

Abstract
In an earlier study we demonstrated that hydroxyapatite nanoparticles coated with chitosan-poly(D,L)-lactide-co-glycolide (HAp/Ch-PLGA) target lungs following their intravenous injection into mice. In this study we utilize an emulsification process and freeze drying to load the composite HAp/Ch-PLGA particles with 17β-hydroxy-17α-picolyl-androst-5-en-3β-yl-acetate (A), a chemotherapeutic derivative of androstane and a novel compound with a selective anticancer activity against lung cancer cells. 1H NMR and 13C NMR techniques confirmed the intact structure of the derivative A following its entrapment within HAp/Ch-PLGA particles. The thermogravimetric and differential thermal analyses coupled with mass spectrometry were used to assess the thermal degradation products and properties of A-loaded HAp/Ch-PLGA. The loading efficiency, as indicated by the comparison of enthalpies of phase transitions in pure A and A-loaded HAp/Ch-PLGA, equaled 7.47 wt.%. The release of A from HAp/Ch-PLGA was sustained, neither exhibiting a burst release nor plateauing after three weeks. Atomic force microscopy and particle size distribution analyses were used to confirm that the particles were spherical with a uniform size distribution of d50 = 168 nm. In vitro cytotoxicity testing of A-loaded HAp/Ch-PLGA using MTT and trypan blue dye exclusion assays demonstrated that the particles were cytotoxic to the A549 human lung carcinoma cell line (46±2%), while simultaneously preserving high viability (83±3%) of regular MRC5 human lung fibroblasts and causing no harm to primary mouse lung fibroblasts. In conclusion, composite A-loaded HAp/Ch-PLGA particles could be seen as promising drug delivery platforms for selective cancer therapies, targeting malignant cells for destruction, while having a significantly lesser cytotoxic effect on the healthy cells.
Keywords: androstane, chitosan, hydroxyapatite, lung cancer, nanoparticle, PLGA
Graphical abstract
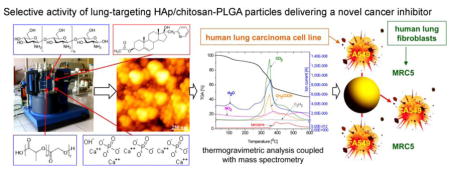
1. Introduction
The potential for application of synthetic calcium phosphates, especially hydroxyapatite (HAp), has become intensely broadened in the past 15 years. It has extended from bone tissue engineering to multiple other fields of biomedicine, e.g., controlled drug delivery, bioactive coatings, gene therapies, wound healing and beyond [1–3]. The enhancement of the properties of HAp in this pursuit of new areas of application has progressed in two major directions: (a) chemical modification and (b) combination with other materials, predominantly polymers. As far as the first direction is concerned, HAp surpasses other biominerals in terms of the capacity to incorporate foreign ions, some of which can be utilized for therapeutic purposes, substituting the role of costlier biomolecules and minimizing the side effects frequently associated with their use [4]. To that end, tailoring of the chemical structure of HAp has been achieved through the substitution of its constitutive ions (Ca2+, PO43− and OH−) with ions such as Mg2+, Zn2+, Cd2+, Y3+, Mn2+, Ni2+, carbonate, selenite, silicate, etc. [5, 6]. Cobalt-substituted HAp, for example, accelerated the process of osseointegration after the reconstruction of bone defects in vivo [7].
Progress in the field of composite nanoparticle synthesis has, on the other hand, enabled the fabrication of a large number of hybrid, multicomponent systems containing HAp. Biocompatible polymeric coatings around HAp nanoparticles have allowed for the development of multifunctional nanosystems as carriers of vitamins, antibiotics and genes [8–13]. The tailoring of the structure of multifunctional nanoparticulate drug carriers is an emerging branch of research, with the goal of enhancing their biocompatibility and therapeutic efficacy [9, 14]. Multifunctional systems exhibit a more convoluted behavior in the course of in vitro and in vivo testing and also demand more versatile techniques for their physicochemical characterization [14]. Nanostructured HAp has been extensively used in the reconstruction of boney tissues and it has also showed promising results as a system with a targeted activity in the treatment of osteosarcoma metastases [15]. HAp nanoparticles can slow down the growth of various cancer cells, particularly human glioma cells [16]. Organ-targeting or tumor-targeting via drug-loaded nanoparticles demonstrated their ability to act on specific receptors [17]. The targeting of receptors by HAp nanocrystals was proposed as the basis for the utilization of HAp doped with rare earth elements for noninvasive medical imaging at the molecular level [18]. Our previous research on nano/micro-particle systems composed of HAp coated with the chitosan-poly(D,L)-lactide-co-glycolide polymer blend (HAp/Ch-PLGA) has justified the use of HAp as a core component of composite drug carriers for organ-targeting therapies [19]. Along with the particle size, shape and composition, the fate of particles injected into the bloodstream is mainly defined by the type and the structure of proteins bound to them [20, 21], which can be controlled by tailoring the composition, the topography and the charge distribution on the particle surface. Coating HAp nanoparticles with different polymers has correspondingly directed HAp to different organs [19]. Specifically, unlike pure HAp and HAp coated with PLGA only, which were being accumulated in liver, spleen and kidneys, HAp/Ch-PLGA particles were overwhelmingly localizing in the lungs of mice following the intravenous injection of the particulate dispersion [19]. Bone defect reparations have been successfully performed using nanoparticulate systems based on PLGA-coated HAp, providing a rationale for the consideration of PLGA as a coating for organ-targeting delivery systems [10–12]. One of the advantages of mixing PLGA with chitosan within nanoparticulate platforms for controlled delivery of chemotherapeutics in the treatment of cancer is an improved permeation of the cell membrane [22]. The enhanced sialylation of malignant cells [23] benefits the idea of using cationic Ch to target cancer cells and may be the reason behind its occasionally observed anticancer properties [24]. The previously observed ability of the positively charged molecules and particles to localize in lungs [25–28] may also be a key to explaining the pronounced lung-targeting potential of HAp/Ch-PLGA particles [19].
Lung cancer is the leading cause of cancer mortality [29]. Estrogen and progesterone are hormones that play an important role in the carcinogenesis of lung cells [30], placing this type of cancer in the category of hormone-dependent cancers. The 17-picolyl, 17-picolinylidene, 17-picolinylidene-16-one and 17-picolyl-16-one androstane derivatives are effective cell inhibitors of hormone-dependent cancers, e.g., adenocarcinoma, prostate cancer, cervix carcinoma, colon cancer, etc. [31–33]. This explains their choice as a chemotherapeutic drug to be delivered to lung cancer cells in this study. As for the choice of the carrier, HAp nanoparticles have been used with success to target tumors through vasculature and extend life expectancy in animal studies. For example, HAp nanoparticles, albeit doped with selenite ions and injected into the bloodstream, extended the lifetime of mice with hepatocellular carcinoma [34]. Also, a direct injection of HAp nanoparticles into a transplanted tumor formed by Bel-7402 human hepatocarcinoma cells in nude mice led to a 50 % reduction in the tumor size [35]. On the other hand, the antiproliferative activity of HAp was twice more intense for cancer cells than for the normal ones, both in vitro and in vivo [35]. The selective cytotoxicity exhibited by HAp was also not due to the release of reactive oxygen species [35] (as is the case with, e.g., carbon nanotubes, quantum dots, Ag nanoparticles), as this would cause unselective cytotoxicity, targeting both cancerous and healthy cells [36]. Rather, nanosized HAp particles get internalized by cancer cells and localized around the endoplasmic reticulum (ER), where they presumably interfere with the translation process by competing for ribosome binding together with mRNA, thus arresting the cell cycle in G0/G1 phase [35]. Interestingly, the androstane derivative chosen as the anticancer drug for this study owes its selective anticancer activity to a similar effect, i.e., the induction of the ER stress response, resulting in calcium release from the ER and the G1 phase cycle arrest [37]. Given the similar mechanism by which HAp and the compound A inhibit the proliferation of cancer cells, a possible therapeutic synergy between the carrier (HAp) and the drug (A) is worth investigating.
In our previous study we demonstrated the ability of nanoparticles composed of HAp coated with Ch-PLGA polymer blend to target lungs following intravenous administration [19]. In this study we describe the fabrication of androstane (17β-hydroxy-17α-picolyl-androst-5-en-3β-yl acetate, A)-loaded HAp/Ch-PLGA particles and the assessment of their anticancer properties against lung cancer cells. The encapsulation efficiency (EE) and the drug loading content (DLC) in multifunctional nanosystems for active or passive targeting are important parameters that define not only the structure and the properties of the system, but also its application efficiency [38, 39]. In the present study we examine the possibility of using differential thermal analysis/thermogravimetric analysis (DTA/TGA) coupled with on-line mass spectrometry to obtain direct information on the efficiency of the androstane derivative encapsulation. The release of the drug is also assessed, knowing that the drug release kinetics defines the effects that the therapy will have on both malignant and healthy cells. Needless to add, anticancer drugs are toxic not only to the cancer cells, but also to the healthy ones. Designing selective anticancer medicaments, having a high activity toward cancer cells but a low activity toward healthy ones, has been an immense challenge in the past twenty years [40, 41]. Tools that employ nanotechnologies may be successfully applied to address these challenges [42, 43]. In this study we test the comparative cytotoxic response of human lung carcinoma (A549) and healthy human lung fibroblasts (MRC5) to their in vitro treatment with the A-loaded HAp/Ch-PLGA composite particles. We have also analyzed the period of recovery of the two types of cells after their treatment with A-loaded HAp/Ch-PLGA.
2. Materials and methods
2.1. Synthesis of 17β-hydroxy-17α-picolyl-androst-5-en-3β-yl acetate-loaded HAp/Ch-PLGA
2.1.1. Synthesis of 17β-hydroxy-17α-picolyl-androst-5-en-3β-yl acetate (A)
The first stage in the preparation of 17β-hydroxy-17α-picolyl-androst-5-en-3β-yl-acetate (A) was the addition of α-picolyllithium to the 17-oxo group of dehydroepiandrosterone, resulting in 17α-picolyl-androst-5-en-3β,17β-diol. This was followed by the acetylation using acetic anhydride in absolute pyridine at room temperature for 3 h [44].
2.1.2. Synthesis of HAp, HAp/Ch-PLGA and A-HAp/Ch-PLGA
An aqueous calcium nitrate (Ca(NO3)2) solution (150 ml; 26.6 wt.%) was added to the solution of ammonium phosphate ((NH4)3PO4) (7 ml H3PO4+165 ml NH4OH+228 ml H2O) at 50°C over the period of 60 minutes, while stirring at the rate of 100 rpm. The solution was then subjected to a heat treatment at 100°C for 60 minutes [45]. The resulting gel was dried at the room temperature in a vacuum drier for 72 h, after which the final product - HAp powder - was obtained. X-ray diffraction run on the HAp powder confirmed its poorly crystalline nature, whereas the PSD technique enabled us to determine the particle size of d50=70 nm [19].
Chitosan (Ch) of a low molecular weight (Aldrich, deacetylation > 75%), dissolved in acetic acid (1 wt.%), was mixed with 17β-hydroxy-17α-picolyl-androst-5-en-3β-yl acetate powder (A) in 2:1 weight ratio while stirring with a magnetic stirrer at 400 rpm. PLGA (50:50, Sigma, USA) dissolved in acetone was mixed with the A-containing Ch solution and HAp gel in 2:3:5 weight ratio. A water solution of poloxamer 188 (polyethylenepolypropylene glycol, 0.1 vol.%) was added dropwise to the resulting mixture, while stirring at 21,000 rpm. The obtained mixture of A, chitosan, PLGA and HAp was slowly poured into a glutaraldehyde solution (Grade I, 25% in H2O), while stirring at 21,000 rpm for 1 h. The obtained mixture was then centrifuged at 3000 rpm and 5°C for 1 h, and the resulting precipitate was subjected to lyophilization at temperatures ranging from −10 to −60°C and pressures ranging from 0.37 mbar to 0.1 mbar for 1 to 8h [19, 46]. The obtained powder was washed with distilled water twice, centrifuged at 1000 rpm and dried again. The final product was a powder composed of HAp particles coated with A-loaded chitosan-poly(D,L)-lactide-co-glycolide (A-HAp/Ch-PLGA). The weight ratio of HAp:Ch:PLGA:A components was 5:2:2:1.
2.2. Characterization
NMR spectra were recorded on a Bruker AVANCE III HD 400 spectrometer operated at 400 MHz (1H) and 100 MHz (13C), and are reported in ppm downfield from the tetramethylsilane internal standard. A thermal analysis was simultaneously performed using a Thermo-Gravimetric Analysis/Differential-Thermal Analysis (TGA/DTA), SETSYS 2400 CS Evolution (Setaram Instrumentation) coupled with a mass spectrometric (MS) gas analysis system (Omni Star, Pfeiffer). Samples containing 10±0.5 mg were analyzed in air by heating (10°C/min) from 28°C to 600°C. Differential scanning calorimetry (DSC) measurements were performed on an Evo 131 (Setaram Instrumentation) differential scanning calorimeter. Samples containing 10±0.5 mg were analyzed in nitrogen by heating (10°C/min) from 25°C to 360°C. Infrared spectroscopy (FT-IR) was done on an Micro-FT-IR Nicolet iN10 with a DTGS room T detector (Thermo Scientific Instruments), using the KBr technique in the spectral range from 400 to 4000 cm−1. The particle size distribution (PSD) was measured on 10 mg/mL of powders dispersed in water using a Mastersizer 2000 (Malvern Instruments Ltd.) and a HydroS dispersion unit for liquid dispersants. Microstructural characterization was performed using an atomic force microscope (AFM, Thermo Microscopes, Autoprobe CP Research). Zeta potential of the particles in suspension was measured in the 1.5–13 pH range on a Zetasizer Nano-ZS (Malvern) dynamic light scattering (DLS) device. Drug release experiments were performed by placing the powder in 50 ml phosphate buffered saline (PBS) in closed conical tubes and incubating at 37°C and 50 rpm for up to three weeks. To estimate the concentration of the released drug, aliquoted solutions were analyzed in duplicates for absorbance on a UV/Vis spectrophotometer (Nanodrop 2000, Thermo Scientific) at λ=263 nm.
2.3 Cell culture
Cytotoxicity assays were performed using two cell lines, human lung carcinoma (A549 ATCC CCL 185) and human lung fibroblasts (MRC-5 ATCC CCL 171) in Dulbecco’s modified Eagle’s medium (DMEM, Gibco BRL, UK) with 4.5g/l glucose, 10% FBS (fetal bovine serum, Sigma) and containing penicillin(100I.U./ml) and streptomycin (100μg/ml). The cell lines were grown at 37°C and in air containing 5 % CO2 (Heracus). The cells were passaged twice a week; those used in the experiments were in the log phase of growth between the third and the tenth passage. Only the viable cells were used. The number of cells and their viability were determined using the dye exclusion test with 0.1% trypan blue.
2.3.1. Trypan blue dye exclusion assay
Cells were seeded at 2×105 cells/ml in 24 well plates (Center well, Falcon), on top of a film made of compressed A-HAp/Ch-PLGA particles and the total concentration of the solid phase equaling 17.6±0.4 mg/ml, i.e., 46.3±1.0 mg/cm2. After the incubation period of 48 hours, the cells were trypsinized, centrifuged at 200 g for 10 minutes and resuspended in DMEM. Ten μl of the cell suspension were removed and mixed with an equal amount of 0.1% trypan blue. Ten μl of the cell/trypan blue solution were pipetted onto a hemocytometer and live cells were counted using an inverted light microscope (REICHERT). Cytotoxicity was expressed in percentages according to the following formula: CI=(1−Ns/Nk)×100%, where Nk was the average number of cells in the control samples, and Ns was the average number of cells in the samples containing the examined substance. All the samples for the dye exclusion test were analyzed in quadruplicates.
2.3.2. MTT cell viability assay
Both lung carcinoma cells and fibroblasts were plated at 2×105 cells/well in a 96-well microtiter plate (Falcon) and grown on top of a film made of compressed HAp/Ch-PLGA or A-HAp/Ch-PLGA particles and the total concentration of the solid phase equaling 17.6±0.4 mg/ml, i.e., 46.3±1.0 mg/cm2. After 48 hours, the MTT reagent was added into all the wells and incubated for 3 hours at 37°C. After 3 hours, 100 μl of 0.04 mol/l HCl in isopropanol were added to each well. Absorbance was read immediately using a microtiter plate reader (Multiscan, MCC/340) at the wavelength of 540 nm and the reference wavelength of 690 nm. Cytotoxicity was expressed in percentages according to the formula: CI=(1−As/Ak)×100%, where Ak was the average absorbance of the control samples and As was the average absorbance of the samples containing the examined substance. All the samples for the colorimetric viability test were analyzed in quadruplicates.
2.3.3. Immunofluorescent staining
Primary mouse lung fibroblasts were isolated from 9-week old C57BL/6 mice. Fibroblasts were grown in DMEM with 10% fetal bovine serum and 5% antibiotic (penicillin/streptomycin) and antimycotic (fungazone). For the immunofluorescence analysis, the cells were seeded onto glass coverslips at 1×105 cells per well in a standard 24-well plate and 1 mg/ml of nanoparticles were added in the powder form to them and swirled to achieve proper dispersion. In addition to dispersed particles, this yielded a moderate amount of particle conglomerates, visually observable in interaction with the cells under a fluorescent optical microscope. Cells were allowed to grow in conjunction with the particles for 4 days at 37°C, 5% CO2. On Day 4, the cells were fixed in 4% paraformaldehyde for 5 minutes, washed thrice in PBS, for 5 minutes each, and stained with Alexa Fluor 568 rhodamine-phalloidin (1:400) for 30 minutes. After staining, the cells were washed thrice, for 5 minutes each, in PBS and incubated for 25 minutes with NucBlue Fixed Cell Ready probe reagent. Cells were then rinsed thrice with PBS, for 5 minutes each, and the coverslips with cells were mounted onto microscope glass slides using Prolong Diamond and cured overnight. Images were acquired on a Zeiss LSM 710 confocal microscope (UIC core imaging facility).
3. Results and discussion
3.1. 1H NMR and 13C NMR analyses of 17β-hydroxy-17α-picolyl-androst-5-en-3β-yl acetate (A)
In the 1H NMR spectra of the androstane derivative (Fig. 1a), compound A, the signals from protons from the heterocyclic ring at C-17 position were detected with a downfield shift, in the 7.20–8.47 ppm range, as a result of the deshielding influence of the nitrogen atom. Two protons from C-20 methylene group were registered at 2.84 and 3.11 ppm. The 13C NMR spectral signals (Fig. 1b) were assigned to carbons from the heterocyclic ring at 122.38, 124.91, 137.00, 147.78 and 160.75 ppm. As expected, the signal from 3-acetoxy group of the compound A was detected in 1H NMR spectra at 2.05 ppm. The corresponding relatively intense 13C NMR signals were observed at 21.46 and 170.60 ppm for methyl and carbonyl atoms from the 3-acetoxy group, respectively.
Figure 1.

Nuclear magnetic resonance (NMR) spectra of 17β-hydroxy-17α-picolyl-androst-5-en-3β-yl acetate (A): a) 1H NMR and b) 13C NMR. 1H NMR (CDCl3, δ, ppm): 0.98 (s, 3H, H-18); 1.07 (s, 3H, H-19); 2.05 (s, 3H, CH3 from A group); 2.84 (d, 1H, Jgem = 14.4 Hz, CH2Py); 3.11 (d, 1H, Jgem = 14.4 Hz, CH2Py); 4.63 (m, 1H, H-3); 5.40 (d, 1H, J = 4.8 Hz, H-6); 7.20 (m, 2H, H-3′ i H-5′, Py); 7.66 (t, 1H, J3′,4′ = J4′,5′ = 7.8 Hz, H-4′, Py); 8.47 (d, 1H, J6′,5′ = 4.8 Hz, H-6′, Py). 13C NMR (CDCl3, δ, ppm): 14.13 (C-18); 19.33 (C-19); 20.75; 21.46 (CH3 from A group); 23.99; 27.77; 31.84; 32.16; 32.70; 35.97; 36.73; 37.09; 38.12; 43.08; 46.37; 50.18; 51.06; 73.93 (C-3); 83.52 (C-17); 121.43 (C-6); 122.38 (C-5′, Py); 124.91 (C-3′, Py); 137.00 (C-4′, Py); 139.80 (C-5); 147.78 (C-6′, Py); 160.75 (C-2′, Py); 170.60 (C=O from A group).
3.2. Differential-Thermal Analysis (DTA)
The DTA thermograms of HAp, Ch, PLGA, A and A-HAp/Ch-PLGA in the temperature range of 25–600°C are shown in Figure 2. In contrast to the one-step thermal degradation of chitin, the thermal degradation of chitosan is characterized by a two-step process entailed by chain scission and repolymerization reactions [47]. After the evaporation of the surface-bound water, yielding the broad endothermic peak at 100°C (Fig. 2a.), one may notice a narrow endothermic peak in the 306–320°C interval. As in agreement with previous findings, this thermal event derives from the degradation and deacetylation of chitosan [48] and the formation of vaporous fragments with a lower molecular weight, including char [47]. A sequence of endothermic peaks in the DTA thermogram of PLGA (Fig. 2a) at temperatures between 328°C and 342°C, as well as two major peaks centered at 364°C and 390°C, originate from the decomposition of the polymer. The decomposition of PLGA at temperatures around 360°C has also been observed by other authors [49]. The suggested mechanism for the thermal degradation of PLGA is based on an interaction between the end hydroxyl groups of the polymeric chains and the resulting formation of cyclic oligomers, lactides, acetaldehydes, methyl glycolates and carbon monoxide. At higher temperatures, the release of carbon dioxide, methyl ketene or formaldehyde occurs [50, 51].
Figure 2.

Differential thermal analysis (DTA) curves for a) HAp, Ch, PLGA and b) A and A-HAp/Ch-PLGA
As seen in Fig. 2b, the DTA thermogram of A-loaded HAp/Ch-PLGA is dominated by peaks characteristic for the pure compound A. The two most prominent endothermic peaks of the compound A, at 204 and 330°C (zone I and II in Fig. 2b, respectively), are detected in A-loaded HAp/Ch-PLGA too. The sequence of endothermic peaks originating from the degradation PLGA is observed at the higher temperature end of the 330°C peak exhibited by the compound A. The major one of these peaks exhibits a shift to lower temperatures, from 365 to 340°C, following the combination of PLGA with HAp in composite HAp/Ch-PLGA, suggesting the surface presence of the polymeric phase in a thinner form, more exposed to the atmospheric heat than the bulkier, pure PLGA particles, thus requiring lesser heat to reach their interior and initiate the degradation process. The semblance of the two thermograms presented in Fig. 2b unequivocally indicates that the method for the encapsulation of the compound A into HAp/Ch-PLGA (section 2.1.1) preserves its native structure.
3.3. TG-MS Gas Analysis
A comparative overview of thermogravimetric (TG) diagrams and the corresponding MS spectra of the gas product analysis of the A-loaded HAp/Ch-PLGA composite and of its four individual components are shown in Figure 3. The TGA curve of HAp demonstrates a total weight loss of about 7% at 600°C [52], being solely due to the evaporation of physically adsorbed water, which, as seen from the corresponding mass spectrogram, is completed by the point the system reaches about 250°C. This observed water loss, centered at around 100°C, agrees with the results of the DT analysis (Fig. 2a).
Figure 3.


Spectra obtained from the thermogravimetric (TG) and mass spectrometric (MS) gas analysis of a) Ch, b) PLGA, c) A and d) A-HAp/Ch-PLGA in the 28–600°C temperature interval and at the heating rate of 10 °C/min.
The moderate weight loss of chitosan (Fig. 3a) up to about 250°C is associated with the loss of adsorbed water. In the subsequent 250–600°C interval, the region extending from 250°C to 300°C stands out with a drastic weight decrease: from 86% to 55%. It is followed by the region extending from 300°C to 450°C, where a milder weight loss is observed, from 55% to 30%, and then the region extending from 450 to 600°C, where the weight drops from 30% down to only 0.6%. Similar TGA results for chitosan have been reported by others as well [53, 54]. A dramatic chitosan weight loss in the 306–320°C temperature interval due to deacetylation (Fig. 2a) is confirmed by the increasing CH3COOH concentration in the MS gas analysis. The release of H2O, CO, CO2 and CH3COOH in the 280–500°C temperature interval has also been confirmed by other authors by means of FT-IR analyses [53]. In the temperature interval between 260 and 400°C, a sudden drop in the PLGA weight occurs (Fig. 3b) and a complete degradation takes place at temperatures higher than 400°C. According to the thermogravimetric analyses of PLGA reported earlier, the pyrolysis of PLGA was detected at temperatures as low as 255°C, whereas its full degradation occurred at temperatures extending from 400°C upwards [49, 55]. In the 260°C–400°C interval, the significant PLGA weight loss is accompanied by the maximal contents of CO2, H2O and CH3COOH in the MS gas analysis. Physicochemical phenomena caused by heating the compound A to 600°C were reflected in 98.71 % total weight loss (Fig. 3c). Detected in the 428–528°C interval, the dissociation of benzene from the rest of the molecule, typical for the MS spectrum of the compound A, indicates the fragmentation of its structure. The TGA curve for A-HAp/Ch-PLGA shown in Figure 3d demonstrates a 56.63 % weight loss at 600°C. The remaining 43.37 % of weight contains only HAp and the compound A, given that the weight ratio of Ch and PLGA at this final stage of the heating process is 0% due to their complete thermal degradation. Every single MS spectrum of the individual constituents of the composite system - HAp, Ch, PLGA and the derivative A - is present in the MS spectrum of A-HAp/Ch-PLGA, which, together with the DTA results, confirms the successful encapsulation of the derivative A.
3.4. Fourier transform infrared spectroscopy (FT-IR) analysis of A and A-HAp/Ch-PLGA
In order to additionally confirm the qualitative content of A-HAp/Ch-PLGA, the material was analyzed using FT-IR spectroscopy. The IR spectra of the compound A and of A-HAp/Ch-PLGA composite particles are shown in Fig. 4a. The wide IR band in the spectrum of A (17β-hydroxy-17α-picolyl-androst-5-en-3β-yl acetate), with the maximum at 3303 cm−1, is assigned to the O-H stretch (17β-OH). Several absorption bands with maxima at 2943 and 2827 cm−1 originate from the alkyl stretches of the C–H group (−CH3 and =CH2). The valence vibrations of the acetate group were observed as a sharp band with the maximum at 1246 cm−1.
Figure 4.

IR spectroscopy and morphology of A-HAp/Ch-PLGA. a) FT-IR spectra, b) the particle size distribution and 2D AFM image
The IR spectrum of A-HAp/Ch-PLGA is additionally characterized by the absorption bands arising from HAp, Ch and PLGA: ν(OH) 3600–3000 cm−1 (HAp, PLGA, Ch; intensive, wide band), ν(C=O) 1750 cm−1 (PLGA; strong intensity), ν(C=O) 1655 cm−1 (amide I band, Ch; strong intensity), δ(HOH) approx. 1640 cm−1 (HAp, PLGA, Ch; weak to medium intensity), δsy(CH) 1380 and δas(CH) 1450 cm−1 (HAp, PLGA, Ch; weak to medium intensity), δ(OH) 1420 cm−1 (HAp, PLGA, Ch; medium intensity), ν(P=O) 1090 cm−1 (HAp; strong intensity), ν(P–O) 1025 cm−1 (HAp; strong intensity overlapped with a band ν(P=O)), δ(CH) and δ(OH) 1000–700 cm−1 (HAp, PLGA, Ch; weak intensity) [19, 45]. The existence of the absorption bands originating exclusively from the compound A in the IR spectrum of A-HAp/Ch-PLGA confirms that the substance was entrapped inside the composite particles. Specifically, the group of absorption bands with maxima at about 2943 and 2827 cm−1 (C–H group from −CH3 and =CH2), =C=O with the maximum at around 1727 cm−1, as well as the valence vibrations of the acetate group at 1246 cm−1 indicate the presence of A in A-HAp/Ch-PLGA.
The cross-linking of chitosan (Ch) by glutaraldehyde (GA) is a complex process, typically leading to inhomogeneous products [56]. Mechanistically, free pendant amine groups of chitosan react with the aldehydic group of GA and yield imine bonds [57], distinguishable as the 1600 cm−1 peak in the corresponding IR spectra and originating from the stretching vibration of the imine C=N bond [58]. This peak was observed in Ch hydrogels formed in a range of different Ch:GA weight ratios (16:1 to 1:1) [59], while the Ch:GA reaction is reported to reach equilibrium at Ch:GA = 1:1 conditions [57]. In this study we used GA as a cross-linker to a fivefold amount compared to the content of Ch (Ch:GA = 5:1). The appearance of a new peak following the initiation of the cross-linking reaction at 1600 cm−1 (Fig. 4a) indicates that the cross-linking occurred in the system.
3.5. Particle size distribution and AFM analysis of A-HAp/Ch-PLGA
The morphology and the size distribution of particles constituting A-HAp/Ch-PLGA powders are shown in Figure 4b. The particles had a uniform distribution of sizes, with the d50 parameter equaling 168 nm. The composite powder contained a small portion of larger and smaller particles; correspondingly, d90=247 nm and d10=135 nm. The surface morphology of A-HAp/Ch-PLGA, analyzed using AFM in a non-contact mode (Fig. 4b), was dominated by oval topographic features, originating from the spherical shape of the individual particles. The coating of nanoparticles of HAp (d50=70 nm) with A-loaded Ch-PLGA in the centrifugal field (section 2.1.2.) yielded spherical particles of a highly similar size and shape as those obtained in our earlier study on the synthesis of A-free HAp/Ch-PLGA [46].
3.6 Differential scanning calorimetry (DSC) and the drug release from A-HAp/Ch-PLGA
Figure 5a shows DSC thermograms of the derivative A and of A-HAp/Ch-PLGA. The endothermic phase transformation observed in the DT analysis of A at 204°C (Fig. 2b) dominates the DSC thermogram with a sharp peak centered at 213.82°C (onset 211.44°C, offset 415.46°C). The same phase transition is noticeable on the thermogram of A-HAp/Ch-PLGA, next to (a) the broad endothermic doublet (onset 53.62°C, offset 131.52°C) corresponding to PLGA and Ch transformations, and (b) the wide exothermic peak corresponding to deacetylation of Ch (onset 290.86°C, offset 312.40°C). The integration of the area under the endothermic peaks paralleling the phase transitions characteristic for the pure and the encapsulated derivative A yielded enthalpy (ΔH) values of 69.704 and 5.207J/g, respectively. Based on the comparison of ΔH for the pure and the encapsulated A, the content of the encapsulated derivative A in A-HAp/Ch-PLGA could be estimated at 7.47 wt.%. Out of the projected drug content of 10 wt.% (sec 2.1.1.), 7.47 wt.% was achieved. The calorimetric method involving the measurement of the enthalpy of the phase transition associated with the melting of the drug within the carrier has been used with success to predict the loading capacity of poorly water-soluble drugs, the category to which the compound A belongs too [60].
Figure 5.

a) Differential scanning calorimetry (DSC) thermograms of A and A-HAp/Ch-PLGA. b) The release of A from A-HAp/Ch-PLGA in PBS at 37 °C over the period of 21 days. Data points represent averages (n=2) and the error bars represent standard deviations.
One of the benefits of polymeric carriers capable of entrapping the hydrophobic drug internally comes from the extension of its therapeutic half-life by preventing the aggregation of the free drug due to hydrophobic forces. Another advantage comes from the sustained release profile, thanks to which the concentration of the drug in the blood and/or the target tissue could be maintained at the therapeutically active level, avoiding the fluctuations between toxic and ineffective levels that are secondary to repetitive intravenous bolus administrations. One such sustained release of A from A-HAp/Ch-PLGA is seen in Fig. 5b. The release follows a relatively linear pattern, with no burst release or plateauing after three weeks of release.
3.7. Zeta potential analysis of HAp/Ch-PLGA and A-HAp/Ch-PLGA particles
At the physiological pH, the zeta potential of A-HAp/Ch-PLGA particles (−8.7±0.5 mV) is significantly more negative than that of A-free HAp/Ch-PLGA particles (−2.8±0.8 mV), the reason being the masking of a portion of the positively charged amine groups of chitosan by the negative charged acetate and hydroxyl groups of A. In contrast, the given hydroxyl and acetate groups are more resistant to protonation under acidic conditions (pHs 3.5 and 5.5) than the amine groups of chitosan, explaining the greater increase of zeta potential in the positive range for HAp/Ch-PLGA than for A-HAp/Ch-PLGA. The isoelectric point of HAp/Ch-PLGA concordantly becomes shifted to lower values, from 6.5 to 5.6, following the entrapment of A.
3.8. Selectivity of the in vitro antitumor activity of A-HAp/Ch-PLGA
Derivatization of 17-picolyl androstane presents a promising starting point for the development of a new group of compounds for the treatment of cancer. Modified androstane derivatives exhibited an antiproliferative activity against several types of human cancer cells, including breast, prostate, cervical, colon and lung adenocarcinoma [37, 61]. Here we report the parallel assessment of cytotoxicity of the compound A, an androstane derivative, delivered using submicron HAp/Ch-PLGA particles as a carrier, on two different cell lines: A549 human lung carcinoma cells and regular MRC5 human lung fibroblasts. After the incubation with A-HAp/Ch-PLGA for 48h, the viability of the cells measured using the trypan blue exclusion test was 83±3% for MRC5 fibroblasts and 65±2% for A549 lung carcinoma cells. The viability for both cell lines was lower than for the corresponding untreated controls as well as for the cells treated with A-free HAp/Ch-PLGA particles (p<0.05), in which case the viability equaled 89±2% for both cell types (data not shown). In contrast, cytotoxicity for the MRC5 fibroblasts treated with A-HAp/Ch-PLGA particles was 17±1%, whereas that for A549 tumor cells was 46±2% (Fig. 6a). Accordingly, the cytotoxic effect of A-HAp/Ch-PLGA particles is almost three times greater on the malignant A549 cell than on the healthy MRC5 fibroblasts. The activity of the pure compound A was equally selective as that of A-HAp/Ch-PLGA, given that the viability of A549 and MRC5 cells treated with the pure compound A equaled 36 % and 83 %, respectively, at [A] = 100 μM. The corresponding IC50 curves are given in the Supplementary Section. When after 48 h of incubation with the tested material, A549 and MRC5 cells were left to recover for another 48 h, the cytotoxicity readings were 23±2% for A549 tumor cells and 11±1% for MRC-5 cells (Fig. 6b). This is to say that following the treatment with A-HAp/Ch-PLGA particles, the malignant cells recovered twice as fast as the healthy ones. This can be explained by the different nature of these two cell lines, A549 and MRC5, with the malignant cells being more aggressive and proliferating more intensely than the regular fibroblasts [62]. The opposite effect, interestingly, is observed for A-free HAp/Ch-PLGA particles, in which case A549 cells did not exhibit any recovery in the same period of time (Fig. 6b), as opposed to MRC5 cells, which recovered almost completely, with the cytotoxicity dropping down to 2%. A similarly selective behavior, targeting cancer cells more intensely than the normal cells, was previously reported for drug-free HAp nanoparticles [35], the core components of composite particles tested in this study.
Figure 6.

Results of the in vitro test of A-HAp/Ch-PLGA on two different cell lines: regular MRC5 human lung fibroblasts and A549 human lung carcinoma cells: a) viability and cytotoxicity estimated using the MTT assay and the dye exclusion test, respectively; b) cytotoxicity following the 48 h recovery period. Data points represent averages (n = 4) and the error bars represent standard deviations.
The immunofluorescent staining of primary fibroblasts interacting with HAp/Ch-PLGA and A-HAp/Ch-PLGA particles demonstrated no negative morphological or proliferative effects on the cells (Fig. 7). No microfilament granulations were observed, and f-actin fibers assume a healthy, striated form in cells in direct contact with both HAp/Ch-PLGA and A-HAp/Ch-PLGA particles. No repulsion of the particles by the cells, previously observed for specific Ch-HAp combinations [63], was observed either. No reduction in the cell density entailed the treatment with A-free and A-loaded HAp/Ch-PLGA particles. In fact, the nuclei count yielded the cell density of 35×103 cells/cm2 for A-HAp/Ch-PLGA as opposed to 24×103 cells/cm2 for A-HAp/Ch-PLGA after 4 days of growth, suggesting that the addition of A to HAp/Ch-PLGA exerted a positive proliferative effect on primary fibroblasts. The main difference observed between HAp/Ch-PLGA and A-HAp/Ch-PLGA lay in the more pronounced uptake of A-free HAp/Ch-PLGA. As seen in Figs. 7c and 7k, a larger number of cells display the internalized particles in the population incubated with A-free HAp/Ch-PLGA. The visually observed reduction in dispersability of HAp/Ch-PLGA following the entrapment of A cannot stem from the modified surface charge of the particles because zeta potential of A-HAp/Ch-PLGA was higher than that of HAp/Ch-PLGA at the physiological pH indicating a lowered surface charge propensity for aggregation following the entrapment of the drug. This effect cannot be either due to a difference in the particle size because there is virtually none: d50 for HAp/Ch-PLGA is 168 nm and d50 for HAp/Ch-PLGA is 163 nm. Both particles also typified by relatively narrowly dispersed spherical morphologies. An increase in the hydrophobicity of the particle surface might be responsible for the larger and less dispersable particle aggregates in the HAp/Ch-PLGA system containing A.
Figure 7.

Primary mouse lung fibroblasts interact with 1 mg/ml HAp/Ch-PLGA (A–D and I–L) and A-HAp/Ch-PLGA particles (E–H) with no cytotoxicity or repulsion. A–D) Primary mouse lung fibroblasts surround HAp/Ch-PLGA particles. E–H) Primary mouse lung fibroblasts surround androstane-loaded HAp/Ch-PLGA particles. I–L) Primary mouse lung fibroblasts endocytose HAp/Ch-PLGA particles. Rhodamine-phalloidin-stained f-actin (red), DAPI-stained nucleus (blue), and auto-fluorescing HAp/Ch-PLGA and A-HAp/Ch-PLGA particles (green).
These data indicate the selectivity with which the androstane derivative A delivered using HAp/Ch-PLGA particles imposes higher cytotoxicity on lung tumor cells than on the healthy ones. The mechanism by which the given androstane derivative exerts a toxic effect on the cancer cells involves the induction of the ER stress response, which results in calcium release from the ER and arrest of the cell in the G1 phase cycle [37]. A carrier capable of further elevating calcium levels as a result of its degradation in the lysosome, alongside localizing itself in the vicinity of the ER, as HAp has been confirmed to do [35], is a logical choice for the delivery vehicle for the given drug. Here we see that the therapeutic selectivity of the compound A becomes preserved following its delivery by means HAp/Ch-PLGA. In vivo delivery and the optimization of the drug/carrier interface will be the subjects of further studies.
4. Conclusion
A novel particulate platform for targeting lung cancer and causing the death of cancer cells in a selective manner was explored. Particles composed of HAp coated with the chitosan-poly(D,L)-lactide-co-glycolide (Ch-PLGA) polymer blend, previously shown to localize in lungs following their intravenous administration, were loaded with an androstane derivative, 17β-hydroxy-17α-picolyl-androst-5-en-3β-yl acetate (A). The results of 1H NMR and 13C NMR analyses were mutually consistent, confirming the entrapment of the synthesized androstane derivative inside HAp/PLGA-Ch particles in a structurally intact form. The thermogravimetric and differential thermal analysis coupled with mass spectrometry was used to qualitatively confirm the drug loading process. A-HAp/Ch-PLGA particles were found to be spherical and had a uniform size distribution of d50=168 nm. By measuring the heat flow associated with phase transitions occurring during heating and calculating the corresponding enthalpies, the drug content of the derivative A in A-HAp/Ch-PLGA particles was estimated at 7.47 wt%.
The results of cytotoxicity and viability assays performed on A549 lung adenocarcinoma cells and on healthy MRC5 fibroblasts demonstrated that the particles of hydroxyapatite/chitosan-poly(D,L)-lactide-co-glycolide loaded with 17β-hydroxy-17α-picolyl-androst-5-en-3β-yl acetate exhibit almost three times higher cytotoxicity towards the carcinoma cells (A549) than towards the healthy cells (MRC5). At the same time, no negative effects were observed on primary lung fibroblasts, indicating the prospect with which this drug delivery system could be further explored as a fine particle platform for a targeted and selective cancer therapy that would impose minimal side effects on the patient.
Supplementary Material
Highlights.
Selective anticancer activity of lung-targeting;
Hydroxyapatite/chitosan-PLGA particles loaded with an cancer inhibitor;
TGA and DTA coupled on-line with MS were used;
No negative effects were observed on primary lung fibroblasts;
Drug delivery platforms for selective cancer therapies
Acknowledgments
Research presented in this paper was supported by the Ministry of Education, Science and Technological Development of the Republic of Serbia (project No. III45004 and 172021) and by the United States National Institutes of Health (R00-DE021416). The authors acknowledge Dušan Škorić, MSc, of the University of Novi Sad for recording the NMR spectra and Shreya Ghosh, MSc, of the University of Illinois in Chicago for assistance with the drug release and zeta potential measurements.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Habraken W, Habibovic P, Epple M, Bohner M. Calcium phosphates in biomedical applications: materials for the future? Mater Today. 2016;19:69–87. [Google Scholar]
- 2.Sharma S, Verma A, Teja BV, Pandey G, Mittapelly N, Trivedi R, Mishra PR. An insight into functionalized calcium based inorganic nanomaterials in biomedicine: Trends and transitions. Colloids Surf B Biointerfaces. 2015;133:120–139. doi: 10.1016/j.colsurfb.2015.05.014. [DOI] [PubMed] [Google Scholar]
- 3.Uskoković V. The role of hydroxyl channel in defining selected physicochemical peculiarities exhibited by hydroxyapatite. RSC Advances. 2015;5:36614–36633. doi: 10.1039/C4RA17180B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bose S, Fielding G, Tarafder S, Bandyopadhyay A. Understanding of dopant-induced osteogenesis and angiogenesis in calcium phosphate ceramics. Trends Biotechnol. 2013;31:594–605. doi: 10.1016/j.tibtech.2013.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ergun C, Webster TJ, Bizios R, Doremus RH. Hydroxyapatite with substituted Mg, Zn, Cd, and Y. I. Structure and microstructure. J Biomed Mater Res. 2002;59:305–311. doi: 10.1002/jbm.1246. [DOI] [PubMed] [Google Scholar]
- 6.Martínez-Casado FJ, Iafisco M, Delgado-López JM, Martínez-Benito C, Ruiz-Perez C, Colangelo D, Oltolina F, Prat M, Gómez-Morales J. Bio-inspired citrate-apatite nanocrystals doped with divalent transition metal ions. Cryst Growth Des. 2016;16:145–153. [Google Scholar]
- 7.Ignjatović N, Ajduković Z, Rajković J, Najman S, Mihailović D, Uskoković D. Enhanced osteogenesis of the nanosize cobalt substituted hydroxyapatite. J Bionic Eng. 2015;12:604–612. [Google Scholar]
- 8.Dorozhkin SV. Calcium orthophosphate-containing biocomposites and hybrid biomaterials for biomedical applications. J Funct Biomater. 2015;6:708–832. doi: 10.3390/jfb6030708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kolmas J, Krukowski S, Laskus A, Jurkitewicz M. Synthetic hydroxyapatite in pharmaceutical applications. Ceram Int. 2016;42:2472–2487. [Google Scholar]
- 10.Ignjatovic NL, Ajdukovic ZR, Savic VP, Uskokovic DP. Size effect of calcium phosphate coated with poly-DL-lactide-co-glycolide on healing processes in bone reconstruction. J Biomed Mater Res B Appl Biomater. 2010;94:108–117. doi: 10.1002/jbm.b.31630. [DOI] [PubMed] [Google Scholar]
- 11.Ignjatović N, Uskoković V, Ajduković Z, Uskoković D. Multifunctional hydroxyapatite and poly(D,L-lactide-co-glycolide) nanoparticles for the local delivery of cholecalciferol. Mater Sci Eng C. 2013;33:943–950. doi: 10.1016/j.msec.2012.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ignjatović NL, Ninkov P, Sabetrasekh R, Uskoković DP. A novel nano drug delivery system based on tigecycline-loaded calciumphosphate coated with poly-DL-lactide-co-glycolide. J Mater Sci Mater Med. 2010;21:231–239. doi: 10.1007/s10856-009-3854-6. [DOI] [PubMed] [Google Scholar]
- 13.Uskoković V, Uskoković D. Nanosized hydroxyapatite and other calcium phosphates: chemistry of formation and application as drug and gene delivery agents. J Biomed Mater Res B Appl Biomater. 2011;96:152–191. doi: 10.1002/jbm.b.31746. [DOI] [PubMed] [Google Scholar]
- 14.Cheng Z, Zaki A, Hui J, Muzykantov V, Tsourkas A. Multifunctional nanoparticles: cost versus benefit of adding targeting and imaging capabilities. Science. 2012;338:903–910. doi: 10.1126/science.1226338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gu W, Wu C, Chen J, Xiao Y. Nanotechnology in the targeted drug delivery for bone diseases and bone regeneration. Int J Nanomedicine. 2013;8:2305–2317. doi: 10.2147/IJN.S44393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chu SH, Feng DF, Ma YB, Li ZQ. Hydroxyapatite nanoparticles inhibit the growth of human glioma cells in vitro and in vivo. Int J Nanomedicine. 2012;7:3659–3666. doi: 10.2147/IJN.S33584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.El-Dakdouki MH, Puré E, Huang X. Development of drug loaded nanoparticles for tumor targeting. Part 1: Synthesis, characterization, and biological evaluation in 2D cell cultures. Nanoscale. 2013;5:3895–3903. doi: 10.1039/c3nr33777d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ashokan A, Menon D, Nair S, Koyakutty M. A molecular receptor targeted, hydroxyapatite nanocrystal based multi-modal contrast agent. Biomaterials. 2010;31:2606–2616. doi: 10.1016/j.biomaterials.2009.11.113. [DOI] [PubMed] [Google Scholar]
- 19.Ignjatović N, Vranješ-Djurić S, Mitić Z, Janković D, Uskoković D. Investigating an organ-targeting platform based on hydroxyapatite nanoparticles using a novel in situ method of radioactive 125Iodine labeling. Mater Sci Eng C. 2014;43:439–446. doi: 10.1016/j.msec.2014.07.046. [DOI] [PubMed] [Google Scholar]
- 20.Saie AA, Ray M, Mahmoudi M, Rotello VM. Engineering the nanoparticle-protein interface for cancer therapeutics. Cancer Treat Res. 2015;166:245–273. doi: 10.1007/978-3-319-16555-4_11. [DOI] [PubMed] [Google Scholar]
- 21.Docter D, Strieth S, Westmeier D, Hayden O, Gao M, Knauer SK, Stauber RH. No king without a crown-impact of the nanomaterial-protein corona on nanobiomedicine. Nanomedicine (Lond) 2015;10:503–519. doi: 10.2217/nnm.14.184. [DOI] [PubMed] [Google Scholar]
- 22.Trif M, Florian PE, Roseanu A, Moisei M, Craciunescu O, Astete CE, Sabliov CM. Cytotoxicity and intracellular fate of PLGA and chitosan-coated PLGA nanoparticles in Madin–Darby bovine kidney (MDBK) and human colorectal adenocarcinoma (Colo 205) cells. J Biomed Mater Res A. 2015;103:3599–3611. doi: 10.1002/jbm.a.35498. [DOI] [PubMed] [Google Scholar]
- 23.Márquez M, Nilsson S, Lennartsson L, Liu Z, Tammela T, Raitanen M, Holmberg AR. Charge-dependent Targeting: Results in six tumor cell lines. Anticancer Res. 2004;24:1347–1352. [PubMed] [Google Scholar]
- 24.Azuma K, Osaki T, Minami S, Okamoto Y. Anticancer and anti-inflammatory properties of chitin and chitosan oligosaccharides. J Funct Biomater. 2015;6:33–49. doi: 10.3390/jfb6010033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Andey T, Marepally S, Patel A, Jackson T, Sarkar S, O’Connell M, Reddy R, Chellappan S, Singh P, Singh M. Cationic lipid guided short-hairpin RNA interference of annexin A2 attenuates tumor growth and metastasis in a mouse lung cancer stem cell model. J Control Release. 2014;184:67–78. doi: 10.1016/j.jconrel.2014.03.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ekrami H, Kennedy AR, Shen WC. Disposition of positively charged Bowman–Birk protease inhibitor conjugates in mice: Influence of protein conjugate charge density and size on lung targeting. J Pharm Sci. 1995;84:456–461. doi: 10.1002/jps.2600840413. [DOI] [PubMed] [Google Scholar]
- 27.Kutscher HL, Chao P, Deshmukh M, Rajan SS, Singh Y, Hu P, Joseph LB, Stein S, Laskin DL, Sinko PJ. Enhanced passive pulmonary targeting and retention of PEGylated rigid microparticles in rats. Int J Pharm. 2010;402:64–71. doi: 10.1016/j.ijpharm.2010.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schiffelers RM, Ansari A, Xu J, Zhou Q, Tang Q, Storm G, Molema G, Lu PY, Scaria PV, Woodle MC. Cancer siRNA therapy by tumor selective delivery with ligand-targeted sterically stabilized nanoparticle. Nucleic Acids Res. 2004;32:el49. doi: 10.1093/nar/gnh140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bender E. Epidemiology: The dominant malignancy. Nature. 2014;513:S2–3. doi: 10.1038/513S2a. [DOI] [PubMed] [Google Scholar]
- 30.Baik CS, Eaton KD. Estrogen signaling in lung cancer: an opportunity for novel therapy. Cancers (Basel) 2012;4:969–988. doi: 10.3390/cancers4040969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gaši KM, Djurenović-Brenesel M, Djurendić E, Sakač M, Čanadi J, Daljev J, Armbruster T, Andrić S, Sladić DM, Božić TT, Novaković IT, Juranić ZD. Synthesis and biological evaluation of some 17-picolyl and 17-picolinylidene androst-5-ene derivatives. Steroids. 2007;72:31–40. doi: 10.1016/j.steroids.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 32.Djurendić E, Daljev J, Sakač M, Čanadi J, Šanta-Jovanović S, Andrić S, Klisurić O, Kojić V, Bogdanović G, Djurenović-Brenesel M, Novaković S, Gaši KP. Synthesis of some epoxy and/or N-oxy 17-picolyl and 17-picolinylidene-androst-5-ene derivatives and evaluation of their biological activity. Steroids. 2008;73:129–138. doi: 10.1016/j.steroids.2007.09.005. [DOI] [PubMed] [Google Scholar]
- 33.Djurendić EA, Ajduković JJ, Sakač MN, Čanadi J, Kojić V, Bogdanović G, Gaši KM. Synthesis and cytotoxic activity of some 17-picolyl and 17-picolinylidene androstane derivatives. Eur J Med Chem. 2012;54:784–792. doi: 10.1016/j.ejmech.2012.06.030. [DOI] [PubMed] [Google Scholar]
- 34.Yanhua W, Hao H, Li Y, Zhang S. Selenium-substituted hydroxyapatite nanoparticles and their in vivo antitumor effect on hepatocellular carcinoma. Colloids Surf B Biointerfaces. 2016;140:297–306. doi: 10.1016/j.colsurfb.2015.12.056. [DOI] [PubMed] [Google Scholar]
- 35.Han Y, Li S, Cao X, Yuan L, Wang Y, Yin Y, Qiu T, Dai H, Wang X. Different inhibitory effect and mechanism of hydroxyapatite nanoparticles on normal cells and cancer cells in vitro and in vivo. Sci Rep. 2014;20:7134. doi: 10.1038/srep07134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nel AE, Mädler L, Velegol D, Xia T, Hoek EMV, Somasundaran P, Klaessig F, Castranova V, Thompson M. Understanding biophysicochemical interactions at the nano-bio interface. Nat Mater. 2009;8:543–557. doi: 10.1038/nmat2442. [DOI] [PubMed] [Google Scholar]
- 37.Ajduković JJ, Djurendić EA, Petri ET, Klisurić OR, Ćelić AS, Sakač MN, Jakimov DS, Penov-Gaši KM. 17(E)-picolinylidene androstane derivatives as potential inhibitors of prostate cancer cell growth: antiproliferative activity and molecular docking studies. Bioorg Med Chem. 2013;21:7257–7266. doi: 10.1016/j.bmc.2013.09.063. [DOI] [PubMed] [Google Scholar]
- 38.Ankrum JA, Miranda OR, Ng KS, Sarkar D, Xu C, Karp JM. Engineering cells with intracellular agent-loaded microparticles to control cell phenotype. Nat Protoc. 2014;9:233–245. doi: 10.1038/nprot.2014.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Long JT, Cheang TY, Zhuo SY, Zeng RF, Dai QS, Li HP, Fang S. Anticancer drug-loaded multifunctional nanoparticles to enhance the chemotherapeutic efficacy in lung cancer metastasis. J Nanobiotechnology. 2014;12:37. doi: 10.1186/s12951-014-0037-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hait WN. Anticancer drug development: the grand challenges. Nat Rev Drug Discov. 2010;9:253–254. doi: 10.1038/nrd3144. [DOI] [PubMed] [Google Scholar]
- 41.Corcos D. Toward a universal treatment for cancer: cell inflation assisted chemotherapy. Cancer Med. 2013;2:421–426. doi: 10.1002/cam4.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wicki A, Witzigmann D, Balasubramanian V, Huwyler J. Nanomedicine in cancer therapy: Challenges, opportunities, and clinical applications. J Control Release. 2015;200:138–157. doi: 10.1016/j.jconrel.2014.12.030. [DOI] [PubMed] [Google Scholar]
- 43.Xu X, Ho W, Zhang X, Bertrand N, Farokhzad O. Cancer nanomedicine: from targeted delivery to combination therapy. Trends Mol Med. 2015;21:223–232. doi: 10.1016/j.molmed.2015.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Miljković DA, Gaši KM. A novel synthetic approach to 3β-acetoxy-17-picolin-ylidene-5-androsten-16-one. Bull Soc Chim Beograd. 1982;47:173–181. [Google Scholar]
- 45.Ignjatović NL, Liu CZ, Czernuszka JT, Uskoković DP. Micro- and nano-injectable composite biomaterials containing calcium phosphate coated with poly(DL-lactide-co-glycolide) Acta Biomater. 2007;3:927–935. doi: 10.1016/j.actbio.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 46.Ignjatović N, Wu V, Ajduković Z, Mihajilov-Krstev T, Uskoković V, Uskoković D. Chitosan-PLGA polymer blends as coatings for hydroxyapatite nanoparticles and their effect on antimicrobial properties, osteoconductivity and regeneration of osseous tissues. Mater Sci Eng C. 2016;60:357–364. doi: 10.1016/j.msec.2015.11.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wanjun T, Cunxin W, Donghua C. Kinetic studies on the pyrolysis of chitin and chitosan. Polym Degrad Stab. 2005;87:389–394. [Google Scholar]
- 48.Qu X, Wirsen A, Albertsson AC. Effect of lactic/glycolic acid side chains on the thermal degradation kinetics of chitosan derivatives. Polymer. 2000;41:4841–4847. [Google Scholar]
- 49.Palacios J, Albano C, González G, Castillo RV, Karam A, Covis M. Characterization and thermal degradation of poly(d,l-lactide-co-glycolide) composites with nanofillers. Polym Eng Sci. 2013;53:1414–1429. [Google Scholar]
- 50.McNeill IC, Leiper HA. Degradation studies of some polyesters and polycarbonates – 2. Polylactide: Degradation under isothermal conditions, thermal degradation mechanism and photolysis of the polymer. Polym Degrad Stab. 1985;11:309–326. [Google Scholar]
- 51.McNeill IC, Leiper HA. Degradation studies of some polyesters and polycarbonates: 3-Polyglycollide. Polym Degrad Stab. 1985;12:373–385. [Google Scholar]
- 52.Kumta PN, Sfeir C, Lee DH, Olton D, Choi D. Nanostructured calcium phosphates for biomedical applications: novel synthesis and characterization. Acta Biomater. 2005;1:65–83. doi: 10.1016/j.actbio.2004.09.008. [DOI] [PubMed] [Google Scholar]
- 53.Corazzari I, Nistico R, Turci F, Faga MG, Franzoso F, Tabasso S, Magnacca G. Advanced physico-chemical characterization of chitosan by means of TGA coupled on-line with FTIR and GCMS: Thermal degradation and water adsorption capacity. Polym Degrad Stab. 2015;112:1–9. [Google Scholar]
- 54.de Britto D, Campana-Filho SP. Kinetics of the thermal degradation of chitosan. Thermochim Acta. 2007;465:73–82. [Google Scholar]
- 55.Erbetta CD’AC, Alves RJ, Resende JM, Freitas RFS, de Sousa RG. Synthesis and characterization of poly(d,l-lactide-co-glycolide) copolymer. J Biomater Nanobiotechnol. 2012;3:208–225. [Google Scholar]
- 56.Kil’deeva NR, Perminov PA, Vladimirov LV, Novikov VV, Mikhailov SN. Mechanism of the reaction of glutaraldehyde with chitosan. Russ J Bioorganic Chem. 2009;35:397–407. [PubMed] [Google Scholar]
- 57.Monteiro OAC, Jr, Airoldi C. Some studies of crosslinking chitosan–glutaraldehyde interaction in a homogeneous system. Int J Biol Macromol. 1999;26:119–128. doi: 10.1016/s0141-8130(99)00068-9. [DOI] [PubMed] [Google Scholar]
- 58.Ramachandran S, Nandhakumar S, Dhanaraju MD. Formulation and characterization of glutaraldehyde cross-linked chitosan biodegradable microspheres loaded with famotidine. Trop J Pharm Res. 2011;10:309–316. [Google Scholar]
- 59.Baldino L, Concilio S, Cardea S, De Marco I, Reverchon E. Complete glutaraldehyde elimination during chitosan hydrogel drying by SC-CO2 processing. J Supercritical Fluids. 2015;103:70–76. [Google Scholar]
- 60.Alskar LC, Porter CJH, Bergstrom CAS. Tools for early prediction of drug loading in lipid-based formulations. Mol Pharmaceutics. 2016;13:251–261. doi: 10.1021/acs.molpharmaceut.5b00704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ajduković JJ, Penov-Gaši KM, Jakimov DS, Jovanović-Šanta SS, Aleksić LD, Djurenović EA. Synthesis, structural analysis and antitumor activity of novel 17α-picolyl and 17 (E)-picolinylidene A-modified androstane derivatives. Bioorg Med Chem. 2015;23:1557–1568. doi: 10.1016/j.bmc.2015.02.001. [DOI] [PubMed] [Google Scholar]
- 62.Farah IO. Differential modulation of intracellular energetic in A549 and MRC-5 cells. Biomed Sci Instrum. 2007;43:110–115. [PubMed] [Google Scholar]
- 63.Uskoković V, Desai TA. In vitro analysis of nanoparticulate hydroxyapatite/chitosan composites as potential drug delivery platforms for the sustained release of antibiotics in the treatment of osteomyelitis. J Pharm Sci. 2014;103:567–579. doi: 10.1002/jps.23824. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.