

Abstract
Purpose
Methamphetamine (METH) abuse is a worldwide drug problem, yet no FDA-approved pharmacological treatments are available for METH abuse. Therefore, we produced an anti- METH single chain antibody fragment (scFv7F9Cys) as a pharmacological treatment for METH abuse. ScFv’s have a short half-life due to their small size, limiting their clinical use. Thus, we examined the pharmacokinetic effects of conjugating poly(ethylene) glycol (-PEG) to scFv7F9Cys to extend its functional half-life.
Methods
The affinity of scFv7F9Cys and PEG conjugates to METH was determined in vitro via equilibrium dialysis saturation binding. Pharmacokinetic and parameters of scFv7F9Cys and scFv7F9Cys-PEG20K (30 mg/kg i.v. each) and their ability to bind METH in vivo were determined in male Sprague-Dawley rats receiving a subcutaneous infusion of METH (3.2 mg/kg/day).
Results
Of three PEGylated conjugates, scFv7F9Cys-PEG20K was determined the most viable therapeutic candidate. PEGylation of scFv7F9Cys did not alter METH binding functionality in vitro, and produced a 27-fold increase in the in vivo half-life of the antibody fragment. Furthermore, total METH serum concentrations increased following scFv7F9Cys or scFv7F9Cys-PEG20K administration, with scFv7F9Cys-PEG20K producing significantly longer changes in METH distribution than scFv7F9Cys.
Conclusions
PEGylation of scFv7F9Cys significantly increase the functional half-life of scFv7F9Cys, suggesting it may be a long-lasting pharmacological treatment option for METH abuse.
Keywords: single chain variable fragments, pharmacokinetics, methamphetamine, poly(ethylene) glycol, drug abuse
Introduction
(+)-Methamphetamine (METH) abuse is a major drug problem in the United States today, as evidenced by a startling 51.5% increase in use between 2007 and 20011 (1,2). Furthermore, in 2005 METH abuse resulted in a total estimated economic burden of over $23 billion in the US, $351 million of which was due to health related costs (3). METH causes a variety of negative systemic effects including hypertension, stroke, and tachycardia. The central nervous system (CNS) is one of the major organ systems subjected to METH’s toxic effects. This can lead to various neurological consequences, including motor slowing, verbal learning impairment, and psychiatric symptoms (4,5). These METH-induced neurological symptoms are in part mediated by a release of dopamine, a down-regulation of the dopamine transporter (6,7), and by an increase in excitotoxic signaling pathways (8,9). Despite the great need for treating these negative medical effects, currently no FDA-approved pharmacological treatments are available for the medical consequences of METH abuse.
High-affinity anti-METH monoclonal antibodies (mAbs) are a potential pharmacological treatment option for METH abuse and toxicity. Anti-METH mAbs show efficacy in reducing METH brain concentrations (10) and cardiac effects (11) by altering the disposition of METH in rodent models of METH use. Antibody binding of METH in the systemic circulation causes redistribution of METH out of tissues and back into the bloodstream. This mechanism, called pharmacokinetic antagonism, moves METH away from its sites of action in the brain, thereby reducing the physiological and rewarding effects of the drug (12). While effective, the mAb format has some limitations, including potential reactions to non-host mAb’s (13). Although mAb7F9 has not been shown to be immunogenic, a smaller antibody fragment lacking the constant region may alleviate potential immune responses.
Therefore, our lab developed a single chain variable fragment (scFv) from the cDNA of a high-affinity anti-METH mAb. An scFv consists of the variable region of the original high-affinity mAb connected by an engineered short chain peptide sequence. Like a mAb, preclinical studies show an anti-METH scFv is capable of altering the disposition of METH in rodents (14). However, the short half-life of the scFv developed in our lab makes it less than ideal for the treatment of chronic conditions like METH addiction, plus it may retain some potential for in vivo immunogenic responses.
Poly(ethylene) glycol (PEG) was first introduced as an immunoprotective agent in 1977, when it was conjugated to bovine serum albumin (15). Since then, PEG has been shown to improve pharmacokinetic properties (e.g. lengthen in vivo half-life), block enzymatic degradation, reduce immunogenicity, and increase the overall safety of many therapeutics (16–18). Due to its association with water molecules, PEG has a larger apparent size in vivo than its actual molecular weight (17). This increase in size reduces renal clearance, thus prolonging the in vivo half-life of the PEG conjugate. Furthermore, the water molecules create a protective shell around the conjugated protein reducing the availability of the protein for enzymatic degradation (17). However, in some studies PEG has been shown to result in vacuolization in both phagocytic and non-phagocytic cells (19). Formation of these vacuoles has not yet been associated with any significant toxicities (19), suggesting further studies are needed to determine if this is a toxic side effect. Despite the formation of vacuoles, PEG conjugation has been employed in the development of multiple FDA-approved drugs, such as Nulasta® and Pegasys® (20–22). Additionally, studies examining the PEGylation of a bacterial anti-cocaine catalytic enzyme (CocE) found a reduced immunogenicity of the bacterial enzyme, as well as protection from a lethal dose of cocaine in vivo (23). Here we will discuss the first example of PEGyation of an anti-METH biologic as a potentially safe and long-lasting treatment for METH abuse.
PEGs are commonly modified by the addition of a functional group, which allows for site-specific conjugations. With this in mind, our scFv (scFv7F9Cys) was designed with a terminal cysteine residue, providing a site for conjugation to a PEG moiety containing a maleimide functional group. By conjugating PEG to scFv7F9Cys our goal was to create an anti-METH therapeutic with an extended half-life more conducive to relapse-prevention treatment rather than overdose. In this manuscript we report the conjugation, purification, and functional properties, both in vitro and in vivo, of scFv7F9Cys and its PEGylated conjugates.
Additionally, we preliminarily investigated the effects of administering scFv7F9Cys and one PEG conjugate, scFv7F9Cys-PEG20K on the effects on protein expression of six METH neuroadaptation markers in the brain. METH-induced neuroadaptation can be observed at the vesicular monoamine transporter 2 (VMAT2), tyrosine hydroxylase (TH) and the dopamine transporter (DAT), ultimately reducing VMAT2 (24,25) and TH (26) function and striatal DAT expression (27) or membrane trafficking (28). Furthermore, a downstream effect of METH-induced dopaminergic toxicity is activation of the glutamatergic system leading to excitotoxicity (29), via upregulation of the α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) and n-methyl-d-aspartate (NMDA) receptors (30). While to our knowledge there are no studies examining the effects of METH on the scaffolding protein, post-synaptic density 95 (PSD-95), it is known to associate with both the AMPA and NMDA receptors (31,32). Additionally, one group (33) found that knocking out PSD-95 function in mice elicited over-activation of the dopaminergic and glutamatergic systems in the presence of cocaine, suggesting that PSD-95 may play a critical role in suppressing neurotoxicity of stimulants, like METH. Therefore, we chose to examine the protective effects of scFv7F9Cys and its PEGylated conjugate, on three markers from the dopaminergic system, DAT, TH, and VMAT2, and the glutamatergic system, NR2A (NMDA subunit), AMPA (GluR2), and PSD-95. We hypothesize that PEGylation of scFv7F9Cys will increase its half-life and provide an increase protection from the neurological effects of METH.
Methods
Chemicals and Drugs
(+)-[2′,6′-3H(n)]Methamphetamine (3H-METH) (39 Ci/mmol) was synthesized at the Research Triangle Institute (Research Triangle Park, NC) and supplied by the National Institute on Drug Abuse (Bethesda, MD). Five, 20, and 40 kDa methoxy-poly (ethylene glycol)-maleimide (Laysan Bio, Inc., Arab, Alabama) were used in scFv-PEG conjugations. All other chemicals were obtained from Fisher Scientific (Fair Lawn, NJ) unless otherwise noted.
Animals
All studies were performed in accordance with the Guide for Care and Use of Laboratory Animals and approved by the UAMS Institutional Animal Care and Use Committee (IACUC). Adult male Sprague-Dawley rats (275–320g) with dual jugular vein catheters surgically implanted were purchased from Charles River (Wilmington, MA). Rats were housed by the UAMS Division of Laboratory Animal Medicine (DLAM) and given ad libitum access water. Food was controlled to maintain animals below 350g in weight.
Cloning and Production of scFv7F9Cys
Murine mAb7F9 was developed as described previously (34,35). As discussed in past reports from this laboratory (14,36), the variable region sequences were organized in a VH-linker-VL order to convert the two heavy and light chain sequences of mAb7F9 to scFv7F9Cys. Sequence elements for a 6-histidine tag and a free cysteine were engineered at the carboxy terminus for purification and site-specific conjugation, as described (14,36). cDNA encoding scFv7F9Cys was synthesized by GenScript (Piscataway, NJ) and cloned into a pUC57 vector (14,36,37). For maintenance the plasmids were transformed into E. coli strain DH5α (Invitrogen, Carlsbad, CA). Plasmids were isolated and tested for fidelity by DNA sequencing (University of Arkansas for Medical Sciences DNA Sequencing Core Facility, Little Rock, AR). Once sequences were confirmed, the plasmids were sent to Catalent Pharma Solutions (Madison, WI), where scFv7F9Cys cDNA was integrated into CHO cells using Catalent’s GPEx retroviral system. Once virus-free, stable cell lines were confirmed, and cells were preserved at −80C for subsequent protein production.
Conjugation of scFv7F9Cys to PEG
ScFv7F9Cys was engineered with a terminal cysteine located away from the METH binding pocket. This served as the conjugation site for PEG functionalized with a maleimide group. ScFv7F9Cys was incubated with a three-fold molar excess TCEP reducing agent (Thermo, Rockford, IL), and subjected to shaking at 22°C for two hrs. Reduced scFv7F9Cys was subsequently incubated with a molar excess of a 5 kDa, 20 kDa, or 40kDa methoxy-poly (ethylene glycol)-maleimide (PEG) shaking at 22°C for two hrs. Samples were analyzed by SDS-PAGE on a 4–12% Bis-Tris gradient polyacrylamine gel (Life Technologies) in MES buffer (Life Technologies). Protein was detected with a Coomaisse-based stain, GelCode Blue (Thermo).
Purification of scFv7F9Cys-PEG20K & -PEG40K
Once conjugated, PEG conjugates were purified using an AKTA Explorer 100 FPLC and a HiTrap 1 mL IMAC FF column (GE Healthcare, Piscataway, NJ). The column was equilibrated with five column volumes of buffer with a low imidazole concentration (20mM NaPO4, 500mM NaCl, 10mM Imidazole). ScFv7F9Cys-PEG20K or scFv7F9Cys-PEG40K was eluted using a five-step gradient (5, 10, 15, 20, 100%) of elution buffer (20mM NaPO4, 500mM NaCl, 500mM Imidazole). All collected fractions were analyzed by SDS-PAGE. Fractions containing the purified PEGylated conjugate were pooled together for future studies.
Determination of METH Binding Affinity
METH binding affinity was determined as described previously (36). Briefly, a teflon reusable base plate (Thermo, Rockford, IL) with rapid equilibrium dialysis (RED) devices was used (Thermo, Rockford, IL). Saturation binding assays were performed to determine the affinity of scFv7F9Cys, scFv7F9Cys-PEG20K, and scFv7F9Cys-PEG40K. Specific binding and non-specific binding were measured with increasing concentrations of 3H-METH and a constant concentration (as determined via a titration assay) of scFv7F9Cys, scFv7F9Cys-PEG20K, or scFv7F9Cys-PEG40K. Non-specific binding was measured using a molar excess of non-radiolabeled METH (10mM). Samples were incubated overnight at room temperature, and shaken gently before being placed in scintillation fluid, vortexed for 30 sec, and analyzed by liquid scintillation spectrophotometry.
Radiolabeling Therapeutic Proteins for Pharmacokinetic Tracers
ScFv7F9Cys-PEG20K (500 μg) was buffer exchanged and concentrated using Pierce Concentrators (9K MWCO, Thermo, Rockford, IL) into 0.1M borate buffer (0.1M Sodium Tetraborate, 0.5M NaCl, pH 8.5). ScFv7F9Cys was buffer exchanged in the same manner into administration buffer (150mM NaPO4, 20mM NaCl, pH 7.5). Five hundred microcuries of 3H-N-succinimidyl-[2,3-3H]propionate (3H-NSP) was placed in a siliconized glass tube and dried under a constant stream of nitrogen gas. After 3H-NSP was completely dried, therapeutic proteins were vortexed with the dried 3H-NSP and mixed on ice for one hr (scFv7F9Cys-PEG20K) or two hrs (scFv7F9Cys). 3H-NSP-labeled scFv7F9Cys and scFv7F9Cys-PEG20K were separated from unreacted 3H-NSP using a Sephadex G-25 column (GE Healthcare). Each collected fraction was analyzed for radioactivity in Scintiverse scintillation fluid using a liquid scintillation spectrophotometer. Fractions that contained radioactivity, and were eluted at levels corresponding to each protein’s size were pooled then dialyzed (Slide-A-Lyzer Dialysis Cassette, Thermo) into administration buffer (pH 6.4). Rats received an intravenous dose of scFv7F9Cys or scFv7F9Cys-PEG20K containing a radioactive tracer of 5 x 106 DPM of 3H-NSP-scFv7F9Cys or 10 x 106 DPM of 3H-NSP-scFv7F9Cys-PEG20K for pharmacokinetic studies.
Pharmacokinetic Studies of scFv7F9Cys and scFv7F9Cys-PEG20K
Two days prior to protein administration, a two-week osmotic pump (Alzet, Cupertino, CA) delivering a constant infusion of 3.2 mg/kg/day of METH or saline was implanted subcutaneously in the mid-back of the rats. Twenty-four hrs after implantation a blood sample was drawn from the right jugular vein catheter to determine the baseline serum concentration of METH. At this time, rats were housed in metabolic cages. ScFv7F9Cys (30 mg/kg plus radioactive tracer), scFv7F9Cys-PEG20K (30 mg/kg plus radioactive tracer) or a volume equivalent of saline was administered through the left jugular vein catheter. A total of four groups were studied: saline+saline, saline+METH, scFv7F9Cys+METH, and scFv7F9Cys-PEG20K+METH. A blood sample (approximately 150 μl) was taken from the right jugular vein catheter prior to protein administration then at 1, 5, 10, 30 mins, 1, 2, 4, 8, 24, 48, and 72 hrs after therapeutic protein administration for scFv7F9Cys-PEG20K and saline groups. Blood sampling was stopped after the 2-hr time point for the scFv7F9Cys group because scFv7F9Cys concentration had returned to baseline levels as determined by the level of radioactive tracer remaining. Hematocrit values were measured at two time points on day zero, prior to administration of the protein dose and four hrs after protein administration. Rats were sacrificed by decapitation under isoflurane anesthesia. Samples were stored at −80°C until analysis was performed.
Due to sampling limitations, two groups of rats were used for scFv7F9Cys, and two groups were used for scFv7F9Cys-PEG20K (n=3 per group). The first groups of scFv7F9Cys or scFv7F9Cys-PEG20K received a radioactive tracer in addition to the 30 mg/kg protein dose and serum samples collected were used for therapeutic protein concentration analysis. The second groups of scFv7F9Cys or scFv7F9Cys-PEG20K were administered 30 mg/kg of therapeutic protein without a radioactive tracer. Serum samples collected from this group were analyzed for METH concentrations.
Pharmacokinetic Analysis of Total METH
METH serum concentrations were analyzed using LC/MS-MS. Solid-phase extraction (SPE) columns (Phenomenex, Torrece, CA), placed on a 24-sample manifold (Phenomenex) were conditioned with methanol and then loading buffer (100mM NaPO4, pH 8.1). After the loading buffer had eluted by gravity, stops were closed on the manifold and loading buffer was added to each column. Next, standards and serum samples were vortexed with 0.2% formic acid in LCMS grade water. Loading buffer containing 1 μg/mL of internal standard was added to each sample. Samples were diluted to between 3 and 25-fold in normal rat serum (PEL-FREEZ, Rogers, AR) to ensure sample was within the generated standard curve. METH standards (stock from Cerilliant, methamphetamine: #M-004, #M-009; amphetamine: #A-019, #A007) used were 0.3, 1, 3, 10, 30, 100, 300, 100, 2000, ng/mL and quality control standards were 3, 10, and 800 ng/mL. Prepared serum samples were then added to the corresponding SPE column and washed with methanol. The columns were dried for 10 mins under vacuum at 9 mPa.
METH was eluted from the SPE columns with four washes of elution buffer (90 mL Acetonitrile, 5 mL NH4OH, and 5 mL THF) into 0.5 N HCl. The columns were dried under vacuum (less than 5 mPa) to ensure recovery of the METH sample. Eluted samples were then dried under a constant stream of N2 at 55°C. Dried samples were reconstituted with 0.1% formic acid in water (LCMS grade), vortexed, and shaken for 30 mins. Samples were centrifuged at 14000 rpm for 5 mins. After centrifugation, samples were transferred to injection vials with pre- slit caps for analysis.
Total METH concentrations (bound plus free) of samples were determined using an Acquity Ultra Performace Liquid Chromatography system and Quattro Premier XE mass spectrometer (Waters Corporation, Milford, MA). Samples were first injected onto an Acquity UPLC BEH C18 1.7 μm column (Waters Corporation, Milford, MA) maintained at 40°C. Samples were separated at a flow rate of 0.3 ml/min with reverse phase chromatography and a linear binary gradient (mobile phase A: 0.1% (v/v) formic acid; mobile phase B: 0.1% (v/v) formic acid in acetonitrile). The column was coupled to an electrospray ionization probe, operating in the positive ion mode. Positive ions for METH and METH-D5 were generated at cone voltages of 18 and 21 volts, respectively. Product ions METH and METH-D5 were generated using argon collision induced disassociation at collision energy of 15 and 21 eV while maintaining a collision cell pressure of 3 x 10−3 torr. Detection was achieved in the multiple-reaction-monitoring (MRM) mode using the precursor→product ions, m/z 149.8→90.8 and 155.2→91.5 for METH and METH-D5 respectively. The lower and upper limit of quantification for METH was 1 and 2000 ng/ml, respectively. All predicted values for standards were within ± 20% of the actual value.
Brain Collection and Preparation
Whole brains were harvested from rats following sacrifice by decapitation under isofluorane anesthesia, then flash frozen in liquid nitrogen and stored at −80°C until processed for qRT-PCR and Western blot analysis. In order to measure both mRNA expression and protein concentration from the same tissue, whole brain samples were pulverized using a large Bessman Tissue Pulverizer (Spectrum Laboratories Inc., Rancho Dominguez, CA). The tissue pulverizer was first cooled in liquid nitrogen. The frozen brain tissue sample was then placed in the pulverizer and the pestle placed on top. The tissue was then pulverized with a lead hammer until it was a powder. The powder was transferred to a 15 mL conical tube and placed at −80°C until later use.
Protein Detection from Brain Tissue
Homogenized brain tissue (approximately 10 mg) was placed in a 1.5 mL eppendorf tube with 500 μl of RIPA buffer (Thermo). Samples were homogenized with a pellet pestle motor and polypropylene pellet pestles, and incubated on ice for 30 mins. After incubation, homogenized tissue samples were sonicated on ice to further break up tissues. Each sample was sonicated for a total of five mins, in a cycle of 10 secs sonication then 10 secs off. Following sonication, the samples were centrifuged at 10,000 x g for 20 mins, and the supernatant collected. The protein concentration was determined via UV280 absorbance. Samples were stored at −80°C until analysis.
Protein concentrations were determined via western blot analysis. Briefly, 150 μg of total protein was loaded per lane of a 4–12% bis-tris gradient polyacrylamide gel (Life Technologies, Carlsbad, CA) for 40 mins at 200V in 1 x MES buffer (Invitrogen, Carlsbad, CA). Protein was then transferred to a nitrocellulose membrane for one hr at 100V in transfer buffer (20% v/v methanol, 192 mM Glycine, 25mM Tris Base). Following transfer, the nitrocellulose membrane was blocked in TBS-T with 5% w/v evaporated milk for one hr. The membrane was then incubated with primary antibody (in TBS-T, 5% w/v BSA) for one hr 25°C (anti-PSD-95, 1:1,000 dilution, NeuroMab, Davis, CA; anti-NR2A, 1:5,000 dilution, Abcam, Cambridge, MA), two hrs at 25°C (anti-VMAT2 [HRP], 1:10,000 dilution, Novus Biologicals, Littleton, CO), or overnight at 4°C (anti-DAT, 1:1,000 dilution, Sigma Aldrich, St. Louis, MO; anti-TH, 1:1,500 dilution, Millipore, Temecula, CA; anti-GluR2, 1:1,000 dilution, NeuroMab; anti-GAPDH, 1:1,000 dilution, Cell Signaling Technology, Danvers, MA). Following incubation in primary antibody, the nitrocellulose membrane was washed three times for 10 mins each in TBS-T. The blot was then incubated with HRP-conjugated secondary antibody (in TBST, 0.2% w/v BSA) for one hr (Goat-anti-rabbit IgG HRP, 1:5,000 dilution, Thermo; Goat-anti-mouse, 1:100 dilution, Thermo) followed by three 10-min washes in TBS-T. Secondary antibody was detected using Immunobilon Western Chemiluminescent HRP Substrate (Millipore).
Data Analysis & Statistics
All values are shown as the average +/− the standard error of the mean (SEM). Saturation binding data and all statistical comparisons for pharmacokinetic data were performed using GraphPad PRISM 6.0d software. Statistical comparisons were determined using unpaired t-tests. Pharmacokinetic data were analyzed via non-compartmental analysis using Phoenix WinNonLin software. ImageJ software was used for a densitometric analysis of western blots. Western blot data statistical analysis was completed using GraphPad PRISM 6.0d software, and all groups were normalized to the housekeeping gene, glyceraldehyde 3-phosphate dehydrogenase (GAPDH), for analysis. Statistical comparisons were determined using one-way ANOVAs with a Tukey post-hoc test.
Results
ScFv7F9Cys was conjugated to three separate poly(ethylene) glycol moieties: PEG5K, PEG20K, and PEG40K. PEGylation was analyzed using SDS-PAGE (Figure 1). SDS-PAGE gels were stained with Gel Code Blue for protein analysis. Purified scFv7F9Cys is shown in the first lane in Figure 1. Incubation of reduced scFv7F9Cys with PEG resulted in higher molecular weight bands for each conjugate: scFv7F9Cys-PEG5K (~35 kDa), scFv7F9Cys-PEG20K (~70 kDa), and scFv7F9Cys-PEG40K (~100 kDa) suggesting all three PEGs were conjugated to scFv7F9Cys. The lower band seen in conjugation lanes (~25 kDa) is unreacted scFv7F9Cys.
Figure 1.
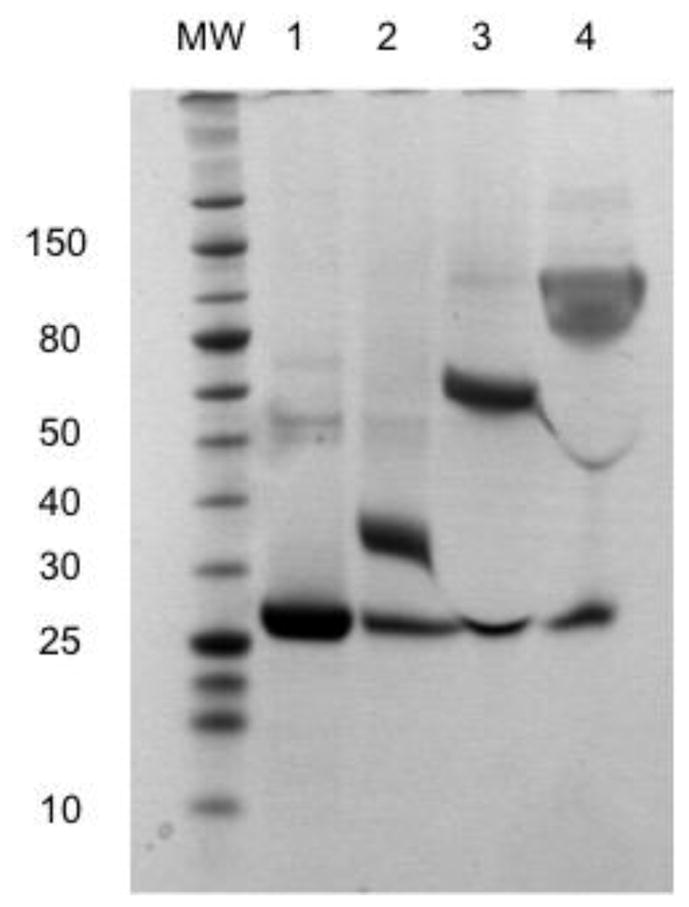
Conjugation of scFv7F9Cys to a linear 5 kDa (lane 2), linear 20 kDa (lane 3), and branched 40 kDa PEG (lane 4). ScFv7F9Cys was incubated with a 20-fold excess of PEG. Each PEG used contained a maleimide functional group that interacted with the free sulfhydryl group of scFv7F9Cys to form a permanent scFv7F9Cys-PEG20K product.
ScFv7F9Cys-PEG5K purification was tested via three different methods: immobilized metal ion affinity chromatography (IMAC), anion exchange chromatography (AEX), and size exclusion chromatography (SEC). However, analysis of each method via SDS-PAGE showed incomplete and inadequate purification of scFv7F9Cys-PEG5K from scFv7F9Cys (data not shown). As such, scFv7F9Cys-PEG5K was eliminated as a possible drug candidate and not tested further (Table I).
Table I.
Development and testing of scFv7F9Cys PEGylated conjugates.
| PEGylation | Purification | METH Binding | |
|---|---|---|---|
| scFv7F9Cys | - | - | 8.7 nM |
| scFv7F9Cys-PEG5K | Yes | No | - |
| scFv7F9Cys-PEG20K | Yes | Yes | 8.0 nM |
| scFv7F9Cys-PEG40K | Yes | Yes | 67.1 nM |
ScFv7F9Cys-PEG20K was purified using a five-step elution profile over an IMAC column (Figure 2A & 2B). SDS-PAGE analysis showed that at low imidazole concentrations, scFv7F9Cys-PEG20K eluted from the column, while high imidazole concentrations were necessary for elution of unreacted scFv7F9Cys. We believe PEG20K sterically hindered how tightly scFv7F9Cys-PEG20K bound to the stationary phase of the column, and thus eluted with a reduced imidazole concentration compared to that necessary for scFv7F9Cys to elute. Fractions containing pure scFv7F9Cys-PEG20K were pooled and used for binding studies.
Figure 2.

IMAC purification of scFv7F9Cys–PEG20K. A nickel affinity column was used. ScFv7F9Cys contains a poly-histidine tag which will bind the nickel on the column. Any bound protein was eluted using imidazole increasing in concentration over 6 steps (0, 5, 10, 15, 20, and 100%). A. Chromatogram of purification: solid line shows UV280 peaks, and dashed line shows the percent of imidazole. B. SDS-PAGE analysis of fractions from IMAC purification. Bars 1, 2, and 3, correspond to fractions analyzed from same section of the chromatogram. C. SDS-PAGE analysis of scFv7F9Cys-PEG40K IMAC purification.
ScFv7F9Cys-PEG40K purification was performed using IMAC (Figure 2C). Similar to scFv7F9Cys-PEG20K, scFv7F9Cys-PEG40K was purified using a five-step elution profile over an IMAC column. Early fractions collected contained a small portion of purified scFv7F9Cys-PEG40K, while later fractions contained the bulk of product with unreacted scFv7F9Cys, as analyzed by SDS-PAGE. Fractions containing purified scFv7F9Cys-PEG40K were pooled for saturation binding studies.
Saturation binding assays were used to determine the affinity of scFv7F9Cys and scFv7F9Cys-PEG20K for METH (Figure 3A). ScFv7F9Cys was found to have a Kd of 8.7 nM. The Kd (8.0 nM) determined for scFv7F9Cys-PEG20K was not significantly different from scFv7F9Cys. Conversely, scFv7F9Cys-PEG40K had a significantly higher Kd of 67.1 nM, suggesting this conjugate was not able to bind METH as tightly as scFv7F9Cys or scFv7F9Cys-PEG20K (Figure 3B). Therefore, scFv7F9Cys-PEG40K was not continued for pharmacokinetic studies, as scFv7F9Cys and scFv7F9Cys-PEG20K were (Table I).
Figure 3.

Saturation binding of scFv7F9Cys and scFv7F9Cys-PEG20K (A), and scFv7F9Cys and scFv7F9Cys-PEG40K (B). A constant concentration of therapeutic protein was incubated with increasing concentrations of 3H-METH. Binding was measured using scintillation counting. Both scFv7F9Cys and scFv7F9Cys-PEG20K were found to have similar KD values for METH, with scFv7F9Cys having a KD of 8.7 nM, while scFv7F9Cys-PEG20K displayed a KD of 8.0 nM. However, scFv7F9Cys-PEG40K did not reach saturation, and was found to have significantly weaker METH binding, with a KD of 67.1 nM. While scFv7F9Cys-PEG40K was capable of binding a higher concentration of METH, we believe that much of the observed binding is not specific interactions with the larger PEG moiety. Open circles correspond to scFv7F9Cys, closed diamonds correspond to scFv7F9Cys-PEG20K, and closed circles correspond to scFv7F9Cys-PEG40K. Error bars are SEM.
To determine the half-life of both scFv7F9Cys and scFv7F9Cys-PEG20K a series of intravenous pharmacokinetic studies (Figure 4) were performed. Based on model-independent analysis of serum concentration-time data of both therapeutic proteins, scFv7F9Cys had an in vivo half-life of approximately 0.9 hrs (~55 mins), and the addition of PEG significantly extended the half-life of scFv7F9Cys by 28-fold to 26 hrs (Figure 5). This extension in half-life was due to a significant 42-fold decrease in systemic clearance of the PEGylated conjugate (4 ml/hr*kg) versus the native single chain (165 ml/hr*kg) (Table II). Additionally, the volume of distribution was decreased slightly for scFv7F9Cys-PEG20K (164 ml/kg) compared to scFv7F9Cys (270 ml/kg). A 43-fold increase in scFv7F9Cys-PEG20K total area under the serum concentration time curve (AUC) (7766428 ng*hr/ml) compared to scFv7F9Cys (182723 ng*hr/ml) was also found.
Figure 4.

Timeline of pharmacokinetic studies. An osmotic pump delivered 3.2 mg/kg/day of METH throughout the course of the study. Prior to administration of the therapeutic protein dose, a blood sample was taken to establish a baseline. ScFv7F9Cys-PEG20K was administered on day 0. Blood samples were taken at 1, 5, 10, 30 mins, 1, 2, 4, 8, 24, 48, 72, and 96 hrs after the protein dose administration. Animals were sacrificed on day 4 of the study.
Figure 5.

Pharmacokinetic analysis serum concentrations of A. scFv7F9Cys and B. scFv7F9Cys-PEG20K. Both therapeutic proteins were labeled with 3H-NSP. ScFv7F9Cys was found to have a half-life of approximately 55 mins, while scFv7F9Cys-PEG20K displayed a 27-fold increase in half-life (25 hrs). There was a 46-fold decrease in Cls with conjugation to PEG (4 ml/hr*kg) compared to scFv7F9Cys (201 ml/hr*kg). Data points are means + SEM. Line displays data points in the terminal elimination phase that were used to calculate half-life.
Table II.
Summary of scFv7F9Cys and scFv7F9Cys-PEG20K pharmacokinetic properties.
| scFv7F9Cys | scFv7F9Cys-PEG | Fold Change | p-value | |
|---|---|---|---|---|
| AUC (ng*hr/ml) | 181672 ± 10848 | 7721296 ± 555294 | 43 | <0.0001 |
| Cls (ml/hr*kg) | 201 ± 9 | 4 ± 0.3 | −46 | <0.0001 |
| Vdbeta (ml/kg) | 270 ± 27 | 164 ± 40 | −2 | <0.05 |
| t½ (hrs) | 0.9 ± 0.08 | 25 ± 0.005 | 27 | <0.0001 |
Changes in METH distribution due to scFv7F9Cys and scFv7F9Cys-PEG20K were also examined (Figure 6). Serum concentrations of total METH (bound plus unbound) were measured using liquid chromatography-tandem mass spectrometry (LC/MS-MS). ScFv7F9Cys increased METH serum concentrations significantly compared to METH controls, but concentrations returned to baseline by the 2-hr time-point. ScFv7F9Cys-PEG20K increased METH serum concentrations significantly over the METH control group through the 48-hr time-point. ScFv7F9Cys-PEG20K increased serum METH concentrations significantly longer than scFv7F9Cys, suggesting that scFv7F9Cys-PEG20K is more efficacious than scFv7F9Cys in favorably altering METH disposition. Previous anti-METH immunotherapy studies have shown a decrease in METH organ and brain concentrations with mAb treatment, and a corresponding increase in METH serum concentrations (38,39). We did not measure brain concentrations, but previous studies suggest that the increases in METH serum concentration observed in this study correspond to decreases in brain METH concentrations (10,38).
Figure 6.

Analysis of METH disposition in the serum in the presence of saline, scFv7F9Cys, or scFv7F9Cys-PEG20K. Data points are means + SEM. A. Total METH serum concentrations over the first 48 hrs of the study. By 2 hrs scFv7F9Cys had returned to baseline, and scFv7F9Cys-PEG20K has reached baseline by 48 hrs. Inset shows METH serum concentrations through the first two hours of the study. Closed diamonds – scFv7F9Cys-PEG20K, open circles – scFv7F9Cys, X – saline control. B. Partial AUC0→2hrs suggested scFv7F9Cys-PEG20K increased total METH concentrations in the serum significantly higher than scFv7F9Cys (p<0.05). However, both scFv7F9Cys (AUC0→2hrs, p<0.005) and scFv7F9Cys-PEG20K (AUC0→2hrs, p<0.005) increased METH serum concentrations significantly compared to saline controls. C. ScFv7F9Cys-PEG20K increased serum METH concentrations significantly higher than saline controls (AUC0→48hrs, p<0.001).
The in vivo activity of both scFv7F9Cys and scFv7F9Cys-PEG20K were calculated by comparing molar concentrations of each therapeutic protein to METH molar concentrations (Figure 7). Overall, the total METH serum concentration was approximately 44% of the scFv7F9Cys serum concentration (Figure 7A). Meanwhile, in the PEGylated conjugate group, total METH serum concentrations were an average of 29% of the therapeutic protein concentration (Figure 7B). Although some METH binding functionality was apparently lost in vivo with the PEGylated conjugate, the AUC0→2hrs of METH concentrations for the scFv7F9Cys-PEG20K group was calculated to be 935 ng*hr/ml, and AUC0→2hrs of the METH serum concentrations was determined to be 340 ng*hr/ml for the scFv7F9Cys group. Therefore, over the first two hrs following i.v. administration, scFv7F9Cys-PEG20K was able to redistribute more METH back into the serum than scFv7F9Cys despite binding less METH overall. Furthermore, scFv7F9Cys-PEG20K was functional through 48 hrs with an AUC0→48hrs value of 4326 ng*hr/ml, showing a 12.7-fold increase in total METH binding activity compared to scFv7F9Cys (active through approximately two hrs). Hence, scFv7F9Cys-PEG20K showed greater METH binding over a longer period of time than scFv7F9Cys despite a slight loss of functionality in vivo.
Figure 7.

Comparison of METH and therapeutic protein serum concentrations (μM). A. Total METH serum concentration was approximately 44% of the scFv7F9Cys serum concentration, and AUC0→2hrs of the METH serum concentrations was determined to be 340 ng*hr/ml, B. In the PEGylated conjugate group, total METH serum concentrations were an average of 29% of the therapeutic protein concentration, with a METH AUC0→2hrs of 935 ng*hr/ml. Total AUC0→48hrs of scFv7F9Cys-PEG20K was 4326 ng*hr/ml. This is a 12.7-fold increase in total METH binding activity compared to scFv7F9Cys, which was only active through two hrs (AUC0→2hrs = 340 ng*hr/ml).
Whole brains collected from the pharmacokinetic studies were pulverized and processed for Western blot analysis. Three markers in the dopaminergic system were measured: the dopamine transporter (DAT), tyrosine hydroxylase (TH), and the vesicular monoamine transporter 2 (VMAT2) (Figure 8A–B). TH was significantly reduced in the presence of scFv7F9Cys-PEG20K compared to all other groups. However, no other differences were seen between groups for the other dopaminergic markers tested. The glutamatergic system was also examined with three markers measured: the NMDA receptor (NR2A), the AMPA receptor (GluR2), and the scaffolding protein PSD-95 (Figure 8C–D). No differences between groups were found for any of the glutamatergic markers. Within both the dopaminergic and glutamatergic systems, there were no significant differences between the saline and METH control groups and the scFv7F9Cys treatment group.
Figure 8.

Whole brain protein levels of METH neurotoxicity markers after treatment with saline, METH, scFv7F9Cys, or scFv7F9Cys-PEG20K. A. Representative Western blots of dopaminergic markers: DAT, TH, and VMAT2. B. Quantification of Western blots for DAT, TH, and VMAT2. C. Representative Western blots of glutamatergic markers: NR2A, GluR2, PSD-95. D. Quantification of Western blots for NR2A, GluR2, and PSD-95. Error bars are SEM. S – Saline, M – METH, 7 – scFv7F9Cys, 7P – scFv7F9Cys-PEG20K, * - compared to saline (p<0.05), # - compared to METH (p<0.05), & - compared to scFv7F9Cys (p<0.05)
Discussion
The studies presented here suggest that conjugation of an anti-METH scFv to a 20 kDa PEG is sufficient to significantly alter scFv pharmacokinetics and METH disposition, while retaining affinity for its soluble, small molecule target (METH). We have shown that, although scFv7F9Cys-PEG20K had slightly diminished METH binding in vivo compared to scFv, the KD value for binding to METH did not differ between the two preparations. As expected, PEGylation of scFv markedly increased the half-life of the antibody fragment. Previous reports of PEGylation of other scFvs have focused on larger targets, such as a solid tumor (40,41), or a non-soluble target, such as a receptor (42). In some cases, scFv PEGylation has resulted in a loss of binding affinity to the target antigen (41,43), while others have reported an increased affinity (40). We found no significant changes in in vitro binding affinity of scFv-PEG to METH compared to scFv binding to METH. We employed site-specific PEGylation via a cysteine-maleimide reaction (40,44) to minimize effects on METH binding. As our analysis via SDS-PAGE suggests, only one PEG was conjugated to each scFv. If multiple PEG moieties were conjugated to a single scFv we would expect to see multiple bands at regular, increasing size increments. However, a single product band was observed in the analysis of each PEGylation reaction performed, suggesting a single PEGylation site per scFv. As PEG readily associates with water molecules, we believe this is why there is a larger difference in observed size between the native scFv and its PEGylated conjugates than would be expected in an anhydrous environment. Other PEGylation approaches have used the more common amine reaction resulting in multiple PEGylated sites on the scFv (45,46). This can result in multiple conjugation sites, leading to hindered binding, thus we chose to conjugate to a single location on the C-terminus. It is possible that, in addition to a small molecule antigen, the cysteine-selective conjugation located away from the binding pocket was responsible for maintaining binding activity of the PEGylated antibody fragment in this study.
The 20 kDa PEG moiety demonstrated the most active binding to METH compared to other molecular weight PEGs that were used for conjugation. Thus, it was selected for use in remainder of the studies. A linear 5 kDa PEG was tested, but unable to be purified to our satisfaction (Table I). Additionally, a branched 40 kDa PEG conjugate was generated, but showed a much lower METH binding affinity (Table I). The linear 20 kDa PEGylated conjugate was purified and did not hinder in vitro binding and was thus chosen to continue through pharmacokinetic studies.
The most significant finding in this study was that the scFv7F9Cys-PEG20K conjugate increased the half-life 27-fold compared to unconjugated scFv. Pharmacokinetic analysis of scFv7F9Cys and scFv7F9Cys-PEG20K suggested that this marked increase in the half-life of the PEGylated conjugate was due to a significant reduction in systemic clearance of the conjugate. Previous reports have shown that PEGs up to 20 kDa in size will reduce renal clearance by increasing the size of the conjugated molecule (17,18,47). However, PEGs larger than 20 kDa are increasingly eliminated via hepatobiliary excretion (47). These results and the size of the PEG used in our studies suggest that renal clearance was reduced by the addition of PEG, thus increasing the half-life of scFv7F9Cys-PEG20K. Further studies will be required to determine the exact elimination route of scFv7F9Cys-PEG20K and scFv7F9Cys.
Based on allometric scaling (48), the predicted half-life in humans for scFv7F9Cys-PEG20K would be approximately 100 hrs, suggesting a dosing regimen of one dose every four days. This is a much more acceptable dosing regimen for relapse prevention than the dosing regimen for scFv7F9Cys, which is predicted to be once every four hrs (based on a predicted half-life in humans of 3.6 hrs). Dosing with a drug once every half-life is a general principle of clinical pharmacology and therapeutics. Dosing at significantly shorter or longer intervals than the half-life can lead to drug accumulation or subtherapeutic drug concentrations, respectively. This is a major reason to modify biologics to extend their half-life. Longer half-lives, if there is no loss of biological function, can reduce the cost, extend the time between dosing, and potentially improve patient compliance.
Finally, we examined the effect of scFv7F9Cys and scFv7F9Cys-PEG20K on neuroadaptive changes that can occur with chronic METH use. To our knowledge, we are the first group to study these effects. Though preliminary, the changes found in whole brain suggest further studies examining specific brain regions (i.e. striatum and hippocampus) and a more complete sampling timeline may provide more insight into the effects of the scFv’s on METH neuroadaptation. No differences between the saline control group and the METH control group were found. There are a few possible explanations for this result, including the analysis method (whole brain rather than specific sections), the timing of brain tissue sampling (after METH serum concentrations had returned to baseline levels), and the dose of METH given was lower than previously seen in chronic METH studies (49–51).
We also observed a reduction in TH found in the scFv7F9Cys-PEG20K group and a slight reduction of TH in the scFv7F9Cys-treated group, suggesting that TH expression could be sensitive to a rapid withdrawal of METH from the brain. This differs from previous studies, which show that TH mRNA and protein expression are increased up to one day following a final METH dose (52,53). However, these studies either did not administer any drug to remove METH from the brain (53), or administered an NMDA antagonist that may elicit off-target effects (52). Additionally, we do not believe that scFv7F9Cys or scFv7F9Cys-PEG have a direct effect in the brain, as a previous biodistribution study from this lab showed that scFv7F9Cys was not detected in brain tissue (54). Therefore, scFv7F9Cys and the PEG20K conjugate likely elicit a rapid removal of METH from the brain without direct effects at other neurological receptors, differing in mechanism from previous studies, and possibly eliciting a new result of reduced TH concentrations. More studies need to be conducted to determine the time course of this effect, as well as whether this alone will have a behavioral effect, but they are beyond the scope of this pharmacokinetic study.
The purpose of conjugating scFv7F9Cys to PEG was to generate a high-affinity pharmacological treatment option for METH abuse through an increase in in vivo half-life. We think, despite a minor loss in in vivo functionality, that scFv7F9Cys-PEG20K warrants further development and testing as a potential pharmacotherapy for METH abuse.
Conclusion
The studies presented here suggest that conjugation of an anti-METH scFv to a 20 kDa PEG is sufficient to significantly improve scFv pharmacokinetics and METH disposition (through an increase in METH serum concentrations), while retaining functional binding capacity for its soluble, small molecule target antigen (METH). We have shown that, although scFv7F9Cys-PEG20K had slightly diminished METH binding in vivo compared to scFv, the measured Kd value for binding to METH did not differ between the two preparations. Most importantly, PEGylation of scFv markedly increased the half-life of the antibody fragment.
Acknowledgments
Disclosures
The authors would like to thank Melinda Gunnell and Sherri Woods for their valuable technical assistance. The UAMS Office of Grants and Scientific Publications provided editing services. This work was funded by NIDA R01 DA026423, DA036600 and NIDA T32 DA022981. S. Michael Owens is Chief Scientific Officer and has financial interests in InterveXion Therapeutics, LLC, a pharmaceutical biotech company, whose main interest is the development of antibody medications for the treatment of human diseases, including drug abuse.
Abbreviations
- METH
methamphetamine
- mAb
monoclonal antibody
- scFv
single chain variable fragment
- LC/MS-MS
liquid chromatography tandem mass spectrometry
- PEG
poly(ethylene) glycol
- t½
half-life
- IMAC
immobilized metal ion affinity chromatography
- Fc
fragment crystallizable
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- AUC
area under the serum concentration-time curve
- LC/MS-MS
liquid chromatography-tandem mass spectrometry
References
- 1.National Drug Intelligence Center (NDIC) National Drug Threat Assessment 2011. 2011 Aug 23;:1–72. [Google Scholar]
- 2.Substance Abuse and Mental Health Services Administration, Center for Behavioral Health Statistics and Quality. The DAWN Report: Emergency Department Visits Involving Methamphetamine: 2007 to 2011. 2014 Jun 4;:1–5. [PubMed] [Google Scholar]
- 3.Nicosia N, Pacula RL, Kilmer B, Lundberg R, Chiesa J. The Economic Cost of Methamphetamine Use in the United States, 2005. 2009 Jan 28;:1–171. [Google Scholar]
- 4.Winslow BT, Voorhees KI, Pehl KA. Methamphetamine abuse. American family physician. 2007;76(8):1169. [PubMed] [Google Scholar]
- 5.Ernst T, Chang L, Leonido-Yee M, Speck O. Evidence for long-term neurotoxicity associated with methamphetamine abuse: A 1H MRS study. Neurology. 2000 Mar 28;54(6):1344–9. doi: 10.1212/wnl.54.6.1344. [DOI] [PubMed] [Google Scholar]
- 6.Thrash B, Karuppagounder SS, Uthayathas S, Suppiramaniam V, Dhanasekaran M. Neurotoxic Effects of Methamphetamine. Neurochem Res. 2009 Aug 21;35(1):171–9. doi: 10.1007/s11064-009-0042-5. [DOI] [PubMed] [Google Scholar]
- 7.Metzger RRR, Haughey HMH, Wilkins DGD, Gibb JWJ, Hanson GRG, Fleckenstein AEA. Methamphetamine-induced rapid decrease in dopamine transporter function: role of dopamine and hyperthermia. J Pharmacol Exp Ther. 2000 Dec 1;295(3):1077–85. [PubMed] [Google Scholar]
- 8.Mark KA, Soghominian J-J, Yamamoto BK. High-Dose Methamphetamine Acutely Activates the Striatonigral Pathway to Increase Striatal Glutamate and Mediate Long-Term Dopamine Toxicity. J Neurosci. 2004 Dec 15;24(50):11449–56. doi: 10.1523/JNEUROSCI.3597-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eisch AJ, O’Dell SJ, Marshall JF. Striatal and cortical NMDA receptors are altered by a neurotoxic regimen of methamphetamine. Synapse. 1996 Mar;22(3):217–25. doi: 10.1002/(SICI)1098-2396(199603)22:3<217::AID-SYN3>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 10.Byrnes-Blake KA, Laurenzana EM, Carroll FI, Abraham P, Gentry WB, Landes RD, et al. Pharmacodynamic mechanisms of monoclonal antibody-based antagonism of (+)-methamphetamine in rats. Eur J Pharmacol. 2003 Feb 14;461(2–3):119–28. doi: 10.1016/s0014-2999(03)01313-x. [DOI] [PubMed] [Google Scholar]
- 11.Gentry WB, Laurenzana EM, Williams DK, West JR, Berg RJ, Terlea T, et al. Safety and efficiency of an anti-(+)-methamphetamine monoclonal antibody in the protection against cardiovascular and central nervous system effects of (+)-methamphetamine in rats. Int Immunopharmacol. 2006 Jun;6(6):968–77. doi: 10.1016/j.intimp.2006.01.008. [DOI] [PubMed] [Google Scholar]
- 12.Owens SM, Atchley WT, Hambuchen MD, Peterson EC, Gentry WB. Monoclonal antibodies as pharmacokinetic antagonists for the treatment of (+)-methamphetamine addiction. CNS Neurol Disord Drug Targets. 2011 Dec 1;10(8):892–8. doi: 10.2174/187152711799219370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peterson E, Owens S, Henry RL. Monoclonal Antibody Form and Function: Manufacturing the Right Antibodies for Treating Drug Abuse. aapsjorg. 2006 May 26;8(2) doi: 10.1007/BF02854909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Peterson EC, Laurenzana EM, Atchley WT, Hendrickson HP, Owens SM. Development and preclinical testing of a high-affinity single-chain antibody against (+)-methamphetamine. Journal of Pharmacology and Experimental Therapeutics. 2008 Apr;325(1):124–33. doi: 10.1124/jpet.107.134395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Abuchowski A, Van Es T, Palczuk N. Alteration of immunological properties of bovine serum albumin by covalent attachment of polyethylene glycol. Journal of Biological Chemistry. 1977;252(11):3578–81. [PubMed] [Google Scholar]
- 16.Veronese F. PEGylation, successful approach to drug delivery. Drug Discovery Today. 2005 doi: 10.1016/S1359-6446(05)03575-0. [DOI] [PubMed] [Google Scholar]
- 17.Harris JM, Martin NE, Modi M. Pegylation: a novel process for modifying pharmacokinetics. Clin Pharmacokinet. 2001;40(7):539–51. doi: 10.2165/00003088-200140070-00005. [DOI] [PubMed] [Google Scholar]
- 18.Bailon P, Berthold W. Polyethylene glycol-conjugated pharmaceutical proteins. Pharmaceutical Science & Technology Today. 1998 Oct 27;1(8):352–6. [Google Scholar]
- 19.Ivens IA, Achanzar W, Baumann A, Brändli-Baiocco A, Cavagnaro J, Dempster M, et al. Toxicologic Pathology. 7. Vol. 43. SAGE Publications; 2015. Oct, PEGylated Biopharmaceuticals: Current Experience and Considerations for Nonclinical Development; pp. 959–83. [DOI] [PubMed] [Google Scholar]
- 20.SAIF2. The Design and Development of Pegfilgrastim (PEG-rmetHuG-CSF, Neulasta. 2004 Feb 24;:1–11. doi: 10.2174/1381612043452613. [DOI] [PubMed] [Google Scholar]
- 21.Ramos EL. Preclinical and Clinical Development of Pegylated Interferon-Lambda 1 in Chronic Hepatitis C. Journal of Interferon & Cytokine Research. 2010 Aug;30(8):591–5. doi: 10.1089/jir.2010.0066. [DOI] [PubMed] [Google Scholar]
- 22.Arvedson T, O’Kelly J, Yang B-B. BioDrugs. 3. Vol. 29. Springer International Publishing; 2015. May 20, Design Rationale and Development Approach for Pegfilgrastim as a Long-Acting Granulocyte Colony-Stimulating Factor; pp. 185–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Park J-B, Kwon YM, Lee T-Y, Brim R, Ko M-C, Sunahara RK, et al. PEGylation of bacterial cocaine esterase for protection against protease digestion and immunogenicity. Journal of Controlled Release. 2010 Mar 3;142(2):174–9. doi: 10.1016/j.jconrel.2009.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kish SJ. Pharmacologic mechanisms of crystal meth. Canadian Medical Association Journal. 2008 Jun 17;178(13):1679–82. doi: 10.1503/cmaj.071675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Riddle EL, Topham MK, Haycock JW, Hanson GR, Fleckenstein AE. Differential trafficking of the vesicular monoamine transporter-2 by methamphetamine and cocaine. Eur J Pharmacol. 2002 Aug 2;449(1–2):71–4. doi: 10.1016/s0014-2999(02)01985-4. [DOI] [PubMed] [Google Scholar]
- 26.Ares-Santos S, Granado N, Espadas I, Martinez-Murillo R, Moratalla R. Methamphetamine Causes Degeneration of Dopamine Cell Bodies and Terminals of the Nigrostriatal Pathway Evidenced by Silver Staining. Neuropsychopharmacology: official publication of the American College of Neuropsychopharmacology. 2013 Oct 30; doi: 10.1038/npp.2013.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sulzer D, Sonders MS, Poulsen NW, Galli A. Mechanisms of neurotransmitter release by amphetamines: A review. Progress in Neurobiology. 2005 Apr;75(6):406–33. doi: 10.1016/j.pneurobio.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 28.Baucum AJ, Rau KS, Riddle EL, Hanson GR, Fleckenstein AE. Methamphetamine increases dopamine transporter higher molecular weight complex formation via a dopamine- and hyperthermia-associated mechanism. J Neurosci. 2004 Mar 31;24(13):3436–43. doi: 10.1523/JNEUROSCI.0387-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lau A, Tymianski M. Glutamate receptors, neurotoxicity and neurodegeneration. Pflugers Arch. 2010 Jul;460(2):525–42. doi: 10.1007/s00424-010-0809-1. [DOI] [PubMed] [Google Scholar]
- 30.Simoes PF, Silva AP, Pereira FC, Marques E, Grade S, Milhazes N, et al. Methamphetamine induces alterations on hippocampal NMDA and AMPA receptor subunit levels and impairs spatial working memory. Neuroscience. 2007 Dec;150(2):433–41. doi: 10.1016/j.neuroscience.2007.09.044. [DOI] [PubMed] [Google Scholar]
- 31.Aarts M. Treatment of Ischemic Brain Damage by Perturbing NMDA Receptor- PSD-95 Protein Interactions. Science. 2002 Oct 25;298(5594):846–50. doi: 10.1126/science.1072873. [DOI] [PubMed] [Google Scholar]
- 32.Ehrlich I, Malinow R. Postsynaptic density 95 controls AMPA receptor incorporation during long-term potentiation and experience-driven synaptic plasticity. Journal of Neuroscience. 2004 Jan 28;24(4):916–27. doi: 10.1523/JNEUROSCI.4733-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang J, Saur T, Duke AN, Grant SGN, Platt DM, Rowlett JK, et al. Motor impairments, striatal degeneration, and altered dopamine-glutamate interplay in mice lacking PSD-95. J Neurogenet. 2014 Mar 1;28(1–2):98–111. doi: 10.3109/01677063.2014.892486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Carroll FI, Blough BE, Pidaparthi RR, Abraham P, Gong PK, Deng L, et al. Synthesis of Mercapto-(+)-methamphetamine Haptens and Their Use for Obtaining Improved Epitope Density on (+)-Methamphetamine Conjugate Vaccines. J Med Chem. 2011 Jul 28;54(14):5221–8. doi: 10.1021/jm2004943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stevens MW, Tawney RL, West CM, Kight AD, Henry RL, Owens SM, et al. Preclinical characterization of an anti-methamphetamine monoclonal antibody for human use. MAbs. 2014 Mar 1;6(2) doi: 10.4161/mabs.27620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nanaware-Kharade N, Gonzalez GA, III, Lay JO, Jr, Hendrickson HP, Peterson EC. Therapeutic Anti-Methamphetamine Antibody Fragment-Nanoparticle Conjugates: Synthesis and in Vitro Characterization. Bioconjugate Chem. 2012 Sep 19;23(9):1864–72. doi: 10.1021/bc300204n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Thakkar S, Nanaware-Kharade N, Celikel R, Peterson EC, Varughese KI. Affinity improvement of a therapeutic antibody to methamphetamine and amphetamine through structure-based antibody engineering. Sci Rep. 2014 Jan 1;4:3673–3. doi: 10.1038/srep03673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Byrnes-Blake KA, Laurenzana EM, Landes RD, Gentry WB, Owens SM. Monoclonal IgG affinity and treatment time alters antagonism of (+)-methamphetamine effects in rats. Eur J Pharmacol. 2005 Oct 3;521(1–3):86–94. doi: 10.1016/j.ejphar.2005.08.016. [DOI] [PubMed] [Google Scholar]
- 39.Laurenzana EM, Byrnes-Blake KA, Milesi-Hallé A, Gentry WB, Williams DK, Owens SM. Use of anti-(+)-methamphetamine monoclonal antibody to significantly alter (+)-methamphetamine and (+)-amphetamine disposition in rats. Drug Metab Dispos. 2003 Nov;31(11):1320–6. doi: 10.1124/dmd.31.11.1320. [DOI] [PubMed] [Google Scholar]
- 40.Lee JI, Eisenberg SP, Rosendahl MS, Chlipala EA, Brown JD, Doherty DH, et al. Site-Specific PEGylation Enhances the Pharmacokinetic Properties and Antitumor Activity of Interferon Beta-1b. Journal of Interferon & Cytokine Research. 2013 Dec;33(12):769–77. doi: 10.1089/jir.2012.0148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kubetzko S, Balic E, Waibel R, Zangemeister-Wittke U, Pluckthun A. PEGylation and Multimerization of the Anti-p185HER-2 Single Chain Fv Fragment 4D5: EFFECTS ON TUMOR TARGETING. Journal of Biological Chemistry. 2006 Nov 10;281(46):35186–201. doi: 10.1074/jbc.M604127200. [DOI] [PubMed] [Google Scholar]
- 42.Nellis DF, Ekstrom DL, Kirpotin DB, Zhu J, Andersson R, Broadt TL, et al. Preclinical Manufacture of an Anti-HER2 scFv-PEG-DSPE, Liposome-Inserting Conjugate. 1. Gram-Scale Production and Purification. Biotechnol Progress. 2008 Sep 5;21(1):205–20. doi: 10.1021/bp049840y. [DOI] [PubMed] [Google Scholar]
- 43.Veronese F. Peptide and protein PEGylation:: a review of problems and solutions. Biomaterials. 2001 doi: 10.1016/s0142-9612(00)00193-9. [DOI] [PubMed] [Google Scholar]
- 44.Natarajan A, Xiong C-Y, Albrecht H, DeNardo GL, DeNardo SJ. Characterization of Site-Specific ScFv PEGylation for Tumor-Targeting Pharmaceuticals. Bioconjugate Chem. 2005 Jan;16(1):113–21. doi: 10.1021/bc0498121. [DOI] [PubMed] [Google Scholar]
- 45.Yamamoto Y, Tsutsumi Y, Yoshioka Y, Nishibata T, Kobayashi K, Okamoto T, et al. Site-specific PEGylation of a lysine-deficient TNF-alpha with full bioactivity. Nat Biotechnol. 2003 May 1;21(5):546–52. doi: 10.1038/nbt812. [DOI] [PubMed] [Google Scholar]
- 46.Monkarsh SP, Ma Y, Aglione A, Bailon P, Ciolek D, DeBarbieri B, et al. Positional isomers of monopegylated interferon alpha-2a: isolation, characterization, and biological activity. Analytical Biochemistry. 1997 May 1;247(2):434–40. doi: 10.1006/abio.1997.2128. [DOI] [PubMed] [Google Scholar]
- 47.Fishburn CS. The pharmacology of PEGylation: Balancing PD with PK to generate novel therapeutics. J Pharm Sci. 2008 Oct;97(10):4167–83. doi: 10.1002/jps.21278. [DOI] [PubMed] [Google Scholar]
- 48.Caldwell GW, Masucci JA, Yan Z, Hageman W. Allometric scaling of pharmacokinetic parameters in drug discovery: can human CL, Vss and t1/2 be predicted from in-vivo rat data? Eur J Drug Metab Pharmacokinet. 2004 Apr;29(2):133–43. doi: 10.1007/BF03190588. [DOI] [PubMed] [Google Scholar]
- 49.Davidson C, Lee TH, Ellinwood EH. Acute and chronic continuous methamphetamine have different long-term behavioral and neurochemical consequences. Neurochem Int. 2005 Feb;46(3):189–203. doi: 10.1016/j.neuint.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 50.Ryan LJ, Linder JC, Martone ME, Groves PM. Histological and ultrastructural evidence thatd-amphetamine causes degeneration in neostriatum and frontal cortex of rats. Brain Research. 1990 Jun;518(1–2):67–77. doi: 10.1016/0006-8993(90)90955-b. [DOI] [PubMed] [Google Scholar]
- 51.Fibiger HC, McGeer EG. Effect of acute and chronic methamphetamine treatment on tyrosine hydroxylase activity in brain and adrenal medulla. Eur J Pharmacol. 1971 Jan 1;16(2):176–80. doi: 10.1016/0014-2999(71)90008-2. [DOI] [PubMed] [Google Scholar]
- 52.Zhang Y, Angulo JA. Contrasting effects of repeated treatment vs. withdrawal of methamphetamine on tyrosine hydroxylase messenger RNA levels in the ventral tegmental area and substantia nigra zona compacta of the rat brain. Synapse. 1996 Nov;24(3):218–23. doi: 10.1002/(SICI)1098-2396(199611)24:3<218::AID-SYN3>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 53.Shepard JD, Chuang DT, Shaham Y, Morales M. Effect of methamphetamine self-administration on tyrosine hydroxylase and dopamine transporter levels in mesolimbic and nigrostriatal dopamine pathways of the rat. Psychopharmacology (Berl) 2006 Mar 23;185(4):505–13. doi: 10.1007/s00213-006-0316-4. [DOI] [PubMed] [Google Scholar]
- 54.Nanaware-Kharade N, Thakkar S, Gonzalez GA, Peterson EC. A Nanotechnology-Based Platform for Extending the Pharmacokinetic and Binding Properties of Anti-methamphetamine Antibody Fragments. Sci Rep. 2015;5:12060. doi: 10.1038/srep12060. [DOI] [PMC free article] [PubMed] [Google Scholar]