

Abstract
Carbon nanotubes (CNTs) are newly developed materials with unique properties and a range of industrial and commercial applications. A rapid expansion in the production of CNT materials may increase the risk of human exposure to CNTs. Studies in rodents have shown that certain forms of CNTs are potent fibrogenic inducers in the lungs to cause interstitial, bronchial, and pleural fibrosis characterized by the excessive deposition of collagen fibers and the scarring of involved tissues. The cellular and molecular basis underlying the fibrotic response to CNT exposure remains poorly understood. Myofibroblasts are a major type of effector cells in organ fibrosis that secrete copious amounts of extracellular matrix proteins and signaling molecules to drive fibrosis. Myofibroblasts also mediate the mechano-regulation of fibrotic matrix remodeling via contraction of their stress fibers. Recent studies reveal that exposure to CNTs induces the differentiation of myofibroblasts from fibroblasts in vitro and stimulates pulmonary accumulation and activation of myofibroblasts in vivo. Moreover, mechanistic analyses provide insights into the molecular underpinnings of myofibroblast differentiation and function induced by CNTs in the lungs.
In view of the apparent fibrogenic activity of CNTs and the emerging role of myofibroblasts in the development of organ fibrosis, we discuss recent findings on CNT-induced lung fibrosis with emphasis on the role of myofibroblasts in the pathologic development of lung fibrosis. Particular attention is given to the formation and activation of myofibroblasts upon CNT exposure and the possible mechanisms by which CNTs regulate the function and dynamics of myofibroblasts in the lungs. It is evident that a fundamental understanding of the myofibroblast and its function and regulation in lung fibrosis will have a major influence on the future research on the pulmonary response to nano exposure, particle and fiber-induced pneumoconiosis, and other human lung fibrosing diseases.
Keywords: Carbon nanotube, Myofibroblast, Lung fibrosis, Animal model, Mechanism, Extracellular matrix
Background
Mammalian lungs are among the most susceptible organs to fibrosis [1]. As the primary respiratory organ, the lungs perform gas exchange between the blood and the inhaled air through a thin layer of alveolar septal structures. These structures are vulnerable to structural and functional alterations, such as interstitial thickening and alveolar destruction, major pathologic features of lung fibrosis. Respiration also exposes the lungs constantly to numerous inhaled fibrogenic agents including toxic chemicals, mycobacteria, and particulate matters [2, 3]. Some chemicals are preferentially uptaken by lung epithelial cells and therefore tend to accumulate in the lungs upon exposure through either respiration or systemic means [4–6]. These exposures can damage the lung tissue to give rise to induced fibrosis. Lung fibrosis also occurs as a common, end stage pathologic development of existing lung diseases caused by infection, chronic inflammation, cardiovascular malfunction, autoimmunity, and idiopathy [7]. In many cases, human lung fibrosis is progressive and refractory to therapy, causing high rates of mortality and disability [8].
The progressive nature and severe outcome of human lung fibrosing diseases are well illustrated by idiopathic pulmonary fibrosis (IPF). IPF initiates insidiously with no known etiology and follows a chronic but progressive course that is ultimately lethal, indicated by the median survival of IPF patients within 2–5 years after diagnosis [9–11]. IPF exhibits a histopathologic pattern of usual interstitial pneumonia, including the occurrence of mild-to-moderate inflammatory infiltration, injury and hyperplasia of alveolar epithelial cells, excessive deposition of the extracellular matrix (ECM), thickening of the alveolar septa, scarring and formation of fibroblastic foci, and temporally heterogeneous fibrotic remodeling of the lung structure, such as honeycombing [9, 11]. No effective therapy is available to the patients with this destructive lung disease except lung transplantation at the present time. Several animal models have been developed and studied for lung fibrosis such as IPF during the past few decades [12, 13]. Among these, the bleomycin-induced lung fibrosis is the most commonly used model. However, bleomycin-induced lung fibrosis differs from IPF in several pathologic aspects. First, bleomycin induces severe alveolar epithelial cell death in the lungs before the onset of fibrosis, whereas extensive alveolar epithelial cell lesions are rarely seen in IPF. Second, bleomycin causes diffusive fibrosis throughout the lung parenchyma with scattered fibrotic foci, while the fibrosis in IPF is predominantly in the form of fibroblastic foci. Third, fibrosis in the bleomycin mouse model resolves spontaneously and thus is reversible, but that in IPF is progressive and irreversible. Overall, the research using the bleomycin and other fibrosis models has not yielded effective translation to the treatment of IPF and other human lung fibrosing diseases including pneumoconiosis caused by exposure to fibrogenic particles and fibers, such as silica and asbestos [4, 14]. As a result, there are increasing efforts to gain improved fundamental understanding of the pathogenesis and development of lung fibrosis, as well as renewed interests in identifying new animal models that mimic human lung fibrosis, in order to achieve better treatment and prevention against human lung fibrosing diseases [8].
Carbon nanotubes (CNTs) are long and hollow nanostructures made of a single layer or concentric multiple layers of one-atom-thick carbon walls, designated as single-walled CNTs (SWCNTs) and multi-walled CNTs (MWCNTs), respectively [15]. As new materials, CNTs have been developed with a variety of industrial and commercial applications for electronic, biomedical, and energy-related uses. The annual production of CNTs has been increased rapidly in recent years [16]. On the other hand, most CNTs are respirable fibers with physicochemical features like nano-scaled diameter, fiber-like shape (high aspect ratio), large surface area, poor solubility, and excessive biopersistence, properties often associated with the fibrogenic and tumorigenic activities of inhaled particles and fibers, thus raising concern over the potential adverse health effects of human exposure to CNTs [17, 18]. Indeed, a marked progress in the understanding of CNT toxicity has been achieved in experimental animal and cell systems during the past decade. Importantly, the research has identified CNTs as a significant fibrogenic inducer in the lungs and the pleural space to cause interstitial, bronchial, and pleural fibrosis in animals [18–27]. The pathologic development and features of CNT-induced pulmonary interstitial fibrosis overlap with those of IPF and pneumoconiosis considerably. From the experimental research point of view, this finding suggests a possible application of CNT-induced lung fibrosis in the study of the human lung fibrosing diseases with regard to their pathogenesis, therapeutic targeting, and biomarkers for exposure and disease monitoring [18, 24, 28]. Recent field studies on CNT-exposed populations demonstrate marked accumulation of inflammatory and fibrotic mediators and biomarkers in the body fluids of workers manufacturing MWCNTs, highlighting a need to protect humans from nano exposure from occupational, environmental, and commercial sources [29–31].
The mechanisms by which CNTs cause lung fibrosis remain unclear, but are believed to involve an exacerbated fibroblastic response [18]1. During fibrosis development, fibroblasts and myofibroblasts act as major effector cells to produce excessive amounts of collagen fibers and other ECM proteins, and to remodel and contract the fibrosing tissues [32–40]. The fibroblastic response in fibrosis bears certain similarity to wound healing following tissue injury. At the early stage of lung fibrosis, resident fibroblasts in the lung interstitial space are activated upon stimulation. Activated fibroblasts migrate and proliferate to result in the accumulation of active fibroblastic cells in regions where injury takes place. At the same time, activated fibroblasts differentiate into myofibroblasts, which are a group of multi-functional mesenchymal cells implicated in wound healing, organ fibrosis, tumorigenesis, and cancer metastasis. Myofibroblasts are characterized by their simultaneous presentation of a high capacity of ECM protein production and their smooth muscle cell-like contractile features obtained through the de novo synthesis of α-smooth muscle actin (α-SMA)-containing stress fibers [34]. In physiologic wound healing, excessive ECM production and remodeling are avoided, as the majority of the α-SMA-expressing myofibroblasts disappear by way of apoptosis upon scar formation. However, during pathologic fibrosis, myofibroblasts become resistant to apoptosis and thereby persist to continuously synthesize and remodel the ECM, which ultimately leads to organ fibrosis and destruction [41]. Understanding the formation, function, and fate of myofibroblasts in tissue remodeling may hold a key to differentiating between physiologic wound healing and the development of organ fibrosis including IPF, pneumoconiosis, and CNT-induced lung pathology.
Several excellent reviews have been published to sum up the biological effects, the overall mode of action, and the interrelations between the physicochemical properties and the bioactivities of CNTs from a toxicological point of view [17–19, 42]. However, the cellular and molecular basis underlying the fibrotic response to CNTs, which is key to understanding the adverse health effects from CNT exposure, remains a topic of considerable challenge. In part, this issue is due to a lack of significant mechanistic insights into organ fibrosis in general. Given the emerging and rapidly advancing research on myofibroblasts and their function in the pathogenesis of fibrosis, we discuss the current literature on CNT-induced rodent lung fibrosis with focus on the formation and role of myofibroblasts in the development of pulmonary fibrosis. Possible mechanisms by which CNTs regulate myofibroblast functions and dynamics during fibrosis development in the lungs will be discussed. We anticipate that such analysis will have a major impact on the future research of the pulmonary response to CNT exposure and therefore will aid in the fundamental understanding, as well as the risk assessment, of lung fibrosis caused by exposure to CNTs, other nanomaterials, and other fibrogenic particles and fibers. The knowledge obtained will also facilitate the identification of new drug targets and biomarkers for the treatment and exposure and disease monitoring of human lung fibrosing diseases.
Carbon nanotube-induced lung fibrosis
CNT-induced lung interstitial fibrosis initiates with a prominent acute inflammatory response, exhibited by recruitment and accumulation of inflammatory cells, including neutrophils, macrophages, and lymphocytes, and elevated secretion of pro-inflammatory and pro-fibrotic cytokines, chemokines, and growth factors, such as TNF-α, IL-1β, IL-6, MCP-1, TGF-β1 and PDGF-A (PDGF subunit A) [22, 24, 25, 43–47]. Along with the acute inflammation, MWCNTs trigger a rapid-onset fibrotic response, indicated by increased deposition of collagen fibers in alveolar septa, detectable as early as day 1 post-exposure [24]. The acute inflammatory and fibrotic responses reach an apex by day 7 post-exposure, after which the pathologic effects transit to chronic fibrosis. At the chronic phase, CNT-induced fibrotic lesions are featured with mild inflammation, thickened alveolar septa, increased deposition of ECM proteins, enhanced expression of fibrosis markers, and formation of fibrotic foci and epithelioid granulomas [27, 48]. Figure 1 depicts the pathologic features and transition of the acute and chronic lung fibrotic lesions induced by CNTs in rodents. The lung fibrotic response to CNT exposure resembles the pulmonary response to deposition of fibrogenic foreign bodies in the lungs. In particular, the pathologic features and dynamics of the pulmonary interstitial fibrosis induced by CNTs display a high similarity to those of pneumoconiosis and IPF, both of which are progressive, incurable, and poorly understood human fibrotic lung diseases [24, 28].
Fig. 1.
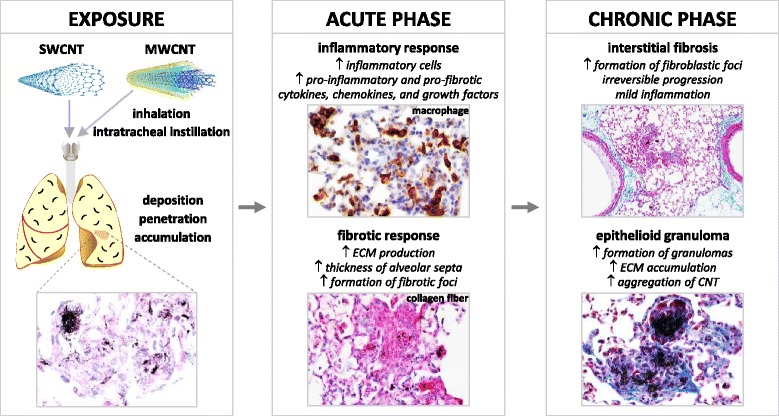
CNT-induced lung fibrosis. CNTs are respirable fibers with a tendency to deposit, penetrate and accumulate in lung tissues (left box). Exposure to CNTs induces acute phase responses including an inflammatory response, represented by the recruitment of Mac2-positive macrophages, and a fibrotic response, shown by Picro-Sirius Red staining for collagens I and III. Acute phase responses start as early as day 1, reach an apex on day 7, and decline after day 7 to significantly lower levels on day 14 post-exposure. In this scenario, day 7 post-exposure may represent an acute-to-chronic transition of CNT-induced pathology in mouse lungs (middle box). CNT-induced chronic phase responses are characterized by interstitial fibrosis and formation of epithelioid granulomas, shown by Masson’s Trichrome staining for collagen fibers on day 28 post-exposure. CNT-induced lung fibrosis appears to be persistent and irreversible in studies for up to 1 year post-exposure (right box)
Notably, CNT-induced lung fibrosis appears to be persistent and irreversible, which differs from bleomycin-induced lung fibrosis. The fibrotic lesions induced by a single dose instillation of bleomycin generally resolve after 28 days post-exposure [12, 13], whereas CNT-induced lung fibrosis was prolonged and was observed 90 days after a single dose intratracheal instillation of SWCNTs in mice, shown by the significant formation of epithelioid granulomas [44]. Similarly, lung fibrosis was observed 90 days after a single dose intratracheal instillation of MWCNTs in rats, demonstrated by thickened alveolar wall and increased collagen deposition [49]. In a more recent study, the long-term effects of CNTs on lung fibrosis were specifically investigated. Either a single dose intratracheal instillation of SWCNTs or an inhalation exposure to SWCNT aerosol (5 h/day for 4 days) induced lung fibrosis in mice 1 year post-exposure, demonstrated by increased levels of collagens in the lungs and the presence of fibrotic histopathological phenotypes [50]. In a separate study with inhalation of MWCNT aerosol (5 h/day for 12 days, 4 times/week for 3 weeks), it was found that lung fibrosis in MWCNT-exposed mice, indicated by increased fibrous collagen in the alveolar region, displayed a progressive increase in the thickness of the alveolar septal connective tissue over time, i.e., 0.17 ± 0.02, 0.22 ± 0.02, 0.26 ± 0.03, 0.25 ± 0.02 and 0.29 ± 0.01 μm on days 1, 14, 84, 168 and 336 post-exposure, which was significantly higher than that of the clean-air control on days 84 and 336 post-exposure [27]. These observations reveal that CNTs induce progressive and persistent fibrosis in the lungs, which is not self-resolved even at 1 year post-exposure. From this prospect, studies on CNT-induced lung fibrosis are promising, as they would, at the very least, supplement the findings from the bleomycin model to better reflect human fibrotic lung diseases, such as IPF and pneumoconiosis.
Some CNTs are capable of inducing bronchial or pleural fibrosis. In the former case, inhalation of MWCNTs in ovalbumin-sensitized mice, a murine asthma model, induced significant airway fibrosis in addition to asthmatic phenotypes [25]. In the latter case, direct instillation of long or short MWCNTs into the pleural cavity induced fiber length-dependent fibrotic responses like asbestos fibers [26]. The long MWCNTs elicited acute inflammation followed by progressive fibrosis on the parietal pleura, whereas the short MWCNTs were rapidly cleared through the stomata of the pleura and as a result, failed to induce pleural fibrosis. In both bronchial and pleural fibrosis, the development of fibrotic lesions involves acute inflammation followed by fibrotic progression similarly to that of interstitial fibrosis described above. The CNT-induced airway and pleural fibrotic lesions may have implications for the study of human asthma and pleural thickening/mesothelioma, respectively, which requires further investigation.
CNTs differ considerably from one another in their physicochemical properties that may impact CNT toxicity in vivo by affecting both the intrinsic pathogenicity and the kinetic behavior of CNTs [18]. Cumulative evidence reveals that the size, shape, surface area, surface reactivity, fiber rigidity, and biopersistence of CNTs are among the important properties to influence their fibrogenic activity in the lungs. The knowledge obtained from these structure-activity relationship studies provides insights into the internal (effective) dose of CNTs in lung fibrosis and hence their risk assessment. The information would also suggest new ways of reducing CNT fibrogenic activity by means of prevention-through-product design.
CNTs exhibit length-dependent activities in causing lung interstitial fibrosis [51, 52], as well as the fibrosis and granuloma formation in the parietal mesothelium [26]. In the latter case, the length-fibrosis correlation can be explained by the observation that long, but not short, CNTs (i.e., >15 μm in length) are retained in the pleural cavity to cause the fibrotic lesions, because the long CNTs are not efficiently cleared off through either the stomatal drain (3–10 μm in diameter) or pleural macrophage phagocytosis (<10–15 μm in diameter), whereas the short CNTs are rapidly eliminated from the pleural cavity through both clearance mechanisms [17]. It is believed that the long CNTs that exceed the diameter of macrophages and therefore are not effectively engulfed by macrophages would trigger the so-called “frustrated phagocytosis”. In this scenario, macrophages that fail to phagocytize the fibers are activated to release an array of bioactive and/or cytotoxic agents, which would cause tissue damage locally, much like the response to long asbestos fibers [17]. It is also possible that CNTs interact with cell surface structures, such as the pattern recognition receptors, to elicit responses from lung cells. In this case, the long CNTs may have a higher capacity of stimulating the cell surface receptors than the short CNT fibers to account for the differential fibrotic effects between long and short CNTs. MWCNTs have been shown to bind to the cell surface bone morphogenetic protein receptor type II (BMPR2) to stimulate the differentiation and to inhibit the apoptosis of mouse myoblast cells [53]. However, evidence supporting a direct interaction of CNTs with cell surface receptors to stimulate lung fibrosis is currently lacking.
It is worth noting that many CNTs are shorter than 5–10 μm and thus would be engulfed by macrophages if not agglomerated or tangled into large masses. Yet these CNTs may still be fibrogenic both in vitro and in vivo. Moreover, the lung parenchyma does not appear to have a “sieve” mechanism like the pleural stomata. Therefore, the length-dependent activities of CNTs in causing lung interstitial fibrosis may differ from those defined in the “fiber length pathogenicity paradigm” derived from mesothelial fibrosis caused by asbestos fibers with regard to both their phenotypes and mechanisms. For instance, the needle-like MWCNTs, i.e., Mitsui XNRI MWNT-7, have a mean length of 3.86 μm and count mean diameter of 49 nm, but are potent in inducing lung fibrosis in vivo, suggesting that properties other than fiber length, in particular, the fiber rigidity in the case of XNRI MWNT-7, play an important role in modulating the fibrogenic activity of nanomaterials [23, 24, 27].
A recent comparison among ten commercial MWCNTs with different morphology, composition, surface area, and functionalization reveals that surface area, fiber length, and surface modifications were better predictors of pulmonary inflammation in vivo [54]. In other studies, the thin film coating of MWCNTs with Al2O3 via atomic layer deposition reduced lung fibrosis in mice [55]; and the surface functionalization of MWCNTs with carboxylation or other covalent modifications demonstrates that strong cationic modifications induced significant lung fibrosis, whereas carboxylation significantly reduced the extent of lung fibrosis, compared with pristine CNTs [56]. Therefore, the surface reactivity and surface charge of CNTs play critical roles in determining their fibrogenicity and toxicity.
Role of myofibroblasts and fibroblasts in lung fibrosis
The fibroblastic response in lung fibrosis is responsible for the fibrotic matrix built-up, ECM remodeling, and tissue contraction to result in lung scarring and destruction. This dynamic process involves both myofibroblasts and fibroblasts that play distinct, but sometimes overlapping, functions; moreover, the interplay between these two types of cells represents an important aspect of fibrosis development, as illustrated in Fig. 2.
Fig. 2.
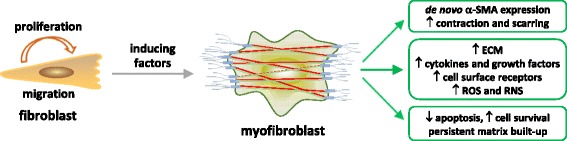
Fibroblasts and myofibroblasts in fibrosis. Fibroblasts and myofibroblasts act as the effector cells in organ fibrosis. Upon fibrogenic stimulation, tissue resident fibroblasts are activated to migrate and proliferate. Activated fibroblasts are the major progenitor cells to differentiate into myofibroblasts, indicated by the de novo expression of α-SMA. Myofibroblasts possess several characteristics, which distinguish them from fibroblasts and render them unique and critical functions in organ fibrosis
Resident fibroblasts are believed to be activated and play important roles in the early phase of wound healing and tissue fibrosis. Activated fibroblasts migrate, proliferate, and secrete certain ECM proteins, cytokines, and growth factors, to promote the fibrosis development at the site of injury. Among these functions, fibroblast proliferation has received increasing attention in fibrosis development. In this regard, the fibroblastic foci in IPF exhibit a prominent accumulation of proliferating fibroblastic cells that are fibroblasts in nature [37, 57]. Fibroblasts isolated from IPF patients show enhanced proliferation in vitro. Furthermore, a number of cellular mechanisms have been identified to account for the pro-proliferative state of these cells. There was an aberrant activation of PI3K/Akt signaling and pathological proliferation in these fibroblasts, which involved a decreased level of the plasma membrane integrin β1/caveolin-1/PTEN complex and a decreased activity of PTEN phosphatase [58]. Activated Akt phosphorylates and inactivates transcription factor FoxO3a (forkhead box O3a) that promotes cell cycle arrest by increasing the expression of CDK inhibitor p27. In a separate study, IPF fibroblasts were shown to have a high level of inactive FoxO3a due to a high Akt activity, which resulted in a reduced level of p27 and, consequently, increased proliferation of the cells [59]. A decreased level of integrin α2β1 and an impaired activity of PP2A phosphatase were also detected in IPF fibroblasts; these changes elevated the levels of inactive, phosphorylated GSK-3β and active nuclear β-catenin, both of which promote fibroblast proliferation [60]. In addition, the IPF fibroblasts display reduced apoptosis. The increased proliferation and reduced apoptosis of fibroblasts expand the fibroblast population and would therefore boost the fibroblast-to-myofibroblast transformation, which ultimately leads to the formation of fibroblastic foci and tissue fibrosis [61–63].
Myofibroblasts represent a heterogeneous group of fibroblastic cells found in tissues undergoing wound healing or organ fibrosis, and within the microenvironment of primary and metastatic cancers [34, 64]. As shown in Fig. 2, myofibroblasts exhibit several notable and unique characteristics that are distinct from those of fibroblasts and are believed to be critical to wound healing, fibrosis, tumorigenesis and cancer metastasis [34, 35, 65–67]. First, myofibroblasts possess a high contractile activity through their de novo synthesis of α-SMA protein. Newly synthesized α-SMA incorporates into intracellular stress fibers that are capable of generating strong force upon contraction, through which myofibroblasts control the cell shape and movement, ECM reorganization, and tissue contraction. Second, myofibroblasts possess a high capacity of protein synthesis and secretion and are believed to be responsible for the production of a major portion of the ECM proteins, such as collagens and fibronectin, for matrix deposition during fibrosis [38, 68–70]. Third, myofibroblasts demonstrate high levels of constitutive and induced expression of cytokines, chemokines, growth factors, and cell surface receptors, which render the cells some properties of inflammatory cells to allow them to respond to a variety of inflammatory, immune, and mechanical signals [71–75]. Fourth, myofibroblasts produce and release reactive oxygen and nitrogen species (ROS, RNS) spontaneously and under stimulation, which contribute to the up-regulation of ECM production and remodeling [76–80]. Last but not least, myofibroblasts in fibrotic tissues show high resistance to apoptosis, possibly due to the persistent activity of TGF-β1 and ECM deposition, leading to a prolonged survival and activity of myofibroblasts during the development of fibrosis [61, 81–83]. Combined, these features of myofibroblasts enable the cells to perform unique, direct, and critical functions in a broad range of physiologic and pathologic processes involving ECM remodeling.
The formation of myofibroblasts involves a complex and as yet not well understood process. In both IPF and bleomycin-induced lung fibrosis, activated interstitial resident fibroblasts are recognized as a major source of myofibroblasts. Myofibroblasts may also derive through trans-differentiation from other types of cells, such as the bone marrow-derived fibrocytes, the pericytes surrounding small blood vessel walls, and the epithelial cells overlying connective tissues. However, the contribution of these trans-differentiated cells to the myofibroblast pool in lung fibrosis remains a subject of debate, as studies examining the role of the trans-differentiated cells in fibrosis development have yielded inconsistent results in different experimental systems [41, 66, 84–88].
The differentiation of myofibroblasts from fibroblasts can be induced by a variety of signals. The transforming growth factor-β (TGF-β; mainly TGF-β1) has been recognized as a central player in driving fibroblasts to differentiate into myofibroblasts in both experimental and clinical settings, though a panel of other factors including ED-A fibronectin, Wnt, NOX4, integrins, ROS, and the stiff ECM have also been shown to promote myofibroblast differentiation, as have been summarized by Hinz and colleagues [41]. TGF-β1 stimulates all characteristics of the differentiated myofibroblasts both in vitro and in vivo [34]. Exaggerated expression and activation of TGF-β are commonly observed in lung fibrotic lesions, such as IPF and bleomycin-induced lung fibrosis [89–93]. Newly synthesized TGF-β is secreted into the ECM in the form of a latent complex [94]. The mechanisms by which TGF-β is induced and activated during fibrosis are not well understood and remain to be a topic of intensive research, which will be discussed in more detail in the section on mediators and mechanisms for CNT-induced lung fibrosis.
Myofibroblasts in carbon nanotube-induced lung fibrosis
Given the critical role of myofibroblasts in lung fibrosis and the predominant pathologic phenotypes of lung fibrosis induced by CNTs described above, the potential effects of CNTs on myofibroblast differentiation and function have drawn attention in recent years.
In vitro studies have provided evidence demonstrating that CNTs stimulate the formation of myofibroblasts from fibroblasts and other types of cells. Treatment of macrophages (RAW264.7) with MWCNTs (average length 10 μm, average outer diameter 8.7 nm, surface area 220 m2/g, total metal 0.2 %) stimulated the cells to produce a range of pro-fibrotic cytokines and growth factors including TNF-α, IL-1β, TGF-β1, and PDGF at both mRNA and protein levels [95]. Induction of the proteins correlated with the activation of the NF-κB signaling pathway. Moreover, the cell-free medium from the treated culture was found capable of stimulating fibroblasts (WI38-VA13) to differentiate into myofibroblasts, as evidenced by significantly elevated expression of α-SMA protein. Similarly, SWCNTs (fiber length 0.5–2 μm, outer diameter 1–2 nm, surface area 480 m2/g, total metal 4.5 %) activated NF-κB and induced the expression of TGF-β1 in macrophages to produce a medium that induced the formation of myofibroblasts from lung fibroblasts [96]. These findings suggest a molecular mechanism by which CNTs stimulate a coordinated response between macrophages and fibroblasts to induce the differentiation of fibroblasts into myofibroblasts via secreted soluble factors, such as TGF-β1 and IL-1β.
CNTs induce the epithelial-mesenchymal transition (EMT), i.e., the trans-differentiation of epithelial cells to mesenchymal cells, to contribute to myofibroblast formation. During this process, epithelial cells gradually acquire a mesenchymal (fibroblast-like) phenotype through the de novo expression of vimentin, FSP-1 (fibroblast specific protein-1), Collagen I, fibronectin, and α-SMA. In a culture of human alveolar epithelial cells (A549), long MWCNTs (length: 5–15 μm) were shown to down-regulate the epithelial cell marker E-cadherin and up-regulate the expression of α-SMA protein, both of which indicate EMT [97]. MWCNTs also induced EMT in rat alveolar type II epithelial cells (RLE-6TN), shown by decreased E-cadherin expression and increased fibronectin expression [98]. These findings reveal that myofibroblasts can derive from epithelial cells through EMT when exposed to CNTs in vitro.
The involvement of myofibroblasts in CNT-induced lung fibrosis in vivo was addressed in a few recent studies. CNTs stimulate the expression of α-SMA mRNA and protein in the lungs of rodents. One study was focused on the effect of SWCNTs on EMT in the lungs wherein the expression of α-SMA and surfactant protein C (SPC, a marker of alveolar epithelial cells) was used as markers to trace cell differentiation and trans-differentiation [99]. Immunofluorescence staining showed that the α-SMA expression occurred in a portion of hyperplastic SPC positive epithelial cells in the lungs on days 28 and 42 post-exposure to SWCNTs; whereas, flow cytometry analysis revealed that about 17, 30, 33, and 29 % of the α-SMA positive cells were SPC positive on days 14, 28, 42 and 56 post-exposure to SWCNTs, respectively. These data demonstrate that a subset of α-SMA positive cells were differentiated from the epithelium-derived fibroblasts under SWCNT exposure. Although the overall effect of SWCNTs on α-SMA expression and myofibroblast differentiation in the lungs was not assessed, this study was the first to report α-SMA induction in CNT-exposed lungs. In a separate study, the toxicity of functionalized SWCNTs in mouse lungs was analyzed [100]. The protein level of α-SMA was significantly increased in lung tissues on day 14 post-exposure to certain surface-modified and functionalized SWCNTs. Therefore, SWCNTs are capable of inducing myofibroblast transformation in the lungs, as shown by increased number of α-SMA positive cells in lung tissues. In another report, the induction of EMT by short (length: 350–700 nm) and long (length: 5–15 μm) MWCNTs was investigated in mouse lungs with α-SMA as a marker of the mesenchymal cells [97]. The percentage of SPC positive cells stained for α-SMA in the lungs was significantly elevated by long, but not short, MWCNTs on days 28 and 56 post-exposure. Thus, MWCNTs induce α-SMA expression in SPC positive cells in the lungs through EMT in a fiber length-dependent manner.
In an attempt to assess the effect of CNTs on myofibroblast transformation during lung fibrosis directly, we conducted a study in mouse lungs exposed to MWCNTs (XNRI MWNT-7, median length 3.86 μm, mean diameter 49 nm, average surface area 26 m2/g, total metal 0.78 %), a potent fibrogenic agent that induces rapid-onset lung fibrosis in mice [23, 24, 43]. We found that a single dose pharyngeal aspiration of MWCNTs (40 μg) significantly induced the expression of α-SMA protein and increased the number of α-SMA positive cells in mouse lungs on days 1, 3, 7 and 14 post-exposure. The increases were especially apparent in the interstitial fibrotic foci where MWCNTs deposited, as revealed by immunohistochemistry and immunofluorescence1. Representative images showing the induced expression of α-SMA on day 7 post-exposure are presented in Fig. 3, which demonstrate, for the first time, that MWCNTs, exemplified by XNRI MWNT-7, can remarkably stimulate myofibroblast transformation for the formation of fibrotic foci during lung fibrosis in vivo.
Fig. 3.
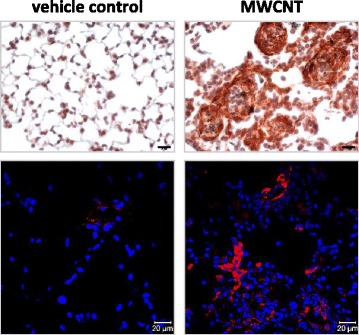
α-SMA expression induced by MWCNTs in mouse lungs. Pulmonary exposure to MWCNTs (XNRI MWNT-7, 40 μg) for 7 days strongly induces α-SMA expression. α-SMA expression in well-formed fibrotic foci is shown by immunohistochemistry (upper panel) and in less well-formed, early stage fibrotic foci by immunofluorescence (lower panel), respectively (scale bar: 20 µm; for immunofluorescence, red: α-SMA staining, blue: nuclear DAPI staining)
Several ECM proteins other than α-SMA typically expressed and produced by myofibroblasts during organ fibrosis display increased expression in CNT-exposed lungs, indicating that the myofibroblasts under CNT exposure are active and functional with regard to fibrosis development. A number of studies using traditional methods of collagen analysis demonstrate remarkably increased amounts of collagen fibers in CNT-exposed lungs, exemplified by the investigations listed in Table 1 with references included therein. Elevated expression and deposition of ECM proteins in the interstitial tissues of CNT-exposed lungs, especially the fibrotic foci where CNTs deposited, were also observed by molecular biology techniques. In one study, long MWCNTs (length: 20–50 μm), but not short MWCNTs (length: 0.5–2 μm), induced the expression of Collagen I mRNA on day 30 and Collagen III mRNA on day 7 and day 30 post-exposure in rat lungs [52]. Certain forms of functionalized SWCNTs significantly increased the levels of Collagen I and Collagen III in mouse lung tissues on day 14 post-exposure [100]. We recently demonstrated that MWCNTs (XNRI MWNT-7, 40 μg) caused significantly elevated mRNA expression of Col1a1 and Col1a2, the genes encoding the pro-alpha1 and pro-alpha2 chains of Collagen I, on days 3 and 7 post-exposure in mouse lungs [24, 101]. The treatment also dramatically increased the production and accumulation of Collagen I in interstitial lung tissues on days 1, 3, 7 and 14 post-exposure. A dose-dependence study showed that the induction of Collagen I accumulation by MWCNTs occurred at low doses, i.e., 5 and 20 μg, on day 7 post-exposure [101]. Additionally, it was detected that the expression of fibronectin mRNA was significantly increased on day 3 and day 7, and the level of fibronectin protein was markedly elevated on day 7, in mouse lungs exposed to MWCNTs (XNRI MWNT-7, 40 μg) [24].
Table 1.
Increased pulmonary collagen deposition by CNTs as revealed by traditional assays
| CNT | Length | Animal | Exposure | Method | Reference |
|---|---|---|---|---|---|
| SWCNT inhalation | 0.1–1 μm | mouse | 1–28 days | Sircol soluble collagen assay | [175] |
| Picro-Sirius Red staining | |||||
| 1–3 μm | mouse | 1 y | Sircol soluble collagen assay | [50] | |
| SWCNT intratracheal injection | 0.5–1.5 μm | mouse | 14 days | Masson’s Trichrome staining | [100] |
| SWCNT pharyngeal aspiration | 1–3 μm | mouse | 7 days | Sircol soluble collagen assay | [176] |
| 5–15 μm | mouse | 14–56 days | Hydroxylproline assay | [99] | |
| Masson’s Trichrome staining | |||||
| 1–3 μm | mouse | 1 y | Sircol soluble collagen assay | [50] | |
| MWCNT inhalation | 3.86 μm | mouse | 2–12 days | Masson’s Trichrome staining | [43] |
| 0.3–50 μm | mouse | 1–98 days | Masson’s Trichrome staining | [47] | |
| 4.3 μm | mouse | 1–336 days | Picro-Sirius Red staining | [27] | |
| 5.5–6.2 μm | rat | 90 days | Masson’s Trichrome staining | [46] | |
| MWCNT intratracheal injection | 20–50 μm | rat | 1–30 days | Picro-Sirius Red staining | [52] |
| 0.7 μm | rat | 60 days | Hydroxylproline assay | [45] | |
| 5.9 μm | Masson’s Trichrome staining | ||||
| MWCNT pharyngeal aspiration | 3.86 μm | mouse | 1–14 days | Masson’s Trichrome staining | [24] |
| Picro-Sirius Red staining | |||||
| 0.5–40 μm | mouse | 28 days | Masson’s Trichrome staining | [55] | |
| 3.86 μm | mouse | 1–56 days | Picro-Sirius Red staining | [23] |
Induction of the platelet-derived growth factor receptor-β (PDGFR-β) expression is another feature of myofibroblast activation during fibrosis in several organs including the liver, kidney and lung [102–105]. By using a double-fluorescent Pdgfrb-Cre reporter mouse strain, it was demonstrated that, in the lungs following bleomycin treatment for 28 days, a marked expansion of the reporter cells occurred in fibrotic regions, in which almost all the reporter cells expressed PDGFR-β and most of the cells expressed α-SMA, indicating that a high percentage of PDGFR-β-expressing cells are lung myofibroblasts [105]. Therefore, an induced expression of PDGFR-β is a marker for myofibroblast activation during fibrogenesis. We examined the level of PDGFR-β in MWCNT-induced lung fibrosis and demonstrated that MWCNTs (XNRI MWNT-7, 40 μg) remarkably increased the PDGFR-β expression and the number of PDGFR-β positive cells in mouse lungs on day 7 post-exposure, especially in interstitial fibrotic foci1. These findings further support the activation of myofibroblasts by CNTs in the lungs in vivo.
Candidate mediators and mechanisms for CNT-induced myofibroblast differentiation and function
A number of signaling molecules and cellular processes induced by CNTs have been suggested to play important roles in myofibroblast differentiation and activation. In this section, we discuss evidence supporting their involvement and their potential mechanisms in the regulation of myofibroblast formation and behaviors in CNT-induced lung fibrosis with the goal to gain insights into the molecular underpinnings of induced lung fibrosis.
TGF-β1
TGF is a superfamily of more than sixty structurally related growth factors that regulate many different physiological and disease processes across species. TGF-β1, along with its isoforms TGF-β2 and 3, is a prototype of the TGF superfamily. TGF-β1 inhibits the proliferation of most types of cells and induces the apoptosis of epithelial cells; conversely, it stimulates mesenchymal cells to proliferate and differentiate into myofibroblasts, which prompts wound healing or tissue fibrosis in various organs [106]. Evidence supports the notion that TGF-β1 is arguably the most predominant pro-fibrotic growth factor in vivo. For instance, TGF-β1 expression is elevated in lung fibrotic lesions, such as IPF and bleomycin-induced lung fibrosis [90–92]; TGF-β1 induces fibroblast activation in vitro and overexpression of active TGF-β1 leads to persisting lung fibrosis in vivo [107, 108]; knockout of the TGF-β1 gene in mice causes severely impaired wound repair alongside severe wasting, generalized inflammation, and tissue necrosis leading to organ failure and death, whereas the epithelium-specific deletion of TGF-β receptor type II protects mice from bleomycin-induced lung fibrosis [109, 110]; lastly, blocking TGF-β1 signaling ameliorates lung fibrosis in animal models [111, 112]. Relevant to this review, TGF-β1 has been recognized as one of the most important endogenous regulator to drive myofibroblast differentiation and activation, because it directly controls the de novo expression of α-SMA and the induced expression of ECM proteins including collagens and fibronectin; moreover many fibrogenic signals activate myofibroblast functions by modulating the expression and/or activation of TGF-β1, or by cross-interacting with TGF-β1 signaling in and outside of the cell (Fig. 4a) [34, 41].
Fig. 4.
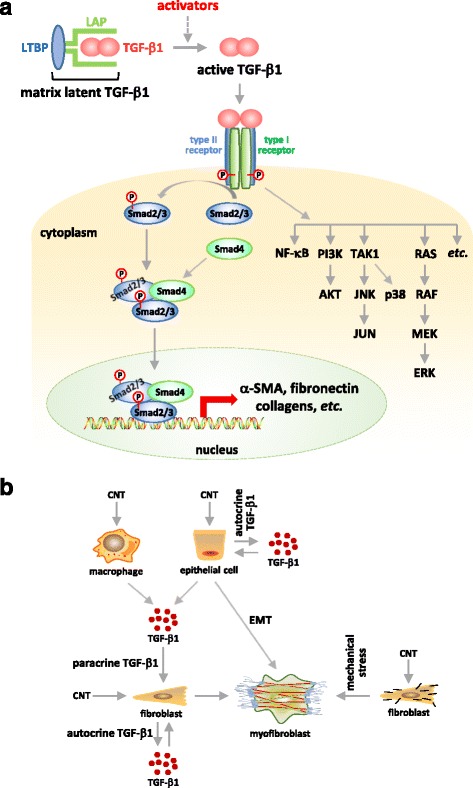
Regulation of myofibroblast formation by TGF-β1. a Schematic presentation of TGF-β1 signaling in myofibroblast formation. Upon stimulation, latent TGF-β1 is activated and active TGF-β1 is released to bind to its receptors on the cell surface to drive the Smad-dependent pathway, which directly up-regulates the transcription of fibrotic genes encoding α-SMA, collagens, and fibronectin. Binding of active TGF-β1 to its receptors also elicits a number of Smad-independent pathways, such as the PI3K-AKT signaling, which may promote myofibroblast differentiation and function. b Role of TGF-β1 in CNT-stimulated myofibroblast differentiation. CNTs induce the production and secretion of TGF-β1 by macrophages and epithelial cells, which serves as a paracrine factor to stimulate fibroblast-to-myofibroblast differentiation. CNTs also directly induce fibroblasts to produce and secrete TGF-β1, which functions as an autocrine factor for fibroblasts to differentiate into myofibroblasts. CNTs may directly promote fibroblast-to-myofibroblast differentiation by mimicking the ECM or intracellular collagen fibers to generate mechanical stress. In addition, CNTs stimulate epithelial cells to produce and secrete TGF-β1, which may induce the trans-differentiation of epithelial cells to myofibroblasts via EMT
As a pro-fibrotic growth factor, TGF-β1 can be induced by injury and other fibrosis-stimulating signals from several types of cells including bone marrow-derived cell lineages, such as macrophages, neutrophils, and T lymphocytes, as well as structural cells, such as airway epithelial cells, endothelial cells, and mesenchymal cells, i.e., fibroblasts and myofibroblasts [34, 90, 113–115]. Newly synthesized TGF-β1 is confined within the latency-associated peptide (LAP) and is further associated with the latent TGF-β binding protein (LTBP). This large latency complex (LLC) is secreted into the ECM where it anchors to the ECM by binding to fibronectin and Fibrillin 1 through LTBP. This configuration keeps TGF-β1 in a latent state that needs to be released from the complex to be activated. Indeed, cumulative evidence reveals that, in addition to induced expression of TGF-β1 mRNA and protein, much of the regulation of TGF-β1 in physiologic and pathologic processes centers on the activation of the latent TGF-β1 [92, 94]. Upon activation, TGF-β1 binds to its receptors to form a complex consisting of a TGF-β1 homodimer, two TGF-β type I receptors, and two TGF-β type II receptors on the cell surface. The formation of the TGF-β1-receptor complex leads to the activation of the Smad-dependent transcription of fibrotic genes encoding α-SMA, collagens, fibronectin, etc., as well as Smad-independent signaling, to drive myofibroblast differentiation from fibroblasts (Fig. 4a).
CNTs have been shown to increase the level of TGF-β1 protein in vitro and in vivo in several types of cells (Fig. 4b). Treatment of mouse leukemic monocyte-macrophage RAW264.7 cells with either MWCNTs or SWCNTs significantly induced the expression of TGF-β1 mRNA and protein; moreover, the cell-free and CNT-free supernatant of a conditioned medium from the treatment stimulated human normal lung fibroblasts (WI38-VA13) to differentiate into myofibroblasts, as indicated by the induced expression of α-SMA [95, 96]. In separate studies, RAW264.7 macrophages exposed to SWCNTs displayed a potent induction of secreted TGF-β1 in the culture medium [22], and RAW264.7 macrophages exposed to long MWCNTs (length: 20–50 μm) had a remarkably increased expression of TGF-β1 mRNA as well as an elevated level of secreted TGF-β1 in the culture medium [52]. Induced expression of TGF-β1 mRNA and protein was also observed in human normal bronchial epithelial cells (BEAS-2B) treated with either MWCNTs or SWCNTs; and the level of secreted TGF-β1 in the culture medium of BEAS-2B cells was increased by SWCNTs [95, 96, 116]. In the above cases, TGF-β1 produced from macrophages and epithelial cells stimulates the differentiation of fibroblasts to myofibroblasts as a paracrine factor.
CNTs also induce the production and secretion of TGF-β1 from fibroblasts directly. SWCNTs induced TGF-β1 expression and promoted rat vascular adventitial fibroblasts to transform to myofibroblasts, indicated by the gained expression of SM22-α (smooth muscle protein 22-α), which is a smooth muscle cell-specific protein and a marker of myofibroblast differentiation [117]. SWCNTs induced TGF-β1 secretion and activation in a dose-dependent manner in human lung fibroblasts (CRL-1490) [118]. Long and short SWCNTs with median lengths of 12.31 and 1.13 μm, respectively, induced TGF-β1 expression and secretion in normal human lung fibroblasts (NHLF); but the induction by the long SWCNTs was significantly more pronounced than that by the short ones [119]. SWCNTs and MWCNTs increased the protein expression of TGF-β1 and Collagen I in CRL-1490 cells as well as the level of secreted TGF-β1 in the culture medium [120]. MWCNTs also induced the expression of α-SMA mRNA and protein in mouse embryonic fibroblasts (NIH 3T3) [98]. In the above cases, CNT exposure activated fibroblasts to produce TGF-β1, which functions as an autocrine factor to induce α-SMA expression and promote the fibroblast-to-myofibroblast differentiation.
Elevation of the TGF-β1 protein level in the bronchoalveolar lavage (BAL) fluid provides a measurement of TGF-β1 induction in vivo. SWCNTs remarkably increased the level of TGF-β1 in the BAL on day 7 post-exposure in mice [22]. SWCNTs and MWCNTs elevated the level of TGF-β1 in the BAL on day 21 post-exposure in mice [56, 116, 121, 122]. Long MWCNTs (length: 20–50 μm) induced an elevated level of TGF-β1 in the BAL on day 1 and in alveolar macrophages on day 7, as well as an increased level of Smad2 phosphorylation, a marker of the activation of TGF-β1 signaling, on day 7 post-exposure in rat lungs [52]. These findings implicate TGF-β1 signaling in CNT-induced lung fibrogenesis. In a separate study, long MWCNTs (length: 5–15 μm), but not short MWCNTs (length: 350–700 nm), increased the level of TGF-β1 in the BAL on days 7 and 28 post-exposure; moreover, the percentage of SPC and α-SMA double positive cells in mouse lungs was significantly elevated by the long MWCNTs, but not the short MWCNTs, on days 28 and 56 post-exposure, suggesting that the induction of TGF-β1 by MWCNTs leads to α-SMA expression in SPC positive cells in the lungs, possibly through the trans-differentiation of alveolar epithelial cells to myofibroblasts [97].
The widely used and well characterized MWCNTs in the study of CNT-induced lung fibrosis, XNRI MWNT-7, are intermediate in length (median length: 3.86 μm) compared with the long and short MWCNTs discussed above. MWNT-7 CNTs have been shown to significantly increase the levels of TGF-β1 protein in mouse BAL on days 3, 7 and 14 post-exposure, and in mouse lung tissues, especially in the regions where fibrosis occurs, on day 7 post-exposure [24]. These findings demonstrate a remarkable induction of TGF-β1 as an acute response to MWCNTs, which might control myofibroblast differentiation and activation, indicated by highly induced expression of α-SMA, Collagen I, and fibronectin during the acute phase response, in MWCNT-exposed lungs [24]1. Taken together, these studies support that TGF-β1 is significantly induced and may play a critical role in promoting myofibroblast differentiation and activation in CNT-triggered lung fibrosis.
A number of mechanisms have been described to account for the activation of the latent TGF-β1 stored in LLC, some of which have implications for CNT-induced lung fibrosis. Activation of the latent TGF-β1 can occur (a) by way of cellular acidification that denatures LAP to release TGF-β1, (b) via ROS that oxidize LAP to perturb the interaction between LAP and TGF-β1, (c) through thrombospondin-1 (TSP-1) that directly interacts with the latent complex to prevent it from binding matured TGF-β1, (d) by proteases including plasmin, MMP-2 and MMP-9, tryptase, elastase, and thrombin that activate TGF-β1 by proteolytic degradation of LLC and then LAP of the latent TGF-β1 complex, and (e) through integrins, such as the epithelial cell-specific αvβ6, which mediates matrix contraction to release active TGF-β1 close to the cell surface, and the fibroblast αvβ8, which presents latent TGF-β1 to a membrane-bound protease (i.e., MT1-MMP) to activate TGF-β1 [123, 124]. It is known that both MWCNTs and SWCNTs stimulate the production of ROS in vitro and in the lungs [95, 96, 101, 125], which would induce the activation of TGF-β1 in a manner analogous to that of asbestos [126, 127]. In recent studies, we have shown that the lung expression of TSP-1 was significantly induced by MWCNTs in vivo [101], whereas that of MMP-2 was induced during lung fibrosis from exposure to silica, paraquat, or bleomycin [28]. Direct evidence supporting a role of these mediators in the activation of latent TGF-β1 in CNT-induced lung fibrosis awaits further investigation.
Certain SWCNTs and MWCNTs have been shown to induce EMT to generate myofibroblasts in the lungs, which is accompanied by the activation of TGF-β1 signaling [97, 99]. These studies suggest that TGF-β1 may induce myofibroblast formation through EMT in CNT-exposed lungs. However, studies on the contribution of myofibroblasts derived via EMT in IPF and in bleomycin-induced lung fibrosis, as well as in fibrosis occurring in other organs such as kidney and liver, are controversial; thus, the role of EMT in fibrosis remains uncertain currently [88, 128]. Evaluation on whether and, if so, how much the TGF-β1-regulated myofibroblast trans-differentiation through EMT contribute to CNT-induced lung fibrosis might provide an answer to this question with regard to the phenotype, mechanism, and function of EMT.
PDGF
The platelet-derived growth factor (PDGF) represents another important pro-fibrotic growth factor that confers multiple functions in organ fibrosis including human and experimental lung fibrosis [128–130]. For instance, overexpression of PDGF-B (PDGF subunit B) in mouse lungs induced severe fibrosis [131]. Instillation of rats with bleomycin caused elevated levels of PDGF-AA (homodimer of PDGF subunit A) and PDGF-BB (homodimer of PDGF subunit B) in the BAL fluid; moreover, the concentrated BAL showed a growth-promoting activity toward lung fibroblasts that can be partially blocked with anti-PDGF-BB (64 %) or anti-PDGF-AA (15 %) antibodies [132]. PDGF is a potent mitogen for cells of a mesenchymal origin, such as fibroblasts, both in vitro and in vivo; and it boosts the recruitment and proliferation of fibroblasts, promotes the differentiation of myofibroblasts from fibroblasts and other types of cells, and increases the production of ECM proteins from myofibroblasts during the fibrosis of various organs [104, 128, 129, 133–135].
PDGF has been shown to be induced by CNTs in the lungs in a number of recent studies. PDGF-AA was significantly increased in mouse BAL on day 21 post-exposure to MWCNTs in a dose-dependent manner [56, 121]. During the early phase response to MWCNTs, PDGF-AA was shown to be significantly increased in the BAL on days 1, 3 and 7, and in lung tissues on day 7, post-exposure to XNRI MWNT-7 by aspiration in mice [24]. In a separate study, PDGF-AA was induced in the BAL on day 1 post-inhalation exposure to MWCNTs in mice [25]. Also, a study in rats revealed that PDGF-AA was induced in the BAL on day 1, and in lung tissues on day 1 and day 21, post-exposure to MWCNTs, which was boosted by co-stimulation with bacterial lipopolysaccharides [136]. These findings suggest the possibility for PDGF to play a role in promoting myofibroblast differentiation and activation in the lungs exposed to CNTs. However, further detailed studies are needed to ascertain this posit.
Th2 cytokines IL-4 and IL-13
The T helper 2 (Th2)-type cytokines IL-4 and IL-13 have been studied intensively in a variety of fibrotic diseases and animal models, and have been demonstrated to function as potent pro-fibrotic mediators to drive fibrosis development [8, 128, 137, 138]. For instance, increased levels of IL-4 were detected in patients with IPF or cryptogenic fibrosing alveolitis [139, 140]; and inhibition of IL-4 by neutralizing antibodies or inhibitors reduced liver fibrosis and dermal fibrosis in mice [141, 142]. IL-13 levels were significantly higher in IPF patients than in normal controls [143]; overexpression of IL-13 in mouse lungs induced subepithelial airway fibrosis [144]; and inhibition of IL-13 by neutralizing antibodies decreased collagen deposition in mouse lungs exposed to bleomycin [145].
IL-4 and IL-13 receptors are located on the cell surface of a number of mouse and human fibroblast subpopulations [146, 147]. In multiple in vitro studies, it was shown that, under the stimulation of IL-4 or IL-13, fibroblasts displayed enhanced proliferation and differentiation, and increased production of α-SMA and ECM proteins, such as type I and type III collagens and fibronectin [146–152], which indicates that IL-4 and IL-13 signaling may promote fibrosis by stimulating fibroblast-to-myofibroblast differentiation and by enhancing tissue remodeling. The Th2-type response may also promote myofibroblast differentiation by activating TGF-β1. It has been reported that IL-13 activates TGF-β1 in two ways: first, IL-13 induces the production of latent TGF-β1 from macrophages [153]; second, IL-13 activates TGF-β1 by increasing the expression of proteins that function in the cleavage of LAP, which keeps TGF-β1 as an inactive form, such as matrix metalloproteinases (MMPs) and cathepsins [153–155]. Taken together, cumulative evidence reveals that IL-4 and IL-13 play critical roles in the initiation and development of fibrosis, which are in part mediated by inducing myofibroblast differentiation.
We recently demonstrated that IL-4 and IL-13 expression and signaling were significantly induced by MWCNTs (XNRI MWNT-7) in mouse lungs [156]. In a genome-wide microarray gene expression study of mouse lung tissues, Th2-driven immune responses were preferentially enriched. In particular, the activation of IL-4 and IL-13 signaling, the center of Th2-type responses, was a dominant effect induced by MWCNTs on day 7 post-exposure. Time-course studies detected that IL-4 was significantly induced by MWCNTs at the mRNA level on days 1, 3, 7 and 14, and at the protein level on days 3, 7 and 14, post-exposure. IL-13 was significantly induced at the mRNA and protein levels on days 3, 7 and 14 post-exposure. In addition, a panel of signature downstream target genes of IL-4/IL-13 signaling, such as Il4i1, Chia, and Ccl11/Eotaxin, were remarkably induced by MWCNTs at both the mRNA and protein levels, further supporting the activation of IL-4/IL-13 signaling. The increased expression of IL-4 and IL-13 during the early phase fibrotic response, i.e., days 1 to 14 post-exposure, strongly suggests the potential for IL-4 and/or IL-13 to play a role in promoting myofibroblast differentiation in MWCNT-exposed lungs to drive fibrosis development.
ROS
Oxidative stress reflects a cellular stress state that occurs when the production of ROS and antioxidant defense are out of balance, which causes multiple damages to the cell, such as DNA strand breaks and DNA mutation, protein peptide chain breaks, and lipid peroxidation, leading to cell death in the severe case [157–159]. ROS have been implicated in promoting fibrosis in multiple organs, such as the lung, liver, and kidney, through a number of mechanisms, as have been discussed in several recent reviews [160–167]. Significantly, it has been established that ROS promote the transformation of fibroblasts to myofibroblasts by interacting with the TGF-β1 signaling pathway [168–172]. ROS can augment the expression and secretion of TGF-β1 and activate the latent TGF-β1 to become active and functional. In a reciprocal manner, TGF-β1 increases ROS production, mainly through the induction of NOX4 expression. The NOX4-dependent production of hydrogen peroxide (H2O2) is essential for TGF-β1-mediated myofibroblast differentiation and ECM production. These findings clearly demonstrate a necessary role of ROS in myofibroblast differentiation and activation.
In the studies on CNT-induced toxicity, numerous observations consistently demonstrate oxidative stress as a predominant mechanism to link CNT exposures to their toxicological and pathological outcomes. A large number of in vitro cell culture studies support that both SWCNTs and MWCNTs directly stimulate ROS production in various types of cells, such as macrophages, fibroblasts, and bronchial and alveolar epithelial cells, as summarized in two recent reviews [18, 173]. For instance, MWCNTs were shown to stimulate the production of ROS in macrophages to activate NF-κB signaling [95], whereas SWCNTs were found to stimulate both fibroblast proliferation and angiogenesis via the induction of ROS production [118]. Importantly, a few in vivo studies have confirmed that CNT exposure results in oxidative stress in tissues. Exposure of mice to SWCNTs led to a dose-dependent accumulation of 4-hydroxy-2-nonenal (4-HNE, a lipid peroxidation biomarker) in the BAL as early as 1 day post-exposure, and a dose- and time-dependent depletion of glutathione (GSH, a major antioxidant) in the lungs, demonstrating the presence of oxidative stress upon exposure to SWCNTs [22]. Another indicator of oxidative stress, heme oxygenase 1 (HO-1), has also been shown to have an increased level in mouse lungs, aorta, and heart on day 7 post-exposure to SWCNTs [174]. In the NADPH oxidase knockout mice that lack the gp91phox (Nox2) subunit of a NOX enzymatic complex and are deficient in ROS production, lung fibrosis induced by SWCNTs or MWCNTs was remarkably attenuated, compared with the wild-type control mice [122, 125, 175, 176].
We analyzed the roles of ROS in MWCNT (XNRI MWNT-7)-induced pathologic effects on the lungs, by using the nuclear factor erythroid 2-related factor 2 (Nrf2)-deficient mice, which have an elevated oxidative stress due to the lack of the defense against oxidative stress mediated by Nrf2 [177, 178]. Under exposure to MWCNTs, several markers indicative of oxidative stress, including ROS production in alveolar macrophages, the levels of DNA oxidation indicators 8-OHdG (8-hydroxy-2′-deoxyguanosine) and γH2AX (phospho-Histone H2A.X (Ser139)) and the level of 4-HNE in lung tissues, were remarkably increased in the lungs; and the increases were markedly more pronounced in Nrf2 knockout lungs than in wild-type lungs. There was also a remarkably higher level of MWCNT-induced lung fibrosis in Nrf2 knockout lungs than in wild-type lungs [101]. Taken together, these in vivo studies strongly support that CNTs induce ROS production in the lungs, and ROS play an important role in the initiation and progression of CNT-induced lung fibrosis, suggesting that CNT-induced ROS can serve as an enhancer to promote myofibroblast differentiation in the lungs.
Role of inflammation and pro-inflammatory cytokines
The role of inflammation in organ fibrogenesis is complex [8]. Acute inflammation precedes and sometimes accompanies fibrosis in most, if not all, lung fibrosis induced by exposure to fibrogenic and cytotoxic agents including chemicals, microbes, and particles and fibers. On the other hand, fibrosis in the absence of apparent tissue injury may occur without a prominent inflammatory phenotype; moreover, anti-inflammation alone does not effectively prevent or block the development of fibrosis. It is believed that the role of inflammation in fibrosis development varies among fibrosing diseases; but once present, increased inflammatory infiltration and secretion create a milieu rich in pro-fibrotic growth factors, cytokines, and chemokines that foster the development of fibrosis, in part mediated by priming or promoting fibroblasts to differentiate into myofibroblasts. In this context, a panel of pro-inflammatory cytokines including TNF-α, IL-1α, IL-1β, and IL-6 have been shown to be pro-fibrotic factors in both mouse and human lung fibrosis models [18, 33].
TNF-α and IL-1β are among the earliest cytokines recognized as pro-fibrotic factors. Overexpression of TNF-α in mouse lungs resulted in spontaneous lung fibrosis [179]. TNF-α appears to play important roles in various fibrosis animal models, such as bleomycin- or silica-induced lung fibrosis and CCl4-induced liver fibrosis, as well as a number of human fibrotic diseases, such as IPF and asbestosis [180–183]. IL-1β and its receptors have been shown to promote fibrosis in different types of organ fibrosis, whereas inhibition of IL-1β signaling reduces the fibrosis development, illustrating a critical role of IL-1β signaling in organ fibrosis [184–191]. The IL-1α-deficient mice exhibited reduced collagen deposition in lung tissues in response to bleomycin treatment [192]. Enhanced IL-6 level was detected in the BAL of IPF patients [193], and IL-6 signaling was found to be key to driving fibrosis in a mouse model of acute peritoneal inflammation [194]. Moreover, a number of studies demonstrate that these cytokines can stimulate mesenchymal cells from multiple organs to express α-SMA [128, 195–199]. How these pro-inflammatory and pro-fibrotic cytokines induce myofibroblast differentiation and activation at the molecular level remains to be delineated.
Upon exposure to CNTs, the lungs elicit an acute inflammatory response as a well-characterized feature of CNT-induced lung toxicity. In many cases, acute inflammation precedes and accompanies CNT-induced lung fibrosis. A number of studies have detected increased expression and production of these pro-inflammatory cytokines in CNT-exposed lungs. The level of TNF-α in the BAL was significantly increased by SWCNTs on day 1 post-exposure in mice [22], by MWCNTs at 12 h or on days 1, 3 and 7 post-exposure in mice [24, 55, 200–202], and by MWCNTs on day 3 post-exposure in rats [45]. The level of IL-1α in the BAL was significantly increased by MWCNTs (XNRI MWNT-7) on days 1, 3, 7 and 14 post-exposure in mice [24]. The level of IL-1β in the BAL was significantly increased by SWCNTs at 40 h or on days 1, 3, 7 and 28 post-exposure in mice [22, 116], and by MWCNTs on days 1, 3 and 21 post-exposure in mice [24, 55, 122, 201]. The level of IL-6 in the BAL was significantly increased by MWCNTs at 12 h or on days 1, 3, 7, 14 and 28 post-exposure in mice [24, 55, 200, 202]. These pro-inflammatory cytokines with increased production from the acute innate immune response may contribute to the induction of myofibroblast differentiation in CNT-exposed lungs.
While induced expression of pro-inflammatory cytokines remains a major mechanism of up-regulation of their signaling, the activation of inflammasome processing of cytokines IL-1β and IL-18 has been increasingly recognized as a critical process for a variety of host responses and diseases including fibrosis [203–205]. Inflammasomes are large protein complexes in the cytoplasm that sense extracellular and intracellular signals to initiate innate immune responses to microbe exposure and tissue injury. Particulate and fibrous materials, such as silica, asbestos, cholesterol crystals, and CNTs, have been shown to activate inflammasomes, mainly the NLRP3 inflammasome, to mediate the proteolytic maturation of IL-1β and IL-18 [188, 205–207]. Activation of the NLRP3 inflammasome increases the formation of myofibroblasts in bleomycin-induced skin fibrosis [204]. Given the broad and critical roles of IL-1β, it is believed that inflammasome activation plays an important role in myofibroblast formation and function in CNT-induced lung fibrosis. However, direct evidence supporting this notion awaits further investigation.
Role of proliferation
During lung fibrosis, fibroblasts undergo an elevated proliferation through multiple mechanisms, which would promote the formation of fibroblastic foci and the destruction of lung tissues. As fibroblasts are the major progenitor cells of myofibroblasts, it is rational to posit that an increase in the number of fibroblasts leads to a higher number of myofibroblasts and the accumulation of myofibroblasts in fibrotic foci.
The induction of fibroblast proliferation by CNTs has been observed in a number of in vitro studies. SWCNTs induced the proliferation of human lung fibroblasts (CRL-1490) in a dose- and time-dependent manner, which was mediated by the ROS-regulated activation of p38 MAPK (mitogen-activated protein kinase) and the induction of TGF-β1 and VEGF (vascular endothelial growth factor) [118, 208]. MWCNTs stimulated the proliferation of multiple types of fibroblasts in tissue culture in a dose- and physicochemical property-dependent manner [51, 209]. Furthermore, MWCNTs directly promoted the proliferation of mouse lung fibroblasts (MLg cells) primed with a low concentration of growth factor TGF-β1 or PDGF, by prolonging the phosphorylation of the extracellular signal-regulated kinase (Erk) 1/2 [210]. CNTs may stimulate fibroblast proliferation by inducing the secretion of soluble factors from epithelial cells, which is NLRP3 inflammasome-dependent, but TGF-β1-independent [211]. These studies demonstrate that certain types of CNTs are capable of stimulating fibroblast proliferation directly and thereby contribute to CNT-induced lung fibrosis.
The tissue inhibitor of metalloproteinase 1 (TIMP1) is highly induced and is secreted into the ECM from macrophages and mesenchymal cells during lung fibrosis. Using the Timp1-deficient mice, we demonstrated that TIMP1 plays a critical role in the development of MWCNT-induced lung fibrosis1. In the lungs of wild-type mice, MWCNTs (XNRI MWNT-7) remarkably increased the proliferation of fibroblasts, indicated by the expression of cell proliferation markers Ki-67 (marker of proliferation Ki-67) and PCNA (proliferating cell nuclear antigen). However, this induction was significantly attenuated in the lungs of Timp1-deficient mice. Accordingly, the MWCNT-induced fibrotic responses, including the formation of fibrotic foci, the differentiation of myofibroblasts, and the production and deposition of ECM proteins, such as Collagen I and fibronectin, were significantly higher in wild-type mice than in Timp1-deficient mice. These findings strongly suggest that MWCNTs stimulate fibroblast proliferation in the lungs and thereby promote myofibroblast differentiation from the enriched fibroblast pool to boost fibrosis. Mechanistic analysis demonstrated that MWCNT-induced fibroblast proliferation might be mediated by the formation of a TIMP1/CD63/integrin β1 complex on the surface of fibroblasts, which promotes the Erk1/2 phosphorylation and activation in fibroblasts in the lungs. These findings establish a direct mechanistic link among MWCNT exposure, fibroblast proliferation, myofibroblast differentiation, and lung fibrosis in vivo.
Role of tissue stiffness, mechanical force, and receptors
In the model of wound healing, the formation of myofibroblasts generally becomes a major event at 1 week after tissue injury, which correlates with a significantly increased tissue tension or stiffness [67]. In fact, it has been shown that the threshold stiffness for the de novo expression of α-SMA in stress fibers ranges around 20,000 Pa, which is about 200 to 2000-fold higher than that of the early wound provisional ECM (i.e., 10–100 Pa) [212]. This increase in tissue stiffness and myofibroblast formation correlates with the increased activation of TGF-β1 protein within the tissue matrix. Mechanistic studies demonstrate that, indeed, the increased stiffness is necessary for the activation of the latent TGF-β1 and the release of activated TGF-β1 from the large latency complex in the vicinity of the site of wound healing or fibrosis [41]. In particular, the mechanical tension generated through the contraction of myofibroblasts is a well-recognized mechanism for the activation of the latent TGF-β1 in the matrix [213]. Based on these findings, it is believed that the CNT-induced tissue injury and fibrotic changes stimulate myofibroblast contraction to increase tissue stiffness, which in turn boosts the differentiation of fibroblasts into myofibroblasts by activating the latent TGF-β1 locally. This chain of events sets in motion a positive feedforward response among myofibroblast formation, contraction, tissue tension, and TGF-β1 activation to drive organ fibrosis. However, a direct measurement of tissue stiffness of the lungs exposed to CNTs in relation to fibrosis and myofibroblast activation is needed to prove this notion.
At the molecular level, CNTs may modulate cellular and ECM mechanical properties through several means. In addition to stimulating the tissue to release soluble factors, such as TGF-β1, discussed above, CNTs may induce the contraction and increase the cellular tension of cells by interfering with the intracellular contractile structures once inside the cell, as some CNT fibers resemble the cytoskeletal or contractile filaments in size and shape. Alternatively, CNT fibers may directly interact with cell surface receptors, such as the pattern recognition receptors. SWCNTs have been observed inside rat vascular adventitial fibroblasts after exposure for 24 and 48 h [117]. MWCNTs were found to be accumulated on the surface of NIH 3T3 fibroblasts 3 h post-exposure, and some MWCNTs entered the cell by way of endocytosis 24 h post-exposure [98]. Moreover, MWCNTs were shown to bind to BMPR2 on the surface of myoblasts to modulate their differentiation [53]. These findings raise the possibility of a direct effect of CNTs on fibroblasts and myofibroblasts by way of mechanical activation of the matrix latent TGF-β1 or biochemical activation of cell surface receptor-mediated intracellular signaling, to stimulate the activation and differentiation of fibroblasts into myofibroblasts. In both scenarios, the physicochemical properties of CNTs, such as the nano-scaled diameter, fiber length, surface area and reactivity, and biopersistence, would be critical parameters to influence their stimulatory activities. Although attractive, these possibilities remain to be proven by direct evidence from studies using multiple cellular, molecular, and biophysical means in the future.
Conclusion
The expanding knowledge on the pathological features and molecular mechanisms of CNT-induced lung fibrosis is in agreement with the overall understanding of lung fibrosis derived from certain human fibrotic lung diseases and experimental animal models in a number of ways. This correlation suggests that CNT-induced lung fibrosis can be used as a new animal disease model for studying the molecular mechanisms underlying human fibrotic lung diseases, such as IPF and pneumoconiosis. Emerging evidence reveals that CNTs potently induce and activate myofibroblasts both in vitro and in vivo. Moreover, CNTs are found capable of inducing and activating a number of critical mediators and cell signaling pathways that have been implicated in myofibroblast function and regulation during fibrosis development, as summarized in Fig. 5. It is clear that the activation of myofibroblasts likely represents a critical and common molecular step toward the development of organ fibrosis, which now includes CNT-induced lung fibrosis.
Fig. 5.
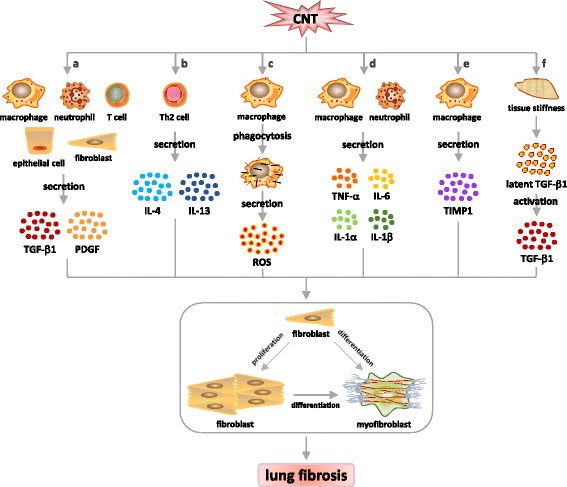
Mediators of CNT-induced myofibroblast differentiation in the lungs. CNTs stimulate multiple mechanisms and mediators capable of promoting myofibroblast formation and function, including a pro-fibrotic growth factors TGF-β1 and PDGF, b Th2 cytokines IL-4 and IL-13, c ROS, d pro-fibrotic cytokines TNF-α, IL-6, IL-1α and IL-1β, e TIMP1, and f tissue stiffness. Activation of these signaling cascades may induce myofibroblast differentiation and activation directly or by boosting fibroblast proliferation to increase the myofibroblast precursor pool, leading to fibrosis in the lungs
Although myofibroblasts have long been recognized as an important group of mesenchymal cells in the development of fibrosis, the contribution of these cells to the fibrotic response induced by fibrogenic particles and fibers, and new materials, such as nanomaterials, has received attention in the field of toxicology only in recent few years. In part, this delayed recognition is due to the difficulty of separating myofibroblasts from fibroblasts during the development of fibrosis with regard to the phenotypes and the available techniques and approaches for analyzing the cells. Indeed, there is a lack of specific markers for identification of myofibroblasts in tissues. Moreover, certain mesenchymal functions during fibrosis are shared between fibroblasts and myofibroblasts, such as the secretion of some ECM proteins. Therefore, it is necessary to use multiple markers, including α-SMA expression, to analyze myofibroblasts and their functions in a specific tissue, time point, and context in which fibrosis takes place. In this respect, the study on CNT-induced lung fibrosis is perhaps advantageous over other lung fibrosis models for the analysis of myofibroblast functions, because the formation of myofibroblasts is exposure-dependent and possibly inducer-specific with respect to the pathological features, mechanisms, and consequences, which can now be readily demonstrated in cultured cells and animals exposed to CNTs.
The fibroblastic response responsible for matrix built-up and scarring is a rather complex and dynamic process, which remains poorly understood to date. From the experimental point of view, the study on myofibroblasts provides an opportunity to unravel the mechanisms underlying the fibroblastic response for the initiation and development of lung fibrosis induced by CNTs, other nanomaterials, and particles and fibers at molecular and cellular levels. From this perspective, the list of mediators and cell signaling pathways summarized above can serve as a reasonable starting point for the mechanistic analysis of myofibroblast regulation and function in fibrosis. Needless to say, further pathological and molecular studies with the aid of genetically engineered mouse strains are required to ascertain the contributions of these candidate mediators and signaling cascades to the onset and pathological outcomes of CNT-induced lung fibrosis in vivo. One caveat to note on this line of research is that these mediators and pathways are not likely to act alone, but work in concert in a highly regulated and time- and context-dependent fashion, to drive the formation and functioning of myofibroblasts leading, ultimately, to fibrosis of the lungs. As such, multiple targets from this myofibroblast-predominant fibroblastic response should be sought after in order to achieve better intervention against lung fibrotic diseases.
Among the identified mediators of myofibroblast activation, TGF-β1 stands out as the most relevant endogenous factor to drive myofibroblast differentiation and function. However, many gaps exist in the understanding of TGF-β1 function and mode of action in the regulation of myofibroblasts and fibrosis induced by CNT exposure. In particular, most toxicological studies on CNT lung fibrosis examined the induction of TGF-β1 mRNA and/or protein expression in cultured cells or in the lungs, which is necessary to establish the involvement of TGF-β1 in CNT toxicity. Several critical questions remain unaddressed. For instance, which signaling pathways and factors mediate the induction of TGF-β1 by CNTs; whether and, if so, how CNTs activate the latent form of TGF-β1 stored in the ECM; and how the activated TGF-β1 controls myofibroblast activation and function upon CNT exposure? The morphologically apparent contractive features of myofibroblasts fittingly explain, at least in part, the inevitable contraction and ultimate scarring of fibrotic tissues in the lungs and other organs. Moreover, the mechano-regulation of matrix remodeling by myofibroblasts appears to be closely correlated with the activation of latent TGF-β1. How this interplay among myofibroblast contraction, tissue stiffness, and TGF-β1 activation occurs to propel CNT-induced lung fibrosis is currently unclear. Apparently, a combination of molecular, biophysical, and genetic approaches is needed to address these questions in future studies.
One implication of the findings from the research on myofibroblasts derives from the notion that organ fibrosis might arise from a failure to suppress the normal repair process of tissue injury to result in the persistent presence and over-functioning of myofibroblasts in fibrotic tissues. It is therefore rational to expect that the information obtained from the study of myofibroblasts and their associated mediators and signaling pathways involved in the pathogenesis of lung fibrosis is likely to generate new insights into both the molecular understanding and the clinical treatment of human fibrotic lung diseases that include IPF, pneumoconiosis, and nanomaterial-induced lung fibrosis.
Acknowledgements
Not applicable.
Funding
The study was supported by a fund to QM from National Institute for Occupational Safety and Health, Health Effects Laboratory Division and the Nanotechnology Research Center.
Availability of data and materials
All data and materials are included in this article.
Authors’ contributions
JD designed and drafted the manuscript. QM revised and finalized the article. Both authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Disclaimer
The findings and conclusions in this report are those of the authors and do not necessarily represent the views of the National Institute for Occupational Safety and Health.
Abbreviations
- 4-HNE
4-hydroxy-2-nonenal
- 8-OHdG
8-hydroxy-2′-deoxyguanosine
- Akt
v-akt murine thymoma viral oncogene homolog
- BAL
Bronchoalveolar lavage
- BMPR2
Bone morphogenetic protein receptor type II
- Ccl11
Chemokine (C-C Motif) ligand 11, or eotaxin
- CDK
Cyclin-dependent kinase
- Chia
Chitinase, acidic, or AMCase
- CNT
Carbon nanotube
- ECM
Extracellular matrix
- ED-A
Extra domain A
- EMT
Epithelial-mesenchymal transition
- Erk
Extracellular signal-regulated kinase
- FoxO3a
Forkhead box O3a
- FSP-1
Fibroblast specific protein-1
- GSH
Glutathione
- GSK-3β
Glycogen synthase kinase-3β
- HO-1
Heme oxygenase 1
- IL
Interleukin
- Il4i1
Interleukin 4 induced 1, or Fig1
- IPF
Idiopathic pulmonary fibrosis
- JNK
c-Jun N-terminal kinase
- Ki-67
marker of proliferation Ki-67
- LAP
Latency-associated protein
- LLC
Large latency complex
- LTBP
Latent TGF-β binding protein
- MAPK
Mitogen-activated protein kinase
- MEK
Mitogen-activated protein kinase kinase
- MMP
Matrix metalloproteinase
- MWCNT
Multi-walled carbon nanotube
- NADPH
Nicotinamide adenine dinucleotide phosphate
- NF-κB
Nuclear factor-κB
- NLRP3
Nucleotide-binding oligomerization domain-like receptor: pyrin domain-containing 3
- NOX
NADPH oxidase
- Nrf2
Nuclear factor erythroid 2-related factor 2
- PCNA
Proliferating cell nuclear antigen
- PDGF
Platelet-derived growth factor
- PDGFR-β
Platelet-derived growth factor receptor-β
- PI3K
Phosphoinositide 3-kinase
- PP2A
Protein phosphatase 2A
- PTEN
Phosphatase and tensin homolog
- RNS
Reactive nitrogen species
- ROS
Reactive oxygen species
- SM22-α
Smooth muscle protein 22-α
- Smad
Sma and Mad related family
- SPC
Surfactant protein C
- SWCNT
Single-walled carbon nanotube
- TAK1
TGF-β-activated kinase 1
- TGF-β1
Transforming growth factor-β1
- Th2
T helper 2
- TIMP1
Tissue inhibitor of metalloproteinase 1
- TNF-α
Tumor necrosis factor-α
- TSP-1
Thrombospondin-1, or Thbs1
- VEGF
Vascular endothelial growth factor
- Wnt
Wingless-type MMTV integration site family
- α-SMA
α-smooth muscle actin
- γH2AX
phospho-Histone H2A.X (Ser139)
Footnotes
Dong J and Ma Q. TIMP1 promotes multi-walled carbon nanotube-induced lung fibrosis by stimulating fibroblast activation and p roliferation. 2016; submitted.
Contributor Information
Jie Dong, Email: wyo6@cdc.gov.
Qiang Ma, Phone: 304-285-6241, Email: qam1@cdc.gov.
References
- 1.Husain AN, Kumar V. The lung. In: Kumar V, Abbas AK, Fausto N, editors. Robbins and Cotran Pathologic Basis of Disease. 7. Philadelphia: Elsevier Saunders; 2005. pp. 711–772. [Google Scholar]
- 2.NIOSH . Health Effects of Occupational Exposure to Respirable Crystalline Silica. DHHS (NIOSH) Publication No. 2002-129. DHHS CDC NIOSH: Cincinnati; 2002. [Google Scholar]
- 3.Morgan WKC, Seaton A. Occupational lung diseases. 3. Philadelphia: W.B. Saunders Company; 1995. [Google Scholar]
- 4.Williamson JD, Sadofsky LR, Hart SP. The pathogenesis of bleomycin-induced lung injury in animals and its applicability to human idiopathic pulmonary fibrosis. Exp Lung Res. 2015;41:57–73. doi: 10.3109/01902148.2014.979516. [DOI] [PubMed] [Google Scholar]
- 5.He X, Wang L, Szklarz G, Bi Y, Ma Q. Resveratrol inhibits paraquat-induced oxidative stress and fibrogenic response by activating the nuclear factor erythroid 2-related factor 2 pathway. J Pharmacol Exp Ther. 2012;342:81–90. doi: 10.1124/jpet.112.194142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dinis-Oliveira RJ, Duarte JA, Sanchez-Navarro A, Remiao F, Bastos ML, Carvalho F. Paraquat poisonings: mechanisms of lung toxicity, clinical features, and treatment. Crit Rev Toxicol. 2008;38:13–71. doi: 10.1080/10408440701669959. [DOI] [PubMed] [Google Scholar]
- 7.Duffield JS, Lupher M, Thannickal VJ, Wynn TA. Host responses in tissue repair and fibrosis. Annu Rev Pathol. 2013;8:241–276. doi: 10.1146/annurev-pathol-020712-163930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wynn TA, Ramalingam TR. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med. 2012;18:1028–1040. doi: 10.1038/nm.2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Meltzer EB, Noble PW. Idiopathic pulmonary fibrosis. Orphanet J Rare Dis. 2008;3:8. doi: 10.1186/1750-1172-3-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eickelberg O, Selman M. Update in diffuse parenchymal lung disease 2009. Am J Respir Crit Care Med. 2010;181:883–888. doi: 10.1164/rccm.201001-0124UP. [DOI] [PubMed] [Google Scholar]
- 11.Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, Brown KK, Colby TV, Cordier JF, Flaherty KR, Lasky JA, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011;183:788–824. doi: 10.1164/rccm.2009-040GL. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moore BB, Hogaboam CM. Murine models of pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2008;294:L152–L160. doi: 10.1152/ajplung.00313.2007. [DOI] [PubMed] [Google Scholar]
- 13.Degryse AL, Lawson WE. Progress toward improving animal models for idiopathic pulmonary fibrosis. Am J Med Sci. 2011;341:444–449. doi: 10.1097/MAJ.0b013e31821aa000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moeller A, Ask K, Warburton D, Gauldie J, Kolb M. The bleomycin animal model: a useful tool to investigate treatment options for idiopathic pulmonary fibrosis? Int J Biochem Cell Biol. 2008;40:362–382. doi: 10.1016/j.biocel.2007.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.De Volder MF, Tawfick SH, Baughman RH, Hart AJ. Carbon nanotubes: present and future commercial applications. Science. 2013;339:535–539. doi: 10.1126/science.1222453. [DOI] [PubMed] [Google Scholar]
- 16.Zhang Q, Huang JQ, Qian WZ, Zhang YY, Wei F. The road for nanomaterials industry: a review of carbon nanotube production, post-treatment, and bulk applications for composites and energy storage. Small. 2013;9:1237–1265. doi: 10.1002/smll.201203252. [DOI] [PubMed] [Google Scholar]
- 17.Donaldson K, Murphy FA, Duffin R, Poland CA. Asbestos, carbon nanotubes and the pleural mesothelium: a review of the hypothesis regarding the role of long fibre retention in the parietal pleura, inflammation and mesothelioma. Part Fibre Toxicol. 2010;7:5. doi: 10.1186/1743-8977-7-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dong J, Ma Q. Advances in mechanisms and signaling pathways of carbon nanotube toxicity. Nanotoxicology. 2015;9:658–676. doi: 10.3109/17435390.2015.1009187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Johnston HJ, Hutchison GR, Christensen FM, Peters S, Hankin S, Aschberger K, Stone V. A critical review of the biological mechanisms underlying the in vivo and in vitro toxicity of carbon nanotubes: The contribution of physico-chemical characteristics. Nanotoxicology. 2010;4:207–246. doi: 10.3109/17435390903569639. [DOI] [PubMed] [Google Scholar]
- 20.Nerl HC, Cheng C, Goode AE, Bergin SD, Lich B, Gass M, Porter AE. Imaging methods for determining uptake and toxicity of carbon nanotubes in vitro and in vivo. Nanomedicine (Lond) 2011;6:849–865. doi: 10.2217/nnm.11.87. [DOI] [PubMed] [Google Scholar]
- 21.Zhao X, Liu R. Recent progress and perspectives on the toxicity of carbon nanotubes at organism, organ, cell, and biomacromolecule levels. Environ Int. 2012;40:244–255. doi: 10.1016/j.envint.2011.12.003. [DOI] [PubMed] [Google Scholar]
- 22.Shvedova AA, Kisin ER, Mercer R, Murray AR, Johnson VJ, Potapovich AI, Tyurina YY, Gorelik O, Arepalli S, Schwegler-Berry D, et al. Unusual inflammatory and fibrogenic pulmonary responses to single-walled carbon nanotubes in mice. Am J Physiol Lung Cell Mol Physiol. 2005;289:L698–L708. doi: 10.1152/ajplung.00084.2005. [DOI] [PubMed] [Google Scholar]
- 23.Porter DW, Hubbs AF, Mercer RR, Wu N, Wolfarth MG, Sriram K, Leonard S, Battelli L, Schwegler-Berry D, Friend S, et al. Mouse pulmonary dose- and time course-responses induced by exposure to multi-walled carbon nanotubes. Toxicology. 2010;269:136–147. doi: 10.1016/j.tox.2009.10.017. [DOI] [PubMed] [Google Scholar]
- 24.Dong J, Porter DW, Batteli LA, Wolfarth MG, Richardson DL, Ma Q. Pathologic and molecular profiling of rapid-onset fibrosis and inflammation induced by multi-walled carbon nanotubes. Arch Toxicol. 2015;89:621–633. doi: 10.1007/s00204-014-1428-y. [DOI] [PubMed] [Google Scholar]
- 25.Ryman-Rasmussen JP, Tewksbury EW, Moss OR, Cesta MF, Wong BA, Bonner JC. Inhaled multiwalled carbon nanotubes potentiate airway fibrosis in murine allergic asthma. Am J Respir Cell Mol Biol. 2009;40:349–358. doi: 10.1165/rcmb.2008-0276OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Murphy FA, Poland CA, Duffin R, Al-Jamal KT, Ali-Boucetta H, Nunes A, Byrne F, Prina-Mello A, Volkov Y, Li S, et al. Length-dependent retention of carbon nanotubes in the pleural space of mice initiates sustained inflammation and progressive fibrosis on the parietal pleura. Am J Pathol. 2011;178:2587–2600. doi: 10.1016/j.ajpath.2011.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mercer RR, Scabilloni JF, Hubbs AF, Battelli LA, McKinney W, Friend S, Wolfarth MG, Andrew M, Castranova V, Porter DW. Distribution and fibrotic response following inhalation exposure to multi-walled carbon nanotubes. Part Fibre Toxicol. 2013;10:33. doi: 10.1186/1743-8977-10-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dong J, Yu X, Porter DW, Battelli LA, Kashon ML, Ma Q. Common and distinct mechanisms of induced pulmonary fibrosis by particulate and soluble chemical fibrogenic agents. Arch Toxicol. 2016;90:385–402. doi: 10.1007/s00204-015-1589-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fatkhutdinova LM, Khaliullin TO, Vasil’yeva OL, Zalyalov RR, Mustafin IG, Kisin ER, Birch ME, Yanamala N, Shvedova AA. Fibrosis biomarkers in workers exposed to MWCNTs. Toxicol Appl Pharmacol. 2016;299:125–131. doi: 10.1016/j.taap.2016.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liou SH, Tsai CS, Pelclova D, Schubauer-Berigan MK, Schulte PA. Assessing the first wave of epidemiological studies of nanomaterial workers. J Nanopart Res. 2015;17:413. doi: 10.1007/s11051-015-3219-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schulte PA, Kuempel ED, Zumwalde RD, Geraci CL, Schubauer-Berigan MK, Castranova V, Hodson L, Murashov V, Dahm MM, Ellenbecker M. Focused actions to protect carbon nanotube workers. Am J Ind Med. 2012;55:395–411. doi: 10.1002/ajim.22028. [DOI] [PubMed] [Google Scholar]
- 32.Clarke DL, Carruthers AM, Mustelin T, Murray LA. Matrix regulation of idiopathic pulmonary fibrosis: the role of enzymes. Fibrogenesis Tissue Repair. 2013;6:20. [DOI] [PMC free article] [PubMed]
- 33.Wynn TA. Cellular and molecular mechanisms of fibrosis. J Pathol. 2008;214:199–210. doi: 10.1002/path.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev Mol Cell Biol. 2002;3:349–363. doi: 10.1038/nrm809. [DOI] [PubMed] [Google Scholar]
- 35.White ES, Lazar MH, Thannickal VJ. Pathogenetic mechanisms in usual interstitial pneumonia/idiopathic pulmonary fibrosis. J Pathol. 2003;201:343–354. doi: 10.1002/path.1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kuhn C, 3rd, Boldt J, King TE, Jr, Crouch E, Vartio T, McDonald JA. An immunohistochemical study of architectural remodeling and connective tissue synthesis in pulmonary fibrosis. Am Rev Respir Dis. 1989;140:1693–1703. doi: 10.1164/ajrccm/140.6.1693. [DOI] [PubMed] [Google Scholar]
- 37.Kuhn C, McDonald JA. The roles of the myofibroblast in idiopathic pulmonary fibrosis. Ultrastructural and immunohistochemical features of sites of active extracellular matrix synthesis. Am J Pathol. 1991;138:1257–1265. [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang K, Rekhter MD, Gordon D, Phan SH. Myofibroblasts and their role in lung collagen gene expression during pulmonary fibrosis. A combined immunohistochemical and in situ hybridization study. Am J Pathol. 1994;145:114–125. [PMC free article] [PubMed] [Google Scholar]
- 39.Katzenstein AL, Myers JL. Idiopathic pulmonary fibrosis: clinical relevance of pathologic classification. Am J Respir Crit Care Med. 1998;157:1301–1315. doi: 10.1164/ajrccm.157.4.9707039. [DOI] [PubMed] [Google Scholar]
- 40.Homer RJ, Elias JA, Lee CG, Herzog E. Modern concepts on the role of inflammation in pulmonary fibrosis. Arch Pathol Lab Med. 2011;135:780–788. doi: 10.5858/2010-0296-RA.1. [DOI] [PubMed] [Google Scholar]
- 41.Hinz B, Phan SH, Thannickal VJ, Prunotto M, Desmouliere A, Varga J, De Wever O, Mareel M, Gabbiani G. Recent developments in myofibroblast biology: paradigms for connective tissue remodeling. Am J Pathol. 2012;180:1340–1355. doi: 10.1016/j.ajpath.2012.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vietti G, Lison D, van den Brule S. Mechanisms of lung fibrosis induced by carbon nanotubes: towards an Adverse Outcome Pathway (AOP) Part Fibre Toxicol. 2016;13:11. doi: 10.1186/s12989-016-0123-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Porter DW, Hubbs AF, Chen BT, McKinney W, Mercer RR, Wolfarth MG, Battelli L, Wu N, Sriram K, Leonard S, et al. Acute pulmonary dose-responses to inhaled multi-walled carbon nanotubes. Nanotoxicology. 2013;7:1179–1194. doi: 10.3109/17435390.2012.719649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lam CW, James JT, McCluskey R, Hunter RL. Pulmonary toxicity of single-wall carbon nanotubes in mice 7 and 90 days after intratracheal instillation. Toxicol Sci. 2004;77:126–134. doi: 10.1093/toxsci/kfg243. [DOI] [PubMed] [Google Scholar]
- 45.Muller J, Huaux F, Moreau N, Misson P, Heilier JF, Delos M, Arras M, Fonseca A, Nagy JB, Lison D. Respiratory toxicity of multi-wall carbon nanotubes. Toxicol Appl Pharmacol. 2005;207:221–231. doi: 10.1016/j.taap.2005.01.008. [DOI] [PubMed] [Google Scholar]
- 46.Kasai T, Umeda Y, Ohnishi M, Kondo H, Takeuchi T, Aiso S, Nishizawa T, Matsumoto M, Fukushima S. Thirteen-week study of toxicity of fiber-like multi-walled carbon nanotubes with whole-body inhalation exposure in rats. Nanotoxicology. 2015;9:413–422. doi: 10.3109/17435390.2014.933903. [DOI] [PubMed] [Google Scholar]
- 47.Ryman-Rasmussen JP, Cesta MF, Brody AR, Shipley-Phillips JK, Everitt JI, Tewksbury EW, Moss OR, Wong BA, Dodd DE, Andersen ME, Bonner JC. Inhaled carbon nanotubes reach the subpleural tissue in mice. Nat Nanotechnol. 2009;4:747–751. doi: 10.1038/nnano.2009.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Park EJ, Roh J, Kim SN, Kang MS, Han YA, Kim Y, Hong JT, Choi K. A single intratracheal instillation of single-walled carbon nanotubes induced early lung fibrosis and subchronic tissue damage in mice. Arch Toxicol. 2011;85:1121–1131. doi: 10.1007/s00204-011-0655-8. [DOI] [PubMed] [Google Scholar]
- 49.Aiso S, Yamazaki K, Umeda Y, Asakura M, Kasai T, Takaya M, Toya T, Koda S, Nagano K, Arito H, Fukushima S. Pulmonary toxicity of intratracheally instilled multiwall carbon nanotubes in male Fischer 344 rats. Ind Health. 2010;48:783–795. doi: 10.2486/indhealth.MS1129. [DOI] [PubMed] [Google Scholar]
- 50.Shvedova AA, Yanamala N, Kisin ER, Tkach AV, Murray AR, Hubbs A, Chirila MM, Keohavong P, Sycheva LP, Kagan VE, Castranova V. Long-term effects of carbon containing engineered nanomaterials and asbestos in the lung: one year postexposure comparisons. Am J Physiol Lung Cell Mol Physiol. 2014;306:L170–L182. doi: 10.1152/ajplung.00167.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vietti G, Ibouraadaten S, Palmai-Pallag M, Yakoub Y, Bailly C, Fenoglio I, Marbaix E, Lison D, van den Brule S. Towards predicting the lung fibrogenic activity of nanomaterials: experimental validation of an in vitro fibroblast proliferation assay. Part Fibre Toxicol. 2013;10:52. doi: 10.1186/1743-8977-10-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang P, Nie X, Wang Y, Li Y, Ge C, Zhang L, Wang L, Bai R, Chen Z, Zhao Y, Chen C. Multiwall carbon nanotubes mediate macrophage activation and promote pulmonary fibrosis through TGF-beta/Smad signaling pathway. Small. 2013;9:3799–3811. doi: 10.1002/smll.201300607. [DOI] [PubMed] [Google Scholar]
- 53.Zhang Y, Mu Q, Zhou H, Vrijens K, Roussel MF, Jiang G, Yan B. Binding of carbon nanotube to BMP receptor 2 enhances cell differentiation and inhibits apoptosis via regulating bHLH transcription factors. Cell Death Dis. 2012;3:e308. doi: 10.1038/cddis.2012.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Poulsen SS, Jackson P, Kling K, Knudsen KB, Skaug V, Kyjovska ZO, Thomsen BL, Clausen PA, Atluri R, Berthing T, et al. Multi-walled carbon nanotube physicochemical properties predict pulmonary inflammation and genotoxicity. Nanotoxicology. 2016;10:1263-75. [DOI] [PMC free article] [PubMed]
- 55.Taylor AJ, McClure CD, Shipkowski KA, Thompson EA, Hussain S, Garantziotis S, Parsons GN, Bonner JC. Atomic layer deposition coating of carbon nanotubes with aluminum oxide alters pro-fibrogenic cytokine expression by human mononuclear phagocytes in vitro and reduces lung fibrosis in mice in vivo. PLoS One. 2014;9:e106870. doi: 10.1371/journal.pone.0106870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li R, Wang X, Ji Z, Sun B, Zhang H, Chang CH, Lin S, Meng H, Liao YP, Wang M, et al. Surface charge and cellular processing of covalently functionalized multiwall carbon nanotubes determine pulmonary toxicity. ACS Nano. 2013;7:2352–2368. doi: 10.1021/nn305567s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.American Thoracic Society Idiopathic pulmonary fibrosis: diagnosis and treatment. International consensus statement. American Thoracic Society (ATS), and the European Respiratory Society (ERS) Am J Respir Crit Care Med. 2000;161:646–664. doi: 10.1164/ajrccm.161.2.ats3-00. [DOI] [PubMed] [Google Scholar]
- 58.Xia H, Khalil W, Kahm J, Jessurun J, Kleidon J, Henke CA. Pathologic caveolin-1 regulation of PTEN in idiopathic pulmonary fibrosis. Am J Pathol. 2010;176:2626–2637. doi: 10.2353/ajpath.2010.091117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nho RS, Hergert P, Kahm J, Jessurun J, Henke C. Pathological alteration of FoxO3a activity promotes idiopathic pulmonary fibrosis fibroblast proliferation on type i collagen matrix. Am J Pathol. 2011;179:2420–2430. doi: 10.1016/j.ajpath.2011.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Xia H, Seeman J, Hong J, Hergert P, Bodem V, Jessurun J, Smith K, Nho R, Kahm J, Gaillard P, Henke C. Low alpha(2)beta(1) integrin function enhances the proliferation of fibroblasts from patients with idiopathic pulmonary fibrosis by activation of the beta-catenin pathway. Am J Pathol. 2012;181:222–233. doi: 10.1016/j.ajpath.2012.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lappi-Blanco E, Soini Y, Paakko P. Apoptotic activity is increased in the newly formed fibromyxoid connective tissue in bronchiolitis obliterans organizing pneumonia. Lung. 1999;177:367–376. doi: 10.1007/PL00007654. [DOI] [PubMed] [Google Scholar]
- 62.Maher TM, Evans IC, Bottoms SE, Mercer PF, Thorley AJ, Nicholson AG, Laurent GJ, Tetley TD, Chambers RC, McAnulty RJ. Diminished prostaglandin E2 contributes to the apoptosis paradox in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2010;182:73–82. doi: 10.1164/rccm.200905-0674OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nho RS, Peterson M, Hergert P, Henke CA. FoxO3a (Forkhead Box O3a) deficiency protects Idiopathic Pulmonary Fibrosis (IPF) fibroblasts from type I polymerized collagen matrix-induced apoptosis via caveolin-1 (cav-1) and Fas. PLoS One. 2013;8:e61017. doi: 10.1371/journal.pone.0061017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gabbiani G, Ryan GB, Majne G. Presence of modified fibroblasts in granulation tissue and their possible role in wound contraction. Experientia. 1971;27:549–550. doi: 10.1007/BF02147594. [DOI] [PubMed] [Google Scholar]
- 65.Thannickal VJ, Toews GB, White ES, Lynch JP, 3rd, Martinez FJ. Mechanisms of pulmonary fibrosis. Annu Rev Med. 2004;55:395–417. doi: 10.1146/annurev.med.55.091902.103810. [DOI] [PubMed] [Google Scholar]
- 66.Hinz B, Phan SH, Thannickal VJ, Galli A, Bochaton-Piallat ML, Gabbiani G. The myofibroblast: one function, multiple origins. Am J Pathol. 2007;170:1807–1816. doi: 10.2353/ajpath.2007.070112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hinz B. The myofibroblast: paradigm for a mechanically active cell. J Biomech. 2010;43:146–155. doi: 10.1016/j.jbiomech.2009.09.020. [DOI] [PubMed] [Google Scholar]
- 68.Ignotz RA, Massague J. Transforming growth factor-beta stimulates the expression of fibronectin and collagen and their incorporation into the extracellular matrix. J Biol Chem. 1986;261:4337–4345. [PubMed] [Google Scholar]
- 69.Selman M, Ruiz V, Cabrera S, Segura L, Ramirez R, Barrios R, Pardo A. TIMP-1, -2, -3, and -4 in idiopathic pulmonary fibrosis. A prevailing nondegradative lung microenvironment? Am J Physiol Lung Cell Mol Physiol. 2000;279:L562–L574. doi: 10.1152/ajplung.2000.279.3.L562. [DOI] [PubMed] [Google Scholar]
- 70.Ramos C, Montano M, Garcia-Alvarez J, Ruiz V, Uhal BD, Selman M, Pardo A. Fibroblasts from idiopathic pulmonary fibrosis and normal lungs differ in growth rate, apoptosis, and tissue inhibitor of metalloproteinases expression. Am J Respir Cell Mol Biol. 2001;24:591–598. doi: 10.1165/ajrcmb.24.5.4333. [DOI] [PubMed] [Google Scholar]
- 71.Heino J, Ignotz RA, Hemler ME, Crouse C, Massague J. Regulation of cell adhesion receptors by transforming growth factor-beta. Concomitant regulation of integrins that share a common beta 1 subunit. J Biol Chem. 1989;264:380–388. [PubMed] [Google Scholar]
- 72.Thannickal VJ, Aldweib KD, Rajan T, Fanburg BL. Upregulated expression of fibroblast growth factor (FGF) receptors by transforming growth factor-beta1 (TGF-beta1) mediates enhanced mitogenic responses to FGFs in cultured human lung fibroblasts. Biochem Biophys Res Commun. 1998;251:437–441. doi: 10.1006/bbrc.1998.9443. [DOI] [PubMed] [Google Scholar]
- 73.Phan SH, Zhang K, Zhang HY, Gharaee-Kermani M. The myofibroblast as an inflammatory cell in pulmonary fibrosis. Curr Top Pathol. 1999;93:173–182. doi: 10.1007/978-3-642-58456-5_18. [DOI] [PubMed] [Google Scholar]
- 74.Finlay GA, Thannickal VJ, Fanburg BL, Paulson KE. Transforming growth factor-beta 1-induced activation of the ERK pathway/activator protein-1 in human lung fibroblasts requires the autocrine induction of basic fibroblast growth factor. J Biol Chem. 2000;275:27650–27656. doi: 10.1074/jbc.M000893200. [DOI] [PubMed] [Google Scholar]
- 75.Mezzano SA, Droguett MA, Burgos ME, Ardiles LG, Aros CA, Caorsi I, Egido J. Overexpression of chemokines, fibrogenic cytokines, and myofibroblasts in human membranous nephropathy. Kidney Int. 2000;57:147–158. doi: 10.1046/j.1523-1755.2000.00830.x. [DOI] [PubMed] [Google Scholar]
- 76.Thannickal VJ, Fanburg BL. Activation of an H2O2-generating NADH oxidase in human lung fibroblasts by transforming growth factor beta 1. J Biol Chem. 1995;270:30334–30338. doi: 10.1074/jbc.270.51.30334. [DOI] [PubMed] [Google Scholar]
- 77.Thannickal VJ, Aldweib KD, Fanburg BL. Tyrosine phosphorylation regulates H2O2 production in lung fibroblasts stimulated by transforming growth factor beta1. J Biol Chem. 1998;273:23611–23615. doi: 10.1074/jbc.273.36.23611. [DOI] [PubMed] [Google Scholar]
- 78.Sambo P, Baroni SS, Luchetti M, Paroncini P, Dusi S, Orlandini G, Gabrielli A. Oxidative stress in scleroderma: maintenance of scleroderma fibroblast phenotype by the constitutive up-regulation of reactive oxygen species generation through the NADPH oxidase complex pathway. Arthritis Rheum. 2001;44:2653–2664. doi: 10.1002/1529-0131(200111)44:11<2653::AID-ART445>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 79.Sugiura H, Liu X, Kobayashi T, Togo S, Ertl RF, Kawasaki S, Kamio K, Wang XQ, Mao L, Shen L, et al. Reactive nitrogen species augment fibroblast-mediated collagen gel contraction, mediator production, and chemotaxis. Am J Respir Cell Mol Biol. 2006;34:592–599. doi: 10.1165/rcmb.2005-0339OC. [DOI] [PubMed] [Google Scholar]
- 80.Wu J, Chitapanarux T, Chen Y, Soon RK, Jr, Yee HF., Jr Intestinal myofibroblasts produce nitric oxide in response to combinatorial cytokine stimulation. J Cell Physiol. 2013;228:572–580. doi: 10.1002/jcp.24164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhang HY, Phan SH. Inhibition of myofibroblast apoptosis by transforming growth factor beta(1) Am J Respir Cell Mol Biol. 1999;21:658–665. doi: 10.1165/ajrcmb.21.6.3720. [DOI] [PubMed] [Google Scholar]
- 82.Horowitz JC, Lee DY, Waghray M, Keshamouni VG, Thomas PE, Zhang H, Cui Z, Thannickal VJ. Activation of the pro-survival phosphatidylinositol 3-kinase/AKT pathway by transforming growth factor-beta1 in mesenchymal cells is mediated by p38 MAPK-dependent induction of an autocrine growth factor. J Biol Chem. 2004;279:1359–1367. doi: 10.1074/jbc.M306248200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Vittal R, Horowitz JC, Moore BB, Zhang H, Martinez FJ, Toews GB, Standiford TJ, Thannickal VJ. Modulation of prosurvival signaling in fibroblasts by a protein kinase inhibitor protects against fibrotic tissue injury. Am J Pathol. 2005;166:367–375. doi: 10.1016/S0002-9440(10)62260-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Phan SH. Biology of fibroblasts and myofibroblasts. Proc Am Thorac Soc. 2008;5:334–337. doi: 10.1513/pats.200708-146DR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Reilkoff RA, Bucala R, Herzog EL. Fibrocytes: emerging effector cells in chronic inflammation. Nat Rev Immunol. 2011;11:427–435. doi: 10.1038/nri2990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Rock JR, Barkauskas CE, Cronce MJ, Xue Y, Harris JR, Liang J, Noble PW, Hogan BL. Multiple stromal populations contribute to pulmonary fibrosis without evidence for epithelial to mesenchymal transition. Proc Natl Acad Sci U S A. 2011;108:E1475–E1483. doi: 10.1073/pnas.1117988108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Moore MW, Herzog EL. Regulation and Relevance of Myofibroblast Responses in Idiopathic Pulmonary Fibrosis. Curr Pathobiol Rep. 2013;1:199–208. doi: 10.1007/s40139-013-0017-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Bartis D, Mise N, Mahida RY, Eickelberg O, Thickett DR. Epithelial-mesenchymal transition in lung development and disease: does it exist and is it important? Thorax. 2014;69:760–765. doi: 10.1136/thoraxjnl-2013-204608. [DOI] [PubMed] [Google Scholar]
- 89.Vyalov SL, Gabbiani G, Kapanci Y. Rat alveolar myofibroblasts acquire alpha-smooth muscle actin expression during bleomycin-induced pulmonary fibrosis. Am J Pathol. 1993;143:1754–1765. [PMC free article] [PubMed] [Google Scholar]
- 90.Khalil N, O’Connor RN, Flanders KC, Unruh H. TGF-beta 1, but not TGF-beta 2 or TGF-beta 3, is differentially present in epithelial cells of advanced pulmonary fibrosis: an immunohistochemical study. Am J Respir Cell Mol Biol. 1996;14:131–138. doi: 10.1165/ajrcmb.14.2.8630262. [DOI] [PubMed] [Google Scholar]
- 91.Coker RK, Laurent GJ, Shahzeidi S, Lympany PA, du Bois RM, Jeffery PK, McAnulty RJ. Transforming growth factors-beta 1, -beta 2, and -beta 3 stimulate fibroblast procollagen production in vitro but are differentially expressed during bleomycin-induced lung fibrosis. Am J Pathol. 1997;150:981–991. [PMC free article] [PubMed] [Google Scholar]
- 92.Khalil N, Parekh TV, O’Connor R, Antman N, Kepron W, Yehaulaeshet T, Xu YD, Gold LI. Regulation of the effects of TGF-beta 1 by activation of latent TGF-beta 1 and differential expression of TGF-beta receptors (T beta R-I and T beta R-II) in idiopathic pulmonary fibrosis. Thorax. 2001;56:907–915. doi: 10.1136/thorax.56.12.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Xu YD, Hua J, Mui A, O’Connor R, Grotendorst G, Khalil N. Release of biologically active TGF-beta1 by alveolar epithelial cells results in pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2003;285:L527–L539. doi: 10.1152/ajplung.00298.2002. [DOI] [PubMed] [Google Scholar]
- 94.Gleizes PE, Munger JS, Nunes I, Harpel JG, Mazzieri R, Noguera I, Rifkin DB. TGF-beta latency: biological significance and mechanisms of activation. Stem Cells. 1997;15:190–197. doi: 10.1002/stem.150190. [DOI] [PubMed] [Google Scholar]
- 95.He X, Young SH, Schwegler-Berry D, Chisholm WP, Fernback JE, Ma Q. Multiwalled carbon nanotubes induce a fibrogenic response by stimulating reactive oxygen species production, activating NF-kappaB signaling, and promoting fibroblast-to-myofibroblast transformation. Chem Res Toxicol. 2011;24:2237–2248. doi: 10.1021/tx200351d. [DOI] [PubMed] [Google Scholar]
- 96.He X, Young SH, Fernback JE, Ma Q. Single-Walled Carbon Nanotubes Induce Fibrogenic Effect by Disturbing Mitochondrial Oxidative Stress and Activating NF-kappaB Signaling. J Clin Toxicol. 2012;S5:005. [DOI] [PMC free article] [PubMed]
- 97.Chen T, Nie H, Gao X, Yang J, Pu J, Chen Z, Cui X, Wang Y, Wang H, Jia G. Epithelial-mesenchymal transition involved in pulmonary fibrosis induced by multi-walled carbon nanotubes via TGF-beta/Smad signaling pathway. Toxicol Lett. 2014;226:150–162. doi: 10.1016/j.toxlet.2014.02.004. [DOI] [PubMed] [Google Scholar]
- 98.Wang P, Wang Y, Nie X, Braini C, Bai R, Chen C. Multiwall carbon nanotubes directly promote fibroblast-myofibroblast and epithelial-mesenchymal transitions through the activation of the TGF-beta/Smad signaling pathway. Small. 2015;11:446–455. doi: 10.1002/smll.201303588. [DOI] [PubMed] [Google Scholar]
- 99.Chang CC, Tsai ML, Huang HC, Chen CY, Dai SX. Epithelial-mesenchymal transition contributes to SWCNT-induced pulmonary fibrosis. Nanotoxicology. 2012;6:600–610. doi: 10.3109/17435390.2011.594913. [DOI] [PubMed] [Google Scholar]
- 100.Zhang Y, Deng J, Zhang Y, Guo F, Li C, Zou Z, Xi W, Tang J, Sun Y, Yang P, et al. Functionalized single-walled carbon nanotubes cause reversible acute lung injury and induce fibrosis in mice. J Mol Med (Berl) 2013;91:117–128. doi: 10.1007/s00109-012-0940-x. [DOI] [PubMed] [Google Scholar]
- 101.Dong J, Ma Q. Suppression of basal and carbon nanotube-induced oxidative stress, inflammation and fibrosis in mouse lungs by Nrf2. Nanotoxicology. 2016;10:699–709. doi: 10.3109/17435390.2015.1110758. [DOI] [PubMed] [Google Scholar]
- 102.Pinzani M, Milani S, Herbst H, DeFranco R, Grappone C, Gentilini A, Caligiuri A, Pellegrini G, Ngo DV, Romanelli RG, Gentilini P. Expression of platelet-derived growth factor and its receptors in normal human liver and during active hepatic fibrogenesis. Am J Pathol. 1996;148:785–800. [PMC free article] [PubMed] [Google Scholar]
- 103.Ikura Y, Morimoto H, Ogami M, Jomura H, Ikeoka N, Sakurai M. Expression of platelet-derived growth factor and its receptor in livers of patients with chronic liver disease. J Gastroenterol. 1997;32:496–501. doi: 10.1007/BF02934089. [DOI] [PubMed] [Google Scholar]
- 104.Chen YT, Chang FC, Wu CF, Chou YH, Hsu HL, Chiang WC, Shen J, Chen YM, Wu KD, Tsai TJ, et al. Platelet-derived growth factor receptor signaling activates pericyte-myofibroblast transition in obstructive and post-ischemic kidney fibrosis. Kidney Int. 2011;80:1170–1181. doi: 10.1038/ki.2011.208. [DOI] [PubMed] [Google Scholar]
- 105.Henderson NC, Arnold TD, Katamura Y, Giacomini MM, Rodriguez JD, McCarty JH, Pellicoro A, Raschperger E, Betsholtz C, Ruminski PG, et al. Targeting of alphav integrin identifies a core molecular pathway that regulates fibrosis in several organs. Nat Med. 2013;19:1617–1624. doi: 10.1038/nm.3282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Leask A, Abraham DJ. TGF-beta signaling and the fibrotic response. FASEB J. 2004;18:816–827. doi: 10.1096/fj.03-1273rev. [DOI] [PubMed] [Google Scholar]
- 107.Sime PJ, Xing Z, Graham FL, Csaky KG, Gauldie J. Adenovector-mediated gene transfer of active transforming growth factor-beta1 induces prolonged severe fibrosis in rat lung. J Clin Invest. 1997;100:768–776. doi: 10.1172/JCI119590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Liu JY, Sime PJ, Wu T, Warshamana GS, Pociask D, Tsai SY, Brody AR. Transforming growth factor-beta(1) overexpression in tumor necrosis factor-alpha receptor knockout mice induces fibroproliferative lung disease. Am J Respir Cell Mol Biol. 2001;25:3–7. doi: 10.1165/ajrcmb.25.1.4481. [DOI] [PubMed] [Google Scholar]
- 109.Kulkarni AB, Karlsson S. Transforming growth factor-beta 1 knockout mice. A mutation in one cytokine gene causes a dramatic inflammatory disease. Am J Pathol. 1993;143:3–9. [PMC free article] [PubMed] [Google Scholar]
- 110.Bottinger EP, Letterio JJ, Roberts AB. Biology of TGF-beta in knockout and transgenic mouse models. Kidney Int. 1997;51:1355–1360. doi: 10.1038/ki.1997.185. [DOI] [PubMed] [Google Scholar]
- 111.Bonniaud P, Margetts PJ, Ask K, Flanders K, Gauldie J, Kolb M. TGF-beta and Smad3 signaling link inflammation to chronic fibrogenesis. J Immunol. 2005;175:5390–5395. doi: 10.4049/jimmunol.175.8.5390. [DOI] [PubMed] [Google Scholar]
- 112.Bonniaud P, Margetts PJ, Kolb M, Schroeder JA, Kapoun AM, Damm D, Murphy A, Chakravarty S, Dugar S, Higgins L, et al. Progressive transforming growth factor beta1-induced lung fibrosis is blocked by an orally active ALK5 kinase inhibitor. Am J Respir Crit Care Med. 2005;171:889–898. doi: 10.1164/rccm.200405-612OC. [DOI] [PubMed] [Google Scholar]
- 113.Assoian RK, Fleurdelys BE, Stevenson HC, Miller PJ, Madtes DK, Raines EW, Ross R, Sporn MB. Expression and secretion of type beta transforming growth factor by activated human macrophages. Proc Natl Acad Sci U S A. 1987;84:6020–6024. doi: 10.1073/pnas.84.17.6020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Grotendorst GR, Smale G, Pencev D. Production of transforming growth factor beta by human peripheral blood monocytes and neutrophils. J Cell Physiol. 1989;140:396–402. doi: 10.1002/jcp.1041400226. [DOI] [PubMed] [Google Scholar]
- 115.Letterio JJ, Roberts AB. Regulation of immune responses by TGF-beta. Annu Rev Immunol. 1998;16:137–161. doi: 10.1146/annurev.immunol.16.1.137. [DOI] [PubMed] [Google Scholar]
- 116.Wang X, Duch MC, Mansukhani N, Ji Z, Liao YP, Wang M, Zhang H, Sun B, Chang CH, Li R, et al. Use of a pro-fibrogenic mechanism-based predictive toxicological approach for tiered testing and decision analysis of carbonaceous nanomaterials. ACS Nano. 2015;9:3032–3043. doi: 10.1021/nn507243w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Lin Z, Liu L, Xi Z, Huang J, Lin B. Single-walled carbon nanotubes promote rat vascular adventitial fibroblasts to transform into myofibroblasts by SM22-alpha expression. Int J Nanomedicine. 2012;7:4199–4206. doi: 10.2147/IJN.S34663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Azad N, Iyer AK, Wang L, Liu Y, Lu Y, Rojanasakul Y. Reactive oxygen species-mediated p38 MAPK regulates carbon nanotube-induced fibrogenic and angiogenic responses. Nanotoxicology. 2013;7:157–168. doi: 10.3109/17435390.2011.647929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Manke A, Luanpitpong S, Dong C, Wang L, He X, Battelli L, Derk R, Stueckle TA, Porter DW, Sager T, et al. Effect of fiber length on carbon nanotube-induced fibrogenesis. Int J Mol Sci. 2014;15:7444–7461. doi: 10.3390/ijms15057444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Mishra A, Stueckle TA, Mercer RR, Derk R, Rojanasakul Y, Castranova V, Wang L. Identification of TGF-beta receptor-1 as a key regulator of carbon nanotube-induced fibrogenesis. Am J Physiol Lung Cell Mol Physiol. 2015;309:L821–L833. doi: 10.1152/ajplung.00002.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wang X, Xia T, Ntim SA, Ji Z, Lin S, Meng H, Chung CH, George S, Zhang H, Wang M, et al. Dispersal state of multiwalled carbon nanotubes elicits profibrogenic cellular responses that correlate with fibrogenesis biomarkers and fibrosis in the murine lung. ACS Nano. 2011;5:9772–9787. doi: 10.1021/nn2033055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Sun B, Wang X, Ji Z, Wang M, Liao YP, Chang CH, Li R, Zhang H, Nel AE, Xia T. NADPH Oxidase-Dependent NLRP3 Inflammasome Activation and its Important Role in Lung Fibrosis by Multiwalled Carbon Nanotubes. Small. 2015;11:2087–2097. doi: 10.1002/smll.201402859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Tatler AL, Jenkins G. TGF-beta activation and lung fibrosis. Proc Am Thorac Soc. 2012;9:130–136. doi: 10.1513/pats.201201-003AW. [DOI] [PubMed] [Google Scholar]
- 124.Hinz B. It has to be the alphav: myofibroblast integrins activate latent TGF-beta1. Nat Med. 2013;19:1567–1568. doi: 10.1038/nm.3421. [DOI] [PubMed] [Google Scholar]
- 125.Shvedova AA, Kisin ER, Murray AR, Kommineni C, Castranova V, Fadeel B, Kagan VE. Increased accumulation of neutrophils and decreased fibrosis in the lung of NADPH oxidase-deficient C57BL/6 mice exposed to carbon nanotubes. Toxicol Appl Pharmacol. 2008;231:235–240. doi: 10.1016/j.taap.2008.04.018. [DOI] [PubMed] [Google Scholar]
- 126.Pociask DA, Sime PJ, Brody AR. Asbestos-derived reactive oxygen species activate TGF-beta1. Lab Invest. 2004;84:1013–1023. doi: 10.1038/labinvest.3700109. [DOI] [PubMed] [Google Scholar]
- 127.Sullivan DE, Ferris M, Pociask D, Brody AR. Tumor necrosis factor-alpha induces transforming growth factor-beta1 expression in lung fibroblasts through the extracellular signal-regulated kinase pathway. Am J Respir Cell Mol Biol. 2005;32:342–349. doi: 10.1165/rcmb.2004-0288OC. [DOI] [PubMed] [Google Scholar]
- 128.Borthwick LA, Wynn TA, Fisher AJ. Cytokine mediated tissue fibrosis. Biochim Biophys Acta. 2013;1832:1049–1060. doi: 10.1016/j.bbadis.2012.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Bonner JC. Regulation of PDGF and its receptors in fibrotic diseases. Cytokine Growth Factor Rev. 2004;15:255–273. doi: 10.1016/j.cytogfr.2004.03.006. [DOI] [PubMed] [Google Scholar]
- 130.Nishioka Y, Azuma M, Kishi M, Aono Y. Targeting platelet-derived growth factor as a therapeutic approach in pulmonary fibrosis. J Med Invest. 2013;60:175–183. doi: 10.2152/jmi.60.175. [DOI] [PubMed] [Google Scholar]
- 131.Yoshida M, Sakuma J, Hayashi S, Abe K, Saito I, Harada S, Sakatani M, Yamamoto S, Matsumoto N, Kaneda Y, et al. A histologically distinctive interstitial pneumonia induced by overexpression of the interleukin 6, transforming growth factor beta 1, or platelet-derived growth factor B gene. Proc Natl Acad Sci U S A. 1995;92:9570–9574. doi: 10.1073/pnas.92.21.9570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Walsh J, Absher M, Kelley J. Variable expression of platelet-derived growth factor family proteins in acute lung injury. Am J Respir Cell Mol Biol. 1993;9:637–644. doi: 10.1165/ajrcmb/9.6.637. [DOI] [PubMed] [Google Scholar]
- 133.Tang WW, Ulich TR, Lacey DL, Hill DC, Qi M, Kaufman SA, Van GY, Tarpley JE, Yee JS. Platelet-derived growth factor-BB induces renal tubulointerstitial myofibroblast formation and tubulointerstitial fibrosis. Am J Pathol. 1996;148:1169–1180. [PMC free article] [PubMed] [Google Scholar]
- 134.Oh SJ, Kurz H, Christ B, Wilting J. Platelet-derived growth factor-B induces transformation of fibrocytes into spindle-shaped myofibroblasts in vivo. Histochem Cell Biol. 1998;109:349–357. doi: 10.1007/s004180050235. [DOI] [PubMed] [Google Scholar]
- 135.Kinnman N, Francoz C, Barbu V, Wendum D, Rey C, Hultcrantz R, Poupon R, Housset C. The myofibroblastic conversion of peribiliary fibrogenic cells distinct from hepatic stellate cells is stimulated by platelet-derived growth factor during liver fibrogenesis. Lab Invest. 2003;83:163–173. doi: 10.1097/01.LAB.0000054178.01162.E4. [DOI] [PubMed] [Google Scholar]
- 136.Cesta MF, Ryman-Rasmussen JP, Wallace DG, Masinde T, Hurlburt G, Taylor AJ, Bonner JC. Bacterial lipopolysaccharide enhances PDGF signaling and pulmonary fibrosis in rats exposed to carbon nanotubes. Am J Respir Cell Mol Biol. 2010;43:142–151. doi: 10.1165/rcmb.2009-0113OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Wynn TA. Fibrotic disease and the T(H)1/T(H)2 paradigm. Nat Rev Immunol. 2004;4:583–594. doi: 10.1038/nri1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Wynn TA. Type 2 cytokines: mechanisms and therapeutic strategies. Nat Rev Immunol. 2015;15:271–282. doi: 10.1038/nri3831. [DOI] [PubMed] [Google Scholar]
- 139.Emura M, Nagai S, Takeuchi M, Kitaichi M, Izumi T. In vitro production of B cell growth factor and B cell differentiation factor by peripheral blood mononuclear cells and bronchoalveolar lavage T lymphocytes from patients with idiopathic pulmonary fibrosis. Clin Exp Immunol. 1990;82:133–139. doi: 10.1111/j.1365-2249.1990.tb05416.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Wallace WA, Ramage EA, Lamb D, Howie SE. A type 2 (Th2-like) pattern of immune response predominates in the pulmonary interstitium of patients with cryptogenic fibrosing alveolitis (CFA) Clin Exp Immunol. 1995;101:436–441. doi: 10.1111/j.1365-2249.1995.tb03131.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Cheever AW, Williams ME, Wynn TA, Finkelman FD, Seder RA, Cox TM, Hieny S, Caspar P, Sher A. Anti-IL-4 treatment of Schistosoma mansoni-infected mice inhibits development of T cells and non-B, non-T cells expressing Th2 cytokines while decreasing egg-induced hepatic fibrosis. J Immunol. 1994;153:753–759. [PubMed] [Google Scholar]
- 142.Ong C, Wong C, Roberts CR, Teh HS, Jirik FR. Anti-IL-4 treatment prevents dermal collagen deposition in the tight-skin mouse model of scleroderma. Eur J Immunol. 1998;28:2619–2629. doi: 10.1002/(SICI)1521-4141(199809)28:09<2619::AID-IMMU2619>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 143.Park SW, Ahn MH, Jang HK, Jang AS, Kim DJ, Koh ES, Park JS, Uh ST, Kim YH, Park JS, et al. Interleukin-13 and its receptors in idiopathic interstitial pneumonia: clinical implications for lung function. J Korean Med Sci. 2009;24:614–620. doi: 10.3346/jkms.2009.24.4.614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Zhu Z, Homer RJ, Wang Z, Chen Q, Geba GP, Wang J, Zhang Y, Elias JA. Pulmonary expression of interleukin-13 causes inflammation, mucus hypersecretion, subepithelial fibrosis, physiologic abnormalities, and eotaxin production. J Clin Invest. 1999;103:779–788. doi: 10.1172/JCI5909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Belperio JA, Dy M, Burdick MD, Xue YY, Li K, Elias JA, Keane MP. Interaction of IL-13 and C10 in the pathogenesis of bleomycin-induced pulmonary fibrosis. Am J Respir Cell Mol Biol. 2002;27:419–427. doi: 10.1165/rcmb.2002-0009OC. [DOI] [PubMed] [Google Scholar]
- 146.Sempowski GD, Beckmann MP, Derdak S, Phipps RP. Subsets of murine lung fibroblasts express membrane-bound and soluble IL-4 receptors. Role of IL-4 in enhancing fibroblast proliferation and collagen synthesis. J Immunol. 1994;152:3606–3614. [PubMed] [Google Scholar]
- 147.Doucet C, Brouty-Boye D, Pottin-Clemenceau C, Canonica GW, Jasmin C, Azzarone B. Interleukin (IL) 4 and IL-13 act on human lung fibroblasts. Implication in asthma. J Clin Invest. 1998;101:2129–2139. doi: 10.1172/JCI741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Fertin C, Nicolas JF, Gillery P, Kalis B, Banchereau J, Maquart FX. Interleukin-4 stimulates collagen synthesis by normal and scleroderma fibroblasts in dermal equivalents. Cell Mol Biol. 1991;37:823–829. [PubMed] [Google Scholar]
- 149.Tiggelman AM, Boers W, Linthorst C, Sala M, Chamuleau RA. Collagen synthesis by human liver (myo)fibroblasts in culture: evidence for a regulatory role of IL-1 beta, IL-4, TGF beta and IFN gamma. J Hepatol. 1995;23:307–317. [PubMed] [Google Scholar]
- 150.Mattey DL, Dawes PT, Nixon NB, Slater H. Transforming growth factor beta 1 and interleukin 4 induced alpha smooth muscle actin expression and myofibroblast-like differentiation in human synovial fibroblasts in vitro: modulation by basic fibroblast growth factor. Ann Rheum Dis. 1997;56:426–431. doi: 10.1136/ard.56.7.426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Hashimoto S, Gon Y, Takeshita I, Maruoka S, Horie T. IL-4 and IL-13 induce myofibroblastic phenotype of human lung fibroblasts through c-Jun NH2-terminal kinase-dependent pathway. J Allergy Clin Immunol. 2001;107:1001–1008. doi: 10.1067/mai.2001.114702. [DOI] [PubMed] [Google Scholar]
- 152.Saito A, Okazaki H, Sugawara I, Yamamoto K, Takizawa H. Potential action of IL-4 and IL-13 as fibrogenic factors on lung fibroblasts in vitro. Int Arch Allergy Immunol. 2003;132:168–176. doi: 10.1159/000073718. [DOI] [PubMed] [Google Scholar]
- 153.Lee CG, Homer RJ, Zhu Z, Lanone S, Wang X, Koteliansky V, Shipley JM, Gotwals P, Noble P, Chen Q, et al. Interleukin-13 induces tissue fibrosis by selectively stimulating and activating transforming growth factor beta(1) J Exp Med. 2001;194:809–821. doi: 10.1084/jem.194.6.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Lanone S, Zheng T, Zhu Z, Liu W, Lee CG, Ma B, Chen Q, Homer RJ, Wang J, Rabach LA, et al. Overlapping and enzyme-specific contributions of matrix metalloproteinases-9 and -12 in IL-13-induced inflammation and remodeling. J Clin Invest. 2002;110:463–474. doi: 10.1172/JCI0214136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Zheng T, Zhu Z, Wang Z, Homer RJ, Ma B, Riese RJ, Jr, Chapman HA, Jr, Shapiro SD, Elias JA. Inducible targeting of IL-13 to the adult lung causes matrix metalloproteinase- and cathepsin-dependent emphysema. J Clin Invest. 2000;106:1081–1093. doi: 10.1172/JCI10458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Dong J, Ma Q. In vivo activation of a T helper 2-driven innate immune response in lung fibrosis induced by multi-walled carbon nanotubes. Arch Toxicol. 2016;90:2231–2248. doi: 10.1007/s00204-016-1711-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Finkel T. Radical medicine: treating ageing to cure disease. Nat Rev Mol Cell Biol. 2005;6:971–976. doi: 10.1038/nrm1763. [DOI] [PubMed] [Google Scholar]
- 158.Ma Q. Transcriptional responses to oxidative stress: pathological and toxicological implications. Pharmacol Ther. 2010;125:376–393. doi: 10.1016/j.pharmthera.2009.11.004. [DOI] [PubMed] [Google Scholar]
- 159.Ma Q. Role of nrf2 in oxidative stress and toxicity. Annu Rev Pharmacol Toxicol. 2013;53:401–426. doi: 10.1146/annurev-pharmtox-011112-140320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Bargagli E, Olivieri C, Bennett D, Prasse A, Muller-Quernheim J, Rottoli P. Oxidative stress in the pathogenesis of diffuse lung diseases: a review. Respir Med. 2009;103:1245–1256. doi: 10.1016/j.rmed.2009.04.014. [DOI] [PubMed] [Google Scholar]
- 161.Kliment CR, Oury TD. Oxidative stress, extracellular matrix targets, and idiopathic pulmonary fibrosis. Free Radic Biol Med. 2010;49:707–717. doi: 10.1016/j.freeradbiomed.2010.04.036. [DOI] [PubMed] [Google Scholar]
- 162.Cheresh P, Kim SJ, Tulasiram S, Kamp DW. Oxidative stress and pulmonary fibrosis. Biochim Biophys Acta. 2013;1832:1028–1040. doi: 10.1016/j.bbadis.2012.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Paik YH, Kim J, Aoyama T, De Minicis S, Bataller R, Brenner DA. Role of NADPH oxidases in liver fibrosis. Antioxid Redox Signal. 2014;20:2854–2872. doi: 10.1089/ars.2013.5619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Crosas-Molist E, Fabregat I. Role of NADPH oxidases in the redox biology of liver fibrosis. Redox Biol. 2015;6:106–111. doi: 10.1016/j.redox.2015.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Liu RM, Desai LP. Reciprocal regulation of TGF-beta and reactive oxygen species: A perverse cycle for fibrosis. Redox Biol. 2015;6:565–577. doi: 10.1016/j.redox.2015.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Okamura DM, Pennathur S. The balance of powers: Redox regulation of fibrogenic pathways in kidney injury. Redox Biol. 2015;6:495–504. doi: 10.1016/j.redox.2015.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Richter K, Konzack A, Pihlajaniemi T, Heljasvaara R, Kietzmann T. Redox-fibrosis: Impact of TGFbeta1 on ROS generators, mediators and functional consequences. Redox Biol. 2015;6:344–352. doi: 10.1016/j.redox.2015.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Barnes JL, Gorin Y. Myofibroblast differentiation during fibrosis: role of NAD(P)H oxidases. Kidney Int. 2011;79:944–956. doi: 10.1038/ki.2010.516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Sampson N, Berger P, Zenzmaier C. Redox signaling as a therapeutic target to inhibit myofibroblast activation in degenerative fibrotic disease. Biomed Res Int. 2014;2014:131737. doi: 10.1155/2014/131737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Siani A, Tirelli N. Myofibroblast differentiation: main features, biomedical relevance, and the role of reactive oxygen species. Antioxid Redox Signal. 2014;21:768–785. doi: 10.1089/ars.2013.5724. [DOI] [PubMed] [Google Scholar]
- 171.Liang S, Kisseleva T, Brenner DA. The Role of NADPH Oxidases (NOXs) in Liver Fibrosis and the Activation of Myofibroblasts. Front Physiol. 2016;7:17. doi: 10.3389/fphys.2016.00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Wang P, Koyama Y, Liu X, Xu J, Ma HY, Liang S, Kim IH, Brenner DA, Kisseleva T. Promising Therapy Candidates for Liver Fibrosis. Front Physiol. 2016;7:47. doi: 10.3389/fphys.2016.00047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Moller P, Christophersen DV, Jensen DM, Kermanizadeh A, Roursgaard M, Jacobsen NR, Hemmingsen JG, Danielsen PH, Cao Y, Jantzen K, et al. Role of oxidative stress in carbon nanotube-generated health effects. Arch Toxicol. 2014;88:1939–1964. doi: 10.1007/s00204-014-1356-x. [DOI] [PubMed] [Google Scholar]
- 174.Li Z, Hulderman T, Salmen R, Chapman R, Leonard SS, Young SH, Shvedova A, Luster MI, Simeonova PP. Cardiovascular effects of pulmonary exposure to single-wall carbon nanotubes. Environ Health Perspect. 2007;115:377–382. doi: 10.1289/ehp.9688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Shvedova AA, Kisin E, Murray AR, Johnson VJ, Gorelik O, Arepalli S, Hubbs AF, Mercer RR, Keohavong P, Sussman N, et al. Inhalation vs. aspiration of single-walled carbon nanotubes in C57BL/6 mice: inflammation, fibrosis, oxidative stress, and mutagenesis. Am J Physiol Lung Cell Mol Physiol. 2008;295:L552–L565. doi: 10.1152/ajplung.90287.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Shvedova AA, Kisin ER, Murray AR, Mouithys-Mickalad A, Stadler K, Mason RP, Kadiiska M. ESR evidence for in vivo formation of free radicals in tissue of mice exposed to single-walled carbon nanotubes. Free Radic Biol Med. 2014;73:154–165. doi: 10.1016/j.freeradbiomed.2014.05.010. [DOI] [PubMed] [Google Scholar]
- 177.Ma Q, He X. Molecular basis of electrophilic and oxidative defense: promises and perils of Nrf2. Pharmacol Rev. 2012;64:1055–1081. doi: 10.1124/pr.110.004333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Ma Q. Advances in mechanisms of anti-oxidation. Discov Med. 2014;17:121–130. [PMC free article] [PubMed] [Google Scholar]
- 179.Miyazaki Y, Araki K, Vesin C, Garcia I, Kapanci Y, Whitsett JA, Piguet PF, Vassalli P. Expression of a tumor necrosis factor-alpha transgene in murine lung causes lymphocytic and fibrosing alveolitis. A mouse model of progressive pulmonary fibrosis. J Clin Invest. 1995;96:250–259. doi: 10.1172/JCI118029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Piguet PF, Collart MA, Grau GE, Kapanci Y, Vassalli P. Tumor necrosis factor/cachectin plays a key role in bleomycin-induced pneumopathy and fibrosis. J Exp Med. 1989;170:655–663. doi: 10.1084/jem.170.3.655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Piguet PF, Collart MA, Grau GE, Sappino AP, Vassalli P. Requirement of tumour necrosis factor for development of silica-induced pulmonary fibrosis. Nature. 1990;344:245–247. doi: 10.1038/344245a0. [DOI] [PubMed] [Google Scholar]
- 182.Hasegawa M, Fujimoto M, Kikuchi K, Takehara K. Elevated serum tumor necrosis factor-alpha levels in patients with systemic sclerosis: association with pulmonary fibrosis. J Rheumatol. 1997;24:663–665. [PubMed] [Google Scholar]
- 183.Bahcecioglu IH, Koca SS, Poyrazoglu OK, Yalniz M, Ozercan IH, Ustundag B, Sahin K, Dagli AF, Isik A. Hepatoprotective effect of infliximab, an anti-TNF-alpha agent, on carbon tetrachloride-induced hepatic fibrosis. Inflammation. 2008;31:215–221. doi: 10.1007/s10753-008-9067-1. [DOI] [PubMed] [Google Scholar]
- 184.Kolb M, Margetts PJ, Anthony DC, Pitossi F, Gauldie J. Transient expression of IL-1beta induces acute lung injury and chronic repair leading to pulmonary fibrosis. J Clin Invest. 2001;107:1529–1536. doi: 10.1172/JCI12568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Zhang Y, Lee TC, Guillemin B, Yu MC, Rom WN. Enhanced IL-1 beta and tumor necrosis factor-alpha release and messenger RNA expression in macrophages from idiopathic pulmonary fibrosis or after asbestos exposure. J Immunol. 1993;150:4188–4196. [PubMed] [Google Scholar]
- 186.Kamari Y, Shaish A, Vax E, Shemesh S, Kandel-Kfir M, Arbel Y, Olteanu S, Barshack I, Dotan S, Voronov E, et al. Lack of interleukin-1alpha or interleukin-1beta inhibits transformation of steatosis to steatohepatitis and liver fibrosis in hypercholesterolemic mice. J Hepatol. 2011;55:1086–1094. doi: 10.1016/j.jhep.2011.01.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Piguet PF, Vesin C, Grau GE, Thompson RC. Interleukin 1 receptor antagonist (IL-1ra) prevents or cures pulmonary fibrosis elicited in mice by bleomycin or silica. Cytokine. 1993;5:57–61. doi: 10.1016/1043-4666(93)90024-Y. [DOI] [PubMed] [Google Scholar]
- 188.Gasse P, Mary C, Guenon I, Noulin N, Charron S, Schnyder-Candrian S, Schnyder B, Akira S, Quesniaux VF, Lagente V, et al. IL-1R1/MyD88 signaling and the inflammasome are essential in pulmonary inflammation and fibrosis in mice. J Clin Invest. 2007;117:3786–3799. doi: 10.1172/JCI32285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Guo J, Gu N, Chen J, Shi T, Zhou Y, Rong Y, Zhou T, Yang W, Cui X, Chen W. Neutralization of interleukin-1 beta attenuates silica-induced lung inflammation and fibrosis in C57BL/6 mice. Arch Toxicol. 2013;87:1963–1973. doi: 10.1007/s00204-013-1063-z. [DOI] [PubMed] [Google Scholar]
- 190.Re SL, Giordano G, Yakoub Y, Devosse R, Uwambayinema F, Couillin I, Ryffel B, Marbaix E, Lison D, Huaux F. Uncoupling between inflammatory and fibrotic responses to silica: evidence from MyD88 knockout mice. PLoS One. 2014;9:e99383. doi: 10.1371/journal.pone.0093532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Girtsman TA, Beamer CA, Wu N, Buford M, Holian A. IL-1R signalling is critical for regulation of multi-walled carbon nanotubes-induced acute lung inflammation in C57Bl/6 mice. Nanotoxicology. 2014;8:17–27. doi: 10.3109/17435390.2012.744110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Suwara MI, Green NJ, Borthwick LA, Mann J, Mayer-Barber KD, Barron L, Corris PA, Farrow SN, Wynn TA, Fisher AJ, Mann DA. IL-1alpha released from damaged epithelial cells is sufficient and essential to trigger inflammatory responses in human lung fibroblasts. Mucosal Immunol. 2014;7:684–693. doi: 10.1038/mi.2013.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Lesur OJ, Mancini NM, Humbert JC, Chabot F, Polu JM. Interleukin-6, interferon-gamma, and phospholipid levels in the alveolar lining fluid of human lungs. Profiles in coal worker’s pneumoconiosis and idiopathic pulmonary fibrosis. Chest. 1994;106:407–413. doi: 10.1378/chest.106.2.407. [DOI] [PubMed] [Google Scholar]
- 194.Fielding CA, Jones GW, McLoughlin RM, McLeod L, Hammond VJ, Uceda J, Williams AS, Lambie M, Foster TL, Liao CT, et al. Interleukin-6 signaling drives fibrosis in unresolved inflammation. Immunity. 2014;40:40–50. doi: 10.1016/j.immuni.2013.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Fan JM, Huang XR, Ng YY, Nikolic-Paterson DJ, Mu W, Atkins RC, Lan HY. Interleukin-1 induces tubular epithelial-myofibroblast transdifferentiation through a transforming growth factor-beta1-dependent mechanism in vitro. Am J Kidney Dis. 2001;37:820–831. doi: 10.1016/S0272-6386(01)80132-3. [DOI] [PubMed] [Google Scholar]
- 196.Doerner AM, Zuraw BL. TGF-beta1 induced epithelial to mesenchymal transition (EMT) in human bronchial epithelial cells is enhanced by IL-1beta but not abrogated by corticosteroids. Respir Res. 2009;10:100. doi: 10.1186/1465-9921-10-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Camara J, Jarai G. Epithelial-mesenchymal transition in primary human bronchial epithelial cells is Smad-dependent and enhanced by fibronectin and TNF-alpha. Fibrogenesis Tissue Repair. 2010;3:2. doi: 10.1186/1755-1536-3-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Rieder F, Kessler SP, West GA, Bhilocha S, de la Motte C, Sadler TM, Gopalan B, Stylianou E, Fiocchi C. Inflammation-induced endothelial-to-mesenchymal transition: a novel mechanism of intestinal fibrosis. Am J Pathol. 2011;179:2660–2673. doi: 10.1016/j.ajpath.2011.07.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Maleszewska M, Moonen JR, Huijkman N, van de Sluis B, Krenning G, Harmsen MC. IL-1beta and TGFbeta2 synergistically induce endothelial to mesenchymal transition in an NFkappaB-dependent manner. Immunobiology. 2013;218:443–454. doi: 10.1016/j.imbio.2012.05.026. [DOI] [PubMed] [Google Scholar]
- 200.Park EJ, Cho WS, Jeong J, Yi J, Choi K, Park K. Pro-inflammatory and potential allergic responses resulting from B cell activation in mice treated with multi-walled carbon nanotubes by intratracheal instillation. Toxicology. 2009;259:113–121. doi: 10.1016/j.tox.2009.02.009. [DOI] [PubMed] [Google Scholar]
- 201.Han SG, Andrews R, Gairola CG. Acute pulmonary response of mice to multi-wall carbon nanotubes. Inhal Toxicol. 2010;22:340–347. doi: 10.3109/08958370903359984. [DOI] [PubMed] [Google Scholar]
- 202.Frank EA, Birch ME, Yadav JS. MyD88 mediates in vivo effector functions of alveolar macrophages in acute lung inflammatory responses to carbon nanotube exposure. Toxicol Appl Pharmacol. 2015;288:322–329. doi: 10.1016/j.taap.2015.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Guo H, Callaway JB, Ting JP. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med. 2015;21:677–687. doi: 10.1038/nm.3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Artlett CM, Sassi-Gaha S, Rieger JL, Boesteanu AC, Feghali-Bostwick CA, Katsikis PD. The inflammasome activating caspase 1 mediates fibrosis and myofibroblast differentiation in systemic sclerosis. Arthritis Rheum. 2011;63:3563–3574. doi: 10.1002/art.30568. [DOI] [PubMed] [Google Scholar]
- 205.Dostert C, Petrilli V, Van Bruggen R, Steele C, Mossman BT, Tschopp J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science. 2008;320:674–677. doi: 10.1126/science.1156995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, Abela GS, Franchi L, Nunez G, Schnurr M, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464:1357–1361. doi: 10.1038/nature08938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Palomaki J, Valimaki E, Sund J, Vippola M, Clausen PA, Jensen KA, Savolainen K, Matikainen S, Alenius H. Long, needle-like carbon nanotubes and asbestos activate the NLRP3 inflammasome through a similar mechanism. ACS Nano. 2011;5:6861–6870. doi: 10.1021/nn200595c. [DOI] [PubMed] [Google Scholar]
- 208.Wang L, Mercer RR, Rojanasakul Y, Qiu A, Lu Y, Scabilloni JF, Wu N, Castranova V. Direct fibrogenic effects of dispersed single-walled carbon nanotubes on human lung fibroblasts. J Toxicol Environ Health A. 2010;73:410–422. doi: 10.1080/15287390903486550. [DOI] [PubMed] [Google Scholar]
- 209.Wang X, Xia T, Ntim SA, Ji Z, George S, Meng H, Zhang H, Castranova V, Mitra S, Nel AE. Quantitative techniques for assessing and controlling the dispersion and biological effects of multiwalled carbon nanotubes in mammalian tissue culture cells. ACS Nano. 2010;4:7241–7252. doi: 10.1021/nn102112b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Vietti G, Ibouraadaten S, Palmai-Pallag M, Yakoub Y, Piret JP, Marbaix E, Lison D, van den Brule S. Towards predicting the lung fibrogenic activity of MWCNT: Key role of endocytosis, kinase receptors and ERK 1/2 signaling. Nanotoxicology. 2016;10:488–500. doi: 10.3109/17435390.2015.1088588. [DOI] [PubMed] [Google Scholar]
- 211.Hussain S, Sangtian S, Anderson SM, Snyder RJ, Marshburn JD, Rice AB, Bonner JC, Garantziotis S. Inflammasome activation in airway epithelial cells after multi-walled carbon nanotube exposure mediates a profibrotic response in lung fibroblasts. Part Fibre Toxicol. 2014;11:28. doi: 10.1186/1743-8977-11-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Goffin JM, Pittet P, Csucs G, Lussi JW, Meister JJ, Hinz B. Focal adhesion size controls tension-dependent recruitment of alpha-smooth muscle actin to stress fibers. J Cell Biol. 2006;172:259–268. doi: 10.1083/jcb.200506179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Wipff PJ, Rifkin DB, Meister JJ, Hinz B. Myofibroblast contraction activates latent TGF-beta1 from the extracellular matrix. J Cell Biol. 2007;179:1311–1323. doi: 10.1083/jcb.200704042. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data and materials are included in this article.