

Abstract
Background
Environmental factors such as inhaled pollutants like cigarette smoke may play a significant role in diseases of the upper airway including chronic rhinosinusitis (CRS). Recent studies have shown that cigarette smoke causes impaired airway epithelial cell barrier function likely through environmental oxidative stress related pathways. The purpose of this study is to explore whether enhancing Nrf2, the body’s master antioxidant system, can ameliorate cigarette smoke induced sinonasal epithelial cell barrier dysfunction.
Methods
Human Sinonasal epithelial cells (HSNECs) were grown from control patients at the air-liquid interface. HSNECs were stimulated with cigarette smoke extract (CSE) with and without pharmacologic activation of Nrf2. HSNECs were then stained for the epithelial cell junctional proteins ZO-1 and JAM-A using confocal microscopy. In addition trans-epithelial electrical resistance (TER) was measured in cultures before and after stimulation with CSE
Results
CSE stimulation caused a global disruption of the epithelial junctional proteins ZO-1 and JAM-A along with an associated decrease in TER levels. Enhancing Nrf2 levels prior to stimulation with CSE was associated with increased localization of ZO-1 and JAM-A levels at the cell surface and statistically significant increases in TER levels.
Conclusions
This is the first study to demonstrate that cigarette smoke induced sinonasal epithelial cell barrier dysfunction is reversible by Nrf2 activation. The Nrf2 antioxidant pathway may represent a potential therapeutic target for cigarette smoke associated sinonasal inflammation.
Keywords: Cigarette smoke, Nrf2
Introduction
Chronic rhinosinusitis (CRS) has multiple etiologic and contributing factors. Associations between CRS and allergy, bacterial colonization, environmental exposures, defects in mucociliary function, dysregulation of the innate immune response, and anatomic variations have all been demonstrated in the literature1–6. Cigarette smoke (CS) is one such environmental exposure that has garnered a lot of attention as an etiologic factor contributing to sinonasal epithelial dysfunction and the development of CRS7–9. A recent review of the literature concluded that there is clear evidence in the literature supporting a contribution to CRS by both active and passive (second hand exposure) smoking10.
The sinonasal epithelial barrier represents one of the most primitive forms of innate immune defense. It provides a physical barrier and serves in mucus production as well as mucociliary clearance. This physical barrier is reinforced with junctional proteins that tightly join adjacent epithelial cells preventing deeper penetration of both microbial and non-microbial elements. Previous studies have demonstrated that CS can induce lower airway epithelial cell permeability and induce oxidative stress11,12.
Nrf2 is a vital transcriptional factor for antioxidant responsive element (ARE)-bearing genes involved in reduction-oxidation reaction (redox) balance13. The importance of normally functioning Nrf2 to combat CS induced oxidative stress in the airways has been demonstrated in knockout mice chronically exposed to CS14,15. Nrf2 agonists have shown protective effects to oxidative stress in epithelial cells from both the lower and upper respiratory tracts indicating a potential for therapeutic intervention13,16.
With an association between CS and CRS and the demonstration of CS induced oxidative stress and subsequent damage to the airway epithelium, we set out to investigate the role of Nrf2 activation on CS induced sinonasal epithelial cell (SNEC) barrier dysfunction. Our working hypothesis was that pharmacologic activation of Nrf2 may exhibit a reparative or protective effect on SNECs exposed to cigarette smoke extract. To our knowledge, this is the first study to evaluate the impact of Nrf2 activation on CS induced epithelial dysfunction from human sinonasal epithelial cells (HSNECs).
Methods
Human subjects
12 control subjects were enrolled in this study. The research protocol was approved through our Institutional Review process, and all subjects gave signed informed consent. Mucosal tissue was collected from the ethmoid sinuses during endoscopic sinus surgery and grown in culture. All control patients were defined as non-smokers without CRS who were undergoing endoscopic sinonasal surgery for dacryocystorhinostomy, cerebrospinal fluid leak repair, or endoscopic skull base surgery. All samples were taken at the start of the case, prior to surgical manipulation of the tissue, and in the case of CSF leak repair or unilateral sinus disease, were taken from the unaffected side. Control subjects were from the same geographic location and thus were exposed to similar levels of environmental pollution. Patient demographics are summarized in Table 1.
Table 1.
Patient Demographics.
| Patient | Age | Gender | Diagnosis |
|---|---|---|---|
| 1 | 68 | F | Grave’s Orbitopathy |
| 2 | 64 | F | Grave’s Orbitopathy |
| 3 | 49 | F | Grave’s Orbitopathy |
| 4 | 45 | F | Grave’s Orbitopathy |
| 5 | 50 | F | Grave’s Orbitopathy |
| 6 | 61 | F | Orbital Hemangioma |
| 7 | 32 | F | Unilateral Ethmoid Mucocele |
| 8 | 66 | F | CSF Leak |
| 9 | 62 | F | Maillary Sinus Fungal ball |
| 10 | 63 | F | Unilateral latrogenic Sinusitis |
| 11 | 72 | M | Sphenoid Sinus Inverted Papilloma |
| 12 | 23 | M | Juvenile Nasopharyngeal Angiofibroma |
SNEC culture at the ALI
Sinonasal epithelial cells were cultured at the air-liquid interface cultures as previously described by our group17–19. Epithelial cells were isolated from tissue samples by enzymatic digestion and grown in cell culture. Once the cells reached confluence, they were trypsinized, suspended in BEGM media containing DMSO, and frozen at −80°C. After thawing, the cells were plated onto 24-well Falcon filter inserts (0.4μm pore size; Becton Dickinson, Franklin Lakes, NJ) coated with human type IV placental collagen (Sigma, Type VI). The P1 cells were grown to confluence with BEGM above (1 ml) and below (2 ml) the filter inserts. When confluent, medium was removed from above the cultures and the medium below the inserts were changed to ALI medium consisting of LHC Basal Medium:DMEM-H (Gibco) (50:50). Each set of cultures came from a separate patient source and was maintained at the air-liquid interface with the apical surfaces remaining free of medium for at least 3 weeks prior to study.
Treatment of SNECs with Cigarette Smoke Extracts
Cigarette smoke extract (CSE) was obtained from Murty Pharmaceuticals, Inc., (Lexington, KY), and consisted of 40mg/ml condensate, and 6% nicotine. According to the manufacturer, CSE was prepared by smoking University of Kentucky’s 3R4F Standard Research Cigarettes on an FTC Smoke Machine. The smoke on the filter is calculated by the weight gain of the filter after smoking. The amount of DMSO is calculated that will dissolve a 4% (40mg/mL) solution. Cells were stimulated apically with 5% or 15% CSE diluted into ALI medium.
Transepithelial Electrical Resistance Measurements
Transepithelial electrical resistance (TER) was monitored using the EVOM2 Voltohmmeter with the STX2 chopstick probe (World Precision Instruments). To measure TER, 500 μL of pre-warmed ALI media was incubated in the apical compartment of the transwell for 30 minutes at 37°C. This media was replaced with the treatment medium and TER was recorded 1 hour and 4 hours post stimulation. TER from each CSE stimulated well was normalized to its pre-stimulation TER.
Immunocytochemistry
Adherent cells at the ALI were washed and fixed with ice-cold 4% paraformaldehyde for 15 minutes at 4°C. The filter membranes were cut from the inserts and divided into 4 quadrants that were placed separately in 24-well plates. After permeabilization with 0.3% Triton-X (EMD Chemicals, Gibbstown, NJ) and blocking of nonspecific binding sites with 10% goat serum, inserts were incubated (4°C, overnight) with AlexaFluor 594 mouse anti-ZO-1 (LifeSpan Biosciences, Seattle, WA) and mouse anti-JAM-A (R&D Systems, Minneapolis, MN). Each sample was counterstained by the nuclear stain, DAPI (Vector Labs, Burlingsgame, CA). After washing, the filter membranes were mounted on glass slides with 95% glycerol. Confocal images were collected with a LSM700 confocal laser-scanning microscope (Zeiss).
RNA extraction and reverse transcription
Total RNA was isolated with the RNeasy Mini kit (Qiagen, Valencia, CA) using the manufacturer’s protocol. RNA was quantified spectrophotometrically and absorbance ratios at 260/280 nm were >1.80 for all samples studied. Five hundred nanograms of total RNA was reverse transcribed in a 20 μL volume with random hexamer primers (Invitrogen), 20 U of RNase inhibitor (Applied Biosystems, Foster City, CA), and the Omniscript RT kit (Qiagen) under conditions provided by the manufacturer.
Real-time polymerase chain reaction
Real time polymerase chain reaction (PCR) was performed in a StepOnePlus (ABI, Foster City, CA) using the SYBR Green PCR Kit (ABI). Primers used in this study are listed in Table 1. The cycle parameters used were 95°C for 20 seconds, then 40 cycles of 95°C for 3 seconds and 60°C for 30 seconds. This was followed by 95°C for 15 seconds, 60°C for 60 seconds, and 95°C for 15 seconds. Amplicon expression in each sample was normalized to its 18S RNA content. The level of expression of target innate immune genes for CD14, Toll-like Receptor 2 (TLR2), and human beta-defensin 1 (HBD1) messenger RNA (mRNA) was determined as the delta cycle threshold (ΔCt), as described. Fold-change was calculated as 2ΔΔCt. Negative controls, consisting of reaction mixtures containing all components except target RNA, were included with each PCR run. Amplified products were sequenced to verify authenticity.
Statistical Analysis
Raw data from real-time PCR were entered into a spreadsheet and statistical analysis was performed using a software program (GraphPad Prism; GraphPad Software, Inc, LaJolla, CA). Data are expressed as mean ± the standard error of mean (SEM). Statistical significance of differences in CD14, TLR2, and HBD1 mRNA expression were determined using the Wilcoxon signed rank test for paired data or unpaired t-test assuming unequal variances. Differences were considered statistically significant at P<0.05.
Results
Cigarette smoke extract induced sinonasal epithelial barrier dysfunction is reversed by activation of the Nrf2 pathway
The particulate phase of cigarette smoke contains several known redox-active components which promote the generation of reactive oxygen species (ROS)20. Recent evidence indicates that the Nrf2 pathway modulates epithelial barrier function by enhancing cellular antioxidant capacity21. To determine the effect of cigarette smoke extracts on sinonasal epithelial barrier, we exposed HSNEC cultures at the ALI to CSE with and without the Nrf2 activator sulforaphane (SFN). Analysis of TER demonstrated that CSE induced disruption of the sinonasal epithelial barrier was reversed by Nrf2 activation (Figure 1).
Figure 1.

TER measurements following exposure to 15% CSE (*p<0.05 Vehicle/Media vs. Vehicle/CSE, **p<0.01 Vehicle/Media vs. Vehicle/CSE, # p<0.05 Vehicle/CSE vs. SFN/CSE). Data are representative of mean±SEM, n=8.
Immunofluorescence staining of the tight junction protein JAM-A and the plaque protein ZO-1 demonstrated marked disruption of the tight junction following CSE exposure. Nrf2 activation demonstrated a reversal in this disruption, indicating a preservation of epithelial barrier integrity (Figure 2).
Figure 2.
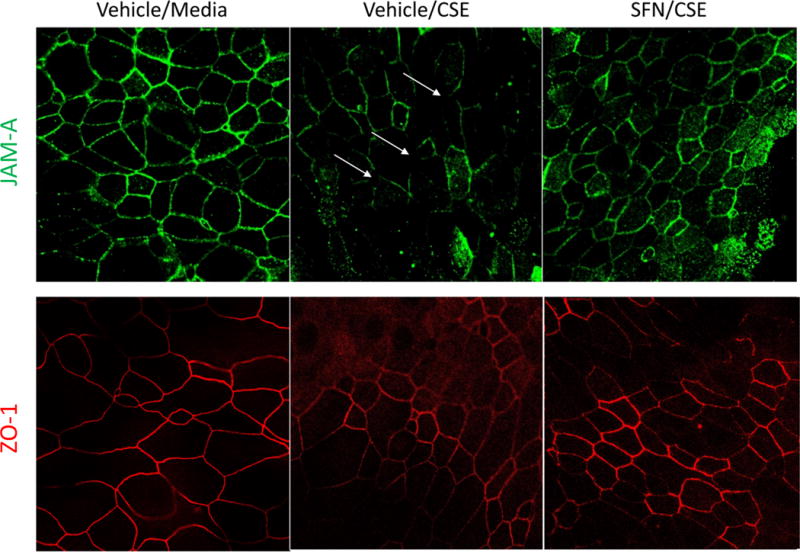
Immunofluorescence staining of JAM-A (green) and ZO-1 (red). Images are representative of four independent experiments.
Cigarette smoke extract increases expression of genes involved in innate immunity
We next sought to determine if this disruption of sinonasal epithelial barrier induces an innate immune response to CSE. To evaluate the effect of CSE on innate immune function in HSNECs, we analyzed mRNA transcript levels of constitutively expressed innate immune genes (Figure 3). CD14 and TLR2 are pattern-recognition receptors involved in sensing microbial components. CSE stimulation induced a statistically significant increase in CD14 and TLR2 mRNA levels in HSNECs (p<0.05). HBD1 is a constitutively expressed antimicrobial peptide secreted by HSNECs. Exposure to CSE induced increased mRNA expression of HBD1 in a dose-dependent manner. SFN was able to reduce the induction of these innate immune genes to some extent.
Figure 3.
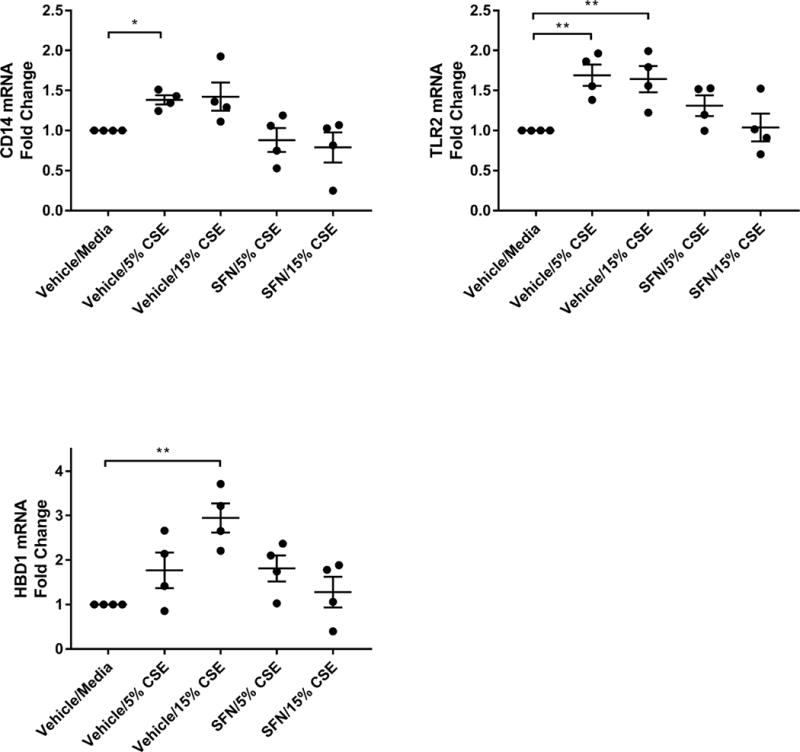
RT-qPCR for mRNA transcripts of CD14, TLR2, and HBD1. *p<0.05, **p<0.01, n=4–6 per group. Data are representative of mean±SEM.
Discussion
Cigarette smoke exposure appears to impact the health and function of HSNECs at several levels. Clinically, exposure to CS has been associated with higher Lund-Mackay scores, alterations of the microbiome22, worsened endoscopic scores after sinus surgery23, and increased rates of revision sinus surgery24. On a cellular level, it has been shown to disrupt the epithelial tight junctions and increase permeability11. Immunologically, it appears to alter innate immune responses including TLR mediated pathways, monocyte-derived macrophage function, and changes in the production of cytokines and effector proteins25. Finally, CS has also been shown to induce oxidative stress, promoting further cellular damage26,27.
The association of CSE exposure and marked disruption of HSNECs tight junctions was demonstrated in this study by both immunofluorescence staining for the junctional proteins JAM-A and ZO-1 and alterations in TER. Interestingly, treatment with SFN, and thus activation of Nrf2, resulted in a reversal of this disruption, as verified by confocal microscopy and TER. This supports the concept that the oxidative stress induced by CSE leads to actual epithelial disruption, our study has shown that this damage is reversible when the appropriate pathways are activated.
Our findings also demonstrated the ability of CSE to stimulate an innate immune response. Gene expression of CD14 and TLR2 significantly increased following HSNECs exposure to CSE. Additionally, the production of the effector protein HBD1 was seen to significantly increase following exposure. Treatment with SFN appears to suppress the innate immune response typically stimulated by CSE.
This is the first study to demonstrate that cigarette smoke induced sinonasal epithelial cell barrier dysfunction and innate immune activation are reversible by Nrf2 pathway activation by SFN. There are several known Nrf2 agonists including synthetic and endogenous compounds13. Some agonists are naturally occurring in various plants and plant oils. Gao et al studied the ability of naturally occurring antioxidants to provide protection to the nasal mucosa against ozone exposure. They found that aerosolized pretreatment with various mixed oil preparations significantly reduced ozone-induced nasal inflammation16. Interestingly, orange oil was shown to induce gene expression of several antioxidant enzymes associated with Nrf216.
This preliminary study provides evidence that activation of Nrf2 may have the potential to reverse epithelial damage caused by environmental irritants such as ozone and CS and may represent a therapeutic target for environmentally associated sinonasal diseases. Further research regarding the role of oxidative stress mechanisms in CRS is warranted.
Table 2.
qPCR primers used in this study.
| Target | Forward | Reverse |
|---|---|---|
| 18s | 5′-GTAACCCGTTGAACCCCA-3′ | 5′-CCATCCAATCGGTAGTAG-3′ |
| CD14 | 5′-GGTTCCTGCTCAGCTACTGG-3′ | 5′-TTAFFTCCTCGAGCGTCAGT-3′ |
| TLR2 | 5′-CCAAGGAAGAATCCTCCAATCA-3′ | 5′-GCTGCCCTTGCAGATACCA-3′ |
| HBD1 | 5-ATGGCCTCAGGTGGTAACTTTC-3′ | 5′-TGACGCAATTGTAATTGTAATGATCAGATCT-3′ |
Acknowledgments
Funding sources: NIH ES020859 (to M.R.)
FAMRI Clinician Innovator Award (to M.R)
Footnotes
Disclosures: No disclosures
Presented at the Spring meeting of the American Rhinologic Society May, 2016 in Chicago, IL
References
- 1.Daines SM, Orlandi RR. Inflammatory cytokines in allergy and rhinosinusitis. Curr Opin Otolaryngol Head Neck Surg. 2010;18:187–190. doi: 10.1097/MOO.0b013e328338206a. [DOI] [PubMed] [Google Scholar]
- 2.Zurak K, Vagic D, Drvis P, et al. Bacterial colonization and granulocyte activation in chronic maxillary sinusitis in asthmatics and non-asthmatics. J Med Microbiol. 2009;58:1231–1235. doi: 10.1099/jmm.0.010579-0. [DOI] [PubMed] [Google Scholar]
- 3.Trevino RJ. Air pollution and its effect on the upper respiratory tract and on allergic rhinosinusitis. Otolaryngol Head Neck Surg. 1996;114:239–41. doi: 10.1016/S0194-59989670174-2. [DOI] [PubMed] [Google Scholar]
- 4.Antunes MB, Cohen NA. Mucociliary clearance-a critical upper airway host defense mechanism and methods of assessment. Curr Opin Allergy Clin Immunol. 2007;7:5–10. doi: 10.1097/ACI.0b013e3280114eef. [DOI] [PubMed] [Google Scholar]
- 5.Lane AP. The role of innate immunity in the pathogenesis of chronic rhinosinusitis. Curr Allergy Asthma Rep. 2009;9:205–212. doi: 10.1007/s11882-009-0030-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kennedy DW. Pathogenesis of chronic rhinosinusitis. Ann Otol Rhinol Laryngol Suppl. 2004;193:6–9. doi: 10.1177/00034894041130s503. [DOI] [PubMed] [Google Scholar]
- 7.Lee WH, Hong SN, Kim HJ, et al. Effects of cigarette smoking on rhinologic diseases: Korean national health and nutrition examination survey 2008–2011. Int Forum Allergy Rhinol. 2015;5:937–943. doi: 10.1002/alr.21553. [DOI] [PubMed] [Google Scholar]
- 8.Tammemagi CM, Davis RM, Benninger MS, et al. Secondhand smoke as a potential cause of chronic rhinosinusitis: a caes-control study. Arch Otolaryngol Head Neck Surg. 2010;136:327–334. doi: 10.1001/archoto.2010.43. [DOI] [PubMed] [Google Scholar]
- 9.Reh DD, Lin SY, Clipp SL, et al. Secondhand tobacco smoke exposure and chronic rhinosinusitis: a population-based case-control study. Am J Rhinol Allergy. 2009;23:562–567. doi: 10.2500/ajra.2009.23.3377. [DOI] [PubMed] [Google Scholar]
- 10.Reh DD, Higgins TS, Smith TL. Impact of tobacco smoke on chronic rhinosinusitis-a review of the literature. Int Forum Allergy Rhinol. 2012;2:362–369. doi: 10.1002/alr.21054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rusznak C, Sapsford RJ, Devalia JL, et al. Cigarette smoke potentiates house dust mite allergen-induced increase in the permeability of human bronchial epithelial cells in vitro. Am J Respir Cell Mol Biol. 1999;20:1238–1250. doi: 10.1165/ajrcmb.20.6.3226. [DOI] [PubMed] [Google Scholar]
- 12.Rusznak C, Mills PR, Devalia JL, et al. Effect of cigarette smoke on the permeability and IL-1 beta and sI-CAM-1 release from cultured human bronchial epithelial cells of never smokers, smokers, and patients with chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol. 2000;23:530–536. doi: 10.1165/ajrcmb.23.4.3959. [DOI] [PubMed] [Google Scholar]
- 13.Cho HY, Kleeberger SR. Association of Nrf2 with airway pathogenesis: lessons learned from genetic mouse models. Arch Toxicol. 2015;89:1931–1957. doi: 10.1007/s00204-015-1557-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Iizuka T, Ishii Y, Itho K, et al. Nrf2-deficient mice are highly susceptible to cigarette smoke-induced emphysema. Genes Cells. 2005;10:1113–1125. doi: 10.1111/j.1365-2443.2005.00905.x. [DOI] [PubMed] [Google Scholar]
- 15.Rangasamy T, Cho CY, Thimmulappa RK, et al. Genetic ablation of Nrf2 enhances susceptibility to cigarette smoke-induced emphysema in mice. J Clin Invest. 2004;114:1248–1259. doi: 10.1172/JCI21146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gao M, Singh A, Macri K, et al. Antioxidant components of naturally-occuring oils exhibit marked anti0inflammatory activity in epithelial cells of the human upper respiratory system. Respiratory Research. 2011;12:92. doi: 10.1186/1465-9921-12-92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ramanathan M, JR, Lane AP. A comparison of experimental methods in molecular chronic rhinosinusitis research. Am J Rhinol. 2007;21:373–377. doi: 10.2500/ajr.2007.21.3034. [DOI] [PubMed] [Google Scholar]
- 18.Kohanski MA, Tharakan A, Lane AP, et al. Bactericidal antibiotics promote reactive oxygen species formation and inflammation in human sinonasal epithelial cells. Int Forum Allergy Rhinol. 2016;6:191–200. doi: 10.1002/alr.21646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Paris G, Pozharskaya T, Asempa T, et al. Damage-associated molecular patterns stimulate interleukin–33 expression in nasal polyp epithelial cells. Int Forum Allergy Rhinol. 2014;4:15–21. doi: 10.1002/alr.21237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Church DF, Pryor WA. Free-radical chemistry of cigarette smoke and its toxicological implications. Environ Health Perspect. 1985;64:111–126. doi: 10.1289/ehp.8564111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sussan TE, Gajghate S, Chatterjee S, et al. Nrf2 reduces allergic asthma in mice through enhanced airway epithelial cytoprotective function. Am J Physiol Lung Cell Mol Physiol. 2015;309:L27–36. doi: 10.1152/ajplung.00398.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Uhliarova B, Adamkov M, Svec M, et al. The effect of smoking on CT score, bacterial colonization and distribution of inflammatory cells in the upper airways of patients with chronic rhinosinusitis. Inhal Toxicol. 2014;26:419–425. doi: 10.3109/08958378.2014.910284. [DOI] [PubMed] [Google Scholar]
- 23.Rudmik L, Mace JC, Smith TL. Smoking and endoscopic sinus surgery: does smoking volume contribute to clinical outcome? Int Forum Allergy Rhinol. 2011;1:145–152. doi: 10.1002/alr.20045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Krzeski A, Galewicz A, CHmielewski R, et al. Influence of cigarette smoking on endoscopic sinus surgery long-term outcomes. Rhinology. 2011;49:577–582. doi: 10.4193/Rhino.10.038. [DOI] [PubMed] [Google Scholar]
- 25.Lee WK, Ramanathan M, JR, Spannhake EW, et al. The cigarette smoke component acrolein inhibits expression of the innate immune components IL-8 and human beta-defensin 2 by sinonasal epithelial cells. Am J Rhinol. 2007;21:658–663. doi: 10.2500/ajr.2007.21.3094. [DOI] [PubMed] [Google Scholar]
- 26.Serikov VB, Leutenegger C, Krutilina R, et al. Cigarette smoke extract inhibits expression of peroxiredoxin V and increases airway epithelial permeability. Inhal Toxicol. 2006;18:79–92. doi: 10.1080/08958370500282506. [DOI] [PubMed] [Google Scholar]
- 27.Li XY, Rahman I, Donaldson K, et al. Mechanisms of cigarette smoke induced increased airspace permeability. Thorax. 1996;51:465–471. doi: 10.1136/thx.51.5.465. [DOI] [PMC free article] [PubMed] [Google Scholar]