

Abstract
Zinc is both an essential and potentially toxic metal. It is widely believed that oral zinc supplementation can reduce the effects of the common cold; however, there is strong clinical evidence that intranasal (IN) zinc gluconate (ZG) gel treatment for this purpose causes anosmia, or the loss of the sense of smell, in humans. Using the rat olfactory neuron cell line, Odora, we investigated the molecular mechanism by which zinc exposure exerts its toxic effects on olfactory neurons. Following treatment of Odora cells with 100 and 200 μM ZG for 0-24 h, RNA-seq and in silico analyses revealed up-regulation of pathways associated with zinc metal response, oxidative stress, and ATP production. We observed that Odora cells recovered from zinc-induced oxidative stress, but ATP depletion persisted with longer exposure to ZG. ZG exposure increased levels of NLRP3 and IL-1β protein levels in a time-dependent manner, suggesting that zinc exposure may cause an inflammasome-mediated cell death, pyroptosis, in olfactory neurons.
Keywords: olfactory neurons, toxicity, Odora, zinc gluconate, pyroptosis
Introduction
Zinc is an essential element for many organisms, and zinc deficiency has been associated with multiple adverse conditions, including impaired immune function (Bonaventura et al., 2015; Brooks et al., 2004; Muller et al., 2001). In response to this observation, over-the-counter zinc lozenges and nasal sprays were introduced for the treatment of the common cold in the United States during the late 1990s. Unfortunately, the scientific and medical community began to report incidences of anosmia, or the loss of the sense of smell, among people who had used intranasal zinc sprays and gels (Alexander and Davidson, 2006; DeCook and Hirsch; Jafek et al., 2004). Due to potential bias in reporting symptoms and actual use of the zinc gluconate (ZG) nasal products, Davidson and Smith evaluated all available evidence at the time concerning the probability of a causal relationship between zinc nasal products and loss of the sense of smell. They concluded that there was evidence of causality between over the counter ZG nasal sprays and impaired sense of smell (Davidson and Smith, 2010). The large number of incidences of anosmia resulted in the removal of intranasal (IN) zinc sprays from store shelves. However, a recent article has reported on several clinical trials which use IN insulin for the treatment of food cravings, forgetfulness in Alzheimer's patients, and diabetes (Dash et al., 2015; Freiherr et al., 2013; Hallschmid et al., 2012; Hamidovic). This is a concern due to the use of zinc to stabilize insulin and to prevent protein aggregation in these formulations (Manallack et al., 1985). While there have not yet been any published reports of anosmia in these trials, possibly due to the relatively small samples size and low zinc concentration, there is the potential for more cases of anosmia in these clinical trials which use IN insulin formulations containing zinc.
Unlike most neurons in the body, olfactory neurons are generated throughout one's lifetime. This process is not synchronized among the neurons, which means that there is a heterogeneous distribution of immature, mature, and dying neurons present in the nose at any given time (Cancalon and Elam, 1980; Farbman, 1994). Thus, it is surprising and alarming that the anosmia in some users of IN ZG spray seem to be permanent, suggesting that the basal cell population, which gives rise to olfactory neurons was destroyed; this very phenomenon was reported in mice treated with 2,6-methylsulfonyl-2,6- dichlorobenzene, a toxicant that destroyed the olfactory epithelium, including basal cells, resulting in respiratory metaplasia, but not recovery of the olfactory epithelium (Bahrami et al., 2000).
There are many proposed mechanisms for zinc-induced toxicity ranging from oxidative stress to impaired ATP production to metal dyshomeostasis. Thus, the elucidation of the mechanism of zinc toxicity is more complicated than it would appear, and is affected by many factors, such as concentration of zinc tested, the length of exposure, the cell type, and the presence of other toxic chemistries. These mechanisms are summarized below in Table 1.
Table 1. Mechanisms of zinc toxicity.
| Mechanism of Zinc Toxicity | Reference |
|---|---|
| Inhibition of glutathione reductase | (Bishop et al., 2007) |
| Production of ROS and/or activation of NADPH oxidase & nitric oxide synthase | (Bishop et al., 2007), (Kim and Koh, 2002), (Lopez et al., 2011) |
| Induction of apoptosis via DNA fragmentation and cytochrome C release | (Rudolf, 2007), (Feng et al., 2002) |
| Induction of necrosis | (Iitaka et al., 2001) |
| Impaired ATP production via inhibition of glycolytic and TCA cycle enzymes | (Rudolf, 2007), (Dineley et al., 2003), (Sheline et al., 2000) |
| Impaired function of the electron transport chain | (Lorusso et al., 1991), (Link and von Jagow, 1995) |
| Inhibition of proton channel (HVCN1), leading to intracellular acidosis | (Iovannisci et al., 2010) |
| Dysregulation of copper homeostasis | (Willis et al., 2005), (Hedera et al., 2009) |
| Dysregulation of calcium homeostasis | (Guo et al., 2014) |
In this study, we used the rat olfactory neuronal cell line, Odora. These cells are an excellent model for olfactory neurons in whole animals because they can properly target odorant receptors to the cell membrane and respond to odorants (Murrell and Hunter, 1999). In addition, as with the intact olfactory mucosa in vivo, Odora cells can be grown as immature undifferentiated neurons or as mature differentiated neurons. In order to elucidate the mechanism underlying zinc-induced anosmia, we performed a series of experiments, beginning with RNA-seq to identify genes and pathways that were up- or down-regulated in response to zinc exposure. After analysis of significantly regulated pathways, zinc-induced changes were confirmed using biochemical assays to measure changes in oxidative stress and ATP production as well as determining the critical amount of time for zinc exposure to irreversibly lead to cell death. Upon finding negative evidence for zinc-induced apoptotic cell death, based on the absence of caspase 3 and 9 upregulation, as well as negative results in an in vitro apoptosis assay, we pursued pyroptosis as a potential mechanism of zinc-induced cell death. Pyroptosis is a caspase-1 mediated mechanism of cell death involving the formation of the inflammsome via recruitment of Nod-like receptor protein 3 (NLRP3) and caspase-1. The activation of caspase-1 results in the activation and release of interleukin-1β, -6, and -18 from the cell into the extracellular space. Characteristics of pyroptosis include loss of plasma membrane integrity, cell swelling, and release of pro-inflammatory intracellular contents (Fink and Cookson, 2005). In vitro studies also identified the important role of caspase-1, the cytokine IL-1β and inflammasome activation, suggesting pyroptosis as a mechanism of cell death.
The goals of this study were to (i) determine what genes and pathways are up- or down-regulated by exposure to ZG in the olfactory neuronal cell line Odora, (ii) determine whether apoptosis occurs in zinc-induced cytotoxicity in Odora cells and (iii) determine whether zinc-induced toxicity in Odora cells can be prevented using anti-oxidants and enzyme inhibitors.
Materials & Methods
Chemicals and reagents
Odora cells were plated in 96-well plates (Falcon, Tewksbury, MA) and 145 × 20 mm tissue culture dishes (Greiner Bio One, Monroe, NC). DMEM, HBSS, and trypsin/EDTA from Hyclone (GE Healthcare Life Sciences, Logan, UT) were used for the cell culture work. Zinc gluconate (MP Biomedicals LLC, Santa Ana, CA) and crystal violet (Allied Chem. Corp., NY, NY) solutions were prepared using distilled water. For the biochemical assays, nitric oxide, and apoptosis work, o-phthalaldehyde (OPA, Alfa Aesar, Ward Hill, MA); luminol, horseradish peroxidase, Nω-Nitro-L-arginine methyl ester hydrochloride (L-NAME), apocynin and sodium arsenite (Sigma-Aldrich, St. Louis, MO); perchloric acid (Thermo Fisher Scientific, Waltham, MA), N-acetylcysteine (NAC, Acros, NJ) were used. The caspase-1 inhibitor, Ac-YVAD-cmk was obtained from Calbiochem (Billerica, MA). All assays utilizing a plate reader were performed with the Spectramax M2e multi-mode plate reader (Molecular Devices, Sunnyvale, CA). Fluorescence measures were performed with the F-4500 Fluorescence Spectrophotometer (Hitachi, Schaumburg, IL), while luminescence measurements were performed with either the Autolumat Plus luminometer (Berthold, Oak Ridge, TN) or Luminometer Model TD-20/20 (Turner BioSystems, Sunnyvale, CA).
Cell viability
Odora cells were obtained from Dr. Dale Hunter and cultured according to published protocols.(Murrell and Hunter, 1999). Immature Odora cells were seeded in 96-well plates (25,000 cells well-1) and allowed to attach and grow for 24 h. Cells were then treated with the ZG at concentrations ranging from 50 μM to 500 μM for times ranging from 1 – 24 h. An equivalent volume of sterile water was added to the media as the vehicle control. After the appropriate length of ZG exposure, crystal violet was used to assess viability as previously described (Kueng et al., 1989) by measuring absorbance at 540 nm. Results were expressed as a percentage of control (water-treated) cells. LC50 values were calculated using the SigmaStat software's 4 parameter logistic equation (sigmoidal) for each experiment. The LC50 from the three biologically independent replicates were averaged together to yield a mean LC50. LC50 studies were similarly conducted in differentiated Odora cells; due to the slower rate of growth of the differentiating Odora cells, these were allowed to grow for 72 h prior to ZG treatment.
RNA-seq analysis
Odora cells were seeded in tissue culture dishes (145 × 20 mm dish, 10ˆ6 cells plate-1) and allowed to attach and grow for 48 h. Cells were then dosed with 100 μM ZG for 6, 12, or 24 h or 200 μM ZG for 6 h. An equivalent amount of water was added in the vehicle control dishes. At the specified time points, RNA was isolated with mirVana miRNA Isolation Kit (Lifetech, Carlsbad, CA). All steps of library construction, cluster generation, and HiSeq (Illumina) sequencing were performed with biological triplicate samples by the Genomics, Epigenomics, and Sequencing Core of the Department of Environmental Health, University of Cincinnati. Differential gene expression analyses between the vehicle control and ZG-treated cells was performed separately at each of the three different exposure time points. Significant genes were selected based on a false-discovery rate– adjusted p-value < 0.01. RNA-seq data was further analyzed using DAVID Bioinformatics Resource 6.7 (Huang da et al., 2009). The gene expression data and results were deposited in GEO (Barrett et al., 2009) and can be accessed at http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE79730.
Protein quantification
Protein concentrations were determined by the bicinchoninic acid method using the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific) and following manufacturer's instructions. The plate was incubated at 37°C for 30 minute before the absorbance was read at 562 nm.
Biochemical Assays
Odora cells were seeded in tissue culture dishes (145 × 20 mm, 10ˆ6 cells plate-1) and allowed to attach and grow for 48 h. Cells were treated with 100 μM ZG for 6, 12, or 24 h (or an equivalent volume of sterile water) before the cells were harvested. GSH and GSSG in Odora cell homogenate were fluorometrically measured following o-phthalaldehyde (OPA) derivatization as previously described (Senft et al., 2000). Hydrogen peroxide concentration was determined using luminol (5-amino-2,3-dihydro-1,4-phthalazinedione) chemiluminescence on a Berthold Autolumat Plus luminometer as described previously (Senft et al., 2002a). ATP levels were determined by harvesting cells washed with ice cold saline (0.9% NaCl), scraped and collected in ice cold 5% perchloric acid, sonicated on ice for 20 sec before an aliquot was neutralized with NaOH in Tris buffer and assayed with the ATP Bioluminescent Assay Kit (Sigma Aldrich) as described previously (Senft et al., 2002b). For all assays, levels were normalized to either mg or μg of protein at the respective time points to take into account any changes in cell number.
Cell death detection ELISA
Odora cells were seeded in a 96-well plate (5,000 cells plate-1) and allowed to attach and grow for 24 h. Cells were then dosed with either water (vehicle control), 60 μM NaAsO2, ZG (100 μM, 150 μM, 200 μM) for 24 h. Manufacturer's instructions were followed for preparation and assaying using the Cell Death Detection ELISA (Roche Diagnostics Corporation, Indianapolis, IN).
Effect of antioxidants, NADPH oxidase inhibitor, and NOS inhibitor on cell viability
Odora cells were plated as described for the cell viability assay. After 24 h, media was removed from the wells and cells were incubated with either the antioxidant or inhibitor solubilized in 100 μL of media for 1 h before the addition of 100 μL of ZG-containing media. Cells were allowed to grow for an additional 24 h. The cells were then fixed, stained, and viability was assessed as described above. The following inhibitors and antioxidants were used: apocynin, 100 μM and 400 μM; NAC, 200 μM; L-NAME), 100 μM. Combination treatments were utilized: apocynin (100 μM) and L-NAME (100 μM); apocynin (100 μM), L-NAME (100 μM) and NAC (200 μM) to see whether this would improve cell viability.
NLRP3 and IL-1β western blot
Odora cells were seeded in tissue culture dishes (145 × 20 mm dish, 600,000 cells dish-1) and allowed to grow for 3 d before exposure to 100 μM ZG for 2, 4, or 6 h. Cells were washed 3× with HBSS before being harvested and homogenized in RIPA buffer (10 mM Tris-Cl, pH 8.0; 1 mM EDTA; 0.5 mM EGTA; 1% Triton X-100; 0.1% sodium deoxycholate; 0.1% SDS; 140 mM NaCl; 1 mM PMSF). Protein was quantified using BCA method as described above. Cell homogenate (20 μg) in sample buffer (10 mM Tris-HCl, pH 7.5; 10 mM ethylenediaminetetraacetic acid; 20% (v/v) glycerol; 1% SDS; 0.005% bromophenol blue; 100 mM dithiothreitol) was heated for 5 min in boiling water then separated on either 8% or 12.5% SDS-acrylamide gels and transferred to PVDF in Tris/glycine transfer buffer with 10% methanol. Protein was transferred at 70V for 45 min, then 95V for an additional 90 min. Membrane was blocked with 5% non-fat milk for 45 min before incubation with NLRP3 antibody (Sigma Aldrich, HPA012878, 1:1000 dilution), IL-1β antibody (Santa Cruz Biotechnology, Dallas, TX, sc-7884, 1:200 dilution), or β-actin (Santa Cruz Biotechnology, sc-47778, 1:10,000 dilution) overnight. Following washing in TBST, blot was incubated with HRP-conjugated secondary antibody (Dako, Carpinteria, CA, 1:2,000). Following 3 washes with TBBS, the blot was visualized using ECL western blot detection system (BioRad, Hercules, CA). Band density was quantitated using ImageJ software (http://search.imagej.net/).
Effect of caspase-1 inhibitor on cell viability
Odora cells were plated as described in the cell viability assay. After 24 h, cells were incubated with caspase-1 inhibitor, Ac-YVAD-cmk, for 1 h before the addition of ZG-containing media. The cells were allowed to grow for an additional 12 h in the presence of the inhibitor. After removing the media, cells were fixed, stained, and viability was assessed as described above.
Statistics
The results from 3 independent experiments were used for calculating significance. Significance was calculated using a 2-way ANOVA, followed by a Holm-Sidak post-test with a p-value less than 0.05 being considering significant.
Results
Cytotoxicity of ZG in Odora cells
As shown in Table 2, the time-dependent effects of zinc toxicity in Odora cells were assayed using the crystal violet assay, and found that there is minimal difference between the 12 and 24 h LC50 values for ZG. No significant cytotoxicity was observed for Odoras incubated with ZG for less than 3 h. As the incubation time increased, there was a concomitant decrease in the ZG LC50. The dose-response was essentially the same in differentiated Odora cells (data not shown), so, due to the similarity in response between differentiated and undifferentiated Odora cells, as well as the more rapid growth rate in undifferentiated Odora cells, all subsequent assays were performed in undifferentiated Odora cells.
Table 2. Odora LC50 for various ZG incubation times.
| Hours of ZG incubation | LC50 |
|---|---|
| 1 hour | > 500 μM |
| 2 hours | > 500 μM |
| 3 hours | 340 μM |
| 4 hours | 285 μM |
| 5 hours | 220 μM |
| 6 hours | 200 μM |
| 12 hours | 170 μM |
| 24 hours | 160 μM |
RNA-seq results
RNA-seq analysis was used to better understand changes in gene expression and to identify potential novel mechanisms of toxicity in olfactory neurons induced by the presence of excess zinc. Based upon the cytotoxicity results, a non-toxic level of ZG (100 μM) was used in order to probe time-dependent changes. At 6 h exposure to 100 μM ZG, genes involved with metal response, such as metallothionein 1a and 2a (Mt1a, Mt2a) and solute carrier 30a1 (Slc30a1) and cell stress response, such as heme oxygenase 1 (Hmox1) and heat shock protein 1a (Hspa1a) were significantly up-regulated. Additionally, genes involved with glutathione metabolism, such as glutamate cysteine ligase, modifier subunit (Gclm) and glutathione reductase (Gsr) were up-regulated by a factor of 2- and 1.8, respectively, compared to control. Genes involved with carbohydrate catabolism such as the aldo-keto reductase family (Akr1b7, Akr1b8, Akr1b10) all showed 6-fold increase in mRNA expression compared to control. Tables showing the most significantly upregulated and downregulated genes at each time point are included as Supplemental Files 1-6.
Using the differentially expressed genes to perform gene ontology and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis, we found a down-regulation in genes involved with tRNA biosynthesis and amino acid metabolism, such as asparagine synthetase (Asns) and cystathionine gamma-lyase (Cth). At 12 h exposure to 100 μM ZG, Mt1a/2a, Hmox1, Slc30a1 along with the aforementioned aldo-keto reductases were still up-regulated. Using pathway analysis, this translated to up-regulation of pathways associated with carbohydrate and pyruvate metabolism. There is also a significant down-regulation of cytoskeletal proteins, such as actin (Actc1), actinin, an actin associated protein (Actn2) and myosin (Myh6, Myh7, Myh2), more then 100-fold compared to control. At 24 h exposure to 100 μM ZG, stress response genes, such as Hmox1 and Hsph1, were still significantly up-regulated along with cell cycle arrest proteins, specifically Growth arrest and DNA-damage-inducible protein (Gadd45a, Gadd45b) and anti-apoptotic proteins, such as Bcl2l1, IAP, Tmbim. Mt1a/2a were no longer up-regulated at this time point. As observed at 12 h of zinc exposure, the cytoskeleton-related genes, such as actin and myosin genes were still significantly down-regulated at 24 h in addition to cell cycle arrest proteins, cyclin D1 (Ccnd1) and cyclin A2 (Ccna2). Also, genes associated with adhesion, such as Cas scaffolding protein family member 4 (Cass4), protocadherin 18 (Pcdh18), chondroadherin (Chad) were also significantly down-regulated. Also, pathways associated with nucleotide metabolism, tRNA biosynthesis, and DNA damage repair were significantly down-regulated. The patterns of differentially expressed genes were virtually identical for the 200 μM ZG for 6 h and 100 μM ZG for 24 h treatments.
Biochemical quantification of GSH, GSSG, H2O2, and ATP
RNA-seq results suggested that the ZG-treated Odora cells were experiencing oxidative stress, resulting in increased GSH production, and increased carbohydrate catabolism to generate ATP. Therefore, biochemical assays to validate the gene expression changes were conducted. GSH levels declined by 25% to reach a minimum at 6 h of ZG exposure and recovered slightly at 12 and 24 h, while GSSG levels did not vary over time (Table 3). H2O2 levels reached a maximum at 6 h of ZG exposure, and returned to near baseline by 24 h (Table 3). There was a steady decline in intracellular ATP over time, with a 20% reduction in ATP levels compared to control cells at 24 h. In contrast to to the gluthatione and H2O2 results, ATP levels did not recover over time (Table 3).
Table 3. Biochemical Measurements in Odora cells exposed to 100 μM ZG for 6, 12, and 24 h.
| Biochemical Measurements | Control | 100 μM ZG | ||
|---|---|---|---|---|
| 6 h | 12 h | 24 h | ||
| GSH (nmol/mg protein) | 66.9 ± 3.8 | 50.0 ± 2.8 * | 57.7 ± 1.5 * | 57.0 ± 1.7 * |
| GSSG (nmol/mg protein) | 15.2 ± 4.0 | 15.4 ± 2.4 | 15.5 ± 2.9 | 14.6 ± 3.0 |
| H2O2 (FU/μg protein) | 0.88 ± 0.06 | 1.15 ± 0.12* | 1.08 ± 0.13 | 0.98 ± 0.09 |
| ATP (μM ATP/μg protein) | 6.25 ± 0.57 | 5.91 ± 0.40 | 5.35 ± 0.65* | 4.90 ± 0.63* |
Data are reported as means ± SEM of 3 biologically independent experiments, with three plates per time point per experiment.
p < 0.05, when compared with control.
Intervention with antioxidants and enzyme inhibitors
Based upon the RNA-seq data, elevation of H2O2, and reduction of GSH, we wanted to investigate whether ZG-induced cytotoxicity could be inhibited using antioxidants or inhibitors of nitric oxide-producing enzymes by measuring cell proliferation with the crystal violet assay. Pre-incubation with apocynin, L-NAME, and NAC all decreased cell death at 200 μM ZG exposure as shown in Figure 1A-C. When L-NAME and NAC were combined, there was increased cell survival at 200 μM ZG exposure, but not at 250 μM ZG exposure as shown in Figure 1D. When apocynin was combined with L-NAME and NAC as shown in Figure 1E, an increase in cell survival was observed at 200 μM ZG exposure but not 250 μM ZG.
Figure 1.
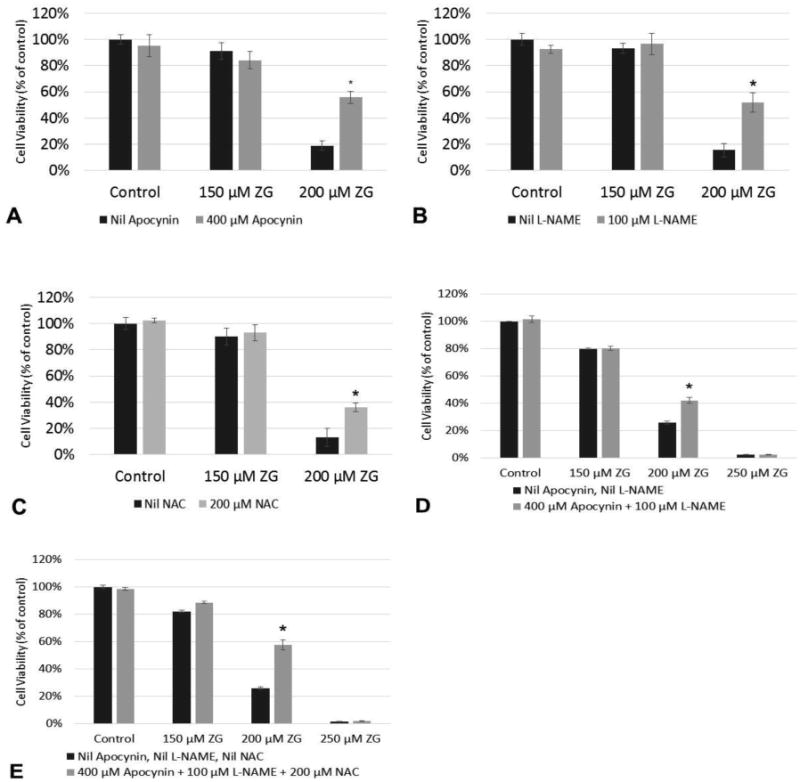
Effects of antioxidants and oxidase inhibitors on ZG-treated Odora cells. Odora cell viability was assessed after incubation with ZG at various concentrations for 24 h in the presence of the antioxidant N-acetylcysteine (NAC) and the oxidase inhibitors apocynin and Nω-Nitro-L-arginine methyl ester hydrochloride (L-NAME), as well as various combinations of these agents. *, p<0.05, when compared with 200 μM ZG exposed cells without any intervention. Data are reported from 3 biologically independent experiments, (each experiment was its own plate), with six wells per treatment condition per experiment. with three plates per treatment condition per experiment.
Zn-induced cell death
Previous literature demonstrated that zinc toxicity is apoptotic in nature19,20. However, as shown in Figure 2, an ELISA which detects histone-associated DNA fragments as a measure of apoptosis did not shown any significant response in Odora cells incubated with 100, 150, or 200 μM ZG compared to vehicle control, while the expected robust response was observed with the positive control.
Figure 2.
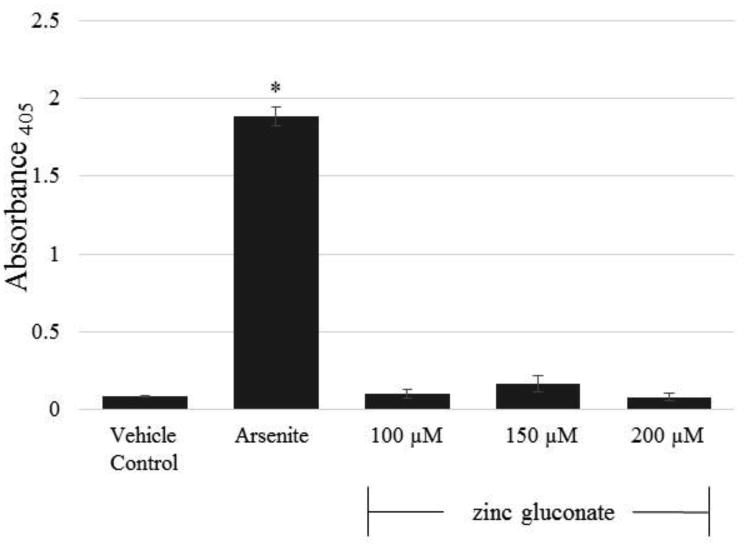
Evaluation of ZG-induced apoptosis in ZG-treated Odora cells. Odora cells incubated with 100 μM, 150 μM, 200 μM ZG do not produce histone fragments suggestive of apoptosis, compared to sodium arsenite (60 μM, positive control) for 24 h, * p < 0.05. Data are reported from 3 biologically independent experiments, with three plates per treatment condition per experiment.
Analysis of NLRP2 and IL1b
ZG-induced production of ROS, which can serve as an activating signal for pyroptosis and inflammasome activation, led us to investigate whether NLRP3 and IL-1b were upregulated in Odora cells in response to zinc exposure, using Western blot analysis.(Harijith et al., 2014) NLRP3 increased over time and reached peak levels at 4 h of exposure to ZG (100 μM), while the inactive precursor form of IL-1β declined slightly at 4 and 6 h. Active IL-1β, which was found at low levels in the control cells, increased after 2 h of zinc exposure, with levels remaining unchanged with prolonged exposure to zinc. β-actin levels did not change with zinc incubation.
Intervention with caspase-1 inhibitor (Ac-YVAD-cmk)
Based upon the increased expression of NLRP3 and IL-1β proteins, which are involved in inflammasome activation, we investigated whether inhibiting caspase-1 could negate ZG-induced cell death in Odora cells. Pre-incubation with 50 μM Ac-YVAD-cmk decreased cell death at 300 μM ZG exposure, as shown in Figure 4. No difference in cell viability as measured using the crystal violet assay was observed between 20, 50, or 100 μM of inhibitor alone (data not shown).
Figure 4.
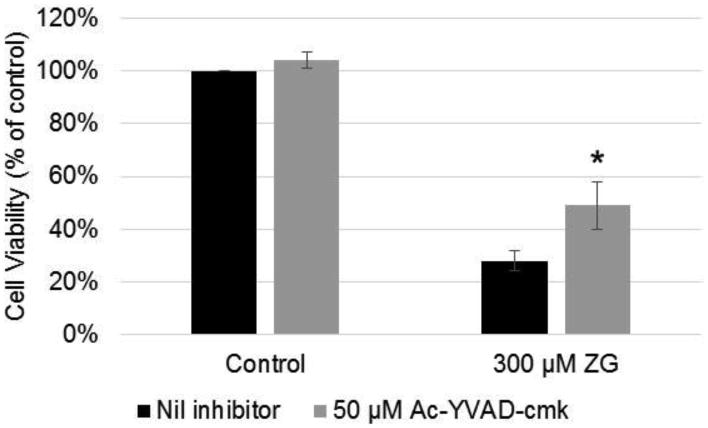
Effects of incubation with caspase-1 inhibitor (Ac-YVAD-cmk) on Odora viability Odora cells were incubated with a toxic dose (300 μM) for 12 hr in the presence of absence (Nil) of the caspase-1 inhibitor (Ac-YVAD-cmk). *, p<0.05, when compared with 300 μM ZG exposed cells without any intervention. Data are reported from 3 biologically independent experiments, (each experiment was its own plate), with six wells per treatment condition per experiment.
Discussion
While the use of RNA-seq and changes in the transcriptome for measuring zinc toxicity in olfactory neurons has not previously been performed, there have been several microarray studies utilizing changes in liver gene expression from excess ionic zinc exposure in in vivo rodent studies. Similar to the RNA-seq work in Odora cells, the changes in gene expression in both rat and mouse liver due to hepatoprotective dose of subcutaneous zinc (100 μmol/kg for 4 d) resulted in up-regulation of metal response genes (e.g. metallothionein (Mt1a, Mt2)), markers of stress (e.g. heat shock protein (Hspaa1), heme oxygenase (Hmox1)), antioxidant response (nuclear factor Nrf2, superoxide dismutase (Sod1), glutamate-cysteine ligase (Gclc)), inflammatory cytokines (interleukin-1β and -6 (Il1b, Il6), and nitric oxide synthase (Nos2)) (Liu et al., 2009). When rats were fed a high zinc diet (347.50 mg Zn/kg basis diet, compared to a normal diet level of 35.94 mg Zn/kg basis diet), up-regulated genes included those involving carbohydrate metabolism (e.g. alcohol dehydrogenase (Adh5), pyruvate dehydrogenase kinase 4 (Pdk4), aldolase C (Aldoc)) (Sun et al., 2007). Another study utilizing intratracheal instillation of zinc sulfate in rats was focused on changes in cardiac gene expression, where up-regulation of respiratory/mitochondrial function genes (pyruvate dehydrogenase kinase 4 (Pdk4), acylcoenzyme A dehydrogenase (Acadvl)), injury/stress response (heme oxygenase 1 (Hmox1), tissue inhibitor of metalloproteinase 3 (Timp3)) was reported, as well as and down-regulation of cytoskeletal genes (alpha actin cardiac 1 (Actc1), beta actin (Actb), Tropomyosin 4 (Tpm4)), checkpoint genes (cyclin D1 (Ccnd1), cyclin D2 (Ccnd2)) (Gilmour et al., 2006).
Despite the fact that these studies were carried out in vivo and focused on different organ systems, there is considerable overlap between pathways of interest in these works and gene expression changes in Odora cells. Similarities between earlier RNA-seq work and the work done in Odora cells include an increase in genes and pathways associated with stress and antioxidant response, energy generation, changes in cytoskeleton maintenance, cell proliferation, and metallothionein and metal response. Differences were observed with the work done in rats given excess dietary zinc and observed for changes in liver gene expression. In these studies, there was an increase in multiple acute-phase response proteins, such as ceruloplasmin, Stat3, egr1, Cxc chemokines, and various heat-shock proteins. With the exception of Hmox1 at 6 and 12 h and Cxc11 at 6 h, these acute-phase responses were not observed until 24 h of exposure in the Odora RNA-seq. Other differences, such as a lack in up-regulation of genes involved with nitrogen and lipid metabolism or ATPase Na+/K+ transporters in the Odora work, may be attributable to the fact that the other studies were performed using whole animals and analysis was done using different tissues, specifically liver and heart, compared to olfactory neurons.
With regard to stress and antioxidant response, up-regulation in Hmox1, Nos2, inflammatory cytokines, and heat shock protein gene expression occurred upon exposure to zinc in both in vitro and in vivo studies, which is an indicator that the cells were undergoing oxidative stress.(Poss and Tonegawa, 1997) In response to the oxidative stress, antioxidant enzymes, including glutathione and superoxide dismutase were upregulated. Others have demonstrated that zinc exposure causes an increase in reactive oxygen species and nitric oxide, while depleting glutathione, and thereby causing cell death (Bishop et al., 2007; Chen and Liao, 2003; Emri et al., 2015; Kim and Koh, 2002; Pong et al., 2002; Seo et al., 2001; Takeyama et al., 1995). Since similar patterns of up-regulated genes were observed in Odora cells exposed to zinc, biochemical assays measuring intracellular glutathione and hydrogen peroxide levels in Odora cells exposed to zinc for 6, 12, and 24 h were performed. Zinc caused elevated hydrogen peroxide after 6 h incubation, but this level plateaued, and then declined over time, suggesting that the cells experienced oxidative stress but were able to degrade the peroxide. It is hypothesized that the hydrogen peroxide and other generated ROS are mitochondrial in origin due to zinc uptake by the mitochondria, where the excess mitochondrial zinc inactivates mitochondrial glutathione reductase and thioredoxin reductase leading to increased damage within the mitochondria (Clausen et al., 2013; Gazaryan et al., 2007). Odora cells were able to control ROS levels via the antioxidant glutathione as demonstrated by the decline in the level of reduced glutathione at 6 h, which recovers to baseline by 24 h. Thus, in Odora cells, ZG caused oxidative stress; however, there are other mechanisms involved since incubation with the antioxidant N-acetyl-cysteine was able to mitigate, but not completely block, zinc-induced cytotoxicity. While the level of nitric oxide was not measured, incubation with apocynin, an NADPH oxidase inhibitor, and L-NAME, a nitric oxide synthase inhibitor, either singly or together did not protect against zinc-mediated cell death. Taken together these data suggests that zinc causes an increase in hydrogen peroxide and nitric oxide; however, these reactive species are not the sole cause of zinc cytotoxicity.
Another potential mechanism for zinc cytotoxicity may be ATP depletion. In the literature, it has been shown that zinc can affect energy production by inhibiting glycolytic enzymes and interfering with the electron transport chain (Gazaryan et al., 2007; Ikeda et al., 1980; Link and von Jagow, 1995; Maret et al., 1999). In cultured cortical neurons, excess intracellular zinc depleted ATP, which could be reversed by the addition of pyruvate (Sheline et al., 2000). Thus, we evaluated the effects of zinc exposure to ATP levels in Odora cells and found that that there was longer exposure to zinc was correlated with decreased levels of intracellular ATP independent of the number of viable cells, which supports the hypothesis that ATP depletion may be a contributing factor to ZG-induced cell death.
The RNA-seq work showing lack of up-regulation of caspase-3 or caspase-9, combined with the negative results from the cell death detection ELISA, as well as lack of DNA laddering in zinc gluconate-treated Odora cells (data not shown), demonstrated that zinc-induced cell death in Odora cells is not apoptotic. Combining the fact that the Odora cells were dying by a non-apoptotic mechanism with the profile of reduced intracellular ATP and elevated ROS suggested the possibility that Odora cells were undergoing pyroptosis (Chavarria-Smith and Vance, 2015; Harijith et al., 2014). Similar to apoptosis, pyroptosis is a programmed mechanism of cell, and involves caspase-1 activation by the inflammasome, a multi-protein complex which includes NOD-like receptor protein 3 (NLRP3), absent in melanoma-2 protein (AIM2), and apoptosis-associated speck-like protein containing a CARD (ASC), which results in the formation and secretion of activated cytokines, such as interleukin-1β and interleukin-18 (IL-1β, IL-18), that trigger an inflammatory response. The triggers for inflammasome activation are quite varied and include reactive oxygen species, extracellular ATP, uric acid crystals, metals, and various viruses and bacteria (de Vasconcelos et al., 2016; Harijith et al., 2014; Martinon et al., 2006; Ogura et al., 2006; Piccini et al., 2008; Simard et al., 2015; Song et al., 2013). We demonstrated via western blot in Odora cells an increase in cytosolic NLRP3 and active IL-1b with increasing duration of exposure to zinc, suggesting the possibility of caspase-1 mediated pyroptosis. However, when the cells were incubated with Ac-YVAD-cmk, a caspase-1 inhibitor, there was a 60% increase in the number of viable cells, but cell death was not completely blocked. This suggests that zinc-induced cell death in Odora cells also involves a caspase-1 independent mechanism of cell death, which needs to be investigated further. Thus, we conclude that zinc-induced toxicity is most likely a combination of several processes including, but not limited to, oxidative stress, inflammasome activation, and ATP depletion.
Supplementary Material
Figure 3.
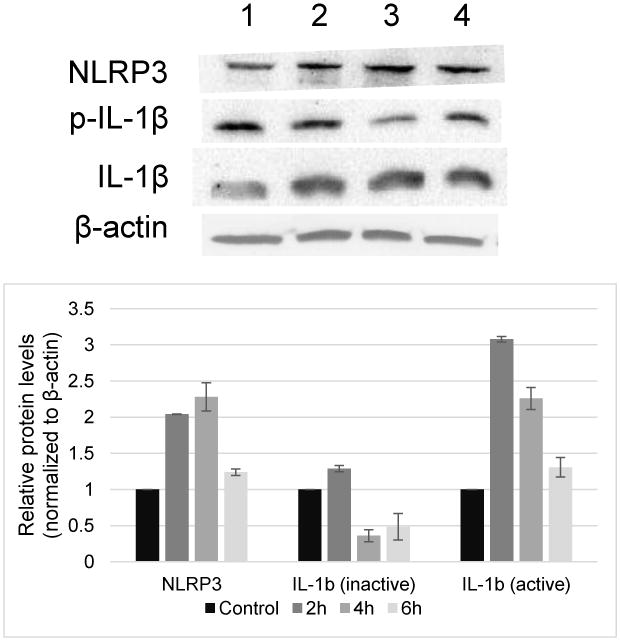
Top panel: Western blot analysis of NLRP3 and IL-1 β protein levels in ZG-treated Odora cells. NLRP3 and IL-1 β protein in Odora cell in responses to 100 μM ZG for 2, 4, and 6 h. Lane 1: cell lysate from untreated, control Odora cells. Lane 2-4: cell lysate from Odora cells exposure to 100 μM ZG for 2, 4, and 6 h, respectively, with 35 μg of protein loaded per lane. Representative gel is shown; results were consistent in two biologically independent experiments. Bottom panel: quantitation of western blot band intensity. (n=2 biologically independent replicates each).
Highlights.
The olfactory neuron cell line Odora was used to study zinc gluconate (ZG) toxicity.
ZG induces oxidative stress, ATP depletion, and cytoskeletal changes in Odora cells.
Metallothionein upregulation was a key response in ZG-treated Odora cells.
ZG-treated Odora cells appear to die via pyroptosis, rather than apoptosis.
Acknowledgments
We gratefully acknowledge the assistance of Dr. Sarah Pixley and Tracy Hopkins for helpful discussions and use of their lab; Dr. Jun Ying for statistical support; Dr. Ravi Panchanathan and Hongzhu Liu for their assistance with the inflammasome work. We also acknowledge the assistance of the University of Cincinnati Genomics, Epigenomics, and Sequencing core, and bioinformatics assistance from Dr. Xiang Zhang, Dr. Mario Medvedovic, Dr. Jingyuan Deng, and Jenny Chen. This research was supported by National Institute of Environmental Health Sciences (NIEHS) Center for Environmental Genetics grant P30-ES06096 and AI 106269 (to GSD)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Literature cited
- Alexander TH, Davidson TM. Intranasal zinc and anosmia: the zinc-induced anosmia syndrome. Laryngoscope. 2006;116:217–220. doi: 10.1097/01.mlg.0000191549.17796.13. [DOI] [PubMed] [Google Scholar]
- Bahrami F, Bergman U, Brittebo EB, Brandt I. Persistent olfactory mucosal metaplasia and increased olfactory bulb glial fibrillary acidic protein levels following a single dose of methylsulfonyl-dichlorobenzene in mice: comparison of the 2,5- and 2, 6-dichlorinated isomers. Toxicol Appl Pharmacol. 2000;162:49–59. doi: 10.1006/taap.1999.8818. [DOI] [PubMed] [Google Scholar]
- Barrett T, Troup DB, Wilhite SE, Ledoux P, Rudnev D, Evangelista C, Kim IF, Soboleva A, Tomashevsky M, Marshall KA, Phillippy KH, Sherman PM, Muertter RN, Edgar R. NCBI GEO: archive for high-throughput functional genomic data. Nucleic Acids Res. 2009;37:D885–890. doi: 10.1093/nar/gkn764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop GM, Dringen R, Robinson SR. Zinc stimulates the production of toxic reactive oxygen species (ROS) and inhibits glutathione reductase in astrocytes. Free Radic Biol Med. 2007;42:1222–1230. doi: 10.1016/j.freeradbiomed.2007.01.022. [DOI] [PubMed] [Google Scholar]
- Bonaventura P, Benedetti G, Albarede F, Miossec P. Zinc and its role in immunity and inflammation. Autoimmun Rev. 2015;14:277–285. doi: 10.1016/j.autrev.2014.11.008. [DOI] [PubMed] [Google Scholar]
- Brooks WA, Yunus M, Santosham M, Wahed MA, Nahar K, Yeasmin S, Black RE. Zinc for severe pneumonia in very young children: double-blind placebo-controlled trial. Lancet. 2004;363:1683–1688. doi: 10.1016/S0140-6736(04)16252-1. [DOI] [PubMed] [Google Scholar]
- Cancalon P, Elam JS. Study of regeneration in the garfish olfactory nerve. J Cell Biol. 1980;84:779–794. doi: 10.1083/jcb.84.3.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavarria-Smith J, Vance RE. The NLRP1 inflammasomes. Immunol Rev. 2015;265:22–34. doi: 10.1111/imr.12283. [DOI] [PubMed] [Google Scholar]
- Chen CJ, Liao SL. Zinc toxicity on neonatal cortical neurons: involvement of glutathione chelation. Journal of Neurochemistry. 2003;85:443–453. doi: 10.1046/j.1471-4159.2003.01691.x. [DOI] [PubMed] [Google Scholar]
- Clausen A, McClanahan T, Ji SG, Weiss JH. Mechanisms of rapid reactive oxygen species generation in response to cytosolic Ca2+ or Zn2+ loads in cortical neurons. PLoS One. 2013;8:e83347. doi: 10.1371/journal.pone.0083347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dash S, Xiao C, Morgantini C, Koulajian K, Lewis GF. Intranasal insulin suppresses endogenous glucose production in humans compared with placebo in the presence of similar venous insulin concentrations. Diabetes. 2015;64:766–774. doi: 10.2337/db14-0685. [DOI] [PubMed] [Google Scholar]
- Davidson TM, Smith WM. The Bradford Hill criteria and zinc-induced anosmia: a causality analysis. Arch Otolaryngol Head Neck Surg. 2010;136:673–676. doi: 10.1001/archoto.2010.111. [DOI] [PubMed] [Google Scholar]
- de Vasconcelos NM, Van Opdenbosch N, Lamkanfi M. Inflammasomes as polyvalent cell death platforms. Cell Mol Life Sci. 2016 doi: 10.1007/s00018-016-2204-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeCook C, Hirsch A. Anosmia due to inhalational zinc: a case report. Chem Senses. 2000;25:593–659. [Google Scholar]
- Dineley KE, Votyakova TV, Reynolds IJ. Zinc inhibition of cellular energy production: implications for mitochondria and neurodegeneration. J Neurochem. 2003;85:563–570. doi: 10.1046/j.1471-4159.2003.01678.x. [DOI] [PubMed] [Google Scholar]
- Emri E, Miko E, Bai P, Boros G, Nagy G, Rozsa D, Juhasz T, Hegeds C, Horkay I, Remenyik E, Emri G. Effects of non-toxic zinc exposure on human epidermal keratinocytes. Metallomics. 2015;7:499–507. doi: 10.1039/c4mt00287c. [DOI] [PubMed] [Google Scholar]
- Farbman AI. Developmental biology of olfactory sensory neurons. Semin Cell Biol. 1994;5:3–10. doi: 10.1006/scel.1994.1002. [DOI] [PubMed] [Google Scholar]
- Feng P, Li TL, Guan ZX, Franklin RB, Costello LC. Direct effect of zinc on mitochondrial apoptogenesis in prostate cells. Prostate. 2002;52:311–318. doi: 10.1002/pros.10128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink SL, Cookson BT. Apoptosis, Pyroptosis, and Necrosis: Mechanistic Description of Dead and Dying Eukaryotic Cells. Infection and Immunity. 2005;73:1907–1916. doi: 10.1128/IAI.73.4.1907-1916.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freiherr J, Hallschmid M, Frey WH, 2nd, Brunner YF, Chapman CD, Holscher C, Craft S, De Felice FG, Benedict C. Intranasal insulin as a treatment for Alzheimer's disease: a review of basic research and clinical evidence. CNS Drugs. 2013;27:505–514. doi: 10.1007/s40263-013-0076-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazaryan IG, Krasinskaya IP, Kristal BS, Brown AM. Zinc irreversibly damages major enzymes of energy production and antioxidant defense prior to mitochondrial permeability transition. J Biol Chem. 2007;282:24373–24380. doi: 10.1074/jbc.M611376200. [DOI] [PubMed] [Google Scholar]
- Gilmour PS, Schladweiler MC, Nyska A, McGee JK, Thomas R, Jaskot RH, Schmid J, Kodavanti UP. Systemic Imbalance of Essential Metals and Cardiac Gene Expression in Rats Following Acute Pulmonary Zinc Exposure. Journal of Toxicology and Environmental Health, Part A. 2006;69:2011–2032. doi: 10.1080/15287390600746173. [DOI] [PubMed] [Google Scholar]
- Guo D, Du Y, Wu Q, Jiang W, Bi H. Disrupted calcium homeostasis is involved in elevated zinc ion-induced photoreceptor cell death. Arch Biochem Biophys. 2014;560:44–51. doi: 10.1016/j.abb.2014.07.014. [DOI] [PubMed] [Google Scholar]
- Hallschmid M, Higgs S, Thienel M, Ott V, Lehnert H. Postprandial administration of intranasal insulin intensifies satiety and reduces intake of palatable snacks in women. Diabetes. 2012;61:782–789. doi: 10.2337/db11-1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamidovic A. Position on zinc delivery to olfactory nerves in intranasal insulin phase I–III clinical trials. Contemporary Clinical Trials. doi: 10.1016/j.cct.2015.08.011. [DOI] [PubMed] [Google Scholar]
- Harijith A, Ebenezer DL, Natarajan V. Reactive oxygen species at the crossroads of inflammasome and inflammation. Frontiers in Physiology. 2014;5:352. doi: 10.3389/fphys.2014.00352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedera P, Peltier A, Fink JK, Wilcock S, London Z, Brewer GJ. Myelopolyneuropathy and pancytopenia due to copper deficiency and high zinc levels of unknown origin II. The denture cream is a primary source of excessive zinc. Neurotoxicology. 2009;30:996–999. doi: 10.1016/j.neuro.2009.08.008. [DOI] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Iitaka M, Kakinuma S, Fujimaki S, Oosuga I, Fujita T, Yamanaka K, Wada S, Katayama S. Induction of apoptosis and necrosis by zinc in human thyroid cancer cell lines. J Endocrinol. 2001;169:417–424. doi: 10.1677/joe.0.1690417. [DOI] [PubMed] [Google Scholar]
- Ikeda T, Kimura K, Morioka S, Tamaki N. Inhibitory effects of Zn2+ on muscle glycolysis and their reversal by histidine. J Nutr Sci Vitaminol (Tokyo) 1980;26:357–366. doi: 10.3177/jnsv.26.357. [DOI] [PubMed] [Google Scholar]
- Iovannisci D, Illek B, Fischer H. Function of the HVCN1 proton channel in airway epithelia and a naturally occurring mutation, M91T. J Gen Physiol. 2010;136:35–46. doi: 10.1085/jgp.200910379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jafek BW, Linschoten MR, Murrow BW. Anosmia after intranasal zinc gluconate use. Am J Rhinol. 2004;18:137–141. [PubMed] [Google Scholar]
- Kim YH, Koh JY. The role of NADPH oxidase and neuronal nitric oxide synthase in zinc-induced poly(ADP-ribose) polymerase activation and cell death in cortical culture. Exp Neurol. 2002;177:407–418. doi: 10.1006/exnr.2002.7990. [DOI] [PubMed] [Google Scholar]
- Kueng W, Silber E, Eppenberger U. Quantification of cells cultured on 96-well plates. Anal Biochem. 1989;182:16–19. doi: 10.1016/0003-2697(89)90710-0. [DOI] [PubMed] [Google Scholar]
- Link TA, von Jagow G. Zinc Ions Inhibit the Qp Center of Bovine Heart Mitochondrial bc1 Complex by Blocking a Protonatable Group. Journal of Biological Chemistry. 1995;270:25001–25006. doi: 10.1074/jbc.270.42.25001. [DOI] [PubMed] [Google Scholar]
- Liu J, Zhou ZX, Zhang W, Bell MW, Waalkes MP. Changes in hepatic gene expression in response to hepatoprotective levels of zinc. Liver International. 2009;29:1222–1229. doi: 10.1111/j.1478-3231.2009.02007.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez V, Foolad F, Kelleher SL. ZnT2-overexpression represses the cytotoxic effects of zinc hyper-accumulation in malignant metallothionein-null T47D breast tumor cells. Cancer Lett. 2011;304:41–51. doi: 10.1016/j.canlet.2011.01.027. [DOI] [PubMed] [Google Scholar]
- Lorusso M, Cocco T, Sardanelli AM, Minuto M, Bonomi F, Papa S. Interaction of Zn2+ with the bovine-heart mitochondrial bc1 complex. European Journal of Biochemistry. 1991;197:555–561. doi: 10.1111/j.1432-1033.1991.tb15944.x. [DOI] [PubMed] [Google Scholar]
- Manallack DT, Andrews PR, Woods EF. Design, synthesis, and testing of insulin hexamer-stabilizing agents. J Med Chem. 1985;28:1522–1526. doi: 10.1021/jm00148a025. [DOI] [PubMed] [Google Scholar]
- Maret W, Jacob C, Vallee BL, Fischer EH. Inhibitory sites in enzymes: Zinc removal and reactivation by thionein. Proceedings of the National Academy of Sciences. 1999;96:1936–1940. doi: 10.1073/pnas.96.5.1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440:237–241. doi: 10.1038/nature04516. [DOI] [PubMed] [Google Scholar]
- Muller O, Becher H, van Zweeden AB, Ye Y, Diallo DA, Konate AT, Gbangou A, Kouyate B, Garenne M. Effect of zinc supplementation on malaria and other causes of morbidity in west African children: randomised double blind placebo controlled trial. BMJ. 2001;322:1567. doi: 10.1136/bmj.322.7302.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murrell JR, Hunter DD. An olfactory sensory neuron line, odora, properly targets olfactory proteins and responds to odorants. J Neurosci. 1999;19:8260–8270. doi: 10.1523/JNEUROSCI.19-19-08260.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogura Y, Sutterwala FS, Flavell RA. The Inflammasome: First Line of the Immune Response to Cell Stress. Cell. 2006;126:659–662. doi: 10.1016/j.cell.2006.08.002. [DOI] [PubMed] [Google Scholar]
- Piccini A, Carta S, Tassi S, Lasiglie D, Fossati G, Rubartelli A. ATP is released by monocytes stimulated with pathogen-sensing receptor ligands and induces IL-1beta and IL-18 secretion in an autocrine way. Proc Natl Acad Sci U S A. 2008;105:8067–8072. doi: 10.1073/pnas.0709684105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pong K, Rong Y, Doctrow SR, Baudry M. Attenuation of zinc-induced intracellular dysfunction and neurotoxicity by a synthetic superoxide dismutase/catalase mimetic, in cultured cortical neurons. Brain Res. 2002;950:218–230. doi: 10.1016/s0006-8993(02)03040-8. [DOI] [PubMed] [Google Scholar]
- Poss KD, Tonegawa S. Reduced stress defense in heme oxygenase 1-deficient cells. Proc Natl Acad Sci U S A. 1997;94:10925–10930. doi: 10.1073/pnas.94.20.10925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolf E. Depletion of ATP and oxidative stress underlie zinc-induced cell injury. Acta Medica (Hradec Kralove) 2007;50:43–49. [PubMed] [Google Scholar]
- Senft AP, Dalton TP, Shertzer HG. Determining glutathione and glutathione disulfide using the fluorescence probe o-phthalaldehyde. Anal Biochem. 2000;280:80–86. doi: 10.1006/abio.2000.4498. [DOI] [PubMed] [Google Scholar]
- Senft AP, Dalton TP, Nebert DW, Genter MB, Hutchinson RJ, Shertzer HG. Dioxin increases reactive oxygen production in mouse liver mitochondria. Toxicol Appl Pharmacol. 2002a;178:15–21. doi: 10.1006/taap.2001.9314. [DOI] [PubMed] [Google Scholar]
- Senft AP, Dalton TP, Nebert DW, Genter MB, Puga A, Hutchinson RJ, Kerzee JK, Uno S, Shertzer HG. Mitochondrial reactive oxygen production is dependent on the aromatic hydrocarbon receptor. Free Radic Biol Med. 2002b;33:1268–1278. doi: 10.1016/s0891-5849(02)01014-6. [DOI] [PubMed] [Google Scholar]
- Seo SR, Chong SA, Lee SI, Sung JY, Ahn YS, Chung KC, Seo JT. Zn2+-induced ERK activation mediated by reactive oxygen species causes cell death in differentiated PC12 cells. Journal of Neurochemistry. 2001;78:600–610. doi: 10.1046/j.1471-4159.2001.00438.x. [DOI] [PubMed] [Google Scholar]
- Sheline CT, Behrens MM, Choi DW. Zinc-induced cortical neuronal death: contribution of energy failure attributable to loss of NAD(+) and inhibition of glycolysis. J Neurosci. 2000;20:3139–3146. doi: 10.1523/JNEUROSCI.20-09-03139.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simard JC, Vallieres F, de Liz R, Lavastre V, Girard D. Silver nanoparticles induce degradation of the endoplasmic reticulum stress sensor activating transcription factor-6 leading to activation of the NLRP-3 inflammasome. J Biol Chem. 2015;290:5926–5939. doi: 10.1074/jbc.M114.610899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song J, Du L, Feng Y, Wu W, Yan Z. Pyroptosis induced by zinc oxide nanoparticles in A549 cells. Wei Sheng Yan Jiu. 2013;42:273–276. [PubMed] [Google Scholar]
- Sun JY, Wang JF, Zl NT, Jing MY, Weng XY. Gene expression profiles analysis of the growing rat liver in response to different zinc status by cDNA microarray analysis. Biological Trace Element Research. 2007;115:169–185. doi: 10.1007/BF02686028. [DOI] [PubMed] [Google Scholar]
- Takeyama Y, Ogino K, Segawa H, Kobayashi H, Uda T, Houbara T. Effects of Zinc on Production of Active Oxygen Species by Rat Neutrophils. Pharmacology & Toxicology. 1995;76:50–55. doi: 10.1111/j.1600-0773.1995.tb00102.x. [DOI] [PubMed] [Google Scholar]
- Willis MS, Monaghan SA, Miller ML, McKenna RW, Perkins WD, Levinson BS, Bhushan V, Kroft SH. Zinc-induced copper deficiency: a report of three cases initially recognized on bone marrow examination. Am J Clin Pathol. 2005;123:125–131. doi: 10.1309/v6gvyw2qtyd5c5pj. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.