

Abstract
Anti-cancer peptides (ACPs) are small cationic and hydrophobic peptides that are more toxic to cancer cells than normal cells. ACPs kill cancer cells by causing irreparable membrane damage and cell lysis, or by inducing apoptosis. Direct-acting ACPs do not bind to a unique receptor, but are rather attracted to several different molecules on the surface of cancer cells. Here we report that an amidated wasp venom peptide, Mastoparan, exhibited potent anti-cancer activities toward leukemia (IC50 ~8-9.2 μM), myeloma (IC50 ~11 μM), and breast cancer cells (IC50 ~20–24 μM), including multidrug resistant and slow growing cancer cells. Importantly, the potency and mechanism of cancer cell killing was related to the amidation of the C-terminal carboxyl group. Mastoparan was less toxic to normal cells than it was to cancer cells (e.g., IC50 to PBMC = 48 μM). Mastoparan killed cancer cells by a lytic mechanism. Moreover, Mastoparan enhanced etoposide-induced cell death in vitro. Our data also suggests that Mastoparan and gemcitabine work synergistically in a mouse model of mammary carcinoma. Collectively, these data demonstrate that Mastoparan is a broad-spectrum, direct-acting ACP that warrants additional study as a new therapeutic agent for the treatment of various cancers.
Keywords: Anti-cancer peptide, leukemia, breast cancer, membrane lysis, synergy, broad-spectrum
Graphical Abstract
Introduction
Over the past 5 years for which there are data, delay-adjusted cancer death rates declined by a modest 1.8 and 1.4% in men and women, respectively[1]. Hence, cancer remains a major health concern, as it represents the second leading cause of death in the United States, and is expected to surpass heart disease as the leading cause of death over the next few years. Therefore, there is a continued need for improvements in cancer screening, diagnosis, and treatment. To this end, anti-cancer peptides (ACPs) represent a potential untapped reservoir of effective adjunctive therapies for the treatment of cancer.
ACPs are small polypeptides (<50 residues) that exhibit preferential toxicity towards cancer cells. ACPs are predominantly composed of cationic and hydrophobic amino acids, giving them an overall positive charge and amphipathic structure that promotes binding to negatively charged cell membranes[2]. In contrast to normal cell membranes, which are largely charge neutral, cancer cell membranes carry a net negative charge due to an abundance of anionic phospholipids (e.g., phosphatidylserine)[3], proteoglycans (e.g., heparan sulfate proteoglycans)[4], O-glycosylated mucins[5], and sialoglycoproteins[6]. Following membrane binding, ACPs kill the cancer cell by causing irreparable membrane damage followed by cell lysis (i.e., direct-acting ACPs), or by initiating a cell death pathway that results in death by apoptosis (i.e., indirect-acting ACPs)[7]. Unlike conventional chemotherapeutic agents, ACPs do not only target rapidly dividing cells; therefore, certain ACPs are also toxic to slow growing cancers[8]. Moreover, because most ACPs act at the level of the cell membrane, ACPs are able to target multidrug-resistant (MDR) cancer cells, including those that overexpress MDR proteins[8,9]. Certain ACPs potentiate killing by conventional chemotherapeutic agents in vitro, suggesting that they hold potential for use as novel adjunctive therapies[8]. Importantly, many ACPs, including direct-acting ACPs, exhibit potent anti-tumour activities toward primary tumours and their metastases without causing harm to normal tissues[10]. Selected direct-acting ACPs can completely eliminate primary tumours, but perhaps more importantly treatment with direct-acting ACPs can initiate an anti-tumour immune response that not only protects the tumour-bearing animal from re-challenge, but can also be adoptively transferred to recipient mice[11,12]. To date there has not been any report suggesting that cancer cells can develop resistance to direct-acting ACPs. This is likely because ACPs are attracted to several different surface molecules rather than a unique targeted receptor. Collectively, these findings suggest that ACPs, particularly those that are direct-acting, have considerable potential for use as novel adjunctive therapies for the treatment of various cancers.
One of the challenges of identifying ACPs is that many peptides with anticancer activity are also toxic towards normal cells. Accordingly, screening approaches to identify novel ACP sequences must be complemented by screening against normal cells to establish whether a given ACP will be selective for tumour cells. Therefore, the purpose of the present study was to screen a small library of short (<20 residues) cationic peptides with various biological activities (e.g., antimicrobial, anti-biofilm, and immune-modulatory properties) to identify novel ACPs that selectively kill cancer cells. The Piscidins NRC-03 and NRC-07 were included as positive controls[8,13]. This screening approach identified amidated Mastoparan as the most potent, selective ACP. Mastoparan is a 14-residue peptide isolated from wasp venom[14]. Recently, non-amidated Mastoparan (i.e., Mastoparan-COOH) was shown to induce apoptosis in melanoma cells[15]. Here, we show that Mastoparan capped with a C-terminal amide (i.e., Mastoparan-NH2), as is observed with the natural peptide, is 8–11-fold more potent than Mastoparan-COOH. Interestingly, it is also shown that Mastoparan amide (herein referred to as Mastoparan) clearly kills cancer cells by a lytic mechanism, which is in contrast to the non-amidated derivative[15]. Mastoparan was toxic to leukemia, myeloma, and breast cancer cells, and showed toxicity towards both slow-growing and MDR cancer cells. Importantly, Mastoparan enhanced etoposide-induced cell death in vitro. Our data also suggests that Mastoparan worked synergistically with gemcitabine in a mouse model of mammary carcinoma. To our knowledge, this is the first study to show that C-terminal amidation affects both ACP potency as well as the mechanism of cancer cell killing. Moreover, this study also shows that ACPs work synergistically with chemotherapeutic agents in vivo. Collectively, these findings demonstrate that Mastoparan warrants consideration as a novel therapeutic agent for the treatment of several different cancers.
Materials and Methods
2.1 Cell culture and conditions
Jurkat and THP-1 human leukemia cells, and HOPC murine myeloma cells were purchased from American Type Culture Collection (Manassas, VA), and were maintained in RPMI 1640 medium (Fisher Scientific, Ottawa, ON) supplemented with 5% fetal bovine serum (FBS), 100 U/ml penicillin, 100 μg/ml streptomycin, 2 mM L-glutamine (Invitrogen, Burlington, ON, Canada). Cells were incubated at 37°C in a 5% CO2 humidified environment for a maximum of three months (passaged as required). While all cell lines were originally obtained from ATCC, MDA-MB-231 breast cancer cells were provided by Dr. S. Drover (Memorial University of Newfound, St. John’s, NL, Canada). T47D breast cancer cells were a gift from Dr. Jonathan Blay (University of Waterloo, Waterloo, ON, Canada). MDA-MB-468, 4T1, and SKBR3 breast carcinoma cells were kindly provided by Drs. Patrick Lee, David Waisman, and Graham Dellaire, respectively, and Dr. Kerry Goralski provided MCF7 and paclitaxel-resistant MCF7-TX400 breast cancer cells (Dalhousie University, Halifax, NS, Canada). All breast carcinoma cells were maintained in DMEM (Fisher) supplemented with 10% FBS, 100 U/ml penicillin, 100 μg/ml streptomycin, and 2 mM L-glutamine, and were incubated at 37°C in a 10% CO2 humidified environment for a maximum of 30 passages. All cell lines were passaged as needed to maintain optimal cell growth, and were routinely tested for mycoplasma contamination using the MycoProbe Mycoplasma Detection Kit (R&D Systems Inc., Minneapolis, MN). IMPACT III testing was performed on 4T1 mouse mammary carcinoma cells by IDEXX BioResearch (Columbia, CO) prior to use in animals. Human mammary epithelial cells (HMECs) were purchased from Lonza Inc. (Mississauga, ON, Canada), and were maintained in Clonetics MEGM (Lonza). HMEC cultures were maintained at 37°C in a 5% CO2 humidified environment for a maximum of six passages. Venous blood was collected from healthy consenting volunteers according to protocols approved by the University of British Columbia Research Ethics Committee. Red blood cells were collected by centrifugation, and peripheral blood mononuclear cells (PBMCs) were isolated by density gradient centrifugation over LymphoPrep (Stemcell Technologies, Vancouver, BC, Canada) as previously described[16].
2.2 Reagents
Mastoparan (INLKALAALAKKIL-NH2) and unamidated Mastoparan (INLKALAALAKKIL-COOH) were purchased from Peptide 2.0 Inc. (Chantilly, VA). Unless otherwise indicated, all experiments were conducted using Mastoparan-NH2, equivalent to natural wasp Mastoparan. Peptide stocks were prepared in sterile water (2 mM) or in saline (1 mg/ml) for in vitro and in vivo experiments, respectively. Paclitaxel, Triton X-100, calcein, dimethylsulfoxide (DMSO), gemcitabine, saline, etoposide, and vinblastine were all purchased from Sigma-Aldrich Canada (Oakville, ON, Canada). Propidium iodide (PI) was from Invitrogen. BOC-D-FMK (pancaspase inhibitor) was purchased from EMD Biosciences (San Diego, CA), and VECTASHIELD (with DAPI) was purchased from Cedarlane Labs (Burlington, ON, Canada).
2.3 MTT Assay
MTT assays were used to assess peptide-mediated cytotoxicity. All adherent cells were seeded (2 × 105 cells/ml) 24 h prior to initiating the experiment to promote cellular adhesion. Leukemia and myeloma cells were seeded (5 × 105 cells/ml) immediately before treatment. All experiments were conducted in flat-well tissue culture plates (Corning, Corning, NY) at a final FBS concentration of 2.5%. Cells were cultured at 37°C in a 5% (leukemia cells, myeloma cells, and PBMCs) or 10% (breast cancer cells and primary mammary epithelial cells) CO2 humidified environment under the indicated conditions for the indicated periods of time. Two hours before the end of the assay, MTT was added to a final concentration of 0.5 μg/ml. Triton X-100 (1% [v/v]) was used as a positive control for 100% cell death, and was added concomitantly with MTT. At the end of the assay, the plates were centrifuged (1400 rpm for 5 min), supernatants were discarded, and the formazan crystals were solubilized in DMSO. Optical density (490 nm) was measured using a microtiter plate reader (Bio-Tek Instruments, Winooski, VT). Percent cytotoxicity was calculated using the following formula: ((E–N)/(P–N)*100), where E, N, and P denote experimental optical density (OD), average OD for the negative control (vehicle), and average OD for the positive control (triton), respectively. Paclitaxel-resistant MCF7-TX400 cells express 2.6-fold more P-glycoprotein than MCF7 cells. P-glycoprotein interferes with the reduction of MTT[8,17]. Thus, trypan blue (Sigma-Aldrich) exclusion counts were used to assess Mastoparan-induced killing of MCF7-TX400 cells.
2.4 Hemolysis Assay
Peptide-mediated hemolysis was quantified using a hemolysis assay. Briefly, red blood cells collected from healthy volunteers, as indicated above, were resuspended (final concentration of 2.5% (v/v) in PBS) in the presence or absence of peptide, and were incubated for 4 h at 37°C in a 5%CO2 humidified environment. Supernatants were collected following centrifugation (1400 rpm), and absorbance (490 nm) was measured using a microtiter plate reader. Percent hemolysis was quantified as described in section 2.3.
2.5 Lactate Dehydrogenase-release Assay
The lactate dehydrogenase (LDH)-release assay was used in lieu of the MTT assay to assess toxicity at early time points. For this, cells were seeded and treated as described in section 2.4. At the end of the assay, at the indicated time points, supernatants were collected, and LDH activity was assessed using the Cytotoxicity Detection Kit (Sigma-Aldrich) as per the manufacturer’s instructions. Percent cytotoxicity was calculated as indicated in section 2.3.
2.6 Propidium Iodide Uptake
Propidium iodide (PI) uptake was used to detect alterations in membrane integrity following peptide treatment. Briefly, Jurkat T leukemia or MDA-MB-231 breast cancer cells (5 × 105 cells) were cultured in the presence or absence of the indicated concentrations of Mastoparan for the indicated periods of time. PI, at a final concentration of 10 μg/ml, was added to the cell suspension 5 min prior to analysis with a FACS Calibur flow cytometer (BD Biosciences, San Jose, CA). FlowJo 7.4 was used to calculate mean channel fluorescence (FlowJo LLC, Ashland, OR).
2.7 DNA Fragmentation Assays
To test for DNA laddering in response to Mastoparan, DNA was isolated from control-, Mastoparan-, and etoposide-treated cells using a kit purchased from Qiagen, Inc. (Mississauga, Ontario, Canada). DNA fragmentation was determined by gel electrophoresis on a 0.8% agarose gel. Alternatively, DNA fragmentation was visualized using the TUNEL assay according to the manufacturer’s instructions (Roche Diagnostics, Laval, QC, Canada). For this, MDA-MB-231 cells were plated (1 × 105 cells) and treated in 8-well chamber slides (VWR International, Mississauga ON, Canada). In contrast, Jurkat T leukemia cells (2.5 × 105 cells) were plated and treated in 24-well tissue culture plates prior to producing cytospins.
2.8 DAPI Staining
DAPI staining was used to assess the nuclear morphology of control-, Mastoparan-, and etoposide-treated cells. For this, MDA-MB-231 and Jurkat cells were grown on multi-chamber slides, or were treated in 24-well tissue culture plates prior to producing cytospins, as described in section 2.7. Following treatment, cells were washed with PBS, fixed with 4% formaldehyde, mounted using VECTASHIELD with DAPI, and were visualized under visible and UV light (x400).
2.9 Calcein Leakage Assay
Calcein leakage from large unilamellar vesicles (LUVs) was performed as described previously[18,19] with slight modifications. Briefly, 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC, powder) and 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-L-serine (POPS, in chloroform) (Avanti Polar Lipids Inc., Alabaster, AL) were solubilized in chloroform to a final concentration of 10 mg/ml. An appropriate volume of each phospholipid was transferred to glass vial to achieve 1 mg total lipid at the indicated lipid composition. The chloroform was evaporated in a stream of nitrogen gas, and the resulting lipid film was stored at −20°C until needed. To prepare calcein encapsulated LUVs, the lipid film was resuspended in buffer (10 mM Tris, 150 mM NaCl, 1mM EDTA, pH 7.4) containing 70 mM calcein. The lipid film was dislodged from the glass vial by vigorous vortexing, and the lipid suspension was subjected to five rounds of freezing and thawing in liquid nitrogen. LUVs were obtained using the Avanti mini-extruder apparatus by passing the lipid suspension 15 times through two 100 nm polycarbonate filters. Finally, calcein encapsulated LUVs were separated from free calcein dye by passing the mixture over a Sephadex G-50 size exclusion column. The lipid concentration in the collected LUV samples was determined using the Ames phosphate assay[20]. Calcein encapsulated LUVs (200 μl) were then diluted in buffer to a final lipid concentration of 10 μM, and were plated in 96 well flat-bottom microplates. Peptide (2 μl) was added to each well to achieve the desired peptide:lipid molar ratio. The samples were incubated at room temperature for ~20 min to allow the peptide and LUVs to equilibrate. Following incubation, the fluorescence emission intensity at 520 nm (490 nm excitation) was measured on a Synergy H1 microplate reader (BioTek, Winooski, VT). The fluorescence intensity resulting from the addition of 2 μl of 1% Triton X-100 or water to calcein LUVs served as positive and negative controls, respectively. Each sample was tested in at least duplicate for each LUV preparation, and the final results presented are the average of three individual experiments.
2.10 Circular Dichroism
Circular dichroism (CD) spectra were acquired on a Jasco J-810 spectropolarimeter (Jasco Inc., Easton, MD) located at the UBC Laboratory for Molecular Biophysics. Far-UV CD spectra were recorded between 190–250 nm using a scan speed of 100nm/min, a response time of 2 seconds and a bandwidth set to 1.0 nm. All samples were run at a final peptide concentration of 25 μM in a 1 mm pathlength quartz Suprasil cuvette (Hellma Analytics, Müllheim, Germany). The final spectra were the accumulated average of five individual scans. Spectra were recorded on peptide samples prepared in buffer (10mM TRIS, pH 7.4) or in the presence of 10 mM dodecylphosphocholine (DPC) or 25 mM sodium dodecyl sulphate (SDS) micelles. Additionally, CD spectra were acquired in the presence of small unilamellar vesicle (SUVs) composed of either POPC or POPS phospholipids at a final lipid concentration of 250 μM. SUVs were prepared by sonicating lipid suspensions and lipid concentrations were determined using the Ames phosphate assay[20]. Finally, spectra were recorded on peptide samples prepared in the presence of structure inducing, 2,2,2-trifluoroethanol (TFE). Raw CD data (in mdeg) was converted to mean residue ellipticity according to Kelly et al[21].
2.11 In Vivo Study
Adult female BALB/c mice were engrafted, in the mammary fat pad, with 4T1 mouse mammary carcinoma cells (1 × 106 cells). Groups of 5 mice were treated, by intraperitoneal (IP) injection, with saline, gemcitabine (60 mg/kg), Mastoparan (6 mg/kg), or gemcitabine and Mastoparan (60 and 6 mg/kg, respectively). Gemcitabine treatment began 5 d after tumour cell inoculation, and occurred every 7 days thereafter (Q7D), whereas Mastoparan treatment began 5 days after tumour cell inoculation, and occurred every 2 d thereafter (Q2D). Tumour size was determined by caliper measurements, and tumour volume was quantified using the equation (L*P2)/2, where L and P denote the longest tumour diameter, and the diameter perpendicular to the longest diameter, respectively. All mice were sacrificed once the first mouse reached its humane endpoint, at which point the tumours were weighed, and photographed.
2.12 Statistical Analyses
All data were analyzed using the unpaired Students t test, or one-way analysis of variance with the Bonferroni multiple comparison post-test, as appropriate.
Results
3.1 Mastoparan induced dose- and time-dependent cytotoxicity in cancer cells
A preliminary screen of 24 short cationic peptides identified Mastoparan as an ACP that was toxic to breast cancer cells but devoid of hemolytic activity (Supplementary Tables 1–3). Subsequent analyses showed that Mastoparan was cytotoxic to Jurkat human T cell acute lymphoblastic leukemia (T-ALL) cells, THP-1 human acute monocytic leukemia (AML) cells, and HOPC mouse myeloma cells (Figure 1A). Mastoparan was also toxic to MDA-MB-231, SKBR3, MDA-MB-468, and T47D human breast cancer cells, as well as 4T1 mouse mammary carcinoma cells (Figure 1B), in addition to prostate, cervical, and ovarian cancer cells (Supplementary Figure 1). In all cases, Mastoparan was nearly twice as potent to cancer cells of hematological origin than it was to cells of epithelial origin. Collectively, these data showed that Mastoparan was a broad-spectrum anti-cancer compound. In addition, trypan blue exclusion assays showed that Mastoparan was as toxic to Paclitaxel-resistant P-glycoprotein-overexpressing MCF7-TX400 cells[8] as it was to parental cells (Figure 1C), indicating that Mastoparan also possessed cytotoxic activity toward MDR cancer cells. It is noteworthy that MCF7-TX400 cells are very slow growing cells in comparison to MCF7 cells, with doubling times of approximately 5.5 and 1.5 days, respectively. Thus, Mastoparan-induced cell death did not depend on the proliferative capacity of the cell, suggesting that Mastoparan may be toxic to indolent or slow growing cancers.
Figure 1. Mastoparan kills several hematopoietic and non hematopoietic cancer cells, including slow growing and multidrug-resistant variants.

(A) Leukemia or myeloma cells, or (B) breast carcinoma cells were cultured in the presence or absence (vehicle control: water) of the indicated concentrations of Mastoparan for 24 h. Percent cytotoxicity was determined by MTT. Data shown represent the mean of 3 independent experiments ± SEM. (C) MCF7 and paclitaxel-resistant MCF7-TX400 cells were cultured in the presence or absence of Mastoparan for 24 h. Cell viability was assessed by Trypan Blue exclusion. All data is plotted as the the mean of 3 independent experiments ± SEM.
3.2 Mastoparan is more toxic to cancer cells than normal cells
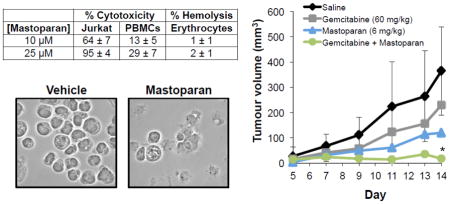
To determine whether Mastoparan was selectively toxic to cancer cells, Mastoparan-induced cytotoxicity was evaluated in normal human PBMCs, human erythrocytes, and primary HMECs. In all cases, Mastoparan was more toxic to cancer cells than it was to normal cells, suggesting that it was somewhat selective for the former (Table 1). To quantify differences in toxicity between cancer cells and normal cells, we calculated the ‘specificity factor’, herein defined to be the fold-difference in the IC50 between cancer cells and normal cells. The specificity factor ranged from 2–6 fold, depending on the cancer cell line, and the normal cell to which it was compared; the exception to this difference being when cancer cell killing was compared to hemolytic activity, owing to the fact that hemolysis never exceeded 15%, even at the highest concentration tested (100 μM). To confirm that reduced toxicity to normal human cells was not a consequence of delayed kinetics, a time course study was performed. As expected, Mastoparan-induced cytotoxicity peaked at 8 h in both primary PBMCs and Jurkat T-ALL cells (Figure 2). It should be noted at concentrations of Mastoparan that induced >50% cytotoxicity toward tumor cell lines, there was <2% lysis of human erythrocytes.
Table 1.
Mastoparan-induced cytotoxicity toward a panel of normal cells and cancer cells
| % Cytotoxicitya,c | % Hemolysisb,c | ||||
|---|---|---|---|---|---|
| [Mastoparan] | Jurkat | MDA-MB-231 | PBMCs | HMECs | Erythrocytes |
| 10 μM | 64 ± 7 | 20 ± 1 | 13 ± 5 | 16 ± 1 | 1 ± 1 |
| 25 μM | 95 ± 4 | 62 ± 2 | 29 ± 7 | 37 ± 7 | 2 ± 1 |
| 50 μM | 97 ± 1 | 96 ± 1 | 49 ± 3 | 67 ± 14 | 7 ± 1 |
Mastoparan-induced cytotoxicity was determined by MTT assay after 24 h.
Hemolytic activity toward human erythrocytes was determined by hemolysis assay after 4 h.
Data shown represent the mean of 3 independent experiments ± SEM.
Figure 2. Mastoparan-induced cell death peaks by 8 h in cancer cells and normal cells.

(A) Jurkat T leukemia cells or (B) normal PBMCs were cultured in the presence or absence of Mastoparan for the indicated periods of time. Percent cytotoxicity was calculated by LDH-release assay. Data shown represent the mean of 3 independent experiments ± SEM.
3.3 Mastoparan is a direct-acting (i.e., lytic) ACP
ACPs kill cells by a direct (i.e., lytic) or indirect (i.e., apoptosis-inducing) mechanisms[7]. Mastoparan triggered lactate dehydrogenase release from leukemia cells in a time-dependent manner (Figure 2), suggesting that Mastoparan functions as a direct-acting ACP. To test this hypothesis, we evaluated PI uptake to determine rapid changes in membrane integrity in response to Mastoparan treatment. Mastoparan caused rapid PI uptake in both Jurkat T-ALL cells (Figure 3A) and MDA-MB-231 breast cancer cells (Supplementary Figure 2A). In contrast to the apoptosis-inducing compound etoposide, Mastoparan did not cause DNA laddering in Jurkat cells (Figure 3B). TUNEL assays also failed to show DNA fragmentation in leukemia or breast cancer cells (data not shown). Moreover, Mastoparan-induced cell death was not reversed by the pan-caspase inhibitor BOC-D-FMK, which significantly reduced etoposide-mediated cytotoxicity (Figure 3C). Similar findings were noted in breast cancer cells (Supplementary Figure 1B). Finally, unlike etoposide, which caused apoptotic body formation in Jurkat T-ALL cells, Mastoparan treatment resulted in the formation of cellular phantoms, suggesting a lytic mechanism (Figure 3D). Similarly, Mastoparan exposure resulted in MDA-MB-231 cell lysis (Supplementary Figure 1C). Collectively, these data show that Mastoparan is a direct-acting ACP in both leukemia and breast cancer cells.
Figure 3. Mastoparan is a direct-acting AC P.

(A) Jurkat T-ALL cells were treated with vehicle (filled peak) or 25 μM Mastoparan (open peak) for 5 or 10 min. Membrane damage was assessed by propidium iodide (PI) uptake. Data shown are from a representative experiment (n=3). Values indicate average mean channel fluorescence (n=3). (B) Jurkat cells were cultured for 4 h as follows: (1) DNA ladder; (2) medium; (3) vehicle; (4) 50 μM etoposide, 4 h; (5) 50 μM etoposide, 24 h, (6) 0.1 μM Mastoparan, 4 h; (7) 10 μM Mastoparan, 4 h; (8) 25 μM Mastoparan, 4 h. After incubation, DNA was isolated and separated on a 0.8% agarose gel. (C) Jurkat cells pretreated with or without the pancaspase inhibitor BOC-D-FMK (40 μM) were cultured in the presence or absence of Mastoparan or etoposide. Cytotoxicity was assessed by MTT assay after 24 h. Data shown represent the mean of 3 independent experiments ± SEM, and are significant (* denotes p <0.05) by Student’s t test. (D) Jurkat cells cultured under the indicated conditions were fixed, stained with DAPI, and were visualized (40x) by UV microscopy. Data shown are from a representative experiment (n=3). Inset images were provided to highlight differences between treatment groups.
3.4 Mastoparan potency is significantly affected by C-terminal amidation
Our findings strongly suggest that Mastoparan is a direct-acting (i.e., lytic) ACP, which is in direct contrast to the recent report by de Azevedo et al[15] who showed, using many of the same cell lines used in this study, that Mastoparan is a less potent and indirect-acting ACP[15]. However, de Azevedo et al studied the anti-cancer properties of non-amidated Mastoparan (i.e., Mastoparan with a C-terminal carboxyl group; Mastoparan-COOH), whereas we studied the natural form of Mastoparan with a C-terminal amide that would neutralize the negative charge of the carboxyl (i.e., Mastoparan-NH2). To determine whether this charge difference explained the drastically different results, we compared the cytotoxic activity of Mastoparan-COOH to Mastoparan-NH2 and found that that the amidated form was indeed significantly more potent than the free carboxyl form of the peptide (Figure 4A). This finding demonstrated that end capping the C-terminus altered the anti-cancer potency of Mastoparan, and likely its mechanism of cancer cell killing. To test this hypothesis, we evaluated differences in the ability of Mastoparan-COOH and Mastoparan-NH2 to cause membrane leakage from LUVs. When LUVs were composed of 100% anionic lipid POPS, both Mastoparan-COOH and Mastoparan-NH2 caused membrane disruption (Figure 4B). In contrast, as the lipid composition became less negatively charged (i.e., greater proportion of zwitterionic lipid POPC), Mastoparan-NH2 induced calcein leakage was largely unchanged while leakage from vesicles in the presence of Mastoparan-COOH was significantly reduced (Figure 4B). Interestingly, Mastoparan-NH2 caused less leakage from pure POPC LUVs compared to POPS containing vesicles, suggesting that part of the selectivity of Mastoparan-NH2 towards cancer cells comes from an enhanced binding to anionic membranes over zwitterionic bilayers.
Figure 4. C-terminal amidation enhances Mastoparan potency.

(A) Jurkat T leukemia cells were cultured in the presence or absence of the indicated concentrations of Mastoparan-NH2 or Mastoparan-COOH for 24 h, at which point cytotoxicity was assessed using the MTT assay. Data shown represent the mean of 3 independent experiments ± SEM and are significant by the Student’s t test (* denotes p < 0.05, in comparison to Mastoparan-NH2-induced cytotoxicity). (B) Calcein encapsulated large unilamellar vesicles (LUVs) of varying composition were incubated with either Mastoparan-NH2 or Mastoparan-COOH at the indicated peptide:lipid ratios for 20 min. Mastoparan-mediated membrane leakage was assessed by measuring the release of calcein dye from LUVs after 20 minutes incubation with the peptide. Data shown represent the mean of 3 independent experiments ± SD. (C) Structural characterization of Mastoparan-NH2 and Mastoparan-COOH by CD spectroscopy. Spectra were acquired on 25 μM peptide samples in 10mM TRIS buffer (pH 7.4), as well as in the presence of 25 mM SDS or 10 mM DPC micelles or SUVs composed of either POPC or POPS phospholipids (lipid concentration of 250 μM). (D) The folding propensity of each peptide was also evaluated by recording CD spectra in the presence of increasing concentrations of TFE (% v/v).
Structural characterization of both forms of Mastoparan by CD spectroscopy revealed that both peptides adopted remarkably similar conformations to each other in buffer and in the presence of micelles or SUVs. The strong negative absorption bands observed at ~200nm revealed that both peptides were unstructured in buffer and in the presence of zwitterionic POPC SUVs (Figure 4C). The presence of SDS or DPC micelles or anionic SUVs composed of POPS phospholipids induced a conformational change in both Mastoparan peptides. The strong minima in the CD spectra at 208 and 222nm as well as the strong maxima at 190 nm for all these samples is characteristic of the peptides adopting an α-helical conformation (Figure 4C).
To further characterize the two Mastoparan peptides, we performed a titration with TFE, which is a known helix-inducing organic solvent[21], to assess whether there were differences in the helical propensities between Mastoparan-NH2 and Mastoparan–COOH (Figure 4D). Interestingly, the amidated form of Mastoparan more readily adopted a helical conformation with characteristic CD signals at ~190, ~208 and ~222nm appearing in spectra recorded on peptide samples prepared with 10% TFE. Conversely, these peaks do not become pronounced in the Mastoparan-COOH spectra until a concentration of 20% TFE was reached, and the intensity of these absorption bands was never as strong as those seen for Mastoparan-NH2.
Together, these data suggest that under physiologically-relevant conditions, C-terminal amidation enhances Mastoparan-mediated membrane lysis and promotes the folding of the peptide into a helical conformation resulting in a more potent, direct-acting ACP.
3.5 Mastoparan enhances killing by chemotherapeutic agents in vitro and in vivo
To assess the therapeutic potential of Mastoparan, we examined whether Mastoparan influenced the cytotoxic activity of other anti-cancer compounds. To this end, we evaluated whether Mastoparan, at its IC10 concentration (4.5 μM), affected etoposide- or vinblastine-induced killing of Jurkat T-ALL cells in vitro. Mastoparan acted as a chemosensitizing agent since low doses of Mastoparan reduced the EC50 of etoposide by approximately 2-fold (Figure 5A). In contrast, Mastoparan did not significantly alter vinblastine-mediated cytotoxicity (i.e., toxicity only increased by 10%; ΔIC50~1.2-fold; Figure 5B). To test whether Mastoparan enhanced the anti-tumour activities of chemotherapy in vivo, we used the 4T1 mammary carcinoma model to explore the potential synergistic relationship between Mastoparan and the antimetabolite gemcitabine in immune competent mice. Gemcitabine was selected for its ability to target myeloid-derived suppressor cells induced by 4T1 tumours grown in the mammary fat pad of BALB/c mice[22]. For this reason, we did not examine synergy between these two compounds in vitro where myeloid-derived suppressor cells were absent. As expected, gemcitabine-treated tumours were smaller than saline-treated tumours; however, the difference in tumour volume and mass were not statistically significant (Figure 6A–B). Similarly, in comparison to saline-treated mice, tumour volume and mass were reduced in Mastoparan-treated mice, although, once again, differences were not statistically significant. In contrast, tumour growth was significantly inhibited in mice that were treated with both gemcitabine and Mastoparan (Figure 6A–B). Indeed, the tumour volume of mice treated with Mastoparan and gemcitabine did not change significantly throughout the duration of the experiment (Figure 6A). Differences in tumour size were apparent in photographs of gross tumours (Figure 6C). Collectively, these data suggest that there may be a synergistic interaction between Mastoparan and gemcitabine in a mouse model of mammary carcinoma.
Figure 5. Mastoparan enhances Jurkat cell killing by etoposide, but not vinblastine.

Jurkat T-ALL cells were cultured in increasing concentrations of (A) etoposide or (B) vinblastine in the presence or absence of Mastoparan (4.5 μM; IC10). Percent cytotoxicity was determined by MTT assay after 72 h. Data shown represent the mean of 3 independent experiments ± SEM.
Figure 6. Mastoparan and Gemcitabine work synergistically in vivo.

4T1 mouse mammary carcinoma cells (1 × 106 cells) were injected into the mammary fat pads of BALB/c mice. Mice were injected (IP) with saline, gemcitabine (60 mg/kg), Mastoparan (6 mg/kg). or a combination of gemcitabine and Mastoparan (60 and 6 mg/kg, respectively). Gemcitabine and Mastoparan treatment began 5 days post injection and continued Q7D and Q2D, respectively. (A) Tumour volume was recorded by caliper measurements every other day until the first mouse reached its humane endpoint At the end of the experiment, the mice were sacrificed, and the tumours were removed, weighed (B), and photographed (C). Data shown in A and B represent the average of 5 mice ± StDev (In panel A, StDev is only shown for saline and Gemcitabine + Mastoparan to illustrate variability in relevant treatment groups), and are significant (* denotes p < 0.05, in comparison to saline-treated mice) by Bonferroni multiple comparison test.
Discussion
Here we demonstrated that the 14-residue wasp venom peptide Mastoparan is a broad-spectrum ACP that is toxic to leukemia, myeloma, and breast cancer cells. No differences in potency were observed between human and mouse cancer cell lines. Mastoparan was also toxic to slow growing and MDR cancer cells (i.e., MCF7-TX400), consistent with a mechanism that involves direct interactions with the cell membrane. As Mastoparan is equally toxic to rapidly dividing and slow growing cells, it may be active toward indolent and slow-growing tumours, giving it an advantage over conventional chemotherapies, which target rapidly dividing cells. Importantly, Mastoparan was significantly more toxic to cancer cell lines than to normal primary cells (selectivity index ~2–6-fold), suggesting that Mastoparan is somewhat selective for the former.
Direct-acting ACPs have several advantages over indirect-acting ACPs, including an ability kill MDR cancer cells, and enhance cell killing by chemotherapeutic agents[8,9]. Direct-acting ACPs exhibit potent anti-tumour activities toward primary tumours and their metastases at doses that will not cause harm to normal tissues[8,10]. Direct-acting ACPs can also induce anti-tumour immune responses that can be adoptively transferred to recipient mice[11,12]. Finally, as direct-acting ACPs are attracted to several different molecules rather than a unique receptor, resistance to direct-acting ACPs is unlikely to occur. Here, we showed that Mastoparan caused PI uptake, and LDH release from cancer cells. Mastoparan-induced cytotoxicity did not result in the formation of a DNA ladder, nor was cytotoxicity reduced in the presence of a broad-spectrum caspase inhibitor. Moreover, Mastoparan exposure did not cause chromatin condensation or apoptotic body formation in leukemia cells; rather, cytospin-isolated Mastoparan-treated cells showed the presence of cellular phantoms. In contrast, Mastoparan exposure significantly reduced the number of intact breast cancer cells, presumably due to cell lysis and cell loss following multiple rounds of washing. In all experiments, consistent findings were observed in breast cancer and leukemia cells. Collectively, these findings suggest that Mastoparan is a broad-spectrum direct-acting ACP.
Our finding that Mastoparan acts as a direct-acting ACP contradicted a previous report that showed that Mastoparan induces mitochondrial-dependent apoptosis in cancer cells[15]. Interestingly, this group tested some of the same cell lines that we assayed, but with very different results. For instance, de Azevedo et al[15], report an IC50 of 77.9 and 251.25 μM in Jurkat T-ALL cells and MDA-MB-231 breast cancer cells, respectively. In contrast, we noted IC50 values of 9.1 and 22 μM, respectively. However, de Azevedo et al evaluated the anti-cancer activity of Mastoparan-COOH, whereas we tested Mastoparan that was capped with a C-terminal amide (Mastoparan-NH2). Therefore, we synthesized both forms of Mastoparan and found that the potency of Mastoparan-COOH was similar to that reported by de Azevedo et al., but that Mastoparan-NH2 was indeed significantly more potent than Mastoparan-COOH. This finding is interesting since, to our knowledge, such a drastic difference in potency as a result of C-terminal amidation has not been reported for ACPs. Thus, the addition of the C-terminal amide not only promotes a direct mechanism of action, but also substantially decreased the amount of peptide required to cause cell death. We examined this concept using an artificial model membrane system. When LUVs were composed exclusively of POPS, we noted no differences between Mastoparan-COOH and Mastoparan-NH2, likely because cationic peptides can more easily disrupt vesicles with a strong negative charge. In contrast, as the lipid composition became more physiologically relevant (less anionic lipid), we noted that Mastoparan-NH2 was significantly more membranolytic. Additionally, CD spectroscopy revealed that Mastoparan-NH2 more readily adopted a helical conformation compared to the free carboxyl form of the peptide. Therefore, the differences in Mastoparan-mediated membrane lysis as well as increased propensity to adopt the active conformation of the peptide might thus explain why C-terminal amidation alters both potency as well as the mechanism of Mastoparan-mediated cytotoxicity. Consistent with this, da Silva et al recently found that C-terminal amidation of related but distinct Mastoparans promoted deeper peptide-membrane interactions[23].
To ascertain the clinical potential of Mastoparan, we tested whether Mastoparan alters cytotoxicity induced by other anti-cancer compounds. For this, we tested whether Mastoparan influences etoposide- and/or vinblastine-mediated killing of leukemia cells in vitro. To this end, we found that Mastoparan decreased the EC50 of etoposide by approximately 2-fold. This finding suggests that Mastoparan either works synergistically with etoposide, or that it may act as a chemo-sensitizing agent. In contrast, Mastoparan increased vinblastine-mediated cytotoxicity by only ~10%, corresponding to background levels of Mastoparan-induced cytotoxicity. These findings indicated that while Mastoparan does not work synergistically with vinblastine, it does not interfere with vinblastine-induced cell death.
We also determined whether Mastoparan altered the anti-tumour activities of chemotherapeutic agents in vivo. For this, we used the 4T1 mammary carcinoma model in BALB/c mice. The animal study was performed in immune-competent mice due to the clinical relevance of this model, and because certain direct-acting ACPs initiate anti-tumour immune responses in immune competent mice[11,12]. As Mastoparan is a direct-acting ACP, Mastoparan might also induce anti-tumour immune responses in tumour-bearing immune-competent mice. Although studying the putative immune-modulatory activities of Mastoparan in vivo was not an objective of these studies, we did not want to rule out this mechanism of tumour clearance when evaluating the anti-tumour potential of Mastoparan in vivo. However, 4T1 mammary carcinomas are highly aggressive and contain high numbers of myeloid-derived suppressor cells, which could have interfered with Mastoparan-induced tumour clearance in vivo[24]. Thus, we tested the potential synergistic relationship between Mastoparan and gemicitabine as the latter drug targets myeloid-derived suppressor cells in vivo[22]. We found that while tumours from mice treated with moderate doses of Mastoparan or gemcitabine appeared to be smaller, there was no significant difference in tumour volume or mass when mice were treated with these compounds alone at the specified doses. In contrast, the combination of both Mastoparan and gemcitabine significantly delayed tumour growth with differences being noted in both tumour volume and mass. Mastoparan and gemictabine were administered IP for this study and future studies will have to address full dose response curves and consider optimization of the dosing schedules for these drugs in this and other relevant tumour models. To the best of our knowledge, this is the first study to report a combination effect that suggests a synergistic relationship between an ACP and a chemotherapeutic agent in vivo. It is also the first to report efficacy of an ACP delivered via IP injection in a mouse model of mammary carcinoma. Thus, we can also conclude that this direct-acting ACP is able to cross the mammary-blood barrier in mice. Future studies should aim to determine the mechanism of synergy between these two compounds in vivo, and investigate whether Mastoparan induces anti-tumour immune responses in immune competent mice.
In summary, we have identified Mastoparan as a direct-acting ACP that exhibits broad-spectrum anti-cancer activities to hematological malignancies as well as breast carcinomas. We noted drastic differences in the potency and membrane disrupting abilities of Mastoparan-COOH and Mastoparan-NH2 that likely explain differences in their mechanism of action in vitro. Finally, we show here that Mastoparan works well when used in combination with chemotherapy in vivo; with results suggesting potential synergistic interactions. These findings highlight the therapeutic potential for the use of Mastoparan, and its derivatives, as a novel adjunctive therapy for the treatment of various cancers.
Supplementary Material
Highlights.
Mastoparan-NH2 is a broad-spectrum, direct-acting, anti-cancer peptide
Mastoparan potency and mechanism of action is related to its C-terminal amidation
Mastoparan enhances etoposide-induced cytotoxicity in vitro
Systemically-delivered Mastoparan and gemcitabine may work synergistically in vivo
Acknowledgments
This work was supported by the Canadian Institute for Health Research (CIHR) [grant reference number: MOP-74493]. The CD spectrometer was provided through grants from the Canada Foundation for Innovation and the Michael Smith Foundation to the Laboratory of Molecular Biophysics. A.L.H and E.F.H were recipients of CIHR postdoctoral fellowships, and A.L.H received postdoctoral support from the Michael Smith Foundation for Health Research (MSFHR). R.E.W.H is the recipient of a Canadian Research Chair.
Abbreviations
- ACP
Anti-cancer peptide
- AML
acute myeloid leukemia
- DMSO
dimethyl sulfoxide
- HMEC
human mammary epithelial cell
- IP
intraperitoneal
- LDH
lactate dehydrogenase
- LUV
large unilamellar vesicle
- MDR
multidrug-resistance
- PBMC
peripheral blood mononuclear cell
- PI
propidium iodide
- POPC
1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine
- POPS
1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-L-serine
- Q2D
every 2 days
- Q7D
every 7 days
- T-ALL
T cell acute lymphoblastic leukemia
Footnotes
Conflict of Interest
The authors declare that there are no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65:5–29. doi: 10.3322/caac.21254. [DOI] [PubMed] [Google Scholar]
- 2.Hoskin DW, Ramamoorthy A. Studies on anticancer activities of antimicrobial peptides. Biochim Biophys Acta. 2008;1778:357–375. doi: 10.1016/j.bbamem.2007.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Utsugi T, Schroit AJ, Connor J, Bucana CD, Fidler IJ. Elevated expression of phosphatidylserine in the outer membrane leaflet of human tumor cells and recognition by activated human blood monocytes. Cancer Res. 1991;51:3062–3066. [PubMed] [Google Scholar]
- 4.Koo CY, Sen YP, Bay BH, Yip GW. Targeting heparan sulfate proteoglycans in breast cancer treatment. Recent Pat Anticancer Drug Discov. 2008;3:151–158. doi: 10.2174/157489208786242278. [DOI] [PubMed] [Google Scholar]
- 5.Bafna S, Kaur S, Batra SK. Membrane-bound mucins: the mechanistic basis for alterations in the growth and survival of cancer cells. Oncogene. n.d;29:2893–2904. doi: 10.1038/onc.2010.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.van Beek WP, Smets LA, Emmelot P. Increased sialic acid density in surface glycoprotein of transformed and malignant cells--a general phenomenon? Cancer Res. 1973;33:2913–2922. [PubMed] [Google Scholar]
- 7.Hilchie AL, Hoskin DW. The application of cationic antimicrobial peptides in cancer treatment: laboratory investigations and clinical potential. In: Chakrabarty A, Rathore AS, editors. Emerging Cancer Therapy: Microbial Approaches and Biotechnological Tools. John Wiley & Sons, Inc; 2010. pp. 309–332. [Google Scholar]
- 8.Hilchie AL, Doucette CD, Pinto DM, Patrzykat A, Douglas S, Hoskin DW. Pleurocidin-family cationic antimicrobial peptides are cytolytic for breast carcinoma cells and prevent growth of tumor xenografts. Breast Cancer Res. 2011;13:R102. doi: 10.1186/bcr3043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Johnstone SA, Gelmon K, Mayer LD, Hancock RE, Bally MB. In vitro characterization of the anticancer activity of membrane-active cationic peptides. I. Peptide-mediated cytotoxicity and peptide-enhanced cytotoxic activity of doxorubicin against wild-type and p-glycoprotein over-expressing tumor cell lines. Anticancer Drug Des. 2000;15:151–160. [PubMed] [Google Scholar]
- 10.Hansel W, Enright F, Leuschner C. Destruction of breast cancers and their metastases by lytic peptide conjugates in vitro and in vivo. Mol Cell Endocrinol. 2007;260–262:183–189. doi: 10.1016/j.mce.2005.12.056. [DOI] [PubMed] [Google Scholar]
- 11.Berge G, Eliassen LT, Camilio KA, Bartnes K, Sveinbjornsson B, Rekdal O. Therapeutic vaccination against a murine lymphoma by intratumoral injection of a cationic anticancer peptide. Cancer Immunol Immunother. n.d;59:1285–1294. doi: 10.1007/s00262-010-0857-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Camilio KA, Berge G, Ravuri CS, Rekdal O, Sveinbjørnsson B. Complete regression and systemic protective immune responses obtained in B16 melanomas after treatment with LTX-315. Cancer Immunol Immunother. 2014;63:601–613. doi: 10.1007/s00262-014-1540-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hilchie AL, Conrad DM, Coombs MRP, Zemlak T, Doucette CD, Liwski RS, et al. Pleurocidin-family cationic antimicrobial peptides mediate lysis of multiple myeloma cells and impair the growth of multiple myeloma xenografts. Leuk Lymphoma. 2013;54:2255–2262. doi: 10.3109/10428194.2013.770847. [DOI] [PubMed] [Google Scholar]
- 14.Moreno M, Giralt E. Three valuable peptides from bee and wasp venoms for therapeutic and biotechnological use: melittin, apamin and mastoparan. Toxins (Basel) 2015;7:1126–1150. doi: 10.3390/toxins7041126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.de Azevedo RA, Figueiredo CR, Ferreira AK, Matsuo AL, Massaoka MH, Girola N, et al. Mastoparan induces apoptosis in B16F10-Nex2 melanoma cells via the intrinsic mitochondrial pathway and displays antitumor activity in vivo. Peptides. 2015;68:113–119. doi: 10.1016/j.peptides.2014.09.024. [DOI] [PubMed] [Google Scholar]
- 16.Nijnik A, Madera L, Ma S, Waldbrook M, Elliott MR, Easton DM, et al. Synthetic cationic peptide IDR-1002 provides protection against bacterial infections through chemokine induction and enhanced leukocyte recruitment. J Immunol. 2010;184:2539–2550. doi: 10.4049/jimmunol.0901813. [DOI] [PubMed] [Google Scholar]
- 17.Vellonen KS, Honkakoski P, Urtti A. Substrates and inhibitors of efflux proteins interfere with the MTT assay in cells and may lead to underestimation of drug toxicity. Eur J Pharm Sci. 2004;23:181–188. doi: 10.1016/j.ejps.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 18.Matsuzaki K, Murase O, Fujii N, Miyajima K. An antimicrobial peptide, magainin 2, induced rapid flip-flop of phospholipids coupled with pore formation and peptide translocation. Biochemistry. 1996;35:11361–11368. doi: 10.1021/bi960016v. [DOI] [PubMed] [Google Scholar]
- 19.Haney EF, Petersen AP, Lau CK, Jing W, Storey DG, Vogel HJ. Mechanism of action of puroindoline derived tryptophan-rich antimicrobial peptides. Biochim Biophys Acta. 2013;1828:1802–1813. doi: 10.1016/j.bbamem.2013.03.023. [DOI] [PubMed] [Google Scholar]
- 20.Ames BN. Academic press, editor. Methods in Enzymology. 1966. Assay of inorganc phosphate, total phosphate and phosphatases; pp. 115–118. [Google Scholar]
- 21.Kelly SM, Jess TJ, Price NC. How to study proteins by circular dichroism. Biochim Biophys Acta. 2005;1751:119–139. doi: 10.1016/j.bbapap.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 22.Le HK, Graham L, Cha E, Morales JK, Manjili MH, Bear HD. Gemcitabine directly inhibits myeloid derived suppressor cells in BALB/c mice bearing 4T1 mammary carcinoma and augments expansion of T cells from tumor-bearing mice. Int Immunopharmacol. 2009;9:900–909. doi: 10.1016/j.intimp.2009.03.015. [DOI] [PubMed] [Google Scholar]
- 23.da Silva AVR, De Souza BM, Dos Santos Cabrera MP, Dias NB, Gomes PC, Neto JR, et al. The effects of the C-terminal amidation of mastoparans on their biological actions and interactions with membrane-mimetic systems. Biochim Biophys Acta. 2014;1838:2357–2368. doi: 10.1016/j.bbamem.2014.06.012. [DOI] [PubMed] [Google Scholar]
- 24.Maenhout SK, Thielemans K, Aerts JL. Location, location, location: functional and phenotypic heterogeneity between tumor-infiltrating and non-infiltrating myeloid-derived suppressor cells. Oncoimmunology. 2014;3:e956579. doi: 10.4161/21624011.2014.956579. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.