

Abstract
Hypoxia is a prominent characteristic of many acute or chronic inflammatory diseases, and exerts significant influence on their progression. Macrophages and neutrophils are major cellular components of innate immunity and contribute not only to O2 deprivation at the site of inflammation, but also alter many of their functions in response to hypoxia to either facilitate or suppress inflammation. Hypoxia stabilizes HIF-αs in macrophages and neutrophils, and these O2-sensitive transcription factors are key regulators of inflammatory responses in myeloid cells. In this review, we will summarize our current understanding of the role of HIF-αs in shaping macrophage and neutrophil functions in the pathogenesis and progression of multiple inflammatory diseases.
Inflammation and hypoxia
Inflammation constitutes the body’s defensive response to injury and/or infection in order to eliminate pathogens and damaged tissue, while initiating tissue repair and healing. Upon tissue injury or pathogen invasion, local sentinel cells such as resident macrophages and mast cells respond to dilate blood vessels, increase vascular permeability, and recruit a variety of leukocytes to the site of inflammation. During the acute phase of inflammatory responses, a major task of such recruited cells is the clearance of damaged tissue or pathogens. Upon transition into the resolution phase, tissue homeostasis is gradually restored. If acute inflammation fails to subside, it progresses into chronic inflammation with potentially serious consequences for the afflicted patient (1, 2). One feature of inflammation sites is low oxygen (O2) tension, termed “hypoxia.” Oxygen tension ranges between 2.5% and 9% in most healthy tissues. However, poor O2 availability resulting from damaged vasculature, high metabolic rates of bacteria and other pathogens, and numerous infiltrating immune cells deprive inflamed tissue of O2, frequently leading to partial O2 pressures (pO2) of less than 1% (3, 4).
The interdependence between inflammation and hypoxia has been evident for many years. Hypoxia is prevalent in multiple inflammatory scenarios, such as inflammatory bowel diseases (IBDs) and rheumatoid arthritis (RA) (5–8). The intestinal mucosa exhibits an O2 gradient from crypt to villus, wherein O2 is highest in the crypts and lowest in the villus tips, which are closest to the anoxic gut lumen (9). This “physiological” hypoxia is largely extended with intestinal inflammation (7, 8). Hypoxia is also a characteristic of inflamed joints in patients with RA. Using a highly sensitive gold microelectrode, investigators accurately measured synovial O2 tension in RA patients (5), demonstrating that RA median O2 tension in synovial tissue (2%–4%) was much lower than that in the noninflamed synovium (9%–12%) (5, 6).
Low O2 tension can also directly contribute to inflammation. In the setting of obesity, hypoxia develops as adipose tissue mass expands, initiating inflammatory responses. Secretion of inflammation-related adipokines (e.g., TNF-α and leptin) increases in hypoxic adipose tissue. Together with additional disruptions in glucose and lipid metabolism, this inflammation can become chronic and systemic, eventually leading to insulin resistance (10, 11). In the lung, alveolar hypoxia can be induced by exposing rats to 10% O2 for up to 8 hours, which triggers macrophage recruitment, enhances expression of HIF-1α and inflammatory cytokines (e.g., macrophage inflammatory protein 1-α [MIP-1α], monocyte chemoattractant protein-1 [MCP-1], and TNF-α), promotes NF-κB activity, and elevates albumin leakage (12). Similar observations were made in mice exposed to 5% O2 for 60 minutes, where levels of IL-6, TNF-α, and IL-1α were elevated in serum and isolated peritoneal macrophages and Kupffer cells (liver macrophages) (13). In humans, hypoxia-induced inflammation is evident in individuals with high-altitude illness. Those who ascend rapidly are at risk of developing high-altitude pulmonary and cerebral edema, caused by hypoxic pulmonary vasoconstriction, high arterial and capillary pressure, and elevated levels of circulating IL-6, IL-1 receptor antagonist (IL-1RA), and C-reactive protein (CRP) (14, 15). All of these examples indicate that the relationship between inflammation and hypoxia exists in many pathological settings, and is a potentially attractive therapeutic target.
Hypoxia and hypoxia-inducible factors
In many of the pathological situations described above (5, 7, 8, 12, 16, 17), HIFs are activated in response to the hypoxic and inflammatory microenvironment. HIFs represent the primary O2-sensing transcription factors (18–21) as heterodimers comprised of an O2-sensitive α subunit (HIF-α) and a constitutively expressed β subunit (HIF-1β or aryl hydrocarbon receptor nuclear translocator [ARNT]). Three α subunits have been discovered thus far: HIF-1α, HIF-2α, and HIF-3α. While HIF-1α and HIF-2α are well characterized, relatively little is known about HIF-3α (22, 23). The HIF3A gene encodes multiple HIF-3α variants, which are structurally distinct from HIF-1α and HIF-2α, as they lack a C-terminal transactivation domain. Divergence in structure and variant diversity allow HIF-3α to have numerous modes of action, regulating a transcriptional program that is distinct from that of HIF-1α (24). In this review, we will focus on HIF-1α and HIF-2α, but additional details about HIF-3α are reviewed elsewhere (23–25). Under normoxia, the O2-sensitive α subunit is hydroxylated on two conserved proline residues (P402/P405 and P564/P531 for HIF-1α/HIF-2α, respectively) within the O2-dependent degradation domain (ODD) by prolyl hydroxylase domain–containing proteins (PHDs) (22, 26). Hydroxylated HIF-α subunits are then polyubiquitinated by the von Hippel-Lindau (VHL) tumor suppressor E3 ubiquitin ligase complex and subsequently degraded via the 26S proteasome (27–29). Under hypoxia, PHDs cannot hydroxylate key HIF-α proline residues due to limited access to their substrate (O2) or redox imbalance (21, 30–32), resulting in HIF-α stabilization. Stabilized HIF-αs translocate into the nucleus, dimerize with their obligate binding partner ARNT, recruit additional coactivators, and bind to hypoxia-response elements (HREs) to enhance transcription of hundreds of genes whose products mediate cellular adaptation to hypoxia. Such pathways include metabolism, angiogenesis, and inflammatory responses.
Other than O2-dependent HIF posttranslational modifications, HIF-α stabilization can be induced by inflammatory stimuli independently of hypoxia. The proinflammatory cytokines TNF-α and IL-1β promote HIF-1α accumulation in an NF-κB–dependent manner (33–35). Bacterial products, such as LPS, can also stabilize HIF-1α under normoxia through multiple pathways, such as NF-κB (36, 37), ROS (38), PHDs (39), and MAPKs (40). On the other hand, hypoxic responses can also be HIF independent. For example, hypoxia suppresses mTOR activity independently of HIF signaling (41), via the mTOR inhibitor REDD1 and the TSC1/TSC2 complex (42). Other hypoxia-responsive pathways include endoplasmic reticulum (ER) stress (43) and NF-κB (37, 44, 45) pathways. One myeloid-specific example is mentioned later in this review (46).
Myeloid cells in inflammation
Macrophages are key cellular components of innate immunity and encompass a highly heterogeneous population of cells with a broad array of phenotypes and functions. Some of these cells are distributed over most of the body, residing in many tissues (e.g., Kupffer cells in the liver, osteoclasts in the bone, and microglia in the brain), while others are differentiated monocytes that infiltrate sites of inflammation to promote adaptive responses or facilitate restoration of tissue homeostasis (47, 48). Upon pathogen invasion or injury, tissue-resident macrophages represent the first responders, recruiting neutrophils via secretion of chemokines (e.g., IL-8 in humans and CXCL1 in mice). Once neutrophils arrive at the compromised site, they release monocyte chemoattractants (e.g., MCP-1) so that large numbers of recruited monocytes/macrophages extend the inflammatory response (49–51). These macrophages normally adopt a proinflammatory or “classically activated” (M1) phenotype, which is often induced by IFN-γ and Toll-like receptor ligands. M1 macrophages elevate their secretion of reactive oxygen and nitrogen species (ROS and NOS) and proinflammatory cytokines, to eliminate pathogens and damaged tissues while recruiting additional immune effector cells. When most of the pathogens or tissue debris are removed, hyperactivation of macrophage bactericidal activity may result in unnecessary destruction of healthy tissue. As highly plastic cells, macrophages then respond to microenvironmental cues (e.g., TH2-type cytokines IL-4 and IL-13) and adopt an “alternatively activated” (M2) phenotype, which suppresses host defenses and facilitates wound healing and tissue remodeling to resolve inflammation and restore homeostasis at the inflamed site (52–55). This oversimplified segregation of macrophage phenotypes was originally applied to in vitro systems and has been widely used to provide a conceptual framework for subsequent research. However, given the complexity of in vivo microenvironments, macrophages exhibit phenotypes across a broad spectrum of activation states, and the simple M1/M2 dichotomy is unlikely to reflect physiological macrophage phenotypes (56–58).
Macrophages are crucial components in the pathogenesis of many inflammatory diseases, including atherosclerosis (59–61), IBDs (62–64), RA (65–68), and airway inflammation/asthma (49, 69–72). Lipid-laden macrophages are typically observed at atherosclerotic plaques. These maladaptive macrophages can induce a nonresolving inflammatory response leading to robust accumulation of cells, lipid, and matrix at the plaque. Defective macrophage efferocytosis (engulfment of dead cells) and enhanced apoptosis contribute to formation of a necrotic plaque core that might eventually rupture, causing platelet aggregation and thrombus formation (59, 60). The functional importance of myeloid cells in atherosclerosis is supported by experimental evidence that interventions to alter monocyte recruitment and/or survival can markedly affect disease progression (73–75). Airway inflammation typically accompanies airway allergic asthma, another disease involving macrophages. The microenvironment in asthma is dominated by type 2–associated cytokines (e.g., IL-4 and IL-13), which preferentially polarize macrophages into the M2 state (49). Elevated numbers of IL-4R+ macrophages have been reported in asthmatic patients with defective lung function (76). Moreover, the presence of IL-4R+ macrophages exacerbates allergen-induced airway inflammation, whereas reduction of IL-4R+ macrophages alleviates this disease (77, 78).
Neutrophils, another major component of the innate immune response, are among the first cells recruited to inflammatory sites. These cells possess multiple means of eliminating invading pathogens, i.e., phagocytosis of microorganisms, degranulation to release antibacterial proteins, and emanation of neutrophil extracellular traps (NETs) (79, 80). Recently, many properties of neutrophils favoring the resolution of inflammation have been revealed (81), including production of annexin A1 (82) and lipid (e.g., LXA4 and 13-series resolvins) proresolution mediators (83, 84), chemokine/cytokine scavenging (e.g., CCL3 and CCL5) (85), and apoptosis-induced macrophage efferocytosis (86, 87). Similarly to macrophages, neutrophils are associated with multiple inflammatory syndromes, such as RA (88–90), chronic obstructive pulmonary disease (91, 92), and IBDs (51, 93). In IBDs, for example, neutrophils contribute to elimination of pathogens and immune cell (e.g., macrophages) recruitment and activation, as well as mucosal wound healing and resolution of inflammation. Of note, precise roles of neutrophils during intestinal inflammation are currently under investigation and are highly debated (51, 94). Some studies using colitis models, either chemically induced (dextran sulfate sodium [DSS] or dinitrobenzene sulfonic acid/trinitrobenzene sulfonic acid [DNBS/TNBS]) or immune system dysregulation–induced (CD4+CD45RBhi T cell transfer), showed that neutrophil depletion exacerbates colitis, suggesting a beneficial role of neutrophils in this setting (95, 96); however, other studies showed a completely opposite phenotype in which neutrophil depletion ameliorates colitis (97, 98). Additionally, the role of neutrophils could also depend on the concomitant presence of monocytes and macrophages (99). Therefore, while neutrophils are clearly associated with intestinal inflammation, whether they exert beneficial or detrimental effects appears to be model dependent and condition dependent.
HIFs in myeloid cells
It is noteworthy that myeloid cells localize predominantly within hypoxic subdomains of tumors and sites of inflammation, and multiple mechanisms have been proposed to explain how hypoxia promotes recruitment and retention of myeloid cells (100). Both HIF-1α and HIF-2α regulate myeloid migratory activity: HIF-1α is recruited to the CXCR4 promoter, stimulating CXCR4 transcription in human monocytes experiencing hypoxia (101). Moreover, CXCR4 is a key chemokine receptor mediating chemotactic responses to CXCL12 ligand, which is upregulated in ischemic tissues such as arthritic joints (102). The HIF-1α/PDK1 axis has been recently shown to contribute to macrophage migratory activity via induction of active glycolysis (103). For HIF-2α, Casazza and colleagues demonstrated a semaphorin 3A/neuropilin1–dependent (SEMA3A/NRP1-dependent) means of macrophage positioning within the tumor. Here, NRP1 repression, which triggers macrophage retention in hypoxic regions, is mediated by HIF-2α–dependent NF-κB activity (104).
Given the preferential localization of myeloid cells in hypoxic regions, significant efforts have defined how HIF-1α and/or HIF-2α promote myeloid cell adaptation to hypoxic environments and mediate inflammation (Figure 1). Cramer and colleagues were the first to demonstrate the importance of HIF-1α in macrophage and neutrophil function in the setting of inflammation (105). HIF-1α was ablated in myeloid cells using lysozyme M (LysM) promoter–driven Cre recombinase, which is specific for the myeloid lineages, i.e., monocytes, macrophages, neutrophils, etc. Myeloid-specific Hif1a deletion results in defective glycolysis and ATP generation, leading to impairment of myeloid cell motility, invasiveness, aggregation, and bacterial killing. Moreover, mice with myeloid-specific HIF-1α deficiency are protected against acute and chronic cutaneous inflammation and arthritis. Subsequent studies investigating HIF-1α specifically in neutrophils demonstrated that hypoxia-induced inhibition of neutrophil apoptosis is dependent on HIF-1α (106), and that HIF-1α is required for phagocytes to fully exert their bactericidal activity (107). More recent attention has focused on myeloid cell immunometabolism (108, 109). Myeloid cells can undergo metabolic reprogramming to adapt to critical changes in the microenvironment. Tannahill and colleagues demonstrated that LPS exposure can alter glutamine-dependent anaplerosis (replenishment of TCA cycle intermediates) to elevate succinate levels, which further stabilize HIF-1α in macrophages, resulting in increased production of IL-1β (110). These findings serve as an excellent example of how HIF-1α–dependent immunometabolism can directly affect cytokine production by macrophages.
Figure 1. Overview of the roles of HIF-1α and HIF-2α in myeloid cells.
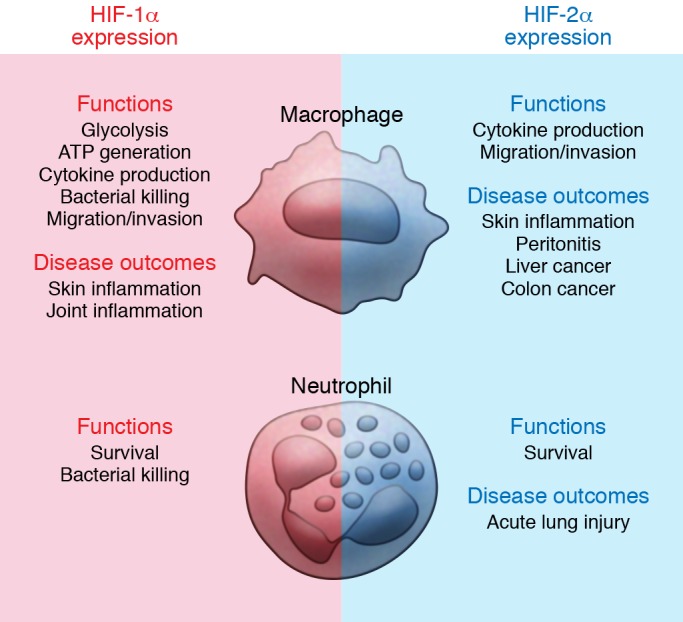
Both HIF-1α and HIF-2α are required for key macrophage functions, such as cytokine production and the ability to migrate and invade. However, macrophage glycolysis, ATP generation, and bactericidal activity have been related to HIF-1α exclusively. Nevertheless, both isoforms contribute to pathogenesis of various acute inflammatory syndromes. Additionally, the roles of myeloid HIF-αs in the setting of tumor inflammation are currently being investigated. As compared with macrophages, less is known about HIF-αs in neutrophils. However, it is very clear that both isoforms are required to inhibit neutrophil apoptosis and elongate their lifespan. While HIF-1α facilitates bacterial killing by neutrophils, many neutrophil functions seem less dependent on HIF-2α, including respiratory burst, chemotaxis, and phagocytosis. Nevertheless, increased neutrophil HIF-2α accumulation correlates with increased neutrophilic inflammation and lung injury in an LPS-induced acute lung injury murine model.
The role of HIF-2α in myeloid cells has also been investigated using the LysM-Cre–mediated deletion strategy. Macrophages lacking HIF-2α exhibit defects in the production of inflammatory cytokines/chemokines in response to hypoxia, migration, and invasion. Myeloid HIF-2α deficiency also protects mice in models of sepsis, cutaneous inflammation, peritonitis, hepatocellular carcinoma, and colitis-associated colorectal cancer (CAC) (111). Like HIF-1α, neutrophil HIF-2α contributes to hypoxia-induced inhibition of apoptosis. HIF-2α deficiency increases neutrophil apoptosis in vivo and ex vivo, leading to suppression of neutrophilic inflammation and inflammatory responses during acute lung injury (112).
One interesting observation from the work of Imtiyaz and colleagues (111) is that, unlike myeloid HIF-1α, HIF-2α deficiency does not alter cellular ATP production. The notion that HIF-1α and HIF-2α exert nonredundant or even opposing functions in macrophages is further supported by a study showing that HIF-1α and HIF-2α differentially regulate NO production by controlling expression of iNOS and arginase 1, respectively (113). Given the complex roles of HIF-1α and HIF-2α in macrophages, pan-HIF inhibition via pharmacological or genetic methods (i.e., Arnt deletion) is warranted. In a murine CAC model, treatment with the HIF inhibitor acriflavine reduces both tumor burden and macrophage infiltration (114). Additionally, myeloid cell–specific ARNT deficiency reduces macrophage proinflammatory cytokine production, and mice lacking myeloid ARNT are protected from cutaneous inflammation and exhibit delayed wound healing (115).
In the following sections, we will discuss the roles of myeloid HIF-αs in the settings of specific inflammatory diseases, as summarized in Table 1.
Table 1. Summary of myeloid HIF’s role in various inflammatory scenarios.

Atherosclerosis.
Atherosclerosis is a chronic inflammatory disease of the arterial vasculature. Retention of apolipoprotein B–containing lipoproteins and accumulation of cholesterol-laden macrophages in the artery wall contribute to this syndrome. Monocytes are first recruited to differentiate into mononuclear phagocytes and ingest lipoproteins; however, lipid buildup in these cells transforms them into foam cells that exhibit dysregulated lipid metabolism and elevated secretion of proinflammatory cytokines (e.g., IL-6 and TNF-α) and macrophage retention factors (i.e., netrin 1 and semaphorin 3E). Foam cells promote the further progression of atherosclerosis (59, 60).
Elevated levels of HIF-1α and HIF-2α are detected in human atherosclerotic carotid plaques compared with normal arteries, where HIF-1α colocalizes with CD68, a macrophage marker (116). Hypoxia has been implicated as a pathogenic factor in atherosclerosis and contributes to the proatherosclerotic functions of macrophages (Figure 2A) (117). Several reports showed that lipid uptake and foam cell formation are dependent on hypoxia and HIF-1α (118–120). Both murine and human macrophage cell lines increase cellular neutral lipid content when cultured under hypoxic conditions; however, this effect is reversed upon HIF-1α depletion. Multiple HIF-1α–dependent mechanisms have been proposed for this phenotype. For example, hypoxia enhances expression of lectin-like oxidized low-density lipoprotein (oxLDL) receptor-1 (LOX-1), which promotes oxLDL uptake in the murine macrophage cell line RAW264.7; silencing of HIF-1α diminishes the upregulation of LOX-1 (119). Hypoxic J774 murine macrophages exhibit elevated sterol accumulation due to (a) enhanced sterol synthesis via increased 3-hydroxy-3-methyl-glutaryl-CoA (HMG-CoA) reductase activity and (b) suppressed cholesterol efflux due to altered subcellular localization of ATP-binding cassette subfamily A member 1 (ABCA1) (120). These phenotypic changes are also HIF-1α dependent. In U937 human monocytes, oxLDL treatment increases the expression of 70 out of 96 key genes that are known to be involved in atherosclerosis, while 57 of these genes (e.g., cyclooxygenase-2 [COX-2], vascular cell adhesion molecule [VCAM-1], and IL-1β) are downregulated with HIF-1α siRNA pretreatment (118). Other proatherosclerotic functions of macrophages, such as promotion of angiogenesis and proteoglycan synthesis, are also dependent on HIF-αs (121, 122). In a coculture system of human monocytes/macrophages and endothelial cells, oxLDL strongly induces HIF1A and VEGFA expression in macrophages, while increasing endothelial cell tube formation. Of note, oxLDL proangiogenic effects are partially lost upon HIF-1α inhibition (121). The notion that myeloid HIF-1α can promote angiogenesis through VEGF upregulation is demonstrated in other studies as well (123). Macrophages can also contribute to pathogenesis by secreting proteoglycans such as versican, which modulate lipoprotein retention and the activity of enzymes, cytokines, and other growth factors in atherosclerotic lesions. Increased versican and perlecan expression is detected in macrophages under hypoxia; versican is coregulated by HIF-1α and HIF-2α, while perlecan is only dependent on HIF-1α (122). Myeloid HIF-1α is also a critical regulator of both glycolytic metabolism and proinflammatory activation of macrophages, and is stabilized by cues in the atherosclerotic microenvironment, such as hypoxia and cytokines. HIF-1α increases transcription of the gene encoding 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 (PFKFB3), a key enzyme in the glycolytic pathway, leading to (a) increased glycolytic flux, (b) increased proinflammatory cytokine production (e.g., TNF-α), and (c) maintenance of macrophage viability (124). Together, these studies reveal that HIF-αs are crucial components in determining macrophage proatherosclerotic functions.
Figure 2. Context-dependent myeloid HIF-α effector functions.

Myeloid HIF-1α and HIF-2α exhibit diverse functions that differ in distinct pathological settings. The two isoforms sometimes work in a similar fashion, but can also oppose each other. (A) In the setting of atherosclerosis, both myeloid HIF-1α and HIF-2α contribute to pathogenesis. HIF-1α promotes lipid uptake in macrophages through induction of LOX-1. Elevation in HMG-CoA reductase activity and surface ABCA1 perinuclear relocation downstream of HIF-1α increases cholesterol synthesis while simultaneously blocking cholesterol efflux. Through VEGF production, myeloid HIF-1α also facilitates oxLDL’s proangiogenic effects. Regulation of PFKFB3 by HIF-1α enhances glycolytic flux and is crucial for both viability and proinflammatory activation of macrophages. Both isoforms contribute to proteoglycan secretion. (B) In adipose tissue of obese subjects, ATM HIF-1α enhances inflammation via induction of hypoxic and proinflammatory genes, while ATM HIF-2α alleviates insulin resistance and adipose tissue inflammation. ATM HIF-2α not only suppresses proinflammatory responses in ATM via induction of arginase 1 (ARG1) expression, but also sensitizes adipocytes to insulin signaling while inhibiting proinflammatory gene transcription. Abbreviations: oxLDL, oxidized low-density lipoprotein; LOX-1, lectin-like oxLDL receptor-1; ABCA1, ATP-binding cassette subfamily A member 1; PFKFPB3, 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3; Glut1, glucose transporter 1; Saa, serum amyloid A.
Unfortunately, in vivo studies do not always provide consistent findings. In a wire-induced vascular injury model, myeloid HIF-1α promotes vascular inflammation and remodeling manifested by increases in TNF-α and IL-6 levels proximal to the injury site and neointimal thickening of injured arteries (125). However, a recent in vivo genetic and drug-based approach suggested the opposite effect in a different mouse model, indicating that HIF-1α and HIF-2α accumulation correlates with reduced atherosclerosis development. The authors inhibited PHD2, resulting in HIF-1α and HIF-2α stabilization, by administering a pharmacological inhibitor (FG-4497) in an LDL receptor–deficient model of atherosclerosis or by crossing Hif-p4h-2 hypomorphic (Hif-p4h-2gt/gt) mice with LDL receptor–deficient mice. PHD2 inhibition led to reductions in levels of atherosclerotic plaque formation, weight gain, insulin resistance, liver and white adipose tissue (WAT) mass, adipocyte size, number of inflammation-associated WAT macrophage aggregates, and high-fat diet–induced increases in serum cholesterol levels (126). The discrepancy with previous findings could be due to non–myeloid-specific inhibition of PHD2 in vivo. As such, the in vivo role of myeloid HIF signaling in atherosclerosis requires further investigation.
Adipose tissue inflammation and obesity.
Adipose tissue hypoxia, chronic inflammation, and macrophage infiltration are key characteristics of obesity (127–130). In lean mice, a majority of the adipose tissue macrophages (ATMs) are alternatively activated M2 macrophages, which suppress proinflammatory responses and maintain adipocyte insulin sensitivity by elevated expression of arginase 1 and IL-10, among other factors. In the setting of obesity, the number of macrophages is increased and their phenotype altered. Many macrophages are in a classically activated (M1) state, which produces NO and secretes proinflammatory cytokines, such as IL-1β, TNF-α, and IL-6. These cytokines potentiate inflammatory responses in adipose tissue that eventually result in insulin resistance (129, 131–133). However, a shift in macrophage polarization in lean versus obese humans is debated. Aron-Wisnewsky and colleagues reported a more M1 than M2 polarization of macrophages (defined by CD40 and CD206 expression, respectively) in obese patients, which shifts to a less proinflammatory profile after weight loss (134). A more recent study, however, demonstrated that even though macrophage numbers increase in adipose tissue of obese patients, most of these ATMs are predominantly M2 macrophages (defined by CD163 and IL-10 expression) (135). These contrasting observations may be a consequence of the oversimplified dichotomy of macrophage polarization (see above). In the work of Wentworth and colleagues, ATMs were found to be positive for both M1 (CD11c) and M2 (CD206) markers, exhibiting a proinflammatory status associated with insulin resistance in obese humans (136).
In obese patients, higher HIF-1α levels are evident in adipose tissue (137), and hypoxia and HIF signaling regulate ATM functions in the setting of obesity (Figure 2B). Fujisaka and colleagues showed that adipose tissue hypoxia induces proinflammatory phenotypes of M1 ATMs, with elevated expression levels of proinflammatory cytokines and hypoxia-related genes (138). In contrast with HIF-1α’s proinflammatory roles in ATMs, macrophage HIF-2α has been suggested to ameliorate adipose tissue inflammation and insulin resistance (139). Choe and colleagues demonstrated that HIF-2α overexpression in macrophages represses NO production and expression of proinflammatory cytokine genes. On the other hand, silencing HIF-2α in palmitate-treated macrophages increases NO production, indicating that HIF-2α is required to downregulate palmitate-induced NO production. Macrophage HIF-2α also regulates the crosstalk between macrophages and adipocytes. Adipocytes cocultured with wild-type macrophages exhibit decreased insulin signaling, while coculture with HIF-2α–deficient macrophages not only reverses the decrease in insulin signaling, but also stimulates adipocyte proinflammatory responses. In a murine model of high-fat diet–induced obesity, HIF-2α haplodeficient (Epas1+/–, “Hif2a+/–” herein) mice were more susceptible to adipose tissue inflammation and became insulin resistant. Upon macrophage depletion, both insulin resistance and adipose tissue inflammation improved in this model (139). In summary, myeloid HIF-1α promotes adipose tissue inflammation by aiding macrophage M1 polarization, while myeloid HIF-2α constrains the inflammatory response and insulin resistance in adipose tissue. These conclusions are consistent with the understanding that different polarization states of macrophages exert opposite effects on adipose tissue inflammation, and are also consistent with the notion that HIF-1α is required for M1 polarization of macrophages, and HIF-2α for M2 polarization (113). However, another study suggests that hypoxia potentiates palmitate-induced expression of the proinflammatory genes IL-6 and IL-1β independently of HIF-1α and HIF-2α in human macrophages. Instead, their induction occurs via activation of JNK and p38 MAPK signaling (46). Another group proposed that insulin resistance and metabolic dysregulation in obese mice are mainly regulated by adipocyte HIF-2α, but not myeloid HIF-2α (140). Clearly, additional effort is needed to determine the extent of HIF-α–mediated regulation of ATM phenotypes and whether HIF-α–dependent ATM phenotypic changes are sufficient to alter adipose tissue inflammation and obesity.
Sepsis.
Sepsis is a life-threatening systemic illness that is normally induced by microbial infection and may result in fatal multi-organ failure in patients. Hyperactivation of the innate immune system is believed to be a key component of this pathophysiology. Macrophages and neutrophils release cytokines, chemokines, and complement-activation mediators soon after the initial microbial stimuli (141–143). LPS, a lipoglycan found in the outer membrane of gram-negative bacteria and often used to induce murine sepsis or endotoxemia, has been shown to stabilize macrophage HIF-1α via p42/44 MAPK and NF-κB signaling pathways (40). HIF-1α subsequently promotes macrophage in vitro production of proinflammatory cytokines such as TNF-α, IL-6, IL-1β, IL-1α, IL-4, and IL-12. When mice with conditional Hif1a deletion in the myeloid lineage are challenged with LPS, they exhibit reduced hypothermia and hypotension, along with enhanced survival compared with mice that express myeloid HIF-1α (39). Myeloid HIF-1α deficiency is also protective in a gram-positive endotoxin-induced murine sepsis model (144). Similar to HIF-1α, deletion of myeloid HIF-2α is also protective against sepsis. Cultured bone marrow–derived macrophages (BMDMs) isolated from mice with myeloid HIF-2α deficiency also exhibit decreased proinflammatory cytokine and increased antiinflammatory cytokine production in response to LPS stimulation. Additionally, myeloid-specific HIF-2α deficiency promotes survival in LPS-challenged mice (111). Collectively, these data show that both HIF-1α and HIF-2α contribute to macrophages’ pathogenic roles in septic pathology. This conclusion is further supported by a more recent study in which 2-methoxyestradiol (2-ME2), a HIF-1α inhibitor (145), protected mice from both LPS- and cecal ligation and puncture–induced (CLP-induced) sepsis. Suppression of cytokines by 2-ME2 was observed in LPS-stimulated peritoneal macrophages, indicating that macrophage phenotypic alterations also contributed to the survival phenotype (146).
Airway allergy and asthma.
Although airway allergy is a chronic inflammatory disease primarily driven by DCs and Th2 T lymphocytes, lung macrophages have also been implicated in airway inflammation and asthma (69–71). In a house dust mite (HDM) antigen–induced experimental model of airway allergy, myeloid HIF-1α deficiency renders mice more susceptible to these stimuli (147). Toussaint and colleagues found that lung macrophage HIF-1α drives expression of immunosuppressive IL-10 to impair DC activation and Th responses (147). A similar protective effect of myeloid HIF-1α is also evident in the setting of pulmonary fungal infections. Shepardson and colleagues found that mice with myeloid HIF-1α deficiency are more susceptible to pulmonary challenge with Aspergillus fumigatus, are defective in fungal clearance, and exhibit decreased lung neutrophil numbers. These phenotypes can be partly attributed to decreased production of CXCL1 and increased neutrophil apoptosis (148). Contradictory to the finding that macrophage HIF-1α prevents airway allergy, a study by Byrne and colleagues suggests that development of airway allergy is dependent on macrophage HIF-1α. They demonstrated that HDM increases HIF-1α abundance in the lung, inducing VEGF and CXCL1 production in primary lung macrophages in a HIF-1α–dependent manner. Pharmacological HIF-1α inhibition in this model suppresses pulmonary allergic inflammation and VEGF and CXCL1 secretion (149). Using an ovalbumin-induced (OVA-induced) asthma model, others have shown that myeloid HIF-1α deficiency reduces airway hyperresponsiveness and eosinophil infiltration. Furthermore, HIF-1α and HIF-2α directly regulate eosinophil chemotaxis in opposing ways (150). Therefore, the role of myeloid HIF-1α in airway diseases remains very complex, and varies in different experimental models.
Gastritis.
Inflammation in the gastric mucosa is most commonly induced by Helicobacter pylori infection in humans. Chronic gastritis may progress to gastrointestinal ulcers or gastric cancer (151). Like other inflammatory diseases, recruitment of immune cells is also evident during gastric inflammation, and macrophage depletion using drug-loaded liposomes has been shown to ameliorate the pathology of H. pylori–induced gastritis (152). A recent report (153) specifically examined the role of myeloid HIF-1α in gastritis. The authors found that HIF-1α levels are positively correlated with the severity of gastritis in patients with H. pylori infections, and HIF-1α is readily observed in macrophages from patient biopsies. In vitro, H. pylori preferentially upregulates Hif1a and downregulates Hif2a transcription in BMDMs, while expression of proinflammatory cytokines is dependent on HIF-1α. Elevated HIF-1α levels also contribute to bactericidal activity of both neutrophils and macrophages. Interestingly, in a murine model of H. pylori–induced gastritis, mice with myeloid-specific HIF-1α deletion failed to exhibit changes in bacterial loads as compared with wild-type animals. Even though myeloid-specific HIF-1α deficiency blocks the induction of proinflammatory gene expression upon H. pylori infection, more severe gastritis is observed in these animals, characterized by worsened histopathological grading, greater immune cell infiltration, and a higher cellular proliferation index compared with infected wild-type animals (153). Overall, myeloid HIF-1α appears to be protective in H. pylori–mediated gastritis; however, additional work is needed to fully explain these counterintuitive phenotypes.
Renal fibrosis and inflammation.
Macrophages represent the dominant infiltrating cell type during progression of chronic kidney disease (CKD), driven partially by low O2 availability in the kidney (154–156). Kobayashi and colleagues addressed the role of myeloid HIF-αs using the typical LysM-Cre strategy in a murine unilateral ureteral obstruction–induced (UUO-induced) kidney injury model. Activation of myeloid HIF via LysM-Cre–driven Vhl deletion attenuates renal inflammation, while deletion of both myeloid Hif1a and Hif2a enhances inflammation, as indicated by increased F4/80+ cell numbers in the kidney. However, the presence of myeloid HIF-αs does not alter renal fibrosis. The authors suggest that hypoxia and/or myeloid HIF-α activation alleviates renal inflammation via suppression of Ccr2 and Ccl2, which are crucial for monocyte recruitment (157). The notion that myeloid HIF-1α regulates UUO-induced nephropathy is further supported by another study using the same LysM-Cre model; however, Tateishi and colleagues reported that myeloid HIF-1α deletion promoted renal fibrosis but did not alter macrophage accumulation in the UUO model. They suggested a different mechanism for the protective role of myeloid HIF-1α in renal fibrosis: suppression of renal connective tissue growth factor (CTGF) within renal cells (158). The discrepancy between the two reports could be due to deletion of two isoforms of HIF-α versus deletion of HIF-1α alone. Nevertheless, both studies suggest a protective role for myeloid HIF-αs in CKD, which partly supports the observation in patients with CDK that elevated renal HIF-1α expression correlates with less severe disease (159).
Cancer-associated inflammation.
A strong link between chronic inflammation and tumor progression has been clearly evident for some time. For example, patients with IBDs are at increased risk of developing colorectal cancer (160–162). Similar to sites of inflammation, the tumor microenvironment is also highly hypoxic. Macrophages predominantly accumulate in hypoxic regions, change their gene expression profiles in response to low O2, and function in response to limited O2 availability (100). Significant effort has delineated the respective roles of myeloid HIF-1α and HIF-2α in the tumor setting, beyond the two examples we will summarize here. In a PyMT model of breast cancer, loss of myeloid HIF-1α significantly decreases tumor mass and inhibits tumor progression, likely through suppression of cytotoxic T cell response to the tumors (163). As for HIF-2α, Imtiyaz and colleagues demonstrated that myeloid HIF-2α deficiency leads to reduced tumor burden and progression in a murine CAC model, while ablating macrophage infiltration of murine hepatocellular carcinoma. The authors suggest that these results could partly be due to defective migration and invasion of macrophages with HIF-2α loss (111). For a more comprehensive discussion of myeloid HIF-αs in cancer, please refer to other reviews within this series (164, 165).
Summary
Overall, hypoxia and inflammation are clearly inextricably linked. Hypoxia can be a strong contributory factor in certain inflammatory diseases; in turn, inflammation sites often exhibit low O2 tension. Myeloid cells are major components of innate immunity that are tightly associated with inflammation in different tissues and found predominantly localized within the hypoxic regions of inflamed tissues. Myeloid cell infiltration on its own can contribute to O2 deprivation at these sites. In response to hypoxia, myeloid cells stabilize HIF-αs, which facilitates their metabolic reprogramming and other adaptations, allowing myeloid cells to take on transient roles in different stages of disease progression. In many types of inflammation described in this review, the roles of myeloid HIF-αs remain incompletely described. In many cases, myeloid HIF-1α and HIF-2α have nonredundant or even opposing effects on myeloid cell functions (20). Therefore, many questions concerning the role of HIF-αs in myeloid cells require further investigation. For example, in specific inflammatory diseases, it is unclear if it would be beneficial or detrimental to target HIF-αs. If targeting of HIF-αs is beneficial, then should a specific HIF-α isoform or both isoforms be targeted? Even if all mechanisms mediated by HIF in inflammatory myeloid cells are elucidated, clinical translation will still be challenging. For example, how can HIF be specifically targeted in myeloid cells? How efficient will these therapies be? Nevertheless, oxygen-sensing pathways in myeloid cells are clearly key determinants of their physiological and pathological functions and these pathways remain attractive therapeutic targets.
Acknowledgments
The authors acknowledge funding from the National Heart, Lung and Blood Institute (grant number HL66310).
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information:J Clin Invest. 2016;126(10):3661–3671. doi:10.1172/JCI84426.
Contributor Information
Nan Lin, Email: nlin@mail.med.upenn.edu.
M. Celeste Simon, Email: celeste2@mail.med.upenn.edu.
References
- 1.Nathan C, Ding A. Nonresolving inflammation. Cell. 2010;140(6):871–882. doi: 10.1016/j.cell.2010.02.029. [DOI] [PubMed] [Google Scholar]
- 2.Fullerton JN, Gilroy DW. Resolution of inflammation: a new therapeutic frontier. Nat Rev Drug Discov. doi: 10.1038/nrd.20. [published online ahead of print March 29, 2016]. doi: 10.1038/nrd.2016.39. [DOI] [PubMed] [Google Scholar]
- 3.Imtiyaz HZ, Simon MC. Hypoxia-inducible factors as essential regulators of inflammation. Curr Top Microbiol Immunol. 2010;345:105–120. doi: 10.1007/82_2010_74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lewis JS, Lee JA, Underwood JC, Harris AL, Lewis CE. Macrophage responses to hypoxia: relevance to disease mechanisms. J Leukoc Biol. 1999;66(6):889–900. doi: 10.1002/jlb.66.6.889. [DOI] [PubMed] [Google Scholar]
- 5.Sivakumar B, Akhavani MA, Winlove CP, Taylor PC, Paleolog EM, Kang N. Synovial hypoxia as a cause of tendon rupture in rheumatoid arthritis. J Hand Surg Am. 2008;33(1):49–58. doi: 10.1016/j.jhsa.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 6.Muz B, Khan MN, Kiriakidis S, Paleolog EM. Hypoxia. The role of hypoxia and HIF-dependent signalling events in rheumatoid arthritis. Arthritis Res Ther. 2009;11(1): doi: 10.1186/ar2568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Colgan SP, Taylor CT. Hypoxia: an alarm signal during intestinal inflammation. Nat Rev Gastroenterol Hepatol. 2010;7(5):281–287. doi: 10.1038/nrgastro.2010.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Taylor CT, Colgan SP. Hypoxia and gastrointestinal disease. J Mol Med. 2007;85(12):1295–1300. doi: 10.1007/s00109-007-0277-z. [DOI] [PubMed] [Google Scholar]
- 9.Zheng L, Kelly CJ, Colgan SP. Physiologic hypoxia and oxygen homeostasis in the healthy intestine. A Review in the Theme: Cellular Responses to Hypoxia. Am J Physiol, Cell Physiol. 2015;309(6):C350–C360. doi: 10.1152/ajpcell.00191.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ye J. Emerging role of adipose tissue hypoxia in obesity and insulin resistance. Int J Obes (Lond) 2009;33(1):54–66. doi: 10.1038/ijo.2008.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Trayhurn P. Hypoxia and adipose tissue function and dysfunction in obesity. Physiol Rev. 2013;93(1):1–21. doi: 10.1152/physrev.00017.2012. [DOI] [PubMed] [Google Scholar]
- 12.Madjdpour C, et al. Decreased alveolar oxygen induces lung inflammation. Am J Physiol Lung Cell Mol Physiol. 2003;284(2):L360–L367. doi: 10.1152/ajplung.00158.2002. [DOI] [PubMed] [Google Scholar]
- 13.Ertel W, Morrison MH, Ayala A, Chaudry IH. Hypoxemia in the absence of blood loss or significant hypotension causes inflammatory cytokine release. Am J Physiol. 1995;269(1 Pt 2):R160–R166. doi: 10.1152/ajpregu.1995.269.1.R160. [DOI] [PubMed] [Google Scholar]
- 14.Bärtsch P, Swenson ER. Acute high-altitude illnesses. N Engl J Med. 2013;369(17):1666–1667. doi: 10.1056/NEJMc1309747. [DOI] [PubMed] [Google Scholar]
- 15.Hartmann G, et al. High altitude increases circulating interleukin-6, interleukin-1 receptor antagonist and C-reactive protein. Cytokine. 2000;12(3):246–252. doi: 10.1006/cyto.1999.0533. [DOI] [PubMed] [Google Scholar]
- 16.He Q, Gao Z, Yin J, Zhang J, Yun Z, Ye J. Regulation of HIF-1{alpha} activity in adipose tissue by obesity-associated factors: adipogenesis, insulin, and hypoxia. Am J Physiol Endocrinol Metab. 2011;300(5):E877–E885. doi: 10.1152/ajpendo.00626.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee KY, Gesta S, Boucher J, Wang XL, Kahn CR. The differential role of Hif1β/Arnt and the hypoxic response in adipose function, fibrosis, and inflammation. Cell Metab. 2011;14(4):491–503. doi: 10.1016/j.cmet.2011.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Majmundar AJ, Wong WJ, Simon MC. Hypoxia-inducible factors and the response to hypoxic stress. Mol Cell. 2010;40(2):294–309. doi: 10.1016/j.molcel.2010.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mucaj V, Shay JE, Simon MC. Effects of hypoxia and HIFs on cancer metabolism. Int J Hematol. 2012;95(5):464–470. doi: 10.1007/s12185-012-1070-5. [DOI] [PubMed] [Google Scholar]
- 20.Keith B, Johnson RS, Simon MC. HIF1α and HIF2α: sibling rivalry in hypoxic tumour growth and progression. Nat Rev Cancer. 2012;12(1):9–22. doi: 10.1038/nrc3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee KE, Simon MC. SnapShot: Hypoxia-Inducible Factors. Cell. 2015;163(5):1288–1288.e1. doi: 10.1016/j.cell.2015.11.011. [DOI] [PubMed] [Google Scholar]
- 22.Kaelin WG, Ratcliffe PJ. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell. 2008;30(4):393–402. doi: 10.1016/j.molcel.2008.04.009. [DOI] [PubMed] [Google Scholar]
- 23.Gu YZ, Moran SM, Hogenesch JB, Wartman L, Bradfield CA. Molecular characterization and chromosomal localization of a third alpha-class hypoxia inducible factor subunit, HIF3alpha. Gene Expr. 1998;7(3):205–213. [PMC free article] [PubMed] [Google Scholar]
- 24.Duan C. Hypoxia-inducible factor 3 biology: complexities and emerging themes. Am J Physiol, Cell Physiol. 2016;310(4):C260–C269. doi: 10.1152/ajpcell.00315.2015. [DOI] [PubMed] [Google Scholar]
- 25.Zhang P, Yao Q, Lu L, Li Y, Chen PJ, Duan C. Hypoxia-inducible factor 3 is an oxygen-dependent transcription activator and regulates a distinct transcriptional response to hypoxia. Cell Rep. 2014;6(6):1110–1121. doi: 10.1016/j.celrep.2014.02.011. [DOI] [PubMed] [Google Scholar]
- 26.Kaelin WG. Proline hydroxylation and gene expression. Annu Rev Biochem. 2005;74:115–128. doi: 10.1146/annurev.biochem.74.082803.133142. [DOI] [PubMed] [Google Scholar]
- 27.Maxwell PH, et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399(6733):271–275. doi: 10.1038/20459. [DOI] [PubMed] [Google Scholar]
- 28.Cockman ME, et al. Hypoxia inducible factor-alpha binding and ubiquitylation by the von Hippel-Lindau tumor suppressor protein. J Biol Chem. 2000;275(33):25733–25741. doi: 10.1074/jbc.M002740200. [DOI] [PubMed] [Google Scholar]
- 29.Jaakkola P, et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292(5516):468–472. doi: 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]
- 30.Chandel NS, et al. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: a mechanism of O2 sensing. J Biol Chem. 2000;275(33):25130–25138. doi: 10.1074/jbc.M001914200. [DOI] [PubMed] [Google Scholar]
- 31.Bell EL, Chandel NS. Mitochondrial oxygen sensing: regulation of hypoxia-inducible factor by mitochondrial generated reactive oxygen species. Essays Biochem. 2007;43:17–27. doi: 10.1042/bse0430017. [DOI] [PubMed] [Google Scholar]
- 32.Lin X, et al. A chemical genomics screen highlights the essential role of mitochondria in HIF-1 regulation. Proc Natl Acad Sci U S A. 2008;105(1):174–179. doi: 10.1073/pnas.0706585104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jung Y, Isaacs JS, Lee S, Trepel J, Liu ZG, Neckers L. Hypoxia-inducible factor induction by tumour necrosis factor in normoxic cells requires receptor-interacting protein-dependent nuclear factor kappa B activation. Biochem J. 2003;370(Pt 3):1011–1017. doi: 10.1042/BJ20021279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhou J, Schmid T, Brüne B. Tumor necrosis factor-alpha causes accumulation of a ubiquitinated form of hypoxia inducible factor-1alpha through a nuclear factor-kappaB-dependent pathway. Mol Biol Cell. 2003;14(6):2216–2225. doi: 10.1091/mbc.E02-09-0598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jung YJ, Isaacs JS, Lee S, Trepel J, Neckers L. IL-1beta-mediated up-regulation of HIF-1alpha via an NFkappaB/COX-2 pathway identifies HIF-1 as a critical link between inflammation and oncogenesis. FASEB J. 2003;17(14):2115–2117. doi: 10.1096/fj.03-0329fje. [DOI] [PubMed] [Google Scholar]
- 36.Fang HY, et al. Hypoxia-inducible factors 1 and 2 are important transcriptional effectors in primary macrophages experiencing hypoxia. Blood. 2009;114(4):844–859. doi: 10.1182/blood-2008-12-195941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rius J, et al. NF-kappaB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1alpha. Nature. 2008;453(7196):807–811. doi: 10.1038/nature06905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nishi K, et al. LPS induces hypoxia-inducible factor 1 activation in macrophage-differentiated cells in a reactive oxygen species-dependent manner. Antioxid Redox Signal. 2008;10(5):983–995. doi: 10.1089/ars.2007.1825. [DOI] [PubMed] [Google Scholar]
- 39.Peyssonnaux C, Cejudo-Martin P, Doedens A, Zinkernagel AS, Johnson RS, Nizet V. Cutting edge: Essential role of hypoxia inducible factor-1alpha in development of lipopolysaccharide-induced sepsis. J Immunol. 2007;178(12):7516–7519. doi: 10.4049/jimmunol.178.12.7516. [DOI] [PubMed] [Google Scholar]
- 40.Frede S, Stockmann C, Freitag P, Fandrey J. Bacterial lipopolysaccharide induces HIF-1 activation in human monocytes via p44/42 MAPK and NF-kappaB. Biochem J. 2006;396(3):517–527. doi: 10.1042/BJ20051839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Arsham AM, Howell JJ, Simon MC. A novel hypoxia-inducible factor-independent hypoxic response regulating mammalian target of rapamycin and its targets. J Biol Chem. 2003;278(32):29655–29660. doi: 10.1074/jbc.M212770200. [DOI] [PubMed] [Google Scholar]
- 42.Brugarolas J, et al. Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev. 2004;18(23):2893–2904. doi: 10.1101/gad.1256804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Koumenis C, et al. Regulation of protein synthesis by hypoxia via activation of the endoplasmic reticulum kinase PERK and phosphorylation of the translation initiation factor eIF2alpha. Mol Cell Biol. 2002;22(21):7405–7416. doi: 10.1128/MCB.22.21.7405-7416.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cummins EP, et al. Prolyl hydroxylase-1 negatively regulates IkappaB kinase-beta, giving insight into hypoxia-induced NFkappaB activity. Proc Natl Acad Sci U S A. 2006;103(48):18154–18159. doi: 10.1073/pnas.0602235103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Safronova O, Pluemsampant S, Nakahama K, Morita I. Regulation of chemokine gene expression by hypoxia via cooperative activation of NF-kappaB and histone deacetylase. Int J Biochem Cell Biol. 2009;41(11):2270–2280. doi: 10.1016/j.biocel.2009.05.003. [DOI] [PubMed] [Google Scholar]
- 46.Snodgrass RG, et al. Hypoxia potentiates palmitate-induced pro-inflammatory activation of primary human macrophages. J Biol Chem. 2016;291(1):413–424. doi: 10.1074/jbc.M115.686709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Davies LC, Jenkins SJ, Allen JE, Taylor PR. Tissue-resident macrophages. Nat Immunol. 2013;14(10):986–995. doi: 10.1038/ni.2705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Davies LC, Taylor PR. Tissue-resident macrophages: then and now. Immunology. 2015;144(4):541–548. doi: 10.1111/imm.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wynn TA, Chawla A, Pollard JW. Macrophage biology in development, homeostasis and disease. Nature. 2013;496(7446):445–455. doi: 10.1038/nature12034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ginhoux F, Jung S. Monocytes and macrophages: developmental pathways and tissue homeostasis. Nat Rev Immunol. 2014;14(6):392–404. doi: 10.1038/nri3671. [DOI] [PubMed] [Google Scholar]
- 51.Fournier BM, Parkos CA. The role of neutrophils during intestinal inflammation. Mucosal Immunol. 2012;5(4):354–366. doi: 10.1038/mi.2012.24. [DOI] [PubMed] [Google Scholar]
- 52.Galli SJ, Borregaard N, Wynn TA. Phenotypic and functional plasticity of cells of innate immunity: macrophages, mast cells and neutrophils. Nat Immunol. 2011;12(11):1035–1044. doi: 10.1038/ni.2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003;3(1):23–35. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- 54.Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. 2012;122(3):787–795. doi: 10.1172/JCI59643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mantovani A, Biswas SK, Galdiero MR, Sica A, Locati M. Macrophage plasticity and polarization in tissue repair and remodelling. J Pathol. 2013;229(2):176–185. doi: 10.1002/path.4133. [DOI] [PubMed] [Google Scholar]
- 56.Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep. 2014;6: doi: 10.12703/P6-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8(12):958–969. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xue J, et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity. 2014;40(2):274–288. doi: 10.1016/j.immuni.2014.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Moore KJ, Tabas I. Macrophages in the pathogenesis of atherosclerosis. Cell. 2011;145(3):341–355. doi: 10.1016/j.cell.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Moore KJ, Sheedy FJ, Fisher EA. Macrophages in atherosclerosis: a dynamic balance. Nat Rev Immunol. 2013;13(10):709–721. doi: 10.1038/nri3520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hansson GK, Hermansson A. The immune system in atherosclerosis. Nat Immunol. 2011;12(3):204–212. doi: 10.1038/ni.2001. [DOI] [PubMed] [Google Scholar]
- 62.Mowat AM, Bain CC. Mucosal macrophages in intestinal homeostasis and inflammation. J Innate Immun. 2011;3(6):550–564. doi: 10.1159/000329099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bain CC, Mowat AM. Macrophages in intestinal homeostasis and inflammation. Immunol Rev. 2014;260(1):102–117. doi: 10.1111/imr.12192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bain CC, Mowat AM. The monocyte-macrophage axis in the intestine. Cell Immunol. 2014;291(1–2):41–48. doi: 10.1016/j.cellimm.2014.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kinne RW, Bräuer R, Stuhlmüller B, Palombo-Kinne E, Burmester GR. Macrophages in rheumatoid arthritis. Arthritis Res. 2000;2(3):189–202. doi: 10.1186/ar86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Maruotti N, Cantatore FP, Crivellato E, Vacca A, Ribatti D. Macrophages in rheumatoid arthritis. Histol Histopathol. 2007;22(5):581–586. doi: 10.14670/HH-22.581. [DOI] [PubMed] [Google Scholar]
- 67.Kinne RW, Stuhlmüller B, Burmester GR. Cells of the synovium in rheumatoid arthritis. Macrophages. Arthritis Res Ther. 2007;9(6): doi: 10.1186/ar2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Davignon JL, et al. Targeting monocytes/macrophages in the treatment of rheumatoid arthritis. Rheumatology (Oxford) 2013;52(4):590–598. doi: 10.1093/rheumatology/kes304. [DOI] [PubMed] [Google Scholar]
- 69.Careau E, Bissonnette EY. Adoptive transfer of alveolar macrophages abrogates bronchial hyperresponsiveness. Am J Respir Cell Mol Biol. 2004;31(1):22–27. doi: 10.1165/rcmb.2003-0229OC. [DOI] [PubMed] [Google Scholar]
- 70.Careau E, Turmel V, Lauzon-Joset JF, Bissonnette EY. Alveolar macrophages reduce airway hyperresponsiveness and modulate cytokine levels. Exp Lung Res. 2010;36(5):255–261. doi: 10.3109/01902140903410757. [DOI] [PubMed] [Google Scholar]
- 71.Bang BR, et al. Alveolar macrophages modulate allergic inflammation in a murine model of asthma. Exp Mol Med. 2011;43(5):275–280. doi: 10.3858/emm.2011.43.5.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Balhara J, Gounni AS. The alveolar macrophages in asthma: a double-edged sword. Mucosal Immunol. 2012;5(6):605–609. doi: 10.1038/mi.2012.74. [DOI] [PubMed] [Google Scholar]
- 73.Lessner SM, Prado HL, Waller EK, Galis ZS. Atherosclerotic lesions grow through recruitment and proliferation of circulating monocytes in a murine model. Am J Pathol. 2002;160(6):2145–2155. doi: 10.1016/S0002-9440(10)61163-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Swirski FK, et al. Monocyte accumulation in mouse atherogenesis is progressive and proportional to extent of disease. Proc Natl Acad Sci U S A. 2006;103(27):10340–10345. doi: 10.1073/pnas.0604260103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Landsman L, et al. CX3CR1 is required for monocyte homeostasis and atherogenesis by promoting cell survival. Blood. 2009;113(4):963–972. doi: 10.1182/blood-2008-07-170787. [DOI] [PubMed] [Google Scholar]
- 76.Melgert BN, ten Hacken NH, Rutgers B, Timens W, Postma DS, Hylkema MN. More alternative activation of macrophages in lungs of asthmatic patients. J Allergy Clin Immunol. 2011;127(3):831–833. doi: 10.1016/j.jaci.2010.10.045. [DOI] [PubMed] [Google Scholar]
- 77.Ford AQ, Dasgupta P, Mikhailenko I, Smith EM, Noben-Trauth N, Keegan AD. Adoptive transfer of IL-4Rα+ macrophages is sufficient to enhance eosinophilic inflammation in a mouse model of allergic lung inflammation. BMC Immunol. 2012;13: doi: 10.1186/1471-2172-13-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Moreira AP, et al. Serum amyloid P attenuates M2 macrophage activation and protects against fungal spore-induced allergic airway disease. J Allergy Clin Immunol. 2010;126(4):712–721.e7. doi: 10.1016/j.jaci.2010.06.010. [DOI] [PubMed] [Google Scholar]
- 79.Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol. 2013;13(3):159–175. doi: 10.1038/nri3399. [DOI] [PubMed] [Google Scholar]
- 80.Wright HL, Moots RJ, Bucknall RC, Edwards SW. Neutrophil function in inflammation and inflammatory diseases. Rheumatology (Oxford) 2010;49(9):1618–1631. doi: 10.1093/rheumatology/keq045. [DOI] [PubMed] [Google Scholar]
- 81.Jones HR, Robb CT, Perretti M, Rossi AG. The role of neutrophils in inflammation resolution. Semin Immunol. 2016;28(2):137–145. doi: 10.1016/j.smim.2016.03.007. [DOI] [PubMed] [Google Scholar]
- 82.Scannell M, Maderna P. Lipoxins and annexin-1: resolution of inflammation and regulation of phagocytosis of apoptotic cells. ScientificWorldJournal. 2006;6:1555–1573. doi: 10.1100/tsw.2006.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Fierro IM, et al. Lipoxin A4 and aspirin-triggered 15-epi-lipoxin A4 inhibit human neutrophil migration: comparisons between synthetic 15 epimers in chemotaxis and transmigration with microvessel endothelial cells and epithelial cells. J Immunol. 2003;170(5):2688–2694. doi: 10.4049/jimmunol.170.5.2688. [DOI] [PubMed] [Google Scholar]
- 84.Dalli J, Chiang N, Serhan CN. Elucidation of novel 13-series resolvins that increase with atorvastatin and clear infections. Nat Med. 2015;21(9):1071–1075. doi: 10.1038/nm.3911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ariel A, et al. Apoptotic neutrophils and T cells sequester chemokines during immune response resolution through modulation of CCR5 expression. Nat Immunol. 2006;7(11):1209–1216. doi: 10.1038/ni1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Leitch AE, Duffin R, Haslett C, Rossi AG. Relevance of granulocyte apoptosis to resolution of inflammation at the respiratory mucosa. Mucosal Immunol. 2008;1(5):350–363. doi: 10.1038/mi.2008.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hallett JM, Leitch AE, Riley NA, Duffin R, Haslett C, Rossi AG. Novel pharmacological strategies for driving inflammatory cell apoptosis and enhancing the resolution of inflammation. Trends Pharmacol Sci. 2008;29(5):250–257. doi: 10.1016/j.tips.2008.03.002. [DOI] [PubMed] [Google Scholar]
- 88.Wipke BT, Allen PM. Essential role of neutrophils in the initiation and progression of a murine model of rheumatoid arthritis. J Immunol. 2001;167(3):1601–1608. doi: 10.4049/jimmunol.167.3.1601. [DOI] [PubMed] [Google Scholar]
- 89.Wright HL, Moots RJ, Edwards SW. The multifactorial role of neutrophils in rheumatoid arthritis. Nat Rev Rheumatol. 2014;10(10):593–601. doi: 10.1038/nrrheum.2014.80. [DOI] [PubMed] [Google Scholar]
- 90.Wittkowski H, et al. Effects of intra-articular corticosteroids and anti-TNF therapy on neutrophil activation in rheumatoid arthritis. Ann Rheum Dis. 2007;66(8):1020–1025. doi: 10.1136/ard.2006.061507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hoenderdos K, Condliffe A. The neutrophil in chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol. 2013;48(5):531–539. doi: 10.1165/rcmb.2012-0492TR. [DOI] [PubMed] [Google Scholar]
- 92.Pesci A, et al. Neutrophils infiltrating bronchial epithelium in chronic obstructive pulmonary disease. Respir Med. 1998;92(6):863–870. doi: 10.1016/S0954-6111(98)90389-4. [DOI] [PubMed] [Google Scholar]
- 93.Campbell EL, et al. Transmigrating neutrophils shape the mucosal microenvironment through localized oxygen depletion to influence resolution of inflammation. Immunity. 2014;40(1):66–77. doi: 10.1016/j.immuni.2013.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Campbell EL, Colgan SP. Neutrophils and inflammatory metabolism in antimicrobial functions of the mucosa. J Leukoc Biol. 2015;98(4):517–522. doi: 10.1189/jlb.3MR1114-556R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kühl AA, et al. Aggravation of different types of experimental colitis by depletion or adhesion blockade of neutrophils. Gastroenterology. 2007;133(6):1882–1892. doi: 10.1053/j.gastro.2007.08.073. [DOI] [PubMed] [Google Scholar]
- 96.Zhang R, Ito S, Nishio N, Cheng Z, Suzuki H, Isobe K. Up-regulation of Gr1+CD11b+ population in spleen of dextran sulfate sodium administered mice works to repair colitis. Inflamm Allergy Drug Targets. 2011;10(1):39–46. doi: 10.2174/187152811794352114. [DOI] [PubMed] [Google Scholar]
- 97.Natsui M, et al. Selective depletion of neutrophils by a monoclonal antibody, RP-3, suppresses dextran sulphate sodium-induced colitis in rats. J Gastroenterol Hepatol. 1997;12(12):801–808. doi: 10.1111/j.1440-1746.1997.tb00375.x. [DOI] [PubMed] [Google Scholar]
- 98.Kankuri E, et al. Suppression of acute experimental colitis by a highly selective inducible nitric-oxide synthase inhibitor, N-[3-(aminomethyl)benzyl]acetamidine. J Pharmacol Exp Ther. 2001;298(3):1128–1132. [PubMed] [Google Scholar]
- 99.Qualls JE, Kaplan AM, van Rooijen N, Cohen DA. Suppression of experimental colitis by intestinal mononuclear phagocytes. J Leukoc Biol. 2006;80(4):802–815. doi: 10.1189/jlb.1205734. [DOI] [PubMed] [Google Scholar]
- 100.Murdoch C, Giannoudis A, Lewis CE. Mechanisms regulating the recruitment of macrophages into hypoxic areas of tumors and other ischemic tissues. Blood. 2004;104(8):2224–2234. doi: 10.1182/blood-2004-03-1109. [DOI] [PubMed] [Google Scholar]
- 101.Schioppa T, et al. Regulation of the chemokine receptor CXCR4 by hypoxia. J Exp Med. 2003;198(9):1391–1402. doi: 10.1084/jem.20030267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hitchon C, Wong K, Ma G, Reed J, Lyttle D, El-Gabalawy H. Hypoxia-induced production of stromal cell-derived factor 1 (CXCL12) and vascular endothelial growth factor by synovial fibroblasts. Arthritis Rheum. 2002;46(10):2587–2597. doi: 10.1002/art.10520. [DOI] [PubMed] [Google Scholar]
- 103.Semba H, et al. HIF-1α-PDK1 axis-induced active glycolysis plays an essential role in macrophage migratory capacity. Nat Commun. 2016;7: doi: 10.1038/ncomms11635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Casazza A, et al. Impeding macrophage entry into hypoxic tumor areas by Sema3A/Nrp1 signaling blockade inhibits angiogenesis and restores antitumor immunity. Cancer Cell. 2013;24(6):695–709. doi: 10.1016/j.ccr.2013.11.007. [DOI] [PubMed] [Google Scholar]
- 105.Cramer T, et al. HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell. 2003;112(5):645–657. doi: 10.1016/S0092-8674(03)00154-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Walmsley SR, et al. Hypoxia-induced neutrophil survival is mediated by HIF-1alpha-dependent NF-kappaB activity. J Exp Med. 2005;201(1):105–115. doi: 10.1084/jem.20040624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Peyssonnaux C, et al. HIF-1alpha expression regulates the bactericidal capacity of phagocytes. J Clin Invest. 2005;115(7):1806–1815. doi: 10.1172/JCI23865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kelly B, O’Neill LA. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res. 2015;25(7):771–784. doi: 10.1038/cr.2015.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.O’Neill LA, Pearce EJ. Immunometabolism governs dendritic cell and macrophage function. J Exp Med. 2016;213(1):15–23. doi: 10.1084/jem.20151570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Tannahill GM, et al. Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature. 2013;496(7444):238–242. doi: 10.1038/nature11986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Imtiyaz HZ, et al. Hypoxia-inducible factor 2alpha regulates macrophage function in mouse models of acute and tumor inflammation. J Clin Invest. 2010;120(8):2699–2714. doi: 10.1172/JCI39506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Thompson AA, et al. Hypoxia-inducible factor 2α regulates key neutrophil functions in humans, mice, and zebrafish. Blood. 2014;123(3):366–376. doi: 10.1182/blood-2013-05-500207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Takeda N, et al. Differential activation and antagonistic function of HIF-{alpha} isoforms in macrophages are essential for NO homeostasis. Genes Dev. 2010;24(5):491–501. doi: 10.1101/gad.1881410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Shay JE, et al. Inhibition of hypoxia-inducible factors limits tumor progression in a mouse model of colorectal cancer. Carcinogenesis. 2014;35(5):1067–1077. doi: 10.1093/carcin/bgu004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Scott C, et al. Reduction of ARNT in myeloid cells causes immune suppression and delayed wound healing. Am J Physiol, Cell Physiol. 2014;307(4):C349–C357. doi: 10.1152/ajpcell.00306.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Pedersen SF, Græbe M, Hag AM, Højgaard L, Sillesen H, Kjær A. (18)F-FDG imaging of human atherosclerotic carotid plaques reflects gene expression of the key hypoxia marker HIF-1α. Am J Nucl Med Mol Imaging. 2013;3(5):384–392. [PMC free article] [PubMed] [Google Scholar]
- 117.Marsch E, Sluimer JC, Daemen MJ. Hypoxia in atherosclerosis and inflammation. Curr Opin Lipidol. 2013;24(5):393–400. doi: 10.1097/MOL.0b013e32836484a4. [DOI] [PubMed] [Google Scholar]
- 118.Jiang G, Li T, Qiu Y, Rui Y, Chen W, Lou Y. RNA interference for HIF-1alpha inhibits foam cells formation in vitro. Eur J Pharmacol. 2007;562(3):183–190. doi: 10.1016/j.ejphar.2007.01.066. [DOI] [PubMed] [Google Scholar]
- 119.Crucet M, Wüst SJ, Spielmann P, Lüscher TF, Wenger RH, Matter CM. Hypoxia enhances lipid uptake in macrophages: role of the scavenger receptors Lox1, SRA, and CD36. Atherosclerosis. 2013;229(1):110–117. doi: 10.1016/j.atherosclerosis.2013.04.034. [DOI] [PubMed] [Google Scholar]
- 120.Parathath S, et al. Hypoxia is present in murine atherosclerotic plaques and has multiple adverse effects on macrophage lipid metabolism. Circ Res. 2011;109(10):1141–1152. doi: 10.1161/CIRCRESAHA.111.246363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Hutter R, et al. Macrophages transmit potent proangiogenic effects of oxLDL in vitro and in vivo involving HIF-1α activation: a novel aspect of angiogenesis in atherosclerosis. J Cardiovasc Transl Res. 2013;6(4):558–569. doi: 10.1007/s12265-013-9469-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Asplund A, et al. Hypoxic regulation of secreted proteoglycans in macrophages. Glycobiology. 2010;20(1):33–40. doi: 10.1093/glycob/cwp139. [DOI] [PubMed] [Google Scholar]
- 123.Ahn GO, et al. Transcriptional activation of hypoxia-inducible factor-1 (HIF-1) in myeloid cells promotes angiogenesis through VEGF and S100A8. Proc Natl Acad Sci U S A. 2014;111(7):2698–2703. doi: 10.1073/pnas.1320243111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Tawakol A, et al. HIF-1α and PFKFB3 mediate a tight relationship between proinflammatory activation and anerobic metabolism in atherosclerotic macrophages. Arterioscler Thromb Vasc Biol. 2015;35(6):1463–1471. doi: 10.1161/ATVBAHA.115.305551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Nakayama T, et al. Role of macrophage-derived hypoxia-inducible factor (HIF)-1α as a mediator of vascular remodelling. Cardiovasc Res. 2013;99(4):705–715. doi: 10.1093/cvr/cvt146. [DOI] [PubMed] [Google Scholar]
- 126.Rahtu-Korpela L, et al. Hypoxia-inducible factor prolyl 4-hydroxylase-2 inhibition protects against development of atherosclerosis. Arterioscler Thromb Vasc Biol. 2016;36(4):608–617. doi: 10.1161/ATVBAHA.115.307136. [DOI] [PubMed] [Google Scholar]
- 127.Xu H, et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest. 2003;112(12):1821–1830. doi: 10.1172/JCI19451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112(12):1796–1808. doi: 10.1172/JCI19246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Wellen KE, Hotamisligil GS. Inflammation, stress, and diabetes. J Clin Invest. 2005;115(5):1111–1119. doi: 10.1172/JCI25102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Olefsky JM, Glass CK. Macrophages, inflammation, and insulin resistance. Annu Rev Physiol. 2010;72:219–246. doi: 10.1146/annurev-physiol-021909-135846. [DOI] [PubMed] [Google Scholar]
- 131.Odegaard JI, Chawla A. Alternative macrophage activation and metabolism. Annu Rev Pathol. 2011;6:275–297. doi: 10.1146/annurev-pathol-011110-130138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Chawla A, Nguyen KD, Goh YP. Macrophage-mediated inflammation in metabolic disease. Nat Rev Immunol. 2011;11(11):738–749. doi: 10.1038/nri3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest. 2007;117(1):175–184. doi: 10.1172/JCI29881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Aron-Wisnewsky J, et al. Human adipose tissue macrophages: m1 and m2 cell surface markers in subcutaneous and omental depots and after weight loss. J Clin Endocrinol Metab. 2009;94(11):4619–4623. doi: 10.1210/jc.2009-0925. [DOI] [PubMed] [Google Scholar]
- 135.Fjeldborg K, Pedersen SB, Møller HJ, Christiansen T, Bennetzen M, Richelsen B. Human adipose tissue macrophages are enhanced but changed to an anti-inflammatory profile in obesity. J Immunol Res. 2014;2014: doi: 10.1155/2014/309548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Wentworth JM, et al. Pro-inflammatory CD11c+CD206+ adipose tissue macrophages are associated with insulin resistance in human obesity. Diabetes. 2010;59(7):1648–1656. doi: 10.2337/db09-0287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Cancello R, et al. Reduction of macrophage infiltration and chemoattractant gene expression changes in white adipose tissue of morbidly obese subjects after surgery-induced weight loss. Diabetes. 2005;54(8):2277–2286. doi: 10.2337/diabetes.54.8.2277. [DOI] [PubMed] [Google Scholar]
- 138.Fujisaka S, et al. Adipose tissue hypoxia induces inflammatory M1 polarity of macrophages in an HIF-1α-dependent and HIF-1α-independent manner in obese mice. Diabetologia. 2013;56(6):1403–1412. doi: 10.1007/s00125-013-2885-1. [DOI] [PubMed] [Google Scholar]
- 139.Choe SS, Shin KC, Ka S, Lee YK, Chun JS, Kim JB. Macrophage HIF-2α ameliorates adipose tissue inflammation and insulin resistance in obesity. Diabetes. 2014;63(10):3359–3371. doi: 10.2337/db13-1965. [DOI] [PubMed] [Google Scholar]
- 140.García-Martín R, et al. Adipocyte-specific hypoxia-inducible factor 2α deficiency exacerbates obesity-induced brown adipose tissue dysfunction and metabolic dysregulation. Mol Cell Biol. 2016;36(3):376–393. doi: 10.1128/MCB.00430-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Rittirsch D, Flierl MA, Ward PA. Harmful molecular mechanisms in sepsis. Nat Rev Immunol. 2008;8(10):776–787. doi: 10.1038/nri2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Fink MP, Warren HS. Strategies to improve drug development for sepsis. Nat Rev Drug Discov. 2014;13(10):741–758. doi: 10.1038/nrd4368. [DOI] [PubMed] [Google Scholar]
- 143.Zinkernagel AS, Johnson RS, Nizet V. Hypoxia inducible factor (HIF) function in innate immunity and infection. J Mol Med. 2007;85(12):1339–1346. doi: 10.1007/s00109-007-0282-2. [DOI] [PubMed] [Google Scholar]
- 144.Mahabeleshwar GH, Qureshi MA, Takami Y, Sharma N, Lingrel JB, Jain MK. A myeloid hypoxia-inducible factor 1α-Krüppel-like factor 2 pathway regulates gram-positive endotoxin-mediated sepsis. J Biol Chem. 2012;287(2):1448–1457. doi: 10.1074/jbc.M111.312702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Mooberry SL. Mechanism of action of 2-methoxyestradiol: new developments. Drug Resist Updat. 2003;6(6):355–361. doi: 10.1016/j.drup.2003.10.001. [DOI] [PubMed] [Google Scholar]
- 146.Yeh CH, et al. Anticancer agent 2-methoxyestradiol improves survival in septic mice by reducing the production of cytokines and nitric oxide. Shock. 2011;36(5):510–516. doi: 10.1097/SHK.0b013e318231866f. [DOI] [PubMed] [Google Scholar]
- 147.Toussaint M, et al. Myeloid hypoxia-inducible factor 1α prevents airway allergy in mice through macrophage-mediated immunoregulation. Mucosal Immunol. 2013;6(3):485–497. doi: 10.1038/mi.2012.88. [DOI] [PubMed] [Google Scholar]
- 148.Shepardson KM, et al. Myeloid derived hypoxia inducible factor 1-alpha is required for protection against pulmonary Aspergillus fumigatus infection. PLoS Pathog. 2014;10(9): doi: 10.1371/journal.ppat.1004378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Byrne AJ, Jones CP, Gowers K, Rankin SM, Lloyd CM. Lung macrophages contribute to house dust mite driven airway remodeling via HIF-1α. PLoS One. 2013;8(7): doi: 10.1371/journal.pone.0069246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Crotty Alexander LE, et al. Myeloid cell HIF-1α regulates asthma airway resistance and eosinophil function. J Mol Med. 2013;91(5):637–644. doi: 10.1007/s00109-012-0986-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Fox JG, Wang TC. Inflammation, atrophy, and gastric cancer. J Clin Invest. 2007;117(1):60–69. doi: 10.1172/JCI30111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Kaparakis M, et al. Macrophages are mediators of gastritis in acute Helicobacter pylori infection in C57BL/6 mice. Infect Immun. 2008;76(5):2235–2239. doi: 10.1128/IAI.01481-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Matak P, et al. Myeloid HIF-1 is protective in Helicobacter pylori-mediated gastritis. J Immunol. 2015;194(7):3259–3266. doi: 10.4049/jimmunol.1401260. [DOI] [PubMed] [Google Scholar]
- 154.Fu Q, Colgan SP, Shelley CS. Hypoxia: the force that drives chronic kidney disease. Clin Med Res. 2016;14(1):15–39. doi: 10.3121/cmr.2015.1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Erwig LP. Macrophages and hypoxia in human chronic kidney disease. Kidney Int. 2008;74(4):405–406. doi: 10.1038/ki.2008.225. [DOI] [PubMed] [Google Scholar]
- 156.Duffield JS. Macrophages and immunologic inflammation of the kidney. Semin Nephrol. 2010;30(3):234–254. doi: 10.1016/j.semnephrol.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Kobayashi H, et al. Myeloid cell-derived hypoxia-inducible factor attenuates inflammation in unilateral ureteral obstruction-induced kidney injury. J Immunol. 2012;188(10):5106–5115. doi: 10.4049/jimmunol.1103377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Tateishi Y, et al. Myeloid HIF-1 attenuates the progression of renal fibrosis in murine obstructive nephropathy. J Pharmacol Sci. 2015;127(2):181–189. doi: 10.1016/j.jphs.2014.12.011. [DOI] [PubMed] [Google Scholar]
- 159.Hung TW, et al. Renal expression of hypoxia inducible factor-1α in patients with chronic kidney disease: a clinicopathologic study from nephrectomized kidneys. Indian J Med Res. 2013;137(1):102–110. [PMC free article] [PubMed] [Google Scholar]
- 160.Eaden JA, Abrams KR, Mayberry JF. The risk of colorectal cancer in ulcerative colitis: a meta-analysis. Gut. 2001;48(4):526–535. doi: 10.1136/gut.48.4.526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Jess T, Rungoe C, Peyrin-Biroulet L. Risk of colorectal cancer in patients with ulcerative colitis: a meta-analysis of population-based cohort studies. Clin Gastroenterol Hepatol. 2012;10(6):639–645. doi: 10.1016/j.cgh.2012.01.010. [DOI] [PubMed] [Google Scholar]
- 162.Danese S, Mantovani A. Inflammatory bowel disease and intestinal cancer: a paradigm of the Yin-Yang interplay between inflammation and cancer. Oncogene. 2010;29(23):3313–3323. doi: 10.1038/onc.2010.109. [DOI] [PubMed] [Google Scholar]
- 163.Doedens AL, et al. Macrophage expression of hypoxia-inducible factor-1 alpha suppresses T-cell function and promotes tumor progression. Cancer Res. 2010;70(19):7465–7475. doi: 10.1158/0008-5472.CAN-10-1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Henze AT, Mazzone M. The impact of hypoxia on tumor-associated macrophages. J Clin Invest. 2016;126(10):3672–3679. doi: 10.1172/JCI84427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Triner D, Shah YM. Hypoxia-inducible factors: a central link between inflammation and cancer. J Clin Invest. 2016;126(10):3689–3698. doi: 10.1172/JCI84430. [DOI] [PMC free article] [PubMed] [Google Scholar]