wasan KM. Lipid formulation strategies for enhancing intestinal transport and absorption of P-glycoprotein (P-gp) substrate drugs: In vitro/in vivo case studies. J Pharm Sci. 2007;96:235–248. doi: 10.1002/jps.20780. [DOI] [PubMed] [Google Scholar]
- 22.Dintaman JM, Silverman JA. Inhibition of P-glycoprotein by D-alpha-tocopheryl polyethylene glycol 1000 succinate (TPGS) Pharm Res. 1999;16:1550–1556. doi: 10.1023/a:1015000503629. [DOI] [PubMed] [Google Scholar]
- 23.Varma MV, Panchagnula R. Enhanced oral paclitaxel absorption with vitamin E-TPGS: Effect on solubility and permeability in vitro, in situ and in vivo. Eur J Pharm Sci. 2005;25:445–453. doi: 10.1016/j.ejps.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 24.Johnson BM, Charman WN, Porter CJ. An in vitro examination of the impact of polyethylene glycol 400, Pluronic P85, and vitamin E d-alphatocopheryl polyethylene glycol 1000 succinate on P-glycoprotein efflux and enterocyte-based metabolism in excised rat intestine. AAPS Pharm Sci. 2002;4:E40. doi: 10.1208/ps040440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Katneni K, Charman SA, Porter CJ. Impact of cremophor-EL and polysorbate-80 on digoxin permeability across rat jejunum: Delineation of thermodynamic and transporter related events using the reciprocal permeability approach. J Pharm Sci. 2007;96:280–293. doi: 10.1002/jps.20779. [DOI] [PubMed] [Google Scholar]
- 26.Zhang H, Yao M, Morrison RA, Chong S. Commonly used surfactant, Tween 80, improves absorption of P-glycoprotein substrate, digoxin, in rats. Arch Pharm Res. 2003;26:768–772. doi: 10.1007/BF02976689. [DOI] [PubMed] [Google Scholar]
- 27.Batrakova EV, Li S, Miller DW, Kabanov AV. Pluronic P85 increases permeability of a broad spectrum of drugs in polarized BBMEC and Caco-2 cell monolayers. Pharm Res. 1999;16:1366–1372. doi: 10.1023/a:1018990706838. [DOI] [PubMed] [Google Scholar]
- 28.Batrakova EV, Miller DW, Li S, Alakhov VY, Kabanov AV, Elmquist WF. Pluronic P85 enhances the delivery of digoxin to the brain: In vitro and in vivo studies. J Pharmacol Exp Ther. 2001;296:551–557. [PubMed] [Google Scholar]
- 29.Zastre J, Jackson J, Burt H. Evidence for modulation of P-glycoprotein-mediated efflux by methoxypolyethylene glycol-block-Polycaprolactone amphiphilic diblock copolymers. Pharm Res. 2004;21:1489–1497. doi: 10.1023/b:pham.0000036925.45002.a2. [DOI] [PubMed] [Google Scholar]
- 30.Regev R, Katzir H, Yeheskely-Hayon D, Eytan GD. Modulation of P-glycoprotein-mediated multidrug resistance by acceleration of passive drug permeation across the plasma membrane. FEBS J. 2007;274:6204–6214. doi: 10.1111/j.1742-4658.2007.06140.x. [DOI] [PubMed] [Google Scholar]
- 31.Batrakova EV, Li S, Vinogradov SV, Alakhov VY, Miller DW, Kabanov AV. Mechanism of pluronic effect on P-glycoprotein efflux system in blood–brain barrier: Contributions of energy depletion and membrane fluidization. J Pharmacol Exp Ther. 2001;299:483–493. [PubMed] [Google Scholar]
- 32.Sarkadi B, Price EM, Boucher RC, Germann UA, Scarborough GA. Expression of the human multidrug resistance cDNA in insect cells generates a high activity drug-stimulated membrane ATPase. J Biol Chem. 1992;267:4854–4858. [PubMed] [Google Scholar]
- 33.Batrakova EV, Li S, Alakhov VY, Miller DW, Kabanov AV. Optimal structure requirements for pluronic block copolymers in modifying P-glycoprotein drug efflux transporter activity in bovine brain microvessel endothelial cells. J Pharmacol Exp Ther. 2003;304:845–854. doi: 10.1124/jpet.102.043307. [DOI] [PubMed] [Google Scholar]
- 34.Sharom FJ, Yu X, Lu P, Liu R, Chu JW, Szabo K, Muller M, Hose CD, Monks A, Varadi A, Seprodi J, Sarakadi B. Interaction of the P-glycoprotein multidrug transporter (MDR1) with high affinity peptide chemosensitizers in isolated membranes, reconstituted systems, and intact cells. Biochem Pharmacol. 1999;58:571–586. doi: 10.1016/s0006-2952(99)00139-2. [DOI] [PubMed] [Google Scholar]
- 35.Batrakova EV, Li S, Li Y, Alakhov VY, Kabanov AV. Effect of pluronic P85 on ATPase activity of drug efflux transporters. Pharm Res. 2004;21:2226–2233. doi: 10.1007/s11095-004-7675-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Garrigos M, Mir LM, Orlowski S. Competitive and non-competitive inhibition of the multidrug-resistance-associated P-glycoprotein ATPase—Further experimental evidence for a multisite model. Eur J Biochem. 1997;244:664–673. doi: 10.1111/j.1432-1033.1997.00664.x. [DOI] [PubMed] [Google Scholar]
- 37.Orlowski S, Selosse MA, Boudon C, Micoud C, Mir LM, Belehradek J, Jr, Garrigos M. Effects of detergents on P-glycoprotein atpase activity: Differences in perturbations of basal and verapamil-dependent activities. Cancer Biochem Biophys. 1998;16:85–110. [PubMed] [Google Scholar]
- 38.Zhang Y, Benet LZ. Characterization of P-glycoprotein mediated transport of K02, a novel vinylsulfone peptidomimetic cysteine protease inhibitor, across MDR1-MDCK and Caco-2 cell monolayers. Pharm Res. 1998;15:1520–1524. doi: 10.1023/a:1011990730230. [DOI] [PubMed] [Google Scholar]
- 39.Williams GC, Knipp GT, Sinko PJ. The effect of cell culture conditions on saquinavir transport through, and interactions with, MDCKII cells over-expressing hM DR1. J Pharm Sci. 2003;92:1957–1967. doi: 10.1002/jps.10458. [DOI] [PubMed] [Google Scholar]
- 40.Schmittgen TD, Zakrajsek BA, Mills AG, Gorn V, Singer MJ, Reed MW. Quantitative reverse transcription-polymerase chain reaction to study mRNA decay: Comparison of endpoint and real-time methods. Anal Biochem. 2000;285:194–204. doi: 10.1006/abio.2000.4753. [DOI] [PubMed] [Google Scholar]
- 41.Chandler B, Detsika M, Owen A, Evans S, Hartkoorn RC, Cane PA, Back DJ, Khoo SH. Effect of transporter modulation on the emergence of nelfinavir resistance in vitro. Antivir Ther. 2007;12:831–834. [PubMed] [Google Scholar]
- 42.Williams GC, Liu A, Knipp G, Sinko PJ. Direct evidence that saquinavir is transported by multidrug resistance-associated protein (MRP1) and canalicular multispecific organic anion transporter (MRP2) Antimicrob Agents Chemother. 2002;46:3456–3462. doi: 10.1128/AAC.46.11.3456-3462.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Honda Y, Ushigome F, Koyabu N, Morimoto S, Shoyama Y, Uchiumi T, Kuwano M, Ohtani H, Sawada Y. Effects of grapefruit juice and orange juice components on P-glycoprotein- and MRP2-mediated drug efflux. Br J Pharmacol. 2004;143:856–864. doi: 10.1038/sj.bjp.0706008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Batrakova EV, Li S, Alakhov VY, Elmquist WF, Miller DW, Kabanov AV. Sensitization of cells overexpressing multidrug-resistant proteins by pluronic P85. Pharm Res. 2003;20:1581–1590. doi: 10.1023/a:1026179132599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Park S, Sinko PJ. P-glycoprotein and mutlidrug resistance-associated proteins limit the brain uptake of saquinavir in mice. J Pharmacol Exp Ther. 2005;312:1249–1256. doi: 10.1124/jpet.104.076216. [DOI] [PubMed] [Google Scholar]
- 46.Miller DW, Batrakova EV, Kabanov AV. Inhibition of multidrug resistance-associated protein (MRP) functional activity with pluronic block copolymers. Pharm Res. 1999;16:396–401. doi: 10.1023/a:1018873702411. [DOI] [PubMed] [Google Scholar]
- 47.Goh LB, Spears KJ, Yao D, Ayrton A, Morgan P, Roland Wolf C, Friedberg T. Endogenous drug transporters in in vitro and in vivo models for the prediction of drug disposition in man. Biochem Pharmacol. 2002;64:1569–1578. doi: 10.1016/s0006-2952(02)01355-2. [DOI] [PubMed] [Google Scholar]
- 48.Tanigawara Y, Okamura N, Hirai M, Yasuhara M, Ueda K, Kioka N, Komano T, Hori R. Transport of digoxin by human P-glycoprotein expressed in a porcine kidney epithelial cell line (LLC-PK1) J Pharmacol Exp Ther. 1992;263:840–845. [PubMed] [Google Scholar]
- 49.Ueda K, Okamura N, Hirai M, Tanigawara Y, Saeki T, Kioka N, Komano T, Hori R. Human P-glycoprotein transports cortisol, aldosterone, and dexamethasone, but not progesterone. J Biol Chem. 1992;267:24248–24252. [PubMed] [Google Scholar]
- 50.Hamada A, Miyano H, Watanabe H, Saito H. Interaction of imatinib mesilate with human P-glycoprotein. J Pharmacol Exp Ther. 2003;307:824–828. doi: 10.1124/jpet.103.055574. [DOI] [PubMed] [Google Scholar]
- 51.Videmann B, Tep J, Cavret S, Lecoeur S. Epithelial transport of deoxynivalenol: Involvement of human P-glycoprotein (ABCB1) and multidrug resistance-associated protein 2 (ABCC2) Food Chem Toxicol. 2007;45:1938–1947. doi: 10.1016/j.fct.2007.04.011. [DOI] [PubMed] [Google Scholar]
- 52.Sachs-Barrable K, Thamboo A, Lee SD, Wasan KM. Lipid excipients Peceol and Gelucire 44/14 decrease P-glycoprotein mediated efflux of rhodamine 123 partially due to modifying P-glycoprotein protein expression within Caco-2 cells. J Pharm Pharm Sci. 2007;10:319–331. [PubMed] [Google Scholar]
- 53.Sriadibhatla S, Yang Z, Gebhart C, Alakhov VY, Kabanov A. Transcriptional activation of gene expression by pluronic block copolymers in stably and transiently transfected cells. Mol Ther. 2006;13:804–813. doi: 10.1016/j.ymthe.2005.07.701. [DOI] [PubMed] [Google Scholar]
- 54.Yang Z, Zhu J, Sriadibhatla S, Gebhart C, Alakhov V, Kabanov A. Promoter- and strain-selective enhancement of gene expression in a mouse skeletal muscle by a polymer excipient Pluronic P85. J Control Release. 2005;108:496–512. doi: 10.1016/j.jconrel.2005.08.015. [DOI] [PubMed] [Google Scholar]
- 55.Batrakova EV, Kelly DL, Li S, Li Y, Yang Z, Xiao L, Alakhova DY, Sherman S, Alakhov VY, Kabanov AV. Alteration of genomic responses to doxorubicin and prevention of MDR in breast cancer cells by a polymer excipient: Pluronic P85. Mol Pharm. 2006;3:113–123. doi: 10.1021/mp050050g. [DOI] [PMC free article] [PubMed] [Google Scholar]