

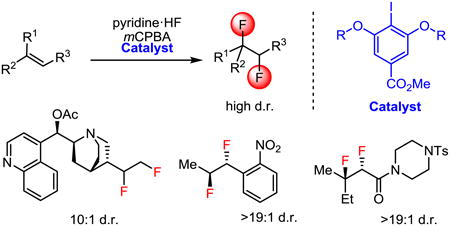
Abstract
We describe a direct, catalytic approach to the 1,2-difluorination of alkenes. The method utilizes a nucleophilic fluoride source and an oxidant in conjunction with an aryl iodide catalyst and is applicable to alkenes with all types of substitution patterns. In general, the vicinal difluoride products are produced with high diastereoselectivities. The observed sense of stereoinduction implicates anchimeric assistance pathways in reactions of alkenes bearing neighboring Lewis basic functionality.
Graphical abstract
In contrast to alkene dichlorination and dibromination, which are among the most general and well-developed reactions in organic chemistry,1,2 analogous 1,2-difluorination reactions present challenges both fundamental and practical. Direct difluorination of alkenes3 may be accomplished with stoichiometric amounts of highly reactive and oxidizing F2,4 XeF2,5 or a combination of reagents such as Selectfluor® or p-iodotoluene difluoride with HF.6,7 Stereocontrol can be difficult to achieve in difluorination reactions utilizing these reagents4b due to the participation of open β-fluorocarbenium intermediates (Scheme 1A)6,8–10 although F2 diluted with N2 has been demonstrated to effect alkene difluorination diastereoselectively at low temperatures.11
Scheme 1. 1,2-Difluorination of Alkenes.
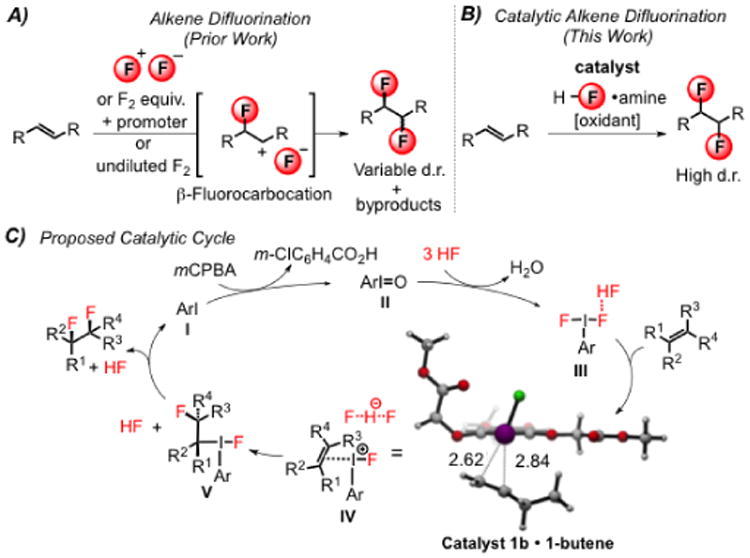
Fluorine incorporation is well-known to modulate a number of important properties of organic molecules, including lipophilicity, bioavailability, and metabolic stability.12 Vicinal difluorides possess the additional interesting characteristic of adopting preferred gauche conformations.13–15 We hypothesized that the extent to which the gauche effect may serve as a useful principle in the design of functional molecules might be increased through improved, direct synthetic access to 1,2-difluoride motifs.16 We report here a new catalytic method for diastereoselective alkene 1,2-difluorination that is applicable to a broad range of alkene substrates.17
Alkene 1,2-difluorination has been demonstrated by Hara and coworkers using stoichiometric p-iodotoluene difluoride with Et3N•5HF as the fluorine source and predominantly isolated terminal alkenes as substrates.7 Based on this seminal report, we envisioned a catalytic method based on a single, readily available nucleophilic fluorination reagent. In this context, aryl iodide catalysis has emerged as a metal-free approach to alkene difunctionalization through the intermediacy of aryliodonium (III) species.18,19 In the proposed catalytic pathway, an aryl iodide (I) could undergo transformation to a reactive iodoarene difluoride (III) through stepwise oxidation to the iodosylarene (II) and subsequent deoxyfluorination with an appropriate HF source.20 Vicinal alkene difluorination can then proceed in a stereospecific manner through the intermediacy of discrete intermediates IV and V.21,22
This approach to the catalytic 1,2-difluorination of alkenes was examined using terminal alkenes 2a and 2b as model substrates (Scheme 2). A systematic evaluation of fluoride sources and oxidants led to the identification of HF–pyridine (pyr•9HF) and m-chloroperbenzoic acid (mCPBA) as the most effective reagent combination.23 Thus, allyl benzene derivative 2a underwent 1,2-difluorination with 1a as catalyst, although a large excess of the HF source (20–100 equiv.) was required to achieve good yields.24 Despite this practical limitation, the reaction mixtures could be worked up safely and in a straightforward manner using basic alumina or aqueous sodium hydroxide to quench excess HF. In the case of isolated alkene 2b (Scheme 2, Reaction 2), added pyridine afforded significant improvements in product yield, most likely by reducing the acidity of the medium and thereby generating a more nucleophilic source of fluoride. The resorcinol derivative 1b proved to be measurably more effective than 1a in catalyzing the 1,2-difluorination of 2b. On the basis of its performance in these and other model reactions (vide infra), as well as the ease of accessing chiral variants for investigation of enantioselectivity (vide infra), catalyst 1b was selected for subsequent studies on the scope of the alkene difluorination reaction.
Scheme 2. Catalyst Identification.

aYields determined by GC with dodecane as an internal standard.
In general, terminal alkenes were found to undergo 1,2-difluorination in moderate-to-good yield (Figure 1). Reactions proceeded to full conversion, with the only identifiable byproducts resulting from competitive epoxidation/ring-opening or from m-chlorobenzoic acid addition to alkyl iodonium intermediates. The difluoride products were readily isolated in pure form by column chromatography. A variety of substituted allylbenzene derivatives were found to display comparable reactivity (2a, 2c–d) and afforded good yields of difluoride products using catalyst 1b and slow addition of alkene on preparative scale. However, phenols and methoxy-substituted arenes were not compatible with the reaction conditions, as expected given the known arene oxidation chemistry of hypervalent iodine species (3l).25 Amine-containing terminal alkenes were protected from oxidation in situ through protonation, allowing for difluorination of substrates 2f–i in good yields. Difluorination of 1,1-disubstituted alkene 2j occurred selectively on the endo face to generate 1,2-difluoride 3j as the major diastereomer.26 Functional group tolerance was further demonstrated with successful difluorination of O-acetylcinchonidine in 63% yield ((–)-3k) and high diastereoselectivity (10:1 d.r.).
Figure 1. Terminal Alkene Substrate Scope.

aReactions were conducted on 1.04 mmol scale, with yields of diastereomerically product isolated after chromatographic purification unless noted otherwise. bReactions conducted with 20 mol% catalyst, with slow addition of substrate over 2 hours. cReactions conducted with 10 mol% catalyst and 6 equivalents of pyridine. dReactions conducted with 20 mol% catalyst. eDetermined by 19F NMR of the crude reaction mixture. fIsolated as a mixture of diastereomers.
In efforts to extend the catalytic 1,2-difluorination protocol to internal alkenes as substrates, we found that symmetrical 1,2-disubstituted alkenes such as E-5-decene and cyclohexene did not provide the desired products; mixtures of unidentified fluorinated and oligomeric products were obtained instead. However, conjugated trisubstituted alkenes such as β,β-dimethylstyrene derivatives 4a–h underwent reaction to afford the corresponding 1,2-difluorides in good yields (Figure 2).27 Subjection of trisubstituted indene 4i to the catalytic reaction conditions provided 5i in 60% yield and 5:1 d.r. favoring the syn diastereomer, as established by 19F NMR and X-ray diffraction analysis of a crystalline derivative.26 Although β-monosubstituted styrenes are susceptible to rearrangement via phenonium intermediates in hypervalent iodine-promoted reactions,28 highly electron-deficient 4j afforded the syn-difluorination product in 19:1 d.r. In contrast, o-nitrostyrene derivatives 4k–m underwent difluorination to give the corresponding anti-addition products with high diastereoselectivity.26 The basis for this intriguing stereochemical reversal is discussed below and in Scheme 3A.
Figure 2. Internal Alkene Substrate Scope.

aReactions were conducted on 0.62–1.94 mmol scale, with yields of diastereomerically pure product isolated after chromatographic purification unless noted otherwise. Diastereomeric ratios (d.r. values) were determined by 19F NMR of crude reaction mixtures. bReactions were conducted with slow addition of substrate over 2 hours and were allowed to progress with stirring for an additional 1 hour. cReaction was conducted at 0 °C with slow addition of substrate over 1 hour and stirring for an additional 1 hour. dReactions were conducted with slow addition of substrate over 2 hours and stirring for an additional 10 hours. eReaction was conducted in neat pyr•9HF for 12 hours. Isolated as a mixture of diastereomers. fReaction was conducted in neat pyr•9HF for 36 hours with a second addition of mCPBA (0.65 equiv) after 24 hours. gReaction was conducted with 20 equiv of HF for 24 hours. Isolated as a mixture of a diastereomers.
Scheme 3. Anti Difluorination via Proposed Anchimeric Assistance.

Alkenes bearing nitrogen-containing heterocyclic substituents (4n–q) were also suitable substrates for the difluorination reactions. The electron-deficient nature of the protonated heterocycles led to diminished reactivity relative to simple styrenes. However, the 1,2-difluoride products (5n–q) could be obtained if the reactions were carried out in neat pyr•9HF consistent with the known activating effect of Brønsted acids on aryl iodide intermediates.29 Amine-containing internal alkene substrates were also compatible with the catalytic reaction conditions. Thus, amitriptyline (4r) underwent difluorination to 5r, and aliphatic internal alkene 4s afforded difluoride 5s with a 10:1 d.r. favoring the endo-diastereomer, albeit in low yield.26, 30
Because of their electron-deficient nature, α, β-unsaturated carbonyl compounds are particularly challenging substrates for electrophilic oxidation reactions. Nonetheless, consistently high product yields and diastereoselectivities (>19:1 d.r.) were obtained in the 1,2-difluorination of a variety of sterically and electronically differentiated acrylamides (Figure 3). In particular, cinnamamides 6a–c and β,β-disubstituted acrylamides 6d–f proved to be effective substrates, and the difluorination of tetrasubstituted alkene 6d was executed on gram scale to afford 7d. Stereoisomers 6e and 6f underwent stereospecific 1,2-difluorination to give diastereomeric difluorides 7e and 7f, respectively. Acrylamides lacking carbocation-stabilizing groups at the β-position (6g–k) were found to be poorly reactive with catalyst 1b. However, modified conditions using catalyst 1c in neat pyr•9HF gave rise to synthetically useful yields (7g–k).
Figure 3. Acrylamide Substrate Scope.

aReactions were conducted on 0.55–5.71 mmol scale, with yields of diastereomerically pure product isolated after chromatographic purification. Diastereomeric ratios (d.r. values) were determined by 19F NMR of crude reaction mixtures. bReactions were conducted using catalyst 1b. cReaction was conducted on 1 gram scale. dReactions were conducted in neat pyr•9HF using catalyst 1c.
In general, diastereoselectivity in the 1,2-difluorination reactions was found to be high, but the identity of the major adduct was highly dependent on substrate structure. Alkenes lacking proximal Lewis basic groups (e.g. 4i–j) underwent addition to give syn-difluorination products. This outcome is consistent with the mechanistic hypothesis outlined in Scheme 1C in which intermediate V undergoes invertive attack by fluoride at the Csp3–iodine(III) bond. Competitive ionization of either a benzylic or tertiary alkyl iodine(III) intermediate could account for formation of the minor diastereomer.31 However, crystallographic characterization of structures of 7a,c,f–g,k revealed that difluorination occurred with anti diastereospecificity,26 in a manner analogous with o-nitrostyrene derivatives as noted above. This stereochemical outcome suggests that reactions of alkenes bearing weakly Lewis basic groups adjacent to the reaction site are subject to anchimeric assistance pathways. We propose that complexes I4k–m and I6 undergo nucleophilic fluorination at the carbon bearing greater positive charge to generate fluoroalkyl iodonium(III) intermediates II4k–m32 and II6 (Scheme 3A and B). Subsequent intramolecular invertive displacement by the nitro group in II4k–m33a and by the carbonyl in II6, affords onium ions III4k–m and oxiraniminium ion III6,33b respectively. These reactive intermediates are proposed to then undergo invertive attack by a fluoride ion to afford anti-difluorinated products 5k–m and 7. The observation of these anchimeric assistance pathways34 highlight the highly electrophilic nature of the aryliodonium(III) species in the catalytic difluorination reactions, and suggest that there may be a rich chemistry associated with trapping the fluorinated intermediates by internal or exogenous nucleophiles.
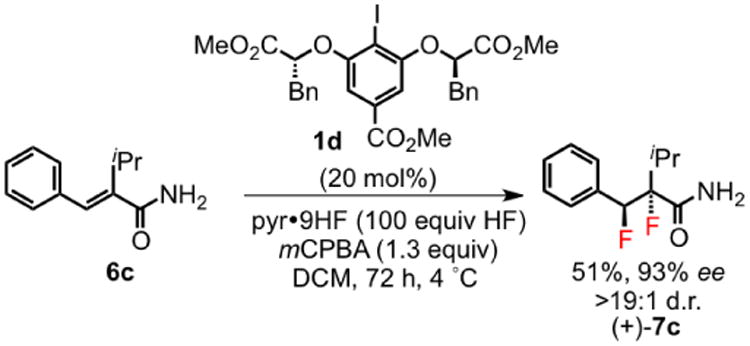
In a preliminary effort to identify asymmetric variants of the difluorination reaction, chiral aryl iodide 1d was prepared and examined in the reaction of cinnamamide 6c (Scheme 4). Reduced reactivity was observed relative to achiral catalyst 1b, but moderate product yields and excellent enantioselectivity were obtained after 72 hours at 4 °C. This result establishes a strong proof-of-concept for the enantioselective catalytic difluorination of alkenes and also confirms the direct participation of hypervalent iodide species in the C–F bond-forming steps of the catalytic reaction.
Scheme 4. Highly Enantioselective 1,2-Difluorination.
This work introduces a new method for catalytic alkene difluorination. Extension of the reactivity principles outlined herein to new alkene 1,2-difunctionalization reactions are under investigation and will be reported in due course.
Supplementary Material
Acknowledgments
This work was supported by the NIH (GM043214), by an NSF pre-doctoral fellowship to S.M.B, and by an NIH post-doctoral fellowship to J.W.M. We thank Dr. Shao-Liang Zheng (Harvard) for determination of all of the X-ray crystal structures.
Funding Sources: No competing financial interests have been declared.
Footnotes
Supporting Information. Experimental procedures and characterization data for new compounds are available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Carey FA, Sundberg RJ. Advanced Organic Chemistry Part B: Re actions and Synthesis. 5. Springer; New York: 2007. pp. 298–305. [Google Scholar]
- 2.For a recent review on catalytic, stereoselective dihalogenation, see Cresswell AJ, Eey STC, Denmark SE. Angew Chem, Int Ed. 2015;54:15642–15682. doi: 10.1002/anie.201507152.
- 3.For an indirect method method, see Olah GA, Welch JT, Vankar YD, Nojima M, Kerekes I, Olah JA. J Org Chem. 1979;44:3872–3881.
- 4.(a) Barton DHR, Lister-James J, Hesse RH, Pechet MM, Rozen S. J Chem Soc, Perkin Trans. 1982;1:1105–1110. [Google Scholar]; (b) Purrington ST, Kagen BS, Patrick TB. Chem Rev. 1986;86:997–1018. [Google Scholar]; (c) Sanford G. J Fluorine Chem. 2007;128:90–104. [Google Scholar]
- 5.(a) Tius MA. Tetrahedron. 1995;51:6605–6634. [Google Scholar]; (b) Tamura M, Takagi T, Quan HD, Sekiya A. J Fluorine Chem. 1999;98:163–166. [Google Scholar]
- 6.Lal GS. J Org Chem. 1993;58:2791–2796. [Google Scholar]
- 7.Hara S, Nakahigashi J, Ishi-I K, Sawaguchi M, Sakai H, Fukuhara T, Yoneda N. Synlett. 1998:495–496. A single example of an internal alkene was also reported to be diastereoselective. [Google Scholar]
- 8.Stavber S, Sotler T, Zupan M. J Org Chem. 1994;59:5891–5894. [Google Scholar]
- 9.For difluorination of alkenes with PhSeF3, PhSeF5, and PhTeF5, see Lermontov SA, Zavorin SI, Bakhtin IV, Pushin NA, Zefirov NS, Stang PJ. J Fluorine Chem. 1998;87:75–83.
- 10.Radical pathways can also be a source for poor diastereocontrol in reactions with undiluted F2, see: Olah G, Molnár A. Hydrocarbon Chemistry. 2. Wiley; Hoboken: 2003. pp. 304–307.
- 11.(a) Rozen S, Brand M. J Org Chem. 1986;51:3607–3611. [Google Scholar]; (b) Vints I, Rozen S. J Org Chem. 2014;79:7261–7265. doi: 10.1021/jo5009542. [DOI] [PubMed] [Google Scholar]; (c) Vints I, Rozen S. Tetrahedron. 2016;72:632–636. [Google Scholar]
- 12.(a) Purser S, Moore PR, Swallow S, Gouverneur V. Chem Soc Rev. 2008;37:320–330. doi: 10.1039/b610213c. [DOI] [PubMed] [Google Scholar]; (b) Müller K, Faeh C, Diederich F. Science. 2007;317:1881–1886. doi: 10.1126/science.1131943. [DOI] [PubMed] [Google Scholar]; (c) Wang J, Sanchez-Rosello M, Acena JL, del Pozo C, Sorochinsky AE, Fustero S, Soloshonok VA, Liu H. Chem Rev. 2014;114:2432–2506. doi: 10.1021/cr4002879. [DOI] [PubMed] [Google Scholar]; (d) Hagmann WK. J Med Chem. 2008;51:4359–4369. doi: 10.1021/jm800219f. [DOI] [PubMed] [Google Scholar]
- 13.For the effect of vicinal 1,2-difluorides on several properties in bioactive molecules, see Huchet QA, Kuhn B, Wagner B, Kratochwil NA, Fischer H, Kansy M, Zimmerli D, Carreira EM, Müller K. J Med Chem. 2015;58:9041–9060. doi: 10.1021/acs.jmedchem.5b01455.
- 14.For a general review of the gauche effect, see O'Hagan D. Chem Soc Rev. 2008;37:308–319. doi: 10.1039/b711844a.Buissonneaud DY, van Mourik T, O'Hagan D. Tetrahedron. 2010;66:2196–2202.Fox SJ, Gourdain S, Coulthurst A, Fox C, Kuprov I, Essex JW, Skylaris CK, Linclau B. Chem Eur J. 2011;17:2340–2343. doi: 10.1002/chem.201405317.
- 15.For a review, see Hunter L. Beilstein J Org Chem. 2010;6:38. doi: 10.3762/bjoc.6.38.Gillis EP, Eastman KJ, Hill MD, Donnelly DJ, Meanwell NA. J Med Chem. 2015;58:8315–8359. doi: 10.1021/acs.jmedchem.5b00258.O'Hagan D. J Org Chem. 2012;77:3689–3699. doi: 10.1021/jo300044q.Wang Z, Hunter L. J Fluorine Chem. 2012;143:143–147.Myers AG, Barbay J, Zhong B. J Am Chem Soc. 2001;123:7207–7219. doi: 10.1021/ja010113y.Yamamoto I, Jordan MJT, Gavande N, Doddareddy MR, Chebib M, Hunter L. Chem Commun. 2012;48:829–831. doi: 10.1039/c1cc15816c.Hunter L, Joliffe KA, Jordan MJT, Jensen P, Macquart RB. Chem Eur J. 2011;17:2340–2343. doi: 10.1002/chem.201003320.Hu XG, Thomas DS, Griffith R, Hunter L. Angew Chem Int, Ed. 2014;53:6176–6179. doi: 10.1002/anie.201403071.O'Hagan D, Rzepa HS, Schüler M, Slawin AMZ. Beilstein J Org Chem. 2006;2:19. doi: 10.1186/1860-5397-2-19.
- 16.For a synthesis and study of multivicinal difluoride compounds, see Hunter L, Kirsch P, Slawin AMZ, O'Hagan D. Angew Chem, Int Ed. 2009;48:5457–5460. doi: 10.1002/anie.200901956. and references therein.
- 17.SiF4 and HF have been shown to catalyze the addition of XeF2 to styrene substrates (reference 5b).
- 18.(a) Ochiai M, Miyamoto K. Eur J Org Chem. 2008:4229–4239. [Google Scholar]; (b) Berthiol F. Synthesis. 2015;47:587–603. [Google Scholar]
- 19.For the seminal reports on the preparation and use of aryl iodine difluorides as fluorinating reagents, see: Weinland RF, Stille W. Chem Ber. 1901;34:2631–2633.Edmunds JJ, Motherwell WB. J Chem Soc, Chem Commun. 1989:881–883.
- 20.(a) Arrica MA, Wirth T. Eur J Org Chem. 2005:395–403. [Google Scholar]; (b) Ye C, Twamley B, Shreeve JM. Org Lett. 2005;7:3961–3964. doi: 10.1021/ol051446t. [DOI] [PubMed] [Google Scholar]
- 21.Gade L, Kang YB. J Am Chem Soc. 2011;133:3658–3667. doi: 10.1021/ja110805b. [DOI] [PubMed] [Google Scholar]
- 22.Computational modeling (M062X/6-31+G(d,p)/SDD for I/CPCM(DCM)) of proposed intermediate IV (Scheme 1) predicts highly unsymmetrical coordination of the alkene to the iodonium fluoride, with the more substituted carbon of the alkene bearing more positive charge. For details, see the supporting information.
- 23.No product was obtained using Et3N•3HF, and reactions with HF (aq.) were impractically slow. Oxidants examined include: tBuOOH, AcOOH, SelectFluor, iodic acid, DDQ, DCC/UHP, sodium peroxydisulfate, Cu(OTf)2, 3,3-dimethyl-2-((4-nitrophenyl)sulfonyl)-1,2-oxaziridine, and oxone.
- 24.The combination of p-iodotoluene difluoride and pyr•9HF was reported to effect the stoichiometric 1,2-difluorination of terminal alkenes in low yields (ref. 7).
- 25.Kita Y, Tohma H, Hatanaka K, Takada T, Fujita S, Mitoh S, Sakurai H, Oka S. J Am Chem Soc. 1994;116:3684–3691. [Google Scholar]
- 26.Confirmed by single crystal X-ray diffraction analysis. For details see the supporting information.
- 27.With substrates 4a–4i, slow addition of alkene was crucial for reducing competitive epoxidation by mCPBA and hydrofluorination. Negligible conversion (<2%) to difluoride products was observed in the absence of catalyst 1b.
- 28.(a) Zupan M, Pollak A. J Chem Soc, Chem Commun. 1975:715–716. [Google Scholar]; (b) Kitamura T, Muta K, Oyamada J. J Org Chem. 2015;80:10431–10436. doi: 10.1021/acs.joc.5b01929. [DOI] [PubMed] [Google Scholar]
- 29.Cotter JL, Andrews LJ, Keefer RM. J Am Chem Soc. 1962;84:793–797. [Google Scholar]
- 30.Competitive rearrangement pathways were minimized with this substrate through the use of lowered amounts of pyr•9HF (20 equivalents of HF).
- 31.(a) Macdonald TL, Narasimhan N, Burka LT. J Am Chem Soc. 1980;102:7760–7765. [Google Scholar]; (b) Cambie RC, Chambers D, Lindsay BG, Rutledge PS, Woodgate PD. J Chem Soc, Perkin Trans. 1980;1:822–827. [Google Scholar]
- 32.Fluoride attack at the homobenzylic position of I4k–m followed by 5-exo displacement of I(III) by the nitro group cannot be rigorously excluded. For literature support of the proposed regioselectivity, see: Fujita M, Yoshida Y, Miyata K, Wakisaka A, Sugimura T. Angew Chem Int Ed. 2010;49:7068–7071. doi: 10.1002/anie.201003503.
- 33.(a) Olah GA, De Member JR. J Am Chem Soc. 1970;92:2562–2564. [Google Scholar]; (b) Walden P. Ber Dtsch Chem Ges. 1896;29:133–138. [Google Scholar]; (c) Winstein S, Lucas HJ. J Am Chem Soc. 1939;61:1576–1581. [Google Scholar]
- 34.Anchimeric assistance has been proposed in dihalogenation reactions that proceed through haliranium intermediates, see: Shibuya GM, Kanady JS, Vanderwal CD. J Am Chem Soc. 2008;130:12514–12518. doi: 10.1021/ja804167v.Nilewski C, Geisser RW, Carreira EM. Nature. 2009;457:573–576. doi: 10.1038/nature07734.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.