

Abstract
Osmium(0) complexes derived from Os3(CO)12 and XPhos (2-dicyclohexylphosphino-2′,4′,6′-triisopropylbiphenyl) catalyze the C-C coupling of α-hydroxy esters 1a-1i, α-ketols 1j-1o or 1,2-diols dihydro-1j-1o with ethylene 2a to form ethylated tertiary alcohols 3a-3o. As illustrated in couplings of 1-octene 2b with vicinally dioxygenated reactants 1a, 1b, 1i, 1j, 1k, 1m, higher α-olefins are converted to adducts 4a, 4b, 4i, 4j, 4k, 4m with complete levels of branched regioselectivity. Oxidation level independent C-C coupling is demonstrated by the reaction of 1-octene 2b with diol dihydro-1k, α-ketol 1k and dione dehydro-1k. Functionalized olefins 2c-2f react with ethyl mandelate 1a to furnish adducts 5a-8a as single regioisomers. The collective data, including deuterium labeling studies, are consistent with a catalytic mechanism involving olefin-dione oxidative coupling to form an oxa-osmacyclopentane, which upon reductive cleavage via hydrogen transfer from the secondary alcohol reactant releases the product of carbinol C-alkylation with regeneration of the ketone. Single crystal X-ray diffraction data of the dinuclear complex Os2(CO)4(O2CR)2(XPhos)2 and the trinuclear complex Os3(CO)11(XPhos) are reported. These studies suggest increased π-backbonding at the stage of the metal-olefin π-complex plays a critical role in facilitating alkene-carbonyl oxidative coupling, as isostructural ruthenium(0) complexes, which are weaker π-donors, do not catalyze the transformations reported herein.
Graphical Abstract
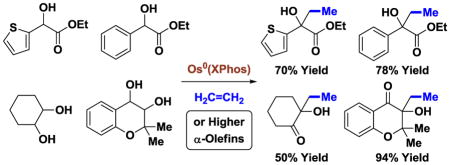
Introduction
α-Olefins are the most abundant petrochemical feedstock beyond alkanes.1 Despite their ubiquity and low cost, the use of α-olefins in the commercial manufacture of commodity chemicals is largely restricted to polymerization,2 hydroformylation3 and alkene metathesis.4 The discovery of alternate classes of byproduct-free catalytic C-C couplings that convert α-olefins to value-added products remains an important yet elusive goal. For example, while intermolecular alkene hydroacylation is attractive, decarbonylation of acylmetal intermediates to form inactive metal carbonyl complexes mandates use of esoteric reactants with chelating groups.5,6 Similarly, intermolecular Prins or carbonyl ene reactions do not extend to the coupling of α-olefins with unactivated aldehydes.7a–c Finally, whereas nickel(0) catalyzes the coupling of α-olefins with simple aldehydes, superstoichiometric quantities of TESOTf and Et3N are required.7d
In connection with the development of C-C bond forming hydrogenations and transfer hydrogenations beyond hydroformylation,8 we recently found that zerovalent ruthenium complexes generated in situ from Ru3(CO)12 and various phosphine ligands catalyze the C-C coupling of vicinally dioxygenated hydrocarbons (1,2-diols, α-ketols, α-hydroxy esters) with diverse π-unsaturated reactants, including dienes,9a–d acrylates9e and alkynes.9g,h,j As in related ruthenium(0) catalyzed Pauson-Khand reactions of vicinal dicarbonyl compounds described by Chatani and Murai,10 these processes are initiated through C=C/C=O oxidative coupling to furnish oxaruthenacycles. Catalytic turnover is achieved via transfer hydrogenolysis of the metalacycle by the alcohol reactant to release product and regenerate the carbonyl partner (Figure 1, top). Based on this mechanism, a ruthenium(0) catalyzed coupling of α-olefins was developed (Figure 1, middle).9f This process, however, was restricted to the use of 3-hydroxy-2-oxindoles, which may be attributed to the exceptional reactivity of the transient isatins. In continuing efforts to broaden the scope of transfer hydrogenative α-olefin coupling, we now demonstrate that osmium(0) catalysts overcome this limitation, enabling the direct C-C coupling of ethylene11 and higher α-olefins with diverse diols, α-ketols and α-hydroxy esters (Figure 1, bottom).
Figure 1.

General mechanism for catalytic C-C coupling of alcohols with π-unsaturated reactants via hydrogen auto-transfer and applications toward the coupling of α-olefins.
Research Design and Methods
The limitations evident in ruthenium(0) catalyzed C-C couplings of α-olefins9f were believed to stem from a high energetic barrier to oxidative coupling. Guided by Hoffmann’s theoretical analysis of the conversion of metal bisolefin complexes to metalacyclopentanes,12 and a large body of experimental evidence,13a the facility of oxidative coupling should be influenced by the degree of backbonding in the preceding metal-olefin π-complex.14 Backbonding confers nucleophilic character to the bound olefin and, in the limiting case, may be viewed as an oxidative addition to the C=C π-bond to form a metalacyclopropane. The Kulinkovich reaction,15 wherein titanium(II)-olefin complexes behave as vicinal dianions, represents a dramatic illustration of this effect. Hence, it was posited that a more strongly reducing metal center should facilitate C=C/C=O oxidative coupling to broaden substrate scope in transfer hydrogenative C-C couplings of α-olefins.
As borne out by the carbonyl stretching frequencies of isostructural ruthenium and osmium complexes HClM(CO)(PPh3)3, M = Os, νco = 1906 cm−1; M = Ru, νco = 1922 cm−1, osmium is a stronger π-donor than ruthenium.16 Indeed, osmium(0) catalysts are effective in couplings of activated secondary alcohols with vinyl acetates in cases where ruthenium(0) catalysts are not.9i For this reason, osmium(0) complexes were assayed in the coupling of racemic ethyl mandelate 1a with ethylene 2a with the goal of generating the ethylated tertiary alcohol 3a (Scheme 1). It was found that monodentate or bidentate triaryl phosphine ligands were ineffective. However, the osmium(0) catalyst modified by PCy3 (tricyclohexylphosphine) provided the desired adduct in 57% yield. Given this promising result, the osmium(0) complex modified by XPhos was eventually identified as the optimal catalyst, delivering the product of carbinol C-H ethylation 3a in 78% yield. Notably, under all conditions evaluated, the corresponding ruthenium(0) catalysts were unable to promote formation of adduct 3a.
Scheme 1.

Selected experiments in the coupling of ethyl mandelate 1a with ethylene 2a to form tertiary alcohol 3a.a
aYields are of material isolated by silica gel chromatography. See Supporting Information for further experimental details.
Under these optimal conditions, aryl- and heteroaryl-substituted α-hydroxy esters 1a-1i were coupled to ethylene 2a to form products of carbinol C-H ethylation 3a-3i (Table 1). As illustrated by the conversion of ethyl 4-bromomandelate 1b to adduct 3b, the osmium(0) catalyst is tolerant of aryl halide functional groups. The transformation of ethyl 4-methoxymandelate 1c and ethyl 4-(trifluoromethyl)mandelate 1d to adducts 3c and 3d, respectively, highlight tolerance of electron rich and as well as electron deficient aryl groups. Substituents at the meta-position of the aryl ring are tolerated, as shown in the formation of 3e and 3f, respectively. Finally, sulfur containing α-hydroxy ester 1g and heteroaromatic α-hydroxy esters 1h and 1i groups are converted to adducts 3g, 3h and 3i, respectively. ortho-Substituted mandelates and alkyl-substituted α-hydroxy esters such as ethyl lactate, were inefficient partners for C-C coupling under these conditions.
Table 1.
Osmium(0) catalyzed coupling of α-hydroxy esters 1a-1i with ethylene 2a to form tertiary alcohols 3a-3i.a

|
Yields are of material isolated by silica gel chromatography. See Supporting Information for further experimental details.
140 °C.
α-Hydroxy esters 1a-1i react by way of transient α-ketoesters for which the vicinal dicarbonyl moieties are electronically differentiated. In corresponding reactions of non-symmetric α-ketols, the vicinal dicarbonyl intermediates are quite similar electronically, rendering the control of regioselectivity uncertain. In the event, application of optimal conditions to the coupling of ethylene 2a with α-ketols 1j-1o delivered the ethylated tertiary alcohols 3j-3o in good to excellent yield (Table 2). Further, in the coupling of α-ketols 1j, 1l-1n, which proceed by way of nonsymmetric diones, the adducts 3j, 3l-3n form as single regioisomers. In addition to the influence of electronic effects on the regioselectivity of oxidative coupling as described by Hoffman12 and in prior work from our laboratory,9e steric effects also play an important role. That is, oxidative coupling will occur such that the osmium center is placed distal to the site of greatest steric demand. α-Ketol 1n is an exception due to the electronic effect associated with the mesomeric effect of the ortho-oxygen atom.
Table 2.
Osmium(0) catalyzed coupling of α-ketols 1j-1o with ethylene 2a to form tertiary alcohols 3j-3o.a

|
Yields are of material isolated by silica gel chromatography. See Supporting Information for further experimental details.
C10H15CO2H (10 mol%)
The coupling of ethylene 2a with α-hydroxy esters 1a-1i or α-ketols 1j-1o to form adducts 3a-3o are redox-neutral transformations. In contrast, the reaction of ethylene 2a with 1,2-diols dihydro-1j-1o represent oxidative processes in which one equivalent of H2 is evolved or transferred to an acceptor (Table 3). The feasibility of such an oxidative process finds precedent in the work of Shvo, who demonstrates that zero-valent ruthenium catalysts derived from Ru3(CO)12 promote oxidative esterifications in which tolane (diphenyl acetylene) serves as H2-acceptor,17 as well as work from our laboratory on oxidative diol-diene [4+2] cycloadditions.9c Initial attempts at the coupling of ethylene 2a with 1,2-diols dihydro-1j-1o using the osmium(0) catalyzed modified by XPhos led to only modest yields of adducts 3j-3o. Given the ability of carboxylic acids to catalyze the hydrogenolysis13 and transfer hydrogenolysis9e of oxametalacycles, these reactions were conducted in the presence of adamantane carboxylic acid (10 mol%). To our delight, the yields of adducts 3j-3o improved considerably and, as observed in couplings conducted from the α-ketol oxidative level, compounds 3j, 3l-3n were again generated as single regioisomers.
Table 3.
Osmium(0) catalyzed coupling of 1,2-diols dihydro-1j-1o with ethylene 2a to form tertiary alcohols 3j-3o.a

|
Yields are of material isolated by silica gel chromatography. See Supporting Information for further experimental details.
Reaction conducted in mesitylene at 150 °C using Os3(CO)12 (3 mol%), XPhos (18 mol%), C10H15CO2H (15 mol%).
To evaluate the applicability of these conditions to higher α-olefins, the coupling of 1-octene 2b with α-hydroxy esters 1a, 1b and 1i and α-ketols 1j, 1k and 1m was attempted (Table 4). Although corresponding reactions of ethylene 2a proceed efficiently in the absence of a carboxylic acid cocatalyst, couplings of 1-octene 2b required the presence of adamantane carboxylic acid (10 mol%) to increase conversion. Additionally, higher concentrations were beneficial, so the reactions were conducted neat. The α-hydroxy esters 1a, 1b and 1i were converted to adducts 4a, 4b and 4i, respectively, with complete levels of branched regioselectivity and good levels of diastereoselectivity. Relative stereochemistry for adducts 4a, 4b and 4i was determined by single crystal X-ray diffraction analysis of a derivative of 4b. A stereochemical model is provided (Scheme 2). α-Ketols 1j, 1k and 1m were converted to adducts 4j, 4k and 4m in a completely regioselective fashion, but with diminished levels of diastereoselectivity.
Table 4.
Osmium(0) catalyzed coupling of 1a, 1b, 1i, 1j, 1k, 1m with 1-octene 2b to form 4a, 4b, 4i, 4j, 4k, 4m.a

|
Yields are of material isolated by silica gel chromatography. See Supporting Information for further experimental details.
Scheme 2.

General mechanism as illustrated in the coupling of ethyl mandelate 1a with 1-octene 2b and stereochemical model.
The present transfer hydrogenative couplings of α-olefins can be conducted in oxidative, redox-neutral or reductive modes. While redox-neutral couplings are most efficient, oxidative and reductive transformations are preparatively useful. The following transformations illustrate this unique capability (eq. 1–3). In the oxidative coupling of 1-octene 2b with diol dihydro-1k, wherein 1-octene serves as hydrogen acceptor, adduct 4k forms in 70% yield (eq. 1). The redox-neutral coupling of 1-octene 2b with α-ketol 1k proceeds in 95% yield (eq. 2). Finally, using 1,4-butanediol as terminal reductant,18 the reductive coupling of 1-octene 2b with the 1,2-dione dehydro-1k proceeds in 68% yield (eq. 3). Such redox-economy allows one to bypass discrete manipulations otherwise required for the adjustment of oxidation level.19
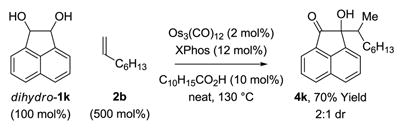 |
(eq. 1) |
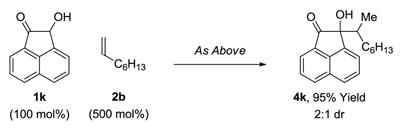 |
(eq. 2) |
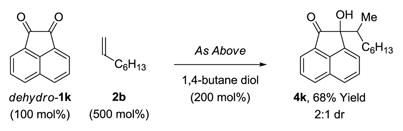 |
(eq. 3) |
To determine the scope of the alkene partner, the coupling of olefins 2a-2f with ethyl mandelate 1a was explored (Table 5). Beyond the previously described couplings of ethylene 2a and 1-octene 2b to form adducts 3a and 4a, respectively, allyl benzene 2c participates in C-C coupling to form tertiary alcohol 5a. For carboxy- and alkoxy-substituted alkenes 2d and 2e, the indicated regioisomers 6a and 7a are formed exclusively. Here, omission of XPhos and adamantane carboxylic acid is required to suppress metalacycle fragmentation en route to products of vinyl transfer (not shown).9i Finally, allyl acetate 2f participates in C-C coupling to form adduct 8a with complete levels of branched regioselectivity. This result is remarkable in view of the fact that ionization of allyl acetate 2f to form π-allyl species in the presence zero-valent osmium does not override the transfer hydrogenative C-C coupling pathway.
Table 5.
Osmium(0) catalyzed coupling of ethyl mandelate 1a with functionalized olefins 2a-2f.a

|
Yields are of material isolated by silica gel chromatography. See Supporting Information for further experimental details.
C10H15CO2H was omitted.
Neat.
C10H15CO2H (30 mol%)
2d (300 mol%).
XPhos was omitted.
Mechanism
With regard to the catalytic mechanism, a simple working model has been proposed as a basis for further refinement (Scheme 2). It is unclear whether the catalyst is mononuclear versus dimetallic or trimetallic. Upon heating toluene solutions of Os3(CO)12 with XPhos in the presence and absence of adamantane carboxylic acid (RCO2H), crystals of the dinuclear complex Os2(CO)4(O2CR)2(XPhos)2 and the trinuclear complex Os3(CO)11(XPhos), respectively, were isolated and characterized by X-ray diffraction (Figure 2). Additionally, the reaction of Os3(CO)12 with 2-(dicyclohexylphosphino)-1-(2-methoxyphenyl)-imidazole, a monophosphine that is structurally related to XPhos, provides the trinuclear osmium complex, Os3(CO)8L2.20b Alternatively, intervention of a mononuclear catalyst finds support in the reaction of Ru3(CO)12 with dppe, bis-(diphenylphosphino)ethane, to provide Ru(CO)3(dppe),20a and the reaction of Ru3(CO)12, 1-adamantanecarboxylic acid and dppp, bis-(diphenylphosphino)propane, to form the catalytically active mononuclear complex Ru(CO)(dppp)(O2CR)2.9e Oxidative coupling of the α-oxoester, dehydro-1a, with 1-octene 2b mediated by zero-valent osmium delivers the oxaosmacycle I.9,10 Related ruthenium(0)-mediated carbonyldiene oxidative couplings deliver isolable metalacycles that are catalytic active and have been shown to form in a reversible manner.9d The oxoester, dehydro-1a, required in the first turnover of the catalytic cycle may be generated via alcohol-olefin hydrogen transfer.17 Direct protonation of oxaosmacycle I by ethyl mandelate 1a to form the osmium alkoxide III requires a 4-centered transition structure and is postulated to be slow compared to protonation of oxaosmacycle I by 1-adamantanecarboxylic acid to form the osmium carboxylate II, which can proceed by way of a 6-centered transition structure.13 Exchange of the carboxylate ligand with 1a to form osmium alkoxide III also may proceed by way of a 6-centered transition structure.13 β-Hydride elimination converts osmium alkoxide III to the osmium alkyl hydride complex IV, which upon C-H reductive elimination releases product 4a and regenerates the osmium(0) catalyst. Beyond the aforesaid electronic effects,9e,12 steric interactions between the n-hexyl side chain of 1-octene 2b and the crowded osmium center contribute to branched regioselectivity.
Figure 2.

Structures of Os2(CO)4(O2CR)2(XPhos)2 (top) and Os3(CO)11(XPhos) (bottom) determined by single crystal X-ray diffraction showing the atom labeling system.a
aDisplacement ellipsoids are scaled to the 50% probability level. Hydrogen atoms have been omitted for clarity.
To challenge the veracity of the proposed mechanism, the following isotopic labelling experiment was performed (eq. 4). The deuterated acenaphthylene ketol deuterio-1k was exposed to ethylene under standard conditions. The pattern of deuterium incorporation in the adduct deuterio-3k was established by 1H and 2H NMR, as well as HRMS analysis. Deuterium incorporated occurs exclusively at the methyl group (22% 2H). The transfer of deuterium from the carbinol position of deuterio-1k to the methyl group of deuterio-3k is consistent with the proposed mechanism (Scheme 2). The relatively low levels of deuterium incorporation may be attributed to exchange with adventitious water or with the hydroxylic proton deuterio-1k.21
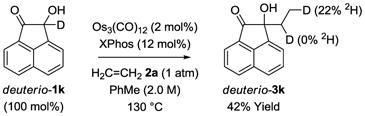 |
(eq. 4) |
Conclusions
In summary, the ability to transform abundant hydrocarbon feedstocks to value-added products in the absence of stoichiometric byproducts is a characteristic shared by nearly all large volume chemical processes. Hence, the discovery and development of byproduct-free transformations applicable to ethylene and α-olefins represents an important objective. Toward this end, we have shown that osmium(0) complexes derived from Os3(CO)12 and XPhos catalyze the transfer hydrogenative C-C coupling of ethylene and higher α-olefins with diverse vicinally dioxygenated hydrocarbons. Coupling may be conducted in a redox-neutral mode using α-ketols or α-hydroxy esters as reactants, or in oxidative or reductive modes using 1,2-diols or 1,2-diones as reactants, respectively. The collective data suggest increased π-backbonding at the stage of the osmium(0)-olefin π-complex plays a critical role in facilitating alkene-carbonyl oxidative coupling, as does the use of transient vicinal dicarbonyl partners, which have relatively low-lying LUMO energies. A challenge associated with the design of transfer hydrogenative coupling of α-olefins with simple primary alcohols will reside in the identification of metal catalysts that are sufficiently electron rich so as to promote oxidative coupling, and whose low-valent forms are accessible through alcohol mediated reduction of the high-valent ions. Indeed, intermolecular catalytic reductive couplings of α-olefins with unactivated carbonyl compounds remain an unmet challenge in chemical research.22
Experimental Section
General Information
All reactions were run under an atmosphere of argon. Os3(CO)12, XPhos, 1-adamantanecarboxylic acid, alkenes 2a-2f, α-hydroxy ester 1a, α-ketol 1o, diol dihydro-1o, and dione dehydro-1k were purchased from commercial suppliers and used as received. α-Hydroxy esters 1b-1i23a were prepared in accordance with the literature procedure. α-Ketols 1j,23b 1k23c 1l,23b 1m,23b 1n23d and diols dihydro-1j,23e dihydro- 1k,23f dihydro-1l,23b 1m23g and dihydro-1n23g were prepared using the cited literature procedures. Pressure tubes were flame dried followed by cooling in a desiccator. Toluene was dried over sodium metal-benzophenone and was distilled immediately prior to use. Anhydrous solvents were transferred by oven-dried syringes. Analytical thin-layer chromatography (TLC) was carried out using 0.25 mm commercial silica gel plates. Infrared spectra were recorded on a Perkin-Elmer 1600 spectrometer. High-resolution mass spectra (HRMS) are reported as m/z (relative intensity). Accurate masses are reported for the molecular ion (M+H, M+Na) or a suitable fragment ion. 1H Nuclear magnetic resonance spectra were recorded using a 400 MHz spectrometer. Coupling constants are reported in Hertz (Hz) for CDCl3 solutions, and chemical shifts are reported as parts per million (ppm) relative to residual CHCl3 δH (7.26 ppm). 13C Nuclear magnetic resonance spectra were recorded using a 100 MHz spectrometer for CDCl3 solutions, and chemical shifts are reported as parts per million (ppm) relative to residual CDCl3 δC (77.16 ppm).
General Procedure A
A resealable pressure tube (15 × 100 mm, 13 mL or 15 × 125 mm, 16 mL) was charged with Os3(CO)12 (5.5 mg, 0.006 mmol, 2 mol%), XPhos (17.1 mg, 0.036 mmol, 12 mol%) and the reactant alcohol (0.30 mmol, 100 mol%). The tube was sealed with a rubber septum and purged with ethylene. Toluene (0.15 mL, 2.0 M) was added and the rubber septum was quickly replaced with a screw cap. The reaction was allowed to stir at the indicated temperature for the stated period of time. After cooling to room temperature, the mixture was evaporated under reduced pressure and the residue was subjected to flash column chromatography (SiO2) under the conditions noted to afford the indicated product.
General Procedure B
A resealable pressure tube (13 × 100 mm, 9 mL) was charged with Os3(CO)12 (3.7 mg, 0.004 mmol, 2 mol%), XPhos (11.4 mg, 0.024 mmol, 12 mol%), AdCO2H (3.6 mg, 0.02 mmol, 10 mol%) and the reactant alcohol (0.20 mmol, 100 mol%). The tube was sealed with a rubber septum and purged with argon. 1-Octene (112.2 mg, 1.0 mmol, 500 mol%) was added via syringe and the rubber septum was quickly replaced with a screw cap. The reaction was allowed to stir at the indicated temperature for the stated period of time. After cooling to room temperature, the mixture was evaporated under reduced pressure and the residue was subjected to flash column chromatography (SiO2) under the conditions noted to afford the indicated product.
Ethyl 2-hydroxy-2-(4-(methylthio)phenyl)acetate (1g)
To a flame-dried 50 mL round-bottom flask charged with ethyl 2-hydroxy-2-(4-(methylthio)phenyl)acetate (1.1 g, 4.9 mmol), was added ethanol (25 mL, 0.2 M). NaBH4 (200 mg, 5.3 mmol) was added portionwise. The reaction mixture was allowed to stir at ambient temperature until the suspension became colorless. Distilled water was added and the reaction mixture was allowed to stir until bubbling stopped. The mixture was extracted with ethyl acetate (3 × 15 mL). The combined organic extracts were washed with brine (1 × 50 mL). The combined organic extracts were dried (MgSO4), filtered and evaporated under reduced pressure. The residue was subjected to column chromatography (SiO2: 20% ethyl acetate in hexanes) to give the title compound (0.93g, 4.1 mmol) in 84% yield as a white solid. 1H NMR (400 MHz, CDCl3): δ 7.34 (d, J = 8.0 Hz, 2H), 7.24 (d, J = 8.0 Hz, 2H), 5.11 (d, J = 8.0 Hz 1H), 4.22 (m, 2H), 3.42 (d, J = 8.0 Hz, 1H), 2.48 (s, 3H), 1.23 (t, J = 7.1 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ 173.6, 138.9, 135.2, 127.0, 126.5, 72.5, 62.3, 15.7, 14.0; HRMS (ESI-MS) Calcd. for C11H14O3S [M+Na]+: 249.0556, Found: 249.0557; FTIR (neat): 3438, 2979, 1726; MP: 91 °C.
Ethyl 2-hydroxy-2-phenylbutanoate (3a).24a
In accordance with general procedure A, 1a (0.3 mmol, 100 mol%) was reacted with ethylene (15 × 100 mm pressure tube, 0.58 mmol, 190 mol%) in toluene (2.0 M) at 130 °C for a 40 hour period. Flash column chromatography (SiO2: 2–5% ether/hexanes) provided the title compound (48.7 mg, 0.23 mmol) as a yellow oil in 78% yield. 1H NMR (400 MHz, CDCl3): δ 7.62–7.59 (m, 2H), 7.37–7.32 (m, 2H), 7.30–7.26 (m, 1H), 4.32–4.16 (m, 2H), 3.78 (d, J = 0.4 Hz, 1H), 2.24 (dqd, J = 14.4, 7.2, 0.8 Hz, 1H), 2.07–1.98 (m, 1H), 1.28 (t, J = 7.2 Hz, 3H), 0.93 (t, J = 7.4 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ 175.5, 142.0, 128.3, 127.7, 125.7, 78.7, 62.5, 32.8, 14.2, 8.2; HRMS (ESI) Calcd. for C12H16O3, [M+Na]+: 231.0992, Found: 231.0998; FTIR (neat): 3504, 2980, 1721.
Ethyl 2-(4-bromophenyl)-2-hydroxybutanoate (3b)
In accordance with Procedure A, 1b (0.3 mmol, 100 mol%) was reacted with ethylene (15 × 100 mm pressure tube, 0.58 mmol, 190 mol%) in toluene (2.0 M) at 130 °C for a 40 hour period. Flash column chromatography (SiO2: 2–4% ether/hexanes) provided the title compound (63.7 mg, 0.22 mmol) as a yellow oil in 74% yield. 1H NMR (400 MHz, CDCl3): δ 7.51–7.44 (m, 4H), 4.32–4.15 (m, 2H), 3.80 (d, J = 0.5 Hz, 1H), 2.24–2.12 (m, 1H), 1.97 (dq, J = 14.7, 7.4 Hz, 1H), 1.27 (t, J = 7.1 Hz, 3H), 0.99 (dd, J = 9.5, 5.2 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ 175.0, 141.0, 131.4, 127.7, 121.9, 78.4, 62.8, 32.9, 14.2, 8.1; HRMS (ESI) Calcd. for C12H15BrO3, [M+Na]+: 309.0097, 311.0077, Found: 309.0104, 311.0085; FTIR (neat): 3499, 2980, 1723.
Ethyl 2-hydroxy-2-(4-methoxyphenyl)butanoate (3c)
In accordance with Procedure A, 1c (0.3 mmol, 100 mol%) was reacted with ethylene (15 × 100 mm pressure tube, 0.58 mmol, 190 mol%) in toluene (2.0 M) at 130 °C for a 40 hour period. Flash column chromatography (SiO2: 50–100% dichloromethane/hexanes to 5% ethyl acetate/hexanes) provided the title compound (43.6 mg, 0.18 mmol) as a yellow oil in 61% yield. 1H NMR (400 MHz, CDCl3): δ 7.54–7.48 (m, 2H), 6.90–6.85 (m, 2H), 4.31–4.14 (m, 2H), 3.80 (s, 3H), 3.73 (s, 1H), 2.26–2.15 (m, 1H), 1.99 (dq, J = 14.6, 7.4 Hz, 1H), 1.27 (t, J = 7.1 Hz, 3H), 0.90 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ 175.7, 159.1, 134.2, 126.9, 113.6, 78.4, 62.5, 55.4, 32.8, 14.3, 8.2; HRMS (ESI) Calcd. for C13H18O4, [M+Na]+: 261.1097, Found: 261.1099; FTIR (neat): 3511, 2970, 1721.
Ethyl 2-hydroxy-2-(4-(trifluoromethyl)phenyl)butanoate (3d)
In accordance with Procedure A, 1d (0.3 mmol, 100 mol%) was reacted with ethylene (15 × 100 mm pressure tube, 0.58 mmol, 190 mol%) in toluene (2.0 M) at 130 °C for a 40 hour period. Flash column chromatography (SiO2: 1–5% ether/hexanes) provided the title compound (63.8 mg, 0.23 mmol) as a yellow oil in 77% yield. 1H NMR (400 MHz, CDCl3): δ 7.79–7.72 (m, 2H), 7.64–7.57 (m, 2H), 4.35–4.17 (m, 2H), 3.87 (s, 1H), 2.23 (dq, J = 14.5, 7.2 Hz, 1H), 2.01 (dq, J = 14.5, 7.4 Hz, 1H), 1.28 (t, J = 6.2 Hz, 3H), 0.92 (t, J = 7.3 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ 174.8, 145.9, 130.0 (q, J = 32.0 Hz), 126.3, 125.2 (q, J = 4.0 Hz), 124.3 (q, J = 271.0 Hz), 78.6, 63.0, 33.1, 14.2, 8.0; 19F NMR (376 MHz, CDCl3): δ -62.6; HRMS (ESI) Calcd. for C13H15F3O3, [M+Na]+: 299.0866, Found: 299.0871; FTIR (neat): 3510, 2985, 1726.
Ethyl 2-hydroxy-2-(3-(trifluoromethyl)phenyl)butanoate (3e)
In accordance with Procedure A, 1e (0.3 mmol, 100 mol%) was reacted with ethylene (15 × 100 mm pressure tube, 0.58 mmol, 190 mol%) in toluene (2.0 M) at 130 °C for a 40 hour period. Flash column chromatography (SiO2: 2–5% ether/hexanes) provided the title compound (63.0 mg, 0.23 mmol) as a yellow oil in 76% yield. 1H NMR (400 MHz, CDCl3): δ 7.92 (s, 1H), 7.86–7.79 (m, 1H), 7.55 (dd, J = 7.7, 0.6 Hz, 1H), 7.46 (dd, J = 7.8, 7.8 Hz, 1H), 4.34–4.19 (m, 2H), 3.90 (d, J = 0.5 Hz, 1H), 2.29–2.18 (m, 1H), 2.01 (dq, J = 14.7, 7.4 Hz, 1H), 1.28 (t, J = 7.2 Hz, 3H), 0.92 (t, J = 7.3 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ 174.9, 143.1, 130.7 (q, J = 32.0 Hz), 129.3, 128.8, 124.6 (q, J = 3.7 Hz), 124.3 (q, J = 271.0 Hz), 122.9 (q, J = 4.0 Hz), 78.5, 63.0, 33.2, 14.2, 8.1; 19F NMR (376 MHz, CDCl3): δ -62.6; HRMS (ESI) Calcd. for C13H15F3O3, [M+Na]+: 299.0866, Found: 299.0873; FTIR (neat): 3513, 2985, 1725.
Ethyl 2-(benzo[d][1,3]dioxol-5-yl)-2-hydroxybutanoate (3f)
In accordance with Procedure A, 1f (0.2 mmol, 100 mol%) was reacted with ethylene (15 × 100 mm pressure tube, 0.58 mmol, 290 mol%) in toluene (2.0 M) at 140 °C for a 40 hour period. Flash column chromatography (SiO2: 3–5% ether/hexanes) provided the title compound (30.8 mg, 0.12 mmol) as a colorless oil in 61% yield. NOTE: Os3(CO)12 (3.6 mg, 0.004 mmol, 2 mol%) and XPhos (11.4 mg, 0.024 mmol, 12 mol%). 1H NMR (400 MHz, CDCl3): δ 7.13–7.05 (m, 2H), 6.77 (dd, J = 7.5, 1.1 Hz, 1H), 6.01–5.92 (m, 2H), 4.33–4.14 (m, H), 3.75 (s, 1H), 2.16 (dq, J = 14.4, 7.2 Hz, 1H), 1.96 (dq, J = 14.7, 7.4 Hz, 1H), 1.27 (t, J = 7.2 Hz, 3H), 0.90 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ 175.5, 147.7, 147.1, 136.1, 119.1, 107.9, 106.7, 101.2, 78.5, 62.6, 32.9, 14.3, 8.1; HRMS (ESI) Calcd. for C13H16O5, [M+Na]+: 275.0890, Found: 275.0899; FTIR (neat): 3507, 2971, 1722.
Ethyl 2-hydroxy-2-(4-(methylthio)phenyl)butanoate (3g)
In accordance with Procedure A, 1g (0.3 mmol, 100 mol%) was reacted with ethylene (15 × 100 mm pressure tube, 0.58 mmol, 190 mol%) in toluene (2.0 M) at 130 °C for a 40 hour period. Flash column chromatography (SiO2: 2–5% ether/hexanes) provided the title compound (30.8 mg, 0.18 mmol) as a yellow oil in 61% yield. 1H NMR (400 MHz, CDCl3): δ 7.57–7.48 (m, 2H), 7.26–7.20 (m, 2H), 4.37–4.10 (m, 2H), 3.76 (s, 1H), 2.48 (s, 3H), 2.25–2.15 (m, 1H), 2.04-1.95 (m, 1H), 1.28 (t, J = 7.1 Hz, 3H), 0.91 (t, J = 7.3 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ 175.2, 138.7, 137.8, 126.2, 126.1 78.3, 62.4, 32.6, 15.7, 14.1, 7.9; HRMS (ESI) Calcd. for C13H18O3S, [M+Na]+: 277.0869, Found: 277.0878; FTIR (neat): 3507, 2979, 1721.
Ethyl 2-(furan-2-yl)-2-hydroxybutanoate (3h)
In accordance with Procedure A, 1h (0.2 mmol, 100 mol%) was reacted with ethylene (15 × 100 mm pressure tube, 0.58 mmol, 290 mol%) in toluene (2.0 M) at 130 °C for a 40 hour period. Flash column chromatography (SiO2: 3–5% ether/hexanes) provided the title compound (25.0 mg, 0.13 mmol) as a yellow oil in 64% yield. 1H NMR (400 MHz, CDCl3): δ 7.36 (d, J = 1.1 Hz, 1H), 6.33 (s, 2H), 4.36–4.14 (m, 2H), 3.82 (s, 1H), 2.21–2.07 (m, 2H), 1.25 (t, J = 7.0 Hz, 3H), 0.93 (t, J = 7.4 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ 173.7, 154.5, 142.5, 110.4, 106.8, 75.5, 62.8, 29.8, 14.3, 7.7; HRMS (ESI) Calcd. for C10H14O4, [M+Na]+: 221.0784, Found: 221.0790; FTIR (neat): 3511, 2970, 1728.
Ethyl 2-hydroxy-2-(thiophen-2-yl)butanoate (3i)
In accordance with Procedure A, 1i (0.3 mmol, 100 mol%) was reacted with ethylene (15 × 100 mm pressure tube, 0.58 mmol, 190 mol%) in toluene (2.0 M) at 130 °C for a 40 hour period. Flash column chromatography (SiO2: 30–50% dichloromethane/hexanes) provided the title compound (45.0 mg, 0.22 mmol) as a colorless oil in 70% yield. 1H NMR (400 MHz, CDCl3): δ 7.22 (dd, J = 5.1, 1.2 Hz, 1H), 7.09 (dd, J = 3.6, 1.2 Hz, 1H), 6.97 (dd, J = 5.1, 3.6 Hz, 1H), 4.35–4.21 (m, 2H), 4.05 (d, J = 0.8 Hz, 1H), 2.26–2.16 (m, 1H), 2.06 (dq, J = 14.7, 7.4 Hz, 1H), 1.31 (t, J = 7.1 Hz, 3H), 0.94 (t, J = 7.3 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ 174.5, 147.1, 127.1, 124.9, 124.1, 77.7, 62.9, 34.4, 14.2, 8.1; HRMS (ESI) Calcd. for C10H14O3S, [M+Na]+: 237.0556, Found: 237.0563; FTIR (neat): 3499, 2979, 1724.
2-Ethyl-2-hydroxy-2,3-dihydro-1H-inden-1-one (3j)
(Using ketol) In accordance with Procedure A, 1j (0.3 mmol, 100 mol%) was reacted with ethylene (15 × 100 mm pressure tube, 0.58 mmol, 190 mol%) in toluene (2.0 M) at 140 °C for a 40 hour period. Flash column chromatography (SiO2: 5–15% ethyl acetate/hexanes) provided the title compound (44.4 mg, 0.25 mmol) as a yellow oil in 84% yield. (Using diol) In accordance with Procedure A, H2-1j (0.15 mmol, 100 mol%) was reacted with ethylene (15 × 100 mm pressure tube, 0.58 mmol, 390 mol%) in toluene (1.5 M) at 140 °C for a 48 hour period. Flash column chromatography (SiO2: 5–15% ethyl acetate/hexanes) provided the title compound (18.8 mg, 0.11 mmol) as a yellow oil in 71% yield. NOTE: Os3(CO)12 (2.7 mg, 0.003 mmol, 2 mol%), XPhos (8.5 mg, 0.018 mmol, 12 mol%). 1H NMR (400 MHz, CDCl3): δ 7.77–7.72 (m, 1H), 7.61 (ddd, J = 7.5, 7.5, 1.2 Hz, 1H), 7.45–7.41 (m, 1H), 7.40–7.34 (m, 1H), 3.27 (d, J = 17.0 Hz, 1H), 3.14 (d, J = 17.0 Hz, 1H), 2.89 (s, 1H), 1.81–1.64 (m, 2H), 0.91 (t, J = 7.5 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ 208.4, 151.7, 135.9, 134.4, 127.9, 126.7, 124.7, 80.3, 39.8, 31.6, 8.0; HRMS (ESI) Calcd. for C11H12O2, [M+Na]+: 199.0730, Found: 199.0736; FTIR (neat): 3413, 2967, 1709.
2-Ethyl-2-hydroxyacenaphthylen-1(2H)-one (3k)
(Using ketol) In accordance with Procedure A, 1k (0.3 mmol, 100 mol%) was reacted with ethylene (15 × 100 mm pressure tube, 0.58 mmol, 190 mol%) in toluene (2.0 M) at 130 °C for a 48 hour period. Flash column chromatography (SiO2: 5–15% ethyl acetate/hexanes) provided the title compound (40.1 mg, 0.18 mmol) as a yellow solid in 63% yield. (Using diol) In accordance with Procedure A, H2-1k (0.15 mmol, 100 mol%) was reacted with ethylene (15 × 125 mm pressure tube, 0.71 mmol, 480 mol%) in toluene (1.5 M) at 140 °C for a 48 hour period. Flash column chromatography (SiO2: 5–15% ethyl acetate/hexanes) provided the title compound (21.3 mg, 0.11 mmol) as a yellow solid in 70% yield. NOTE: Os3(CO)12 (2.7 mg, 0.003 mmol, 2 mol%), XPhos (8.5 mg, 0.018 mmol, 12 mol%) and AdCO2H (2.7 mg, 0.015mmol, 10 mol%). 1H NMR (400 MHz, CDCl3): δ 8.13 (d, J = 8.2 Hz, 1H), 7.95 (d, J = 7.0 Hz, 1H), 7.89 (dd, J = 7.9, 1.2 Hz, 1H), 7.74 (dd, J = 8.1, 7.1 Hz, 1H), 7.71–7.62 (m, 2H), 2.86 (s, 1H), 2.17–1.99 (m, 2H), 0.76 (t, J = 7.5 Hz, 3H); 13C NMR (100 MHz, CDCl3−): δ 206.2, 141.8, 139.5, 132.1, 131.2, 130.8, 128.9, 128.4, 125.4, 122.0, 120.5, 80.9, 31.6, 8.2; HRMS (ESI) Calcd. for C14H12O2, [M+Na]+: 235.0730, Found: 235.0737; FTIR (neat): 3369, 2970, 2931, 1716; MP: 92.7–93.1 °C
2-Ethyl-2-hydroxy-3,4-dihydronaphthalen-1(2H)-one (3l).24b
(Using ketol) In accordance with Procedure A, 1l (0.3 mmol, 100 mol%) was reacted with ethylene (15 × 100 mm pressure tube, 0.58 mmol, 190 mol%) in toluene (2.0 M) at 140 °C for a 40 hour period. Flash column chromatography (SiO2: 5–7% ethyl acetate/hexanes) provided the title compound (47.4 mg, 0.25 mmol) as a brown oil in 83% yield. NOTE: AdCO2H (5.4 mg, 0.03mmol, 10 mol%). (Using diol) In accordance with Procedure A, H2-1l (0.15 mmol, 100 mol%) was reacted with ethylene (15 × 125 mm pressure tube, 0.71 mmol, 480 mol%) in toluene (1.5 M) at 140 °C for a 48 hour period. Flash column chromatography (SiO2: 5–7% ethyl acetate/hexanes) provided the title compound (20.3 mg, 0.11 mmol) as a brown oil in 71% yield. NOTE: Os3(CO)12 (2.7 mg, 0.003 mmol, 2 mol%), XPhos (8.5 mg, 0.018 mmol, 12 mol%) and AdCO2H (2.7 mg, 0.015mmol, 10 mol%). 1H NMR (400 MHz, CDCl3): δ 8.01 (dd, J = 7.8, 1.2 Hz, 1H), 7.51 (ddd, J = 7.5, 7.5, 1.4 Hz, 1H), 7.33 (dd, J = 7.6, 7.6 Hz, 1H), 7.27–7.21 (m, 1H), 3.81 (s, 1H), 3.15–2.94 (m, 2H), 2.34 (ddd, J = 13.5, 5.1, 2.3 Hz, 1H), 2.21–2.10 (m, 1H), 1.78–1.60 (m, 2H), 0.93 (t, J = 7.4 Hz, 3H); 13C NMR (100 MHz, CDCl3−): δ 202.1, 143.6, 134.1, 130.4, 129.1, 128.0, 127.0, 75.9, 33.7, 28.5, 26.6, 7.3; HRMS (ESI) Calcd. for C12H14O2, [M+Na]+: 213.0886, Found: 213.0892; FTIR (neat): 3488, 2931, 1681.
3-Ethyl-3-hydroxychroman-4-one (3m)
(Using ketol) In accordance with Procedure A, 1m (0.3 mmol, 100 mol%) was reacted with ethylene (15 × 100 mm pressure tube, 0.58 mmol, 190 mol%) in toluene (2.0 M) at 130 °C for a 48 hour period. Flash column chromatography (SiO2: 2–4% ethyl acetate/hexanes) provided the title compound (49.6 mg, 0.26 mmol) as a yellow oil in 86% yield. (Using diol) In accordance with Procedure A, H2-1m (0.15 mmol, 100 mol%) was reacted with ethylene (15 × 125 mm pressure tube, 0.71 mmol, 480 mol%) in toluene (1.5 M) at 140 °C for a 48 hour period. Flash column chromatography (SiO2: 2–4% ethyl acetate/hexanes) provided the title compound (17.3 mg, 0.09 mmol) as a yellow oil in 60% yield. NOTE: Os3(CO)12 (2.7 mg, 0.003 mmol, 2 mol%), XPhos (8.5 mg, 0.018 mmol, 12 mol%) and AdCO2H (2.7 mg, 0.015mmol, 10 mol%). 1H NMR (400 MHz, CDCl3): δ 7.89–7.85 (m, 1H), 7.51 (ddd, J = 8.4, 7.2, 1.8 Hz, 1H), 7.06 (ddd, J = 8.0, 7.2, 1.0 Hz, 1H), 6.97 (dd, J = 8.4, 0.6 Hz, 1H), 4.39 (d, J = 11.3 Hz, 1H), 4.16 (d, J = 11.3 Hz, 1H), 3.62 (s, 1H), 1.80 (q, J = 7.5 Hz, 2H), 0.94 (t, J = 7.5 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ 196.9, 161.5, 136.7, 127.6, 121.9, 118.5, 118.0, 73.1, 72.9, 27.8, 7.0; HRMS (CI) Calcd. for C11H12O3, [M+H]+: 215.0679, Found: 215.0686; FTIR (neat): 3466, 2973, 2936, 1691, 1607.
3-Ethyl-3-hydroxy-2,2-dimethylchroman-4-one (3n)
(Using ketol) In accordance with Procedure A, 1n (0.3 mmol, 100 mol%) was reacted with ethylene (15 × 100 mm pressure tube, 0.58 mmol, 190 mol%) in toluene (2.0 M) at 140 °C for a 40 hour period. Flash column chromatography (SiO2: 2–4% ether/hexanes) provided the title compound (56.2 mg, 0.26 mmol) as a yellow oil in 85% yield. (Using diol) In accordance with Procedure A, H2-1n (0.15 mmol, 100 mol%) was reacted with ethylene (15 × 100 mm pressure tube, 0.58 mmol, 390 mol%) in toluene (1.5 M) at 140 °C for a 48 hour period. Flash column chromatography (SiO2: 2–4% ethyl acetate/hexanes) provided the title compound (31.1 mg, 0.14 mmol) as a yellow oil in 94% yield. NOTE: Os3(CO)12 (2.7 mg, 0.003 mmol, 2 mol%), XPhos (8.5 mg, 0.018 mmol, 12 mol%). 1H NMR (400 MHz, CDCl3): δ 7.78 (dd, J = 7.8, 1.7 Hz, 1H), 7.48 (ddd, J = 8.6, 7.2, 1.8 Hz, 1H), 6.98 (dt, J = 12.0, 2.5 Hz, 1H), 6.89 (dd, J = 8.4, 0.5 Hz, 1H), 3.89 (s, 1H), 1.92–1.80 (m, 2H), 1.52 (s, 3H), 1.26 (s, 3H), 0.68 (t, J = 7.5 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ 197.8, 159.3, 136.7, 126.9, 121.1, 118.7, 118.3, 84.6, 78.7, 25.3, 22.2, 20.4, 7.3; HRMS (ESI) Calcd. for C13H16O3, [M+Na]+: 243.0992, Found: 243.0993; FTIR (neat): 3484, 2976, 1690.
2-Ethyl-2-hydroxycyclohexan-1-one (3o).24c
(Using ketol) In accordance with Procedure A, 1o (0.3 mmol, 100 mol%) was reacted with ethylene (15 × 100 mm pressure tube, 0.58 mmol, 190 mol%) in toluene (2.0 M) at 130 °C for a 48 hour period. Flash column chromatography (SiO2: 5–10% ether/hexanes) provided the title compound (31.1 mg, 0.22 mmol) as a colorless oil in 73% yield. (Using diol) In accordance with Procedure A, H2-1o (0.15 mmol, 100 mol%) was reacted with ethylene (15 × 125 mm pressure tube, 0.71 mmol, 480 mol%) in meistylene (2.0 M) at 150 °C for a 48 hour period. Flash column chromatography (SiO2: 5–10% ether/hexanes) provided the title compound (10.7 mg, 0.15 mmol) as a colorless oil in 50% yield. NOTE: Os3(CO)12 (4.1 mg, 0.0045 mmol, 3 mol%), XPhos (13.2 mg, 0.027 mmol, 18 mol%) and AdCO2H (4.1 mg, 0.023 mmol, 15 mol%). 1H NMR (400 MHz, CDCl3): δ 3.93 (s, 1H), 2.51–2.40 (m, 2H), 2.18 (ddd, J = 13.1, 5.8, 3.0 Hz, 1H), 2.14–2.03 (m, 1H), 1.91 (dq, J = 14.7, 7.4 Hz, 1H), 1.84–1.54 (m, 5H), 0.90–0.75 (m, 3H); 13C NMR (100 MHz, CDCl3): δ 214.6, 79.4, 40.7, 38.2, 30.2, 28.0, 22.9, 7.0; HRMS (ESI) Calcd. for C8H14O2, [M+Na]+: 142.0994, Found: 142.0994; FTIR (neat): 3485, 2938, 1707.
Ethyl 2-hydroxy-3-methyl-2-phenylnonanoate (4a)
In accordance with Procedure B, 1a (0.2 mmol, 100 mol%) was reacted with 1-octene (13 × 100 mm pressure tube, 0.15 mL, 1.0 mmol, 500 mol%) at 130 °C for a 40 hour period. Flash column chromatography (SiO2: 2% ether/hexanes) provided the title compound (31.1 mg, 0.22 mmol, d.r. = 5:1) as a colorless oil in 62% yield. 1H NMR (400 MHz, CDCl3): δ (major) 7.67–7.62 (m, 2H), 7.37–7.30 (m, 2H), 7.29–7.24 (m, 1H), 4.33–4.13 (m, 2H), 3.68 (d, J = 0.6 Hz, 1H), 2.47–2.35 (m, 1H), 1.51–1.16 (m, 13H), 0.90 (dd, J = 8.9, 4.9 Hz, 3H), 0.68 (d, J = 6.8 Hz, 3H). (minor) 7.67–7.62 (m, 2H), 7.37–7.30 (m, 2H), 7.29–7.24 (m, 1H), 4.33–4.13 (m, 2H), 3.74 (d, J = 0.6 Hz, 1H), 2.47–2.35 (m, 1H), 1.51–1.16 (m, 13H), 0.97 (d, J = 6.6 Hz, 3H), 0.82 (t, J = 7.1 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ (major) 176.0, 141.5, 128.1, 127.5, 126.1, 81.7, 62.5, 40.8, 31.9, 31.8, 29.5, 27.7, 22.8, 14.3, 14.2, 12.8. (minor) 175.9, 141.3, 128.1, 127.5, 126.2, 81.6, 62.6, 40.6, 31.9, 29.6, 29.3, 27.6, 22.7, 14.2, 14.1, 12.8; HRMS (ESI) Calcd. for C18H28O3, [M+Na]+: 315.1931, Found: 315.1940; FTIR (neat): 3514, 2928, 2857, 1721.
Ethyl 2-(4-bromophenyl)-2-hydroxy-3-methylnonanoate (4b)
In accordance with Procedure B, 1b (0.2 mmol, 100 mol%) was reacted with 1-octene (13 × 100 mm pressure tube, 0.15 mL, 1.0 mmol, 500 mol%) at 130 °C for a 40 hour period. Flash column chromatography (SiO2: 20–35% dichloromethane/hexanes) provided the title compound (45.4 mg, 0.12 mmol, d.r. = 4:1) as a colorless oil in 61% yield. 1H NMR (400 MHz, CDCl3): δ (major) 7.54–7.50 (m, 2H), 7.48–7.43 (m, 2H), 4.33–4.14 (m, 2H), 3.67 (s, 1H), 2.38–2.28 (m, 1H), 1.48–0.97 (m, 13H), 0.89 (d, J = 6.9 Hz, 3H), 0.66 (d, J = 6.8 Hz, 3H). (minor) 7.54–7.50 (m, 2H), 7.48–7.43 (m, 2H), 4.33–4.14 (m, 2H), 3.73 (s, 1H), 2.38–2.28 (m, 1H), 1.48–0.97 (m, 13H), 0.95 (d, J = 6.6 Hz, 3H), 0.83 (t, J = 7.1 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ (major) 175.5, 140.6, 131.3, 128.1, 121.7, 81.5, 62.8, 40.9, 31.9, 31.7, 29.5, 27.6, 22.8, 14.3, 14.2, 12.8. (minor) 175.4, 140.6, 131.3, 128.1, 121.7, 81.4, 62.9, 40.7, 31.9, 29.6, 29.3, 27.6, 22.7, 14.2, 14.1, 12.8; HRMS (ESI) Calcd. for C18H27BrO3, [M+Na]+: 393.1036, Found: 393.1043; FTIR (neat): 3507, 2927, 2856, 1723.
Ethyl 2-hydroxy-3-methyl-2-(thiophen-2-yl)nonanoate (4i)
In accordance with Procedure B, 1i (0.2 mmol, 100 mol%) was reacted with 1-octene (13 × 100 mm pressure tube, 0.15 mL, 1.0 mmol, 500 mol%) at 130 °C for a 40 hour period. Flash column chromatography (SiO2: 20–40% dichloromethane/hexanes) provided the title compound (47.8 mg, 0.16 mmol, d.r. = 5:1) as a colorless oil in 80% yield. 1H NMR (400 MHz, CDCl3): δ (major) 7.21 (dd, J = 5.2, 1.2 Hz, 1H), 7.09 (dd, J = 3.6, 1.2 Hz, 1H), 6.98 (dd, J = 5.2, 3.6 Hz, 1H), 4.38–4.19 (m, 2H), 3.95 (d, J = 0.5 Hz, 1H), 2.33–2.20 (m, 1H), 1.48–1.05 (m, 14H), 0.91–0.83 (m, 2H), 0.81 (d, J = 6.8 Hz, 3H). (minor) 7.22 (dd, J = 5.2, 1.2 Hz, 1H), 7.09 (dd, J = 3.6, 1.2 Hz, 1H), 6.99–6.97 (m, 1H), 4.38–4.19 (m, 2H), 4.00 (d, J = 0.5 Hz, 1H), 2.33–2.20 (m, 1H), 1.48–1.05 (m, 14H), 0.93 (d, J = 6.6 Hz, 3H), 0.91–0.83 (m, 2H); 13C NMR (100 MHz, CDCl3): δ (major) 175.0, 146.9, 127.1, 124.8, 124.3, 81.0, 62.8, 42.6, 31.9, 31.6, 29.5, 27.6, 22.8, 14.22, 14.15, 12.8. (minor) 174.9, 146.7, 127.1, 124.9, 124.4, 80.9, 62.9, 42.5, 31.9, 29.6, 29.4, 27.7, 22.7, 14.2, 14.0, 12.8; HRMS (ESI) Calcd. for C16H26O3S, [M+Na]+: 321.1495, Found: 321.1502; FTIR (neat): 3502, 2929, 2857, 1725.
2-Hydroxy-2-(octan-2-yl)-2,3-dihydro-1H-inden-1-one (4j)
In accordance with Procedure B, 1j (0.2 mmol, 100 mol%) was reacted with 1-octene (13 × 100 mm pressure tube, 0.15 mL, 1.0 mmol, 500 mol%) at 130 °C for a 40 hour period. Flash column chromatography (SiO2: 2–3% ether/hexanes) provided the title compound (44.3 mg, 0.17 mmol, d.r. = 1:1) as a colorless oil in 85% yield. 1H NMR (400 MHz, CDCl3): δ (A) 7.75 (d, J = 7.7 Hz, 1H), 7.61 (dd, J = 10.8, 4.1 Hz, 1H), 7.43 (d, J = 7.7 Hz, 1H), 7.37 (dd, J = 7.5, 7.5 Hz, 1H), 3.28 (d, J = 17.4 Hz, 1H), 2.98 (d, J = 17.4 Hz, 1H), 2.51 (s, 1H), 1.93–1.72 (m, 15H), 1.48–0.99 (m, 9.5H), 0.87 (t, J = 6.8 Hz, 3H), 0.67 (d, J = 6.9 Hz, 3H). (B) 7.75 (d, J = 7.7 Hz, 1H), 7.61 (dd, J = 10.8, 4.1 Hz, 1H), 7.43 (d, J = 7.7 Hz, 1H), 7.37 (dd, J = 7.5 Hz, 7.5 Hz, 1H), 3.28 (d, J = 17.4 Hz, 1H), 2.98 (d, J = 17.4 Hz, 1H), 2.53 (s, 1H), 1.93–1.72 (m, 1.5 H), 1.48–0.99 (m, 12.5H), 0.82 (t, J = 6.8 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ (A) 209.1, 152.8, 135.8, 135.4, 127.8, 126.6, 124.5, 82.3, 40.6, 36.8, 32.0, 30.5, 29.6, 27.8, 22.8, 14.5, 13.5. (B) 209.0, 152.6, 135.8, 135.5, 127.8, 126.7, 124.5, 82.3, 40.6, 37.1, 31.9, 31.5, 29.4, 27.7, 22.7, 14.22, 14.16; HRMS (ESI) Calcd. for C17H24O2, [M+Na]+: 283.1669, Found: 283.1679; FTIR (neat): 3447, 2926, 1709.
2-Hydroxy-2-(octan-2-yl)acenaphthylen-1(2H)-one (4k)
(Using dihydro-1k) In accordance with Procedure B, dihydro-1k (0.2 mmol, 100 mol%) was reacted with 1-octene (13 × 100 mm pressure tube, 0.15 mL, 1.0 mmol, 500 mol%) at 130 °C for a 40 hour period. Flash column chromatography (SiO2: 5–7% ethyl acetate/hexanes) provided the title compound (41.5 mg, 0.14 mmol, d.r. = 2:1) as a light green solid in 70% yield. (Using 1k) In accordance with Procedure B, 1k (0.2 mmol, 100 mol%) was reacted with 1-octene (13 × 100 mm pressure tube, 0.15 mL, 1.0 mmol, 500 mol%) at 130 °C for a 40 hour period. Flash column chromatography (SiO2: 5–7% ethyl acetate/hexanes) provided the title compound (56.3 mg, 0.19 mmol, d.r. = 2:1) as a light green solid in 95% yield. (Using dehydro-1k) In accordance with Procedure B, dehydro-1k (0.2 mmol, 100 mol%) was reacted with 1-octene (13 × 100 mm pressure tube, 0.15 mL, 1.0 mmol, 500 mol%) at 130 °C for a 40 hour period. Flash column chromatography (SiO2: 5–7% ethyl acetate/hexanes) provided the title compound (40.3 mg, 0.14 mmol, d.r. = 2:1) as a light green solid in 68% yield. NOTE: The reaction was conducted in the presence of 1,3-butane diol (36.0 mg, 0.4 mmol, 200 mol%). 1H NMR (400 MHz, CDCl3): δ (major) 8.11 (dd, J = 8.1, 0.5 Hz, 1H), 7.92 (ddd, J = 4.0, 2.0, 2.0 Hz, 1H), 7.90–7.85 (m, 1H), 7.72 (ddd, J = 8.1, 7.1, 1.0 Hz, 1H), 7.68–7.61 (m, 2H), 2.84 (d, J = 2.4 Hz, 1H), 2.27–1.94 (m, 1H), 1.49–0.96 (m, 10H), 0.91–0.83 (m, 3H), 0.58 (t, J = 6.2 Hz, 3H). (minor) 8.11 (dd, J = 8.1, 0.5 Hz, 1H), 7.92 (ddd, J = 4.0, 2.0, 2.0 Hz, 1H), 7.90–7.85 (m, 1H), 7.72 (ddd, J = 8.1, 7.1, 1.0 Hz, 1H), 7.68–7.61 (m, 2H), 2.84 (d, J = 2.4 Hz, 1H), 2.27–1.94 (m, 1H), 1.49–0.96 (m, 13H), 0.79 (t, J = 7.1 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ (major) 207.2, 142.5, 138.4, 132.01, 131.99, 130.7, 128.7, 128.3, 125.4, 121.7, 121.6, 83.1, 41.6, 32.0, 30.2, 29.6, 27.9, 22.8, 14.4, 14.2. (minor) 207.2, 142.4, 138.7, 132.0, 131.9, 130.8, 128.7, 128.3, 125.4, 121.6, 121.4, 82.9, 41.4, 31.8, 31.3, 29.2, 27.5, 22.6, 14.1, 13.3; HRMS (ESI) Calcd. for C20H24O2, [M+Na]+: 319.1669, Found: 319.1678; FTIR (neat): 3423, 2924, 1708; MP: 79.8–81.1 °C.
3-Hydroxy-3-(octan-2-yl)chroman-4-one (4m)
In accordance with Procedure B, 1m (0.2 mmol, 100 mol%) was reacted with 1-octene (13 × 100 mm pressure tube, 0.15 mL, 1.0 mmol, 500 mol%) at 130 °C for a 40 hour period. Flash column chromatography (SiO2: 2–3% ether/hexanes) provided the title compound (34.8 mg, 0.13 mmol, d.r. = 1:1) as a pale yellow solid in 63% yield. 1H NMR (400 MHz, CDCl3): δ (A) 7.84 (dd, J = 7.8, 1.7 Hz, 1H), 7.56–7.47 (m, 1H), 7.05 (ddd, J = 8.2, 1.9, 1.0 Hz, 1H), 6.96 (ddd, J = 8.4, 3.0, 0.6 Hz, 1H), 4.58 (dd, J = 21.0, 11.7 Hz, 1H), 4.06 (dd, J = 11.7, 5.6 Hz, 1H), 3.56 (s, 1H), 1.97–1.87 (m, 1H), 1.76–1.64 (m, 0.5H), 1.49–0.94 (m, 12.5H), 0.88 (dd, J = 8.4, 5.0 Hz, 3H). (B) 7.84 (dd, J = 7.8, 1.7 Hz, 1H), 7.56–7.47 (m, 1H), 7.05 (ddd, J = 8.2, 1.9, 1.0 Hz, 1H), 6.96 (ddd, J = 8.4, 3.0, 0.6 Hz, 1H), 4.58 (dd, J = 21.0, 11.7 Hz, 1H), 1.76–1.64 (m, 0.5H), 1.49–0.94 (m, 9.5H), 0.80 (t, J = 7.0 Hz, 3H), 0.74 (d, J = 6.9 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ (A) 209.1, 152.8, 135.8, 135.4, 127.8, 126.6, 124.5, 82.3, 40.6, 36.8, 32.0, 31.5, 29.6, 27.7, 22.8, 14.5, 14.2. (B) 209.1, 152.6, 135.8, 135.4, 127.8, 126.7, 124.5, 82.3, 40.6, 37.1, 31.9, 30.5, 29.4, 27.7, 22.7, 14.2, 13.5; HRMS (ESI) Calcd. for C17H24O3, [M+Na]+: 299.1618, Found: 299.1623; FTIR (neat): 3453, 2927, 1684; MP: 67.8–68.0 °C.
Ethyl 2-hydroxy-3-methyl-2,4-diphenylbutanoate (5a)
In accordance with Procedure B, 1a (0.2 mmol, 100 mol%) was reacted with 2c (13 × 100 mm pressure tube, 0.13 mL, 1.0 mmol, 500 mol%) in toluene (2.0 M) at 130 °C for a 40 hour period. Flash column chromatography (SiO2: 30–60% dichloromethane/hexanes) provided the title compound (34.6 mg, 0.11 mmol, d.r. = 5:1) as colorless oil in 58% yield. NOTE: AdCO2H (30 mol%). 1H NMR (400 MHz, CDCl3): δ (major) 7.71–7.65 (m, 2H), 7.38–7.26 (m, 5H), 7.22 (d, J = 7.3 Hz, 3H), 4.33–4.15 (m, 2H), 3.84 (d, J = 0.5 Hz, 1H), 2.82–2.67 (m, 2H), 2.57 (dd, J = 13.4, 10.5 Hz 1H), 1.33 (t, J = 7.1 Hz, 3H), 0.62 (t, J = 6.8 Hz, 3H). (minor) 7.81–7.77 (m, 2H), 7.42 (dd, J = 10.5, 4.9 Hz, 2H), 7.38–7.26 (m, 2H), 7.18 (dd, J = 11.2, 4.3 Hz, 2H), 7.05 (d, J = 7.1 Hz, 2H), 4.33–4.15 (m, 2H), 3.87 (d, J = 0.6 Hz, 1H), 2.82–2.67 (m, 1H), 2.50 (d, J = 13.7 Hz, 1H), 2.21 (dd, J = 13.6 Hz, 11.6 Hz, 1H), 1.28 (t, J = 7.2 Hz, 3H), 0.90 (d, J = 6.5 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ (major) 175.8, 141.3, 141.0, 129.4, 128.4, 128.3, 127.6, 126.1, 126.0, 81.3, 62.8, 43.2, 38.6, 14.3, 12.7. (minor) 175.5, 141.4, 141.1, 129.2, 128.4, 128.3, 127.8, 126.2, 125.9, 81.2, 62.8, 43.4, 36.4, 14.2, 13.6; HRMS (ESI) Calcd. for C19H22O3, [M+Na]+: 321.1461, Found: 321.1466; FTIR (neat): 3505, 2976, 2361, 2342, 1715.
Ethyl 2-hydroxy-2-phenyl-3-(pivaloyloxy)butanoate (6a)
In accordance with Procedure B, 1a (0.2 mmol, 100 mol%) was reacted with 2d (13 × 100 mm pressure tube, 0.09 mL, 0.6 mmol, 300 mol%) in toluene (2.0 M) at 130 °C for a 24 hour period. Flash column chromatography (SiO2: 2–4% ethyl acetate/hexanes) provided the title compound (51.8 mg, 0.25 mmol, d.r. = 1.5:1) as a pale yellow oil in 84% yield. NOTE: XPhos and AdCO2H was omitted. 1H NMR (400 MHz, CDCl3−): δ (major) 7.47–7.28 (m, 5H), 6.09 (q, J = 5.2 Hz, 1H), 5.19 (s, 1H), 4.25–4.06 (m, 2H), 1.53 (d, J = 5.2 Hz, 3H), 1.23–1.17 (m, 3H), 1.11 (s, 9H). (minor) 7.47–7.28 (m, 5H), 5.84 (q, J = 5.2 Hz, 1H), 5.14 (s, 1H), 4.25–4.06 (m, 2H), 1.44 (d, J = 5.3 Hz, 3H), 1.23–1.17 (m, 3H), 1.21 (s, 9H); 13C NMR (100 MHz, CDCl3): δ (major) 178.3. 170.6. 136.4. 128.7. 128.6. 127.3. 95.5. 79.0. 61.5. 38.9. 27.0. 20.9. 14.2. (minor) 178.3, 170.1, 136.0, 129.0, 128.8, 127.5, 95.0, 79.3, 61.4, 39.0, 27.1, 20.8, 14.1; HRMS (ESI) Calcd. for C17H24O5, [M+Na]+: 331.1516, Found: 331.1520; FTIR (neat): 2979, 1789, 1174.
Ethyl 2-hydroxy-2-phenyl-2-(tetrahydrofuran-2-yl)acetate (7a)
In accordance with Procedure B, 1a (0.2 mmol, 100 mol%) was reacted with 2e (13 × 100 mm pressure tube, 0.05 mL, 0.6 mmol, 300 mol%) in toluene (2.0 M) at 140 °C for a 24 hour period. Flash column chromatography (SiO2: 2–5% ethyl acetate/hexanes) provided the title compound (39.0 mg, 0.23 mmol, d.r. = 1:1) as a pale yellow oil in 78% yield. NOTE: XPhos and AdCO2H was omitted. 1H NMR (400 MHz, CDCl3): δ (A) 7.44 (ddd, J = 7.8, 7.8, 1.3 Hz, 2H), 7.39–7.27 (m, 3H), 5.36 (d, J = 3.6 Hz, 1H), 5.26 (s, 1H), 4.25–4.06 (m, 2H), 4.01–3.94 (m, 1H), 3.89–3.83 (m, 1H), 2.19–1.80 (m, 4H), 1.21 (t, J = 7.1 Hz, 3H). (B) 7.44 (ddd, J = 7.8, 7.8, 1.3 Hz, 2H), 7.39–7.27 (m, 3H), 5.17 (d, J = 4.4 Hz, 1H), 5.09 (s, 1H), 4.25–4.06 (m, 2H), 3.89–3.83 (m, 1H), 3.81–3.77 (m, 1H), 2.19–1.80 (m, 4H), 1.21 (t, J = 7.1 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ (A) 171.4, 137.2, 128.5, 128.4, 127.2, 102.7, 75.8, 67.5, 61.3, 32.6, 23.3, 14.2. (B) 171.4, 136.6, 128.64, 128.56, 127.4, 103.2, 77.4, 67.7, 61.1, 32.5, 23.6, 14.2; HRMS (ESI) Calcd. for C14H18O4, [M+Na]+: 273.1097, Found: 273.1108; FTIR (neat): 2981, 1747.
Ethyl 4-acetoxy-2-hydroxy-3-methyl-2-phenylbutanoate (8a)
In accordance with Procedure B, 1a (0.2 mmol, 100 mol%) was reacted with 2f (13 × 100 mm pressure tube, 0.11 mL, 1.0 mmol, 500 mol%) in toluene (2.0 M) at 140 °C for a 40 hour period. Flash column chromatography (SiO2: 5–7% ethyl acetate/hexanes) provided the title compound (33.6 mg, 0.12 mmol, d.r. = 1.6:1) as a pale yellow oil in 60% yield. 1H NMR (400 MHz, CDCl3): δ (major) 7.62 (ddd, J = 3.4, 1.9, 1.9 Hz, 2H), 7.35 (ddd, J = 11.8, 4.6 Hz, 3H), 4.33–4.15 (m, 3H), 4.07 (dd, J = 11.0, 6.5 Hz, 1H), 3.80 (s, 1H), 2.96–2.86 (m, 1H), 2.04 (s, 3H), 1.28 (t, J = 7.2 Hz, 3H), 0.70 (d, J = 6.9 Hz, 3H). (minor) 7.66 (ddd, J = 3.4, 1.9, 1.9 Hz, 2H), 7.31–7.25 (m, 3H), 4.33–4.20 (m, 2H), 3.90–3.81 (m, 2H), 3.80 (s, 1H), 2.96–2.86 (m, 1H), 1.81 (s, 3H), 1.29 (t, J = 7.0 Hz, 3H), 1.04 (d, J = 6.6 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ (major) 175.3, 170.8, 140.8, 128.3, 127.8, 126.0, 78.7, 66.0, 62.7, 40.0, 21.0, 14.1, 11.6. (minor) 174.7, 171.1, 140.2, 128.4, 127.9, 125.9, 79.6, 65.5, 62.9, 40.1, 20.8, 14.2, 12.6; HRMS (ESI) Calcd. for C15H20O5, [M+Na]+: 303.1203, Found: 303.1215; FTIR (neat): 3494, 2981, 1727, 1231.
2-(4-bromophenyl)-2-hydroxy-3-methylnonyl 4-bromobenzenesulfonate (4b Derivative)
An ethereal solution (5 mL) of 4b (1.39 g, 3.7 mmol) was added dropwise to a 100 mL round-bottom flask charged with an ethereal (30 mL, 0.12 M) suspension of LAH (709 mg, 18.7 mmol) at 0°C. The reaction was removed from the ice-bath and was allowed to stir for a 1 hour period. Distilled water (1 mL) was added slowly. Distilled water (3 ml) and 15% NaOH aqueous (1 mL) were added to the reaction mixture. To the vigorously stirred solution was added portions of MgSO4 until the reaction mixture solidified. The reaction mixture was filtered through a fritted glass funnel with the aid of ether. The filtrate was evaporated under reduced pressure and was used in the next step without further purification. To the crude diol (1.16 g, 3.5 mmol) was added dichloromethane (30 ml, 1.1 M), 4-bromobenzenesulfonyl chloride (996 mg, 3.9 mmol), DMAP (42 mg, 0.35 mmol) and Et3N (1 mL, 7.1 mmol). The reaction was allowed to stir at ambient temperature for a 1 hour period. NaHCO3 (10 mL) and distilled water (10 ml) were added to the reaction mixture. The mixture was transferred to a separatory funnel and extracted with ethyl acetate (3 × 20 mL). The combined organic extracts were washed with brine (1 × 50 mL). The combined organic extracts were dried (MgSO4), filtered and evaporated under reduced pressure. The crude 4b derivative residue was subjected to column chromatography (SiO2: 20% ethyl acetate/hexanes) to give the title compound (1.7 g, 3.3 mmol) in 90% yield as a white solid. 1H NMR (400 MHz, CDCl3): δ 7.65 (d, J = 8.7 Hz, 2H), 7.58 (d, J = 8.7 Hz, 2H), 7.39. (d, J = 8.6, 2H), 7.12 (d, J = 8.6 Hz, 2H), 4.34 (s, 2H), 2.11 (s, 1H), 1.86–1.76 (m, 1H), 1.55–1.46 (m, 1H), 1.37–1.05 (m, 8H), 0.91–0.80 (m, 4H), 0.74 (d, J = 6.9 Hz, 2H); 13C NMR (100 MHz, CDCl3): δ 140.3, 134.4, 132.6, 131.1, 129.2, 127.6, 74.7, 40.5, 31.8, 30.2, 29.4, 27.6, 22.6, 14.0, 13.8; HRMS (ESI-MS) Calcd. for C22H28Br2O4S [M+Na]+: 568.9967, Found: 568.9983; FTIR (neat): 3610, 2927, 1727, 1577; MP: 98–100 °C.
2-hydroxyacenaphtylen-1(2H)-one-2-d (deuterio-1k)
To a flame-dried 50 mL round-bottom flask charged with 2H-spiro[acenaphthylene-1,2′-[1,3]dioxolan]-2-one (891 mg, 3.9 mmol) was added ethanol (20 ml, 0.2 M). NaBD4 (180 mg, 4.3 mmol) was added portionwise. The reaction mixture was allowed to stir at ambient temperature for 30 min. Distilled water was added and the reaction mixture was allowed to stir until bubbling stopped. The mixture was extracted with ethyl acetate (3 × 15 mL). The combined organic extracts were washed with brine (1 × 50 mL). The combined organic extracts were dried (MgSO4), filtered and evaporated under reduced pressure. Without further purification the crude alcohol residue was added ethanol (20 mL) and 6.0 M HCl aqueous (15 ml). The reaction mixture was allowed to stir at ambient temperature for the stated time. Distilled water was added and the mixture was extracted with ethyl acetate (3 × 15 mL). The combined organic extracts were washed with brine (1 × 50 mL). The combined organic extracts were dried (MgSO4), filtered and evaporated under reduced pressure. The crude solid was subjected to column chromatography (SiO2: 15% ethyl acetate in hexanes) to give the title compound (0.69 g, 3.7 mmol) in 95% yield as a white solid. 1H NMR (400 MHz, CDCl3): δ 8.15 (d, J = 8.1 Hz, 1H), 7.98 (d, J = 7.1 Hz, 1H), 7.92. (dd, J = 8.1, 1.1 Hz, 1H), 7.79–7.66 (m, 3H), 3.12 (s, 1H); 2H NMR (77 MHz, CHCl3): δ 5.37 (s, 1D); HRMS (ESI-MS) Calcd. for C12H7DO2 [M+Na]+: 208.0479, Found: 208.0460; FTIR (neat): 3400, 3064, 1702; MP: 160–162 °C.
2-(ethyl-1,2-d2)-2-hydroxyacenaphthylen-1(2H)-one (deuterio-3k)
In accordance with Procedure A, deuterio-1k (0.2 mmol, 100 mol%) was reacted with ethylene (15 × 125 mm pressure tube, 0.82 mmol, 410 mol%) in toluene (2.0 M) at 130 °C for a 48 hour period. Flash column chromatography (SiO2: 5–15% ethyl acetate/hexanes) provided the title compound (18.0 mg, 0.08 mmol) in 42% yield as a white solid. 1H NMR (400 MHz, CDCl3): δ 8.14 (d, J = 8.2 Hz, 1H), 7.97 (d, J = 7.0 Hz, 1H), 7.91 (dd, J = 7.9, 1.2 Hz, 1H), 7.79–7.64 (m, 3H), 2.72 (s, 1H), 2.19–2.01 (m, 2H), 0.78 (t, J = 7.5 Hz, 2.78H); 2H NMR (77 MHz, CHCl3): δ 0.77 (s, 0.22H); HRMS (ESI-MS) Calcd. for C14H11DO2 [M+Na]+: 236.0792, Found: 236.0781; FTIR (neat): 3370, 2973, 2927, 1716; MP: 92–93 °C.
Supplementary Material
Acknowledgments
The Robert A. Welch Foundation (F-0038) and the NIH-NIGMS (RO1-GM069445) and the Center for Green Chemistry and Catalysis for partial support of this research.
Footnotes
Supporting Information. Spectroscopic data for all new compounds (1H NMR, 13C NMR, IR, HRMS), including images of NMR spectra. Single crystal X-ray diffraction data for a derivative of 4b and the osmium complexes Os2(CO)4(O2CR)2(XPhos)2 and Os3(CO)11(XPhos). This material is available free of charge via the internet at http://pubs.acs.org
References
- 1.For reviews, see: Lappin GR, Sauer JD, editors. Alpha Olefins Applications Handbook. M. Dekker; New York: 1989.
- 2.For recent reviews, see: Brintzinger HH, Fischer D, Mülhaupt R, Rieger B, Waymouth RM. Angew Chem, Int Ed Engl. 1995;34:1143.Mecking S. Angew Chem, Int Ed. 2001;40:534.Boehm LL. Angew Chem, Int Ed. 2003;42:5010. doi: 10.1002/anie.200300580.Eisch JJ. Organometallics. 2012;31:4917.McInnis JP, Delferro M, Marks TJ. Acc Chem Res. 2014;47:2545. doi: 10.1021/ar5001633.Klosin J, Fontaine PP, Figueroa R. Acc Chem Res. 2015;48:2004. doi: 10.1021/acs.accounts.5b00065.
- 3.For recent reviews, see: Beller M, Cornils B, Frohning CD, Kohlpaintner CW. J Mol Catal A. 1995;104:17.Frohning CD, Kohlpaintner CW, Bohnen H-W. In: In Applied Homogeneous Catalysis with Organometallic Compounds. Cornils B, Herrmann WA, editors. Vol. 1. Wiley-VCH; Weinheim: 1996. pp. 29–104.Rhodium Catalyzed Hydroformylation; van Leeuwen PWNM, Claver C, editors. Kluwer Academic Publishers; Norwell, MA: 2000. Breit B, Seiche W. Synthesis. 2001:1.Weissermel K, Arpe H-J. Industrial Organic Chemistry. 4. Wiley-VCH; Weinheim: 2003. pp. 127–144.van Leeuwen PWNM, editor. Homogeneous Catalysis: Understanding the Art. Kluwer Academic Publishers; Dordrecht: 2004.
- 4.For recent reviews, see: Grubbs RH. Tetrahedron. 2004;60:7117.Chikkali S, Mecking S. Angew Chem, Int Ed. 2012;51:5802. doi: 10.1002/anie.201107645.Czaban J, Torborg C, Grela K. In: In Sustainable Catalysis. Hii KK, Williams MT, Dunn PJ, Krische MJ, editors. John Wiley and Sons; New York: 2013. pp. 163–214.Popoff N, Mazoyer E, Pelletier J, Gauvin RM, Taoufik M. Chem Soc Rev. 2013;42:9035. doi: 10.1039/c3cs60115c.
- 5.For a recent review, see: Leung JC, Krische MJ. Chem Sci. 2012;3:2202.
- 6.For hydroacylation via catalytic reductive coupling of anhydrides with α-olefins or styrenes, see: Hong YT, Barchuk A, Krische MJ. Angew Chem Int Ed. 2006;128:6885. doi: 10.1002/anie.200602377.Bandar JS, Ascic E, Buchwald SL. J Am Chem Soc. 2016;138:5821. doi: 10.1021/jacs.6b03086.
- 7.For recent reviews, see: Koichi M, Masaki S. Chem Rev. 1992;92:1021.Pastor IM, Yus M. Curr Org Chem. 2007;11:925.Clarke ML, France MB. Tetrahedron. 2008;64:9003.Ho CY, Schleicher KD, Chan CW, Jamison TF. Synlett. 2009:2565. doi: 10.1055/s-0029-1217747.
- 8.For a recent review, see: Ketcham JM, Shin I, Montgomery TP, Krische MJ. Angew Chem Int Ed. 2014;53:9142. doi: 10.1002/anie.201403873.
- 9.(a) Leung JC, Geary LM, Chen TY, Zbieg JR, Krische MJ. J Am Chem Soc. 2012;134:15700. doi: 10.1021/ja3075049. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Chen TY, Krische MJ. Org Lett. 2013;15:2994. doi: 10.1021/ol401184k. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Geary LM, Glasspoole BW, Kim MM, Krische MJ. J Am Chem Soc. 2013;135:3796. doi: 10.1021/ja400691t. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Park BY, Montgomery TP, Garza VJ, Krische MJ. J Am Chem Soc. 2013;135:16320. doi: 10.1021/ja4087193. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) McInturff EL, Mowat J, Waldeck AR, Krische MJ. J Am Chem Soc. 2013;135:17230. doi: 10.1021/ja410533y. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Yamaguchi E, Mowat J, Luong T, Krische MJ. Angew Chem Int Ed. 2013;52:8428. doi: 10.1002/anie.201303552. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) McInturff EL, Nguyen KD, Krische MJ. Angew Chem Int Ed. 2014;53:3232. doi: 10.1002/anie.201311130. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Saxena A, Perez F, Krische MJ. J Am Chem Soc. 2015;137:5883. doi: 10.1021/jacs.5b02755. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Park BY, Luong T, Sato H, Krische MJ. J Am Chem Soc. 2015;137:7652. doi: 10.1021/jacs.5b04688. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Saxena A, Perez F, Krische MJ. Angew Chem Int Ed. 2016;55:1493. doi: 10.1002/anie.201509646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Chatani N, Tobisu M, Asaumi T, Fukumoto Y, Murai S. J Am Chem Soc. 1999;121:7160. [Google Scholar]; (b) Tobisu M, Chatani N, Asaumi T, Amako K, Ie Y, Fukumoto Y, Murai S. J Am Chem Soc. 2000;122:12663. [Google Scholar]
- 11.For a recent review on metal catalyzed C-C couplings of ethylene, see: Saini V, Stokes BJ, Sigman MS. Angew Chem Int Ed. 2013;52:11206. doi: 10.1002/anie.201303916.
- 12.Stockis A, Hoffmann R. J Am Chem Soc. 1980;102:2952. [Google Scholar]
- 13.(a) Ngai MY, Barchuk A, Krische MJ. J Am Chem Soc. 2007;129:280. doi: 10.1021/ja0670815. [DOI] [PubMed] [Google Scholar]; (b) Williams VM, Kong JR, Ko BJ, Mantri Y, Brodbelt JS, Baik MH, Krische MJ. J Am Chem Soc. 2009;131:16054. doi: 10.1021/ja906225n. and references cited therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.(a) Dewar MJS. Bull Soc Chim Fr. 1951:C71. [Google Scholar]; (b) Chatt J, Duncanson LA. J Chem Soc. 1953:2939. [Google Scholar]; (c) Dewar MJS, Ford GP. J Am Chem Soc. 1979;101:783. [Google Scholar]
- 15.Kulinkovich OG, Sviridov SV, Vasilevskii DA, Pritytskaya TS. Zh Org Khim. 1989;25:2244.For a review, see: Breit B. J Prakt Chem. 2000;342:211.
- 16.Parshall GW. Complexes of Ruthenium, Osmium, Rhodium, and Iridium Containing Hydride Carbonyl, or Nitrosyl Ligands. In: Ahmad N, Levison JJ, Robinson SD, Uttley MF, editors. Inorganic Syntheses. Vol. 15. McGraw-Hill, Inc; 1974. pp. 45–64. [Google Scholar]
- 17.Blum Y, Reshef D, Shvo Y. Tetrahedron Lett. 1981;22:1541. [Google Scholar]
- 18.Maytum HC, Tavassoli B, Williams JMJ. Org Lett. 2007;9:4387. doi: 10.1021/ol702029n. [DOI] [PubMed] [Google Scholar]
- 19.For a review on redoxeconomy, see: Burns NZ, Baran PS, Hoffmann RW. Angew Chem Int Ed. 2009;48:2854. doi: 10.1002/anie.200806086.
- 20.(a) Sanchez-Delgado RA, Bradley JS, Wilkinson G. J Chem Soc Dalton Trans. 1976:399. [Google Scholar]; (b) Wu L, Liu Q, Spannenberg A, Jackstell R, Beller M. Chem Commun. 2015;51:3080. doi: 10.1039/c4cc05626d. [DOI] [PubMed] [Google Scholar]
- 21.Tse SKS, Xue P, Lin Z, Jia G. Adv Synth Catal. 2010;352:1512. [Google Scholar]
- 22.For intramolecular reductive coupling of unactivated olefins with unactivated carbonyl partners, see: Kablaoui NM, Buchwald SL. J Am Chem Soc. 1995;117:6785.Crowe WE, Rachita MJ. J Am Chem Soc. 1995;117:6787.Kablaoui NM, Buchwald SL. J Am Chem Soc. 1996;118:3182.
- 23.(a) Ianni A, Waldvogel SR. Synthesis. 2006:2103. [Google Scholar]; (b) Lunardi I, Cazetta T, Conceição GJA, Moran PJS, Rodrigues JAR. Adv Synth Catal. 2007;349:925. [Google Scholar]; (c) Merz A, Dietl F, Tomahogh R, Weber G, Sheldrick GM. Tetrahedron. 1984;40:665. [Google Scholar]; (d) Patonay T, Lévai A, Nemes C, Timár T, Tóth G, Adam W. J Org Chem. 1996;61:5375. [Google Scholar]; (e) Emmanuvel L, Mahammad T, Shaikh A, Sudalai A. Org Lett. 2005;7:5071. doi: 10.1021/ol052080n. [DOI] [PubMed] [Google Scholar]; (f) Platt KL, Oesch F. Synthesis. 1982:459. [Google Scholar]; (g) Wang ZM, Kakiuchi K, Sharpless KB. J Org Chem. 1994;59:6895. [Google Scholar]
- 24.(a) Hatano M, Ito O, Suzuki S, Ishihara K. J Org Chem. 2010;75:5008. doi: 10.1021/jo100563p. [DOI] [PubMed] [Google Scholar]; (b) Arai T, Takasugi H, Sato T, Noguchi H, Kanoh H, Kaneko K, Yanagisawa A. Chem Lett. 2005;34:1590. [Google Scholar]; (c) Yan J, Travis BR, Borhan B. J Org Chem. 2004;69:9299. doi: 10.1021/jo048665x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.