

Abstract
Hepatocyte death, which can be apoptosis or necrosis depending on the context, is a prominent feature of liver disease. The lectin concanavalin A (ConA) activates immune cells, resulting in inflammatory liver injury and hepatocyte necrosis. In this issue of the JCI, Günther et al. demonstrate that the pseudokinase mixed lineage kinase domain–like protein (MLKL) participates in hepatocyte death in ConA injury and that MLKL-mediated death is independent of the receptor-interacting protein kinase RIPK3. RIPK3 was absent in hepatocytes, and MLKL-deficient mice, but not RIPK3-deficient mice, were protected from ConA-induced liver injury. The authors also present evidence that an unidentified kinase activates MLKL, as RIPK1 bound MLKL but did not phosphorylate it. Moreover, ConA rapidly induced MLKL, mediated by the IFN-γ/STAT1 pathway, while activation and translocation to the plasma membrane required TNF. Increased phospho-MLKL staining in liver biopsies from patients with autoimmune hepatitis suggests a role for MLKL in this disease. This study describes a previously unrecognized form of cell death in the liver that should be further explored as a potential therapeutic target in immune-mediated liver injury.
Cell death pathways
Cell death plays a central role in nearly all diseases, determining the extent of tissue destruction and subsequent organ function. Additionally, factors released from dying cells are associated with secondary inflammatory responses and fibrosis.
Apoptosis and necrosis are distinct forms of cell death involving different signal transduction pathways. Apoptosis is dependent on caspase activation, and in certain cell types, such as hepatocytes, apoptosis requires mitochondrial participation and involves selective mitochondrial release of death amplifiers that are regulated by the BCL2 family of proteins. Until the last decade, necrosis had been viewed as an unregulated, accidental form of cell death that occurs in the setting of bioenergetic collapse, leading to cell lysis and subsequent activation of proinflammatory responses by the aberrant releases of the contents of dying cells. However, recent evidence has shown that mitochondrial permeability transition pore opening, which is regulated by cyclophilin D and signal transduction pathways, such as JNK, that are induced by mitochondrial ROS, leads to regulated necrosis in certain contexts (1, 2). More recently, a type of death receptor-mediated necrosis, which occurs in the presence of caspase inhibition (with or without cIAP inhibition) has been discovered (referred to as necroptosis). During necroptosis, the receptor-interacting protein kinases RIPK1 and RIPK3 and the pseudokinase mixed lineage kinase domain–like protein (MLKL) form a complex referred to as the necrosome (3), which promotes RIPK3-mediated phosphorylation of MLKL, thereby triggering a conformational change that results in MLKL oligomerization, translocation to plasma membrane, and subsequent lethal permeation of lipid bilayer (4). RIPK3 is the only activator of MLKL that has been described to date. Moreover, the role of natural necroptosis in disease is controversial, with a recent study refuting previously published studies that suggested a role for necroptosis in a number of disease models of necrosis in vivo (5). Additionally, interpretation of many studies is complicated by potential off-target effects of inhibitors, such as the RIPK1 kinase inhibitor necrostatin-1s (nec-1) (6); non-death-related functions of RIPK1 and RIPK3 (7); and technical issues, including the specificity of RIPK1- and RIPK3-targeting antisera and poor strain matching of control and knockout mice (8). The field has only recently shifted from evaluating necroptosis in mice with global deletions to studying animals harboring conditional deletions of Ripk1 and Ripk3. Importantly, both RIPK1 and RIPK3 have been shown to regulate apoptosis in certain contexts (7). Furthermore, the roles of these mediators are very complex and depend on both their kinase activity and platform functions and are influenced by posttranslational modifications, such as phosphorylation and ubiquitination, resulting in extremely complex, context-specific scenarios (7).
What pathway killed the hepatocyte?
Inflammatory liver disease is caused by a variety of factors, including viral infection, autoimmunity, and toxin-mediated injury. In most models of hepatitis, apoptosis seems to be the dominant form of cell death. In contrast, acetaminophen-induced (APAP-induced) liver injury results in necrotic hepatocyte death that occurs even in animals lacking RIPK3 or MLKL. The lectin concanavalin A (ConA) has also been widely used to study liver necrosis; it induces an acute hepatitis that is associated with T and NKT cell–mediated inflammation. It is not clear how ConA induces hepatic necrosis; however, previous reports have shown that ConA alone causes necrotic cell death on histology, with little caspase-3 activity in the liver, although this has been somewhat controversial (9–12).
In this issue, Günther et al. further evaluate the role of regulated necrosis in inflammatory liver disease (13), confirm previous reports that necrotic hepatocyte death in APAP-induced liver injury is independent of RIPK3 and MLKL, and reveal that ConA-mediated hepatocyte death is mediated by MLKL through a RIPK3-independent mechanism (8, 13). The latter result is intriguing, as RIPK3, the only identified kinase capable of activating MLKL, is not detectable in normal hepatocytes (8, 13). RIPK3 induction has been suggested to occur in chronic liver injury, such as alcoholic and nonalcoholic steatohepatitis; however, actual necroptosis due to putative induction of RIPK3 in the liver has not been convincingly demonstrated (14–16). Hepatocytes do express MLKL, though at lower levels than nonparenchymal cells, making MLKL-induced cell death a possibility (8).
Using Mlkl–/– mice and strain-matched controls, Günther et al. clearly demonstrate a hepatocyte-intrinsic, RIPK3-independent role for MLKL in ConA-mediated liver injury (13). Mlkl–/– mice were markedly protected from ConA-induced damage, while Ripk3–/– animals were not. MLKL activation, as evidenced by membrane translocation in hepatocytes in response to ConA treatment, occurred in the absence of RIPK3-mediated phosphorylation. While RIPK3 expression was not detectible in hepatocytes following ConA administration, both RIPK1 and MLKL were induced in this model. Despite evidence of RIPK1/MLKL complex formation, direct activation of MLKL by RIPK1 was excluded. Together, these results suggest the existence and participation of another MLKL-activating kinase in ConA-mediated liver injury; however, such a kinase has yet to be identified. The importance of the observed increase in RIPK1 levels is not certain, but it may be required for the appropriate assembly of a complex with MLKL and other components, including the unidentified MLKL activating kinase.
Günther and colleagues initially explored canonical necroptosis in ConA-induced hepatocyte death and showed that the RIPK1 inhibitor nec-1s impedes ConA toxicity by blocking MLKL translocation to plasma membrane; however, nec-1s did not prevent MLKL induction in response to ConA toxicity (13). At face value, nec-1s protection in the ConA model suggests that RIPK1 kinase activity is required for MLK1-dependent cell death. However, the necrostatins, which are allosteric kinase inhibitors, may also affect protein folding, binding, and catalytic activity-independent functions of RIPK1; therefore, future assessment of ConA-mediated injury in kinase-dead RIPK1 knockin mice would be extremely informative. The platform function and kinase activity of RIPK1 play complex roles in cell death pathways. For example, conditional RIPK1 deficiency in the intestine or knockdown of RIPK1 in the liver sensitizes these organs to TNF-induced apoptosis; however, such susceptibility is not seen in kinase-dead RIPK1 knockin mice (17, 18), indicating that the platform function of RIPK1 is important for inhibiting apoptosis. Interestingly, RIPK1-dependent apoptosis inhibition seems to be independent of canonical NF-κB activation and target gene expression. Instead, this process may involve the regulation and turnover of cFLIP and cIAP1/2 (17). Indeed, efficient RIPK1 knockdown was recently shown to exacerbate ConA-, LPS-, and α-galactosylceramide–mediated NKT cell–induced liver injury. Moreover, sensitization that occurred in response to RIPK1 knockdown was prevented by caspase inhibition, indicating elevated apoptosis in these models. Enhanced apoptosis did not occur in kinase-dead RIPK1 knockin mice (17). Thus, it appears that the kinase activity of RIPK1 is required for MLKL-induced necrosis that occurs in response to ConA, while the platform function of RIPK1 inhibits apoptosis.
An important aspect of the work by Günther et al. is the demonstration that ConA promotes a robust and rapid induction of MLKL expression, which was elegantly shown to be mediated by IFN-γ signaling through STAT1 (13). Isolated increase in MLKL levels, without its activating by phosphorylation to induce conformational change and translocation to plasma membrane, is insufficient to induce necrosis. As induction of MLKL expression was required for cell death (ConA did not mediate injury in Stat1–/– mice), it appears that the basal levels of MLKL in hepatocytes are insufficient to support necrosis. The data indicate that a critical threshold level of MLKL must be achieved in the liver to support MLKL-mediated necrosis. While induction of MLKL required IFN-γ/STAT1 signaling, activation required TNF signaling. Tnfr1–/– mice were markedly protected against ConA necrosis, despite MLKL induction; however, loss of TNF signaling in these animals prevented MLKL translocation to the cell membrane. Therefore, both induction (via IFN-γ/STAT1) and activation, as reflected by translocation of MLKL to the cell membrane (via TNF signaling), are required for hepatocyte death in the ConA model. It is unclear whether TNF signaling is required to activate RIPK1, or another intermediary protein, such as the unidentified MLKL-activating kinase (Figure 1).
Figure 1. ConA-induced liver necrosis.
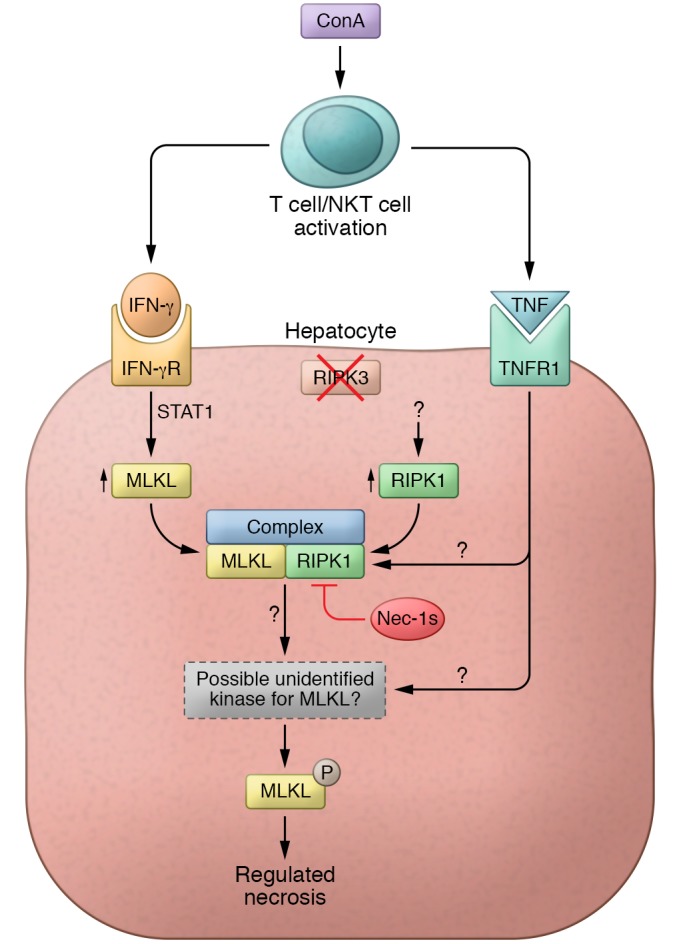
ConA activates T cells and NKT cells, which secrete cytokines, such as IFN-γ and TNF, that are known to be critical for liver injury in this model. Günther et al. discovered that IFN-γ signaling induces MLKL above a critical threshold (13). This increase in MLKL sensitizes hepatocytes to necrosis but alone is insufficient to promote necrosis and requires activation of MLKL. RIPK3 is the only kinase known to activate MLKL; however, RIPK3 is not expressed in hepatocytes or induced by ConA. Ripk3–/– mice given ConA developed liver injury, but Mlkl–/– mice were resistant to ConA. The RIPK1 inhibitor nec-1s was protective in ConA-treated mice, and this protection was upstream of MLKL activation. RIPK1 expression increased in this model and was independent of TNF or IFN-γ, occurring via an unknown mechanism. RIPK1 was found in a complex with MLKL, presumably with other not-yet-identified proteins; however, RIPK1 did not directly phosphorylate MLKL but possibly leads to the activation of the unidentified kinase activator of MLKL. As TNF signaling is required for cell death, it must lead to the activation of either RIPK1 or the unidentified kinase to activate MLKL. IFN-γR, IFN-γ receptor; TNFR, TNF receptor.
There are some apparent discrepancies between the findings of Günther and colleagues and prior publications. For example, Kaufmann et al. previously showed that ConA toxicity was mitigated by liver-specific deletion of caspase-8 and demonstrated a contribution of the kinase JNK and the regulator BIM (19). Kang et al. showed that deletion of Ripk3 in myeloid cells, but not hepatocytes, protected mice against ConA toxicity (20). Some of the discrepancies among these reports may be due to off-target effects of inhibitors, the dose of inhibitors and ConA used, strain-specific variations in the mice, and inconsistency in the use of littermate controls. In addition, FasL, TRAIL, and PARP-1 have also been suggested to be involved in ConA toxicity (12, 21) but were not explored by Günther et al.
Outstanding questions
Günther et al. also explored the possibility that their findings in the mouse ConA immune-mediated toxicity model are relevant to human autoimmune hepatitis (AIH), which is characterized by an influx of adaptive immune cells (lymphocytes and plasma cells) in the liver and extensive hepatocyte death (13). Indeed, immunohistochemistry of liver sections from patients with AIH revealed increased pMLKL expression and plasma membrane localization. It would be informative to perform cleaved caspase-3 staining to assess the relative contribution of apoptosis to AIH, as adaptive immune-mediated liver injury is likely to involve FasL, granzyme/perforin, and other apoptosis mediators. The translational implications of the study by Günther et al. are currently uncertain and will require a better understanding of the mechanisms of MLKL activation and the relative contribution of noncanonical necroptosis and apoptosis to AIH. While Günther et al. show that IFN-γ or IFN-β, but not IFN-α, induces the expression of MLKL, this induction was insufficient to cause necrosis in the absence of a second signal, likely TNF. These findings raise the possibility that this mechanism may not only contribute to the occurrence of acute liver injury, but to the unmasking of AIH, which is sometimes seen in patients treated with IFN (22).
Therapeutic approaches targeting IFN-γ/STAT1 induction of MLKL may have potential to limit immune-mediated liver injury; however, the effect of inhibiting one death pathway on other death pathways will need to be taken into consideration. While inhibition of necroptosis did not enhance apoptosis in the ConA model, it might prove to be an issue in adaptive immune-mediated injury, such as AIH, in which apoptosis might be enhanced by inhibition of necroptosis. It will also be important to determine if MLKL-mediated hepatocyte death is involved in other common liver diseases, such as alcoholic and nonalcoholic steatohepatitis, acute and chronic viral hepatitis, and idiosyncratic immune-mediated drug-induced liver injury.
The study by Günther et al. not only reveals that IFN-γ/STAT1 signaling induces MLKL, but also provides compelling evidence for a RIPK3-independent MLKL activation pathway, which might be referred to as noncanonical necroptosis (13). While the mechanism for MLKL induction is clearly defined, the mechanism(s) for phospho-activation of MLKL and the precise role of RIPK1 in this form of necroptosis will require additional work. Regardless, findings of Günther and colleagues provide findings that advance the field of immune-mediated liver injury. The mystery of ConA-initiated necrosis is nearly solved. We now know that MLKL did it with RIPK1 and help from an unidentified kinase. The investigation continues.
Acknowledgments
This work was supported by NIH grant RO1DK067215 (to NK), P30DK48522 (to NK), RO1DK078586 (to ZXL), K08DK109141 (to LD), and U01AA021857 (to ZXL).
Footnotes
Conflict of interest: N. Kaplowitz has been a paid expert witness for cases related to acetaminophen toxicity.
Reference information:J Clin Invest. 2016;126(11):4068–4071. doi:10.1172/JCI90830.
See the related article beginning on page 4346.
Contributor Information
Lily Dara, Email: lily.dara@usc.edu.
Zhang-Xu Liu, Email: zxliu@usc.edu.
Neil Kaplowitz, Email: kaplowit@hsc.usc.edu.
References
- 1.Ramachandran A, Lebofsky M, Baines CP, Lemasters JJ, Jaeschke H. Cyclophilin D deficiency protects against acetaminophen-induced oxidant stress and liver injury. Free Radic Res. 2011;45(2):156–164. doi: 10.3109/10715762.2010.520319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Win S, Than TA, Min RW, Aghajan M, Kaplowitz N. c-Jun N-terminal kinase mediates mouse liver injury through a novel Sab (SH3BP5)-dependent pathway leading to inactivation of intramitochondrial Src. Hepatology. 2016;63(6):1987–2003. doi: 10.1002/hep.28486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Linkermann A, Green DR. Necroptosis. N Engl J Med. 2014;370(5):455–465. doi: 10.1056/NEJMra1310050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hildebrand JM, et al. Activation of the pseudokinase MLKL unleashes the four-helix bundle domain to induce membrane localization and necroptotic cell death. Proc Natl Acad Sci U S A. 2014;111(42):15072–15077. doi: 10.1073/pnas.1408987111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Newton K, et al. RIPK3 deficiency or catalytically inactive RIPK1 provides greater benefit than MLKL deficiency in mouse models of inflammation and tissue injury. Cell Death Differ. 2016;23(9):1565–1576. doi: 10.1038/cdd.2016.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Takahashi N, et al. Necrostatin-1 analogues: critical issues on the specificity, activity and in vivo use in experimental disease models. Cell Death Dis. 2012;3: doi: 10.1038/cddis.2012.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ismaili N. Treatment trends and outcomes of small-cell carcinoma of the bladder: in regard to Koay et al. (Int J Radiat Oncol Biol Phys 2011 Oct 20) Int J Radiat Oncol Biol Phys. 2012;82(4):1319–1320. doi: 10.1016/j.ijrobp.2011.11.079. [DOI] [PubMed] [Google Scholar]
- 8.Dara L, et al. Receptor interacting protein kinase 1 mediates murine acetaminophen toxicity independent of the necrosome and not through necroptosis. Hepatology. 2015;62(6):1847–1857. doi: 10.1002/hep.27939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Künstle G, Hentze H, Germann PG, Tiegs G, Meergans T, Wendel A. Concanavalin A hepatotoxicity in mice: tumor necrosis factor-mediated organ failure independent of caspase-3-like protease activation. Hepatology. 1999;30(5):1241–1251. doi: 10.1002/hep.510300517. [DOI] [PubMed] [Google Scholar]
- 10.Ni HM, Chen X, Ding WX, Schuchmann M, Yin XM. Differential roles of JNK in ConA/GalN and ConA-induced liver injury in mice. Am J Pathol. 2008;173(4):962–972. doi: 10.2353/ajpath.2008.080358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Takeda K, Hayakawa Y, Van Kaer L, Matsuda H, Yagita H, Okumura K. Critical contribution of liver natural killer T cells to a murine model of hepatitis. Proc Natl Acad Sci U S A. 2000;97(10):5498–5503. doi: 10.1073/pnas.040566697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tiegs G, Hentschel J, Wendel A. A T cell-dependent experimental liver injury in mice inducible by concanavalin A. J Clin Invest. 1992;90(1):196–203. doi: 10.1172/JCI115836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Günther C, et al. The pseudokinase MLKL mediates programmed hepatocellular necrosis independent of RIPK3 during hepatitis. J Clin Invest. 2016;126(11):4346–4360. doi: 10.1172/JCI87545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dara L, Kaplowitz N. The many faces of RIPK3: What about NASH? Hepatology. doi: 10.1002/hep.28700. [published online ahead of print June 24, 2016]. doi: 10.1002/hep.28700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Roychowdhury S, McMullen MR, Pisano SG, Liu X, Nagy LE. Absence of receptor interacting protein kinase 3 prevents ethanol-induced liver injury. Hepatology. 2013;57(5):1773–1783. doi: 10.1002/hep.26200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Roychowdhury S, et al. Receptor interacting protein 3 protects mice from high-fat diet-induced liver injury. Hepatology. doi: 10.1002/hep.28676. [published online ahead of print June 15, 2016]. doi: 10.1002/hep.28676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Suda J, et al. Knockdown of RIPK1 markedly exacerbates murine immune-mediated liver injury through massive apoptosis of hepatocytes, independent of necroptosis and inhibition of NF-κB. J Immunol. doi: 10.4049/jimmunol.1600690. [published online ahead of print September 7, 2016]. doi: 10.4049/jimmunol.1600690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Takahashi N, et al. RIPK1 ensures intestinal homeostasis by protecting the epithelium against apoptosis. Nature. 2014;513(7516):95–99. doi: 10.1038/nature13706. [DOI] [PubMed] [Google Scholar]
- 19.Kaufmann T, et al. Fatal hepatitis mediated by tumor necrosis factor TNFα requires caspase-8 and involves the BH3-only proteins Bid and Bim. Immunity. 2009;30(1):56–66. doi: 10.1016/j.immuni.2008.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kang YJ, et al. Regulation of NKT cell-mediated immune responses to tumours and liver inflammation by mitochondrial PGAM5-Drp1 signalling. Nat Commun. 2015;6: doi: 10.1038/ncomms9371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jouan-Lanhouet S, et al. TRAIL induces necroptosis involving RIPK1/RIPK3-dependent PARP-1 activation. Cell Death Differ. 2012;19(12):2003–2014. doi: 10.1038/cdd.2012.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Francis GS, et al. Hepatic reactions during treatment of multiple sclerosis with interferon-β-1a: incidence and clinical significance. Drug Saf. 2003;26(11):815–827. doi: 10.2165/00002018-200326110-00006. [DOI] [PubMed] [Google Scholar]