

CONSPECTUS
The functions of metal complexes are directly linked to the local environment in which they are housed; modifications to the local environment (or secondary coordination sphere) are known to produce changes in key properties of the metal centers that can affect reactivity. Non-covalent interactions are the most common and influential forces that regulate the properties of secondary coordination spheres, which leads to complexities in structure that are often difficult to achieve in synthetic systems. Using key architectural features from the active sites of metalloproteins as inspiration, we have developed molecular systems that enforce intramolecular hydrogen bonds (H-bonds) around a metal center via incorporation of H-bond donors and acceptors into rigid ligand scaffolds. We have utilized these molecular species to probe mechanistic aspects of biological dioxygen activation and water oxidation.
This Account describes the stabilization and characterization of unusual M–oxo and heterobimetallic complexes. These types of species have been implicated in a range of oxidative processes in biology but are often difficult to study because of their inherent reactivity. Our H-bonding ligand systems allowed us to prepare an FeIII–oxo species directly from the activation of O2 that was subsequently oxidized to form a monomeric FeIV–oxo species with an S = 2 spin state, similar to those species proposed as key intermediates in non-heme monooxygenases. We also demonstrated that a single MnIII–oxo center that was prepared from water could be converted to a high spin MnV–oxo species via stepwise oxidation—a process that mimics the oxidative charging of the oxygen-evolving complex (OEC) of photosystem II.
Current mechanisms for photosynthetic O–O bond formation invoke a MnIV–oxyl species rather than the isoelectronic MnV–oxo system as the key oxidant based on computational studies. However, there is no experimental information to support the existence of an Mn–oxyl radical. We therefore probed the amount of spin density on the oxido ligand of our complexes using EPR spectroscopy in conjunction with oxygen-17 labeling. Our findings showed that there is a significant amount of spin on the oxido ligand, yet the M–oxo bonds are best described as highly covalent and there is no indication that an oxyl radical is formed. These results offer the intriguing possibility that high spin M–oxo complexes are involved in O–O bond formation in biology.
Ligand redesign to incorporate H-bond accepting units (sulfonamido groups) simultaneously provided a metal ion binding pocket, adjacent H-bond acceptors, and an auxiliary binding site for a second metal ion. These properties allowed us to isolate a series of heterobimetallic complexes of FeIII and MnIII in which a group II metal ion was coordinated within the secondary coordination sphere. Examination of the influence of the second metal ion on the electron transfer properties of the primary metal center revealed unexpected similarities between CaII and SrII ions—a result with relevance to the OEC. In addition, the presence of a second metal ion was found to prevent intramolecular oxidation of the ligand with an O-atom transfer reagent.
The Importance of the Secondary Coordination Sphere
Location matters… This old saying is used in variety of different contexts but it is never more appropriate than in describing factors that govern the reactivity of a metal complex. No molecular species operates in isolation without interacting with its local environment. There is a growing body of evidence that local environments, or secondary coordination spheres, have profound effects on function that range from the modulation of physical properties (e.g. redox potentials) to the delivery of reactants and removal of products. It is thus possible to understand function, and dysfunction, of a metal-containing species within the context of properties associated with their local environments, regardless of system type or length-scale that range from angstroms to tens of nanometers.
The interplay between metal centers and local environments is best exemplified by active sites within metalloproteins, where emerging structure-function relationships show how environmental factors govern key physical properties (Figure 1). For instance, work by Lu on biological electron transfer processes demonstrated that changes in the composition of amino acids that surround the redox-active copper centers in azurin alter redox potentials over a 1 V range.1 Miller and Brunold found similar results in superoxide dismutases in which a single point mutation proximal to the active site changed the enzyme’s redox potential over 0.6 V.2 Interactions between metal sites and protein-derived secondary coordination spheres also have importance in biocatalysis by metalloeznymes. For example, cyctochrome C oxidase reduces an inherently stable compound (O2) to two equivalents of H2O via a mechanism that requires the precise transfer of protons and electrons during turnover.3 This is facilitated by a variety of non-covalent interactions between the metal complex and nearby functional groups.
Figure 1.

Structure of the OEC cluster (PDB: 3ARC) illustrating the H-bonding network (dashed lines). Sphere key: purple (Mn), light blue (Ca2+), red (oxido/hydroxo ligands), blue (nitrogen), white (water).
Protein active sites have evolved to incorporate a complex network of weak non-covalent interactions to promote efficient and selective function. The level of structural complexity found within these active sites is often difficult to duplicate in synthetic systems – it has even been problematic to produce synthetic systems whose secondary coordination spheres contain just a single structural aspect of a protein active site. The problems are rooted in limits in molecular designs, which are often focused on controlling the structural properties of the primary coordination sphere. One notable exception is the work of DuBois, who built synthetic electrocatalysts for proton reduction that place amine sites within the secondary coordination sphere: these basic groups are necessary to achieve remarkable rate enhancements for this reaction.4
Early synthetic systems that utilized rigid organic scaffolds to build steric bulk around a metal center suggested a method for positioning functional groups within the secondary coordination sphere to affect function.5,6 Our group was one of the first to use this concept of a rigid ligand framework to demonstrate that intramolecular hydrogen bonds (H-bonds) can be designed into synthetic systems and used to promote the activation of small molecules.7 We have developed a bioinspired program that seeks to incorporate structural factors that promote intramolecular H-bonds within metal complexes and to examine the influence of the H-bonding interactions on structure and function. Our goal in this Account is to illustrate the use of our systems that contain intramolecular H-bonds in probing fundamental questions of reactivity for biologically relevant metal complexes. The emphasis will be on the chemistry of high spin metal-oxo complexes that are prevalent in non-heme iron oxygenases and possibly within the active form of the oxygen-evolving complex of photosystem II. A series of synthetic metal complexes will be introduced and their chemical and physical properties discussed as they relate to biological function.
Hydrogen Bonding Networks within Transition Metal Complexes
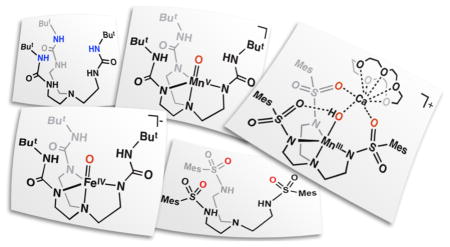
Over the last 15 years, we have introduced several tripodal ligands and demonstrated their ability to support intramolecular H-bonding networks around transition metal ions. The design concepts for many of these ligands have been described previously,9–11 and only those principles used to develop our prototypical urea-based tripod, [H3buea]3-, will be described here. The parent compound, H6buea, can be selectively deprotonated to produce a trianionic metal binding pocket with an N4 primary coordination environment (Figure 2A). The binding of a metal ion within this pocket positions the remaining components of the urea arms perpendicular to the trigonal plane formed by the three deprotonated nitrogen atoms to create a rigid scaffolding around the metal ion. The resulting cavity formed by the urea arms installs the three remaining NH groups proximal to the metal ion such that they can form intramolecular H-bonds to exogenous ligands on the metal center. This design allows the secondary coordination sphere of the metal ion, and thus its local environment, to be regulated by the intramolecular H-bonds that are supported by the [H3buea]3- ligand. An additional consequence of the structural constraints of the ligand is the enforcement of local C3 symmetry around the bonded metal ion, which has proven useful for enforcing high spin electronic configurations, as discussed below.
Figure 2.

Depiction of an H-bonding ligand (A) and M–oxo complexes (B, M = Fe, Mn).
In one of our first studies with [H3buea]3-, we examined the activation of O2 using FeII and MnII complexes of [H3buea]3- to produce a unique pair of monomeric MIII–oxo complexes (MIIIH3buea(O)]2-, MIII = Fe, Mn, Figure 2B).7,12 The complexes were structurally, spectroscopically, and computationally characterized, and all the data support intramolecular H-bonds between the M–oxo units and the [H3buea]3- ligand. The isolation of the [MIIIH3buea(O)]2- complexes was important in the field of metal–oxo chemistry because they represented the first examples of terminal oxido ligands to either a MnIII or an FeIII center; all previously characterized M–oxo complexes of Fe or Mn had the oxido ligand bridging between at least two metal centers because of the strong thermodynamic driving force to form MIII–(μ-O)n–MIII units (n = 1, Fe and n = 2, Mn). Our ability to prepare and isolate the monomeric versions was undoubtedly a consequence, in part, of the constrained environment that surrounds the M–oxo unit and prevents formation of multinuclear species. The [MIIIH3buea(O)]2- complexes also provided valuable information on the influence of H-bonds on the bonding within a M–oxo unit. Since the seminal work of Gray,13 molecular orbital theories have described the bonding of an oxido ligand as a combination of σ and π bonds, but in the [MIIIH3buea(O)]2- complexes, the bonding interactions were best described as single bonds. Three strong intramolecular H-bonds between the oxido ligand and the NH groups of [H3buea]3- compensate for the loss of π-bonds in the complexes, with ~24 kcal/mol in additional energy gained from the H-bonds in [FeIIIH3buea(O)]2-.14 These findings illustrated that H- bonds can, in some cases, be used in place of π-bonds to stabilize metal complexes with terminal oxido ligands.
High Valent M–Oxo Complexes: FeIV–Oxo
High valent Fe–oxo complexes have been proposed as the kinetically competent oxidants in both heme and non-heme oxygenases. Heme systems, such as the cytochrome P450s,15 contain an FeIV–oxo unit with an additional ligand radical, whereas the non-heme counterparts are only oxidized to FeIV–oxo species.16 Both of these FeIV–oxo species are transient intermediates that are often difficult to detect and study in biological systems. Nevertheless, we have learned much about both these types of Fe–oxo centers in proteins in the last 10 years, but we will focus on non-heme systems for this Account.
The high reactivity of non-heme FeIV–oxo species in biology motivated synthetic chemists to target similar types of complexes. The first examples came from the work of Que and Nam, who showed that aza-macrocycles can stabilize high valent Fe–oxo complexes.17–19 However, these and all of the other early examples of non-heme FeIV–oxo complexes have S = 1 spin ground states, which some have argued could account for the lower reactivity of these synthetic complexes compared to oxygenases. We therefore sought to prepare FeIV–oxo complexes with S=2 spin ground states that were amenable to structural and spectroscopic characterization. Our [FeIIIH3buea(O)]2- complex stood out as a suitable synthon because the arrangement of d orbitals enforced by the local C3 symmetry favors high spin FeIV complexes, and electrochemical studies showed an obtainable oxidation event at a potential of −0.90 V vs [Cp2Fe]+/0 that we assigned to the FeIII/FeIV couple. After numerous attempts to chemically oxidize [FeIIIH3buea(O)]2- by one electron, treatment with the mild oxidant [FeIIICp2]+ afforded the desired [FeIVH3buea(O)]- complex.20 To our surprise, the same FeIV–oxo complex could be prepared from the proton-coupled oxidation of [FeIIIH3buea(OH)]– with [FeIIICp2]+ (Scheme 1). The presence of the oxido ligand in the FeIV–oxo product was confirmed by FTIR studies, which showed a strong peak at 799 cm−1 that shifted to 768 cm−1 in the Fe–18O isotopomer, consistent with the shift predicted from the simple harmonic oscillator model. The potassium salt of [FeIVH3buea(O)]− was sufficiently stable at −35°C for the growth of single crystals, from which the molecular structure was determined by XRD methods (Figure 3A). The metrical parameters of the structure agree with the formulation of this complex as an FeIV=O species, in particular the Fe–O bond length of 1.680(1) Å (Table 1). This bond distance is a contraction of 0.133 Å relative to the FeIII–oxo analog. In addition, the Fe–O unit is pulled toward the trigonal plane formed by the coordinated nitrogen atoms (a change of nearly 0.2 Å). This results in an out-of-plane displacement of the oxido ligand from the NH donors of [H3buea]3-, which weakens the H-bonding interactions, as evident in the sharpening of the NH vibrational bands and a shift to higher energy.
Scheme 1.

Figure 3.

Molecular structure (A) and EPR spectrum of [FeIVH3buea(O)]– (B). Asterisks in B indicate signals from small amounts of impurities.
Table 1.
Comparison of key physical properties of [MnH3buea(O)]m complexes.
| FeIII–O | FeIV=O | MnIII–O | MnIV–O | MnV=O | |
|---|---|---|---|---|---|
| S | 5/2 | 2 | 2 | 3/2 | 1 |
| ν(M–O)a | 671 | 798 | 700 | 737 | 737, 754 |
| M–Ob | 1.813(3) | 1.680(1) | 1.771(5) | 1.76c | 1.68c |
| g | 6.0, 2.0 | 8.19, 4.06 | 8.08 | 5.26, 2.38, 1.62 | 4.01 |
| Da | −1 | 4.0(5) | 2.0(2) | 3.0 | 5.0(5) |
| E/D | 0.005 | 0.03 | 0.055 | 0.26 | 0.005 |
| E1/2d | −0.90 | – | −1.0 | −0.076 | – |
| ρπ | – | 0.56 | 0.30 | – | 0.45 |
cm−1,
Å,
from EXAFS data.
V vs [FeCp]+/0
In order to determine the spin state of [FeIVH3buea(O)]-, we probed its electronic structure by EPR spectroscopy. Prior to our work, no EPR spectrum of an FeIV–oxo species had ever been obtained because most conventional EPR spectrometers are only sensitive to half-integer spin systems. With [FeIVH3buea(O)]-, we detected the first signal for a nonheme Fe(IV)–oxo complex using a modified EPR technique in which the microwave frequency is aligned parallel to the magnetic field (parallel mode), which allows integer spin systems to be detected. The signal of [FeIVH3buea(O)]- contains two features at g values of 8.2 and 4.1 that were simulated to fit a species with an S = 2 spin state, D = +4.7 cm−1, E/D = 0.03, and gz = 2.02 (Figure 3B). These signals arise from the |±2〉 and |±1〉 excited state doublets within the S = 2 spin manifold. The small degree of rhombic distortion, indicated by the E/D value, suggests that [FeIVH3buea(O)]- remains C3 symmetric in solution, and quantification of the signal indicated 80% conversion of [FeIIIH3buea(OH)]- to [FeIVH3buea(O)]-. At the same time that we were working on [FeIV(O)buea]2-, Que published a related system that also utilized C3 symmetry to give a high spin FeIV–oxo complex.21,22 This design concept was later used by Chang, who prepared a high spin FeIV–oxo complex using a pyrrolide tripodal ligand with C3 symmetry.23
The discovery of the parallel mode EPR spectrum for [FeIVH3buea(O)]– indicated that EPR spectroscopy might be useful for interrogating high spin FeIV–oxo units in all kinds of systems. For example, both FeIV–oxo systems of Que24 and Chang23 have now been characterized by parallel mode EPR spectroscopy and have similar signals as those found for [FeIVH3buea(O)]–, which confirms their S=2 spin states. However, no related EPR signals have been observed for non-heme metalloproteins, even though theory predicts that the intensity of parallel mode EPR signals should be greater in more rhombic ligand fields (intensity is proportional to (E/D)4), such as those within proteins active sites. The search thus continues to find these types of signals in proteins.
High Valent M–Oxo Complexes: MnV–Oxo
Concurrent with our studies on FeIV–oxo complexes, we also pursued the related high valent Mn–oxo species. Our initial reason for these studies was centered on the fundamental problem of whether a high spin MnV–oxo complex could be prepared. At the time, nearly all of the d2 M–oxo complexes were diamagnetic, which was consistent with the established molecular orbital theory of Gray that predicted low-spin (S=0) systems for complexes having approximately C4 symmetry. One exception was the high spin CrIV–oxo complexes of Theopold whose unusual spin states were attributed to the local C3 symmetry.25 We reasoned that a MnV–oxo complex of our [H3buea]3- ligand should have a similar electronic structure and that this complex could be obtained via sequential oxidation of the [MnIIIH3buea(O)]2- complex that we had already isolated (Scheme 2). This premise was supported by cyclic voltammatry experiments on [MnIIIH3buea(O)]2- that showed two reversible one-electron redox processes at −1.0 V and 0.076 V vs [FeCp2]0/+. These low potentials allowed us to access both [MnIVH3buea(O)]- and [MnVH3buea(O)] with the mild oxidant [FeIIICp2]+ (Scheme 2). Full characterization of [MnIVH3buea(O)]- and [MnVH3buea(O)] support the step-wise oxidation of [MnIIIH3buea(O)]2- to these high valent analogs (Table 1).26,27 A key method that illustrates this sequential conversion is EPR spectroscopy, in which the observed signal alternates between perpendicular mode and parallel mode as the spin state converts between half-integer and integer spin (Figure 4). For the MnIII–oxo complex, no signal is observed in perpendicular mode, but two features are found in parallel mode at g values of 8.08 and 4.5 that are consistent with an S=2 spin ground state. In addition, a six-line hyperfine splitting pattern is observed on the g = 8.08 signal, indicating that the complex is mononuclear. Upon oxidation with one equivalent of [FeIIICp2]+, this parallel mode signal disappears, and a new spectrum appears in perpendicular mode that is indicative of a mononuclear, S=3/2 MnIV–oxo complex. Final oxidation with a second equivalent of [FeIIICp2]+ afforded a loss of the MnIV signal with concomitant growth of a new parallel mode signal at g=4.01. Simulations of this parallel mode signal fit an S=1 spin state that supports a MnV–oxo formulation, and the hyperfine splitting pattern shows that it remains mononuclear. Together, these results confirmed that a high spin MnV–oxo complex could be formed by enforcing C3 symmetry around the metal center.
Scheme 2.

Figure 4.

EPR data for the stepwise oxidation of [MnIIIH3buea(O)]2- (A) to [MnIVH3buea(O)]- (B) and [MnVH3buea(O)] (C). The signal at g = 4.4 in C is unreacted [FeIIICp2]+.
Spin Density Within High Valent M–oxo Units
During the course of our investigations on the series of high valent Mn–oxo complexes discussed above, we recognized the possible relationship of these complexes to the oxygen evolving complex (OEC) in photosystem II. This active site consists of a Mn4CaO5 cluster that can be described as a distorted Mn3CaO4 cube with an external Mn center supported by bridging oxido ligands (Figure 1).8,28 Through four photo-induced oxidations of the cluster, coordinated substrate water molecules are deprotonated and coupled to produce dioxygen. A network of H-bonds surrounding the active site assists in bringing in substrate water molecules and shuttling the resulting protons out of the active site. The most oxidized state of the cluster that leads to O–O bond formation has yet to be directly detected and characterized, but studies of the states leading up to it have lead to the proposed formulation as Mn3IVMnVO or Mn3IVMnIVO•, with the latter form currently having the greatest acceptance.29,30 Our [MnVH3buea(O)] complex contains several of the same characteristics as the most oxidized form of the cluster, including an intramolecular H-bonding network surrounding a high valent, high spin Mn–oxo unit. We have therefore proposed that our MnV–oxo complex provides a simplified construct with which to examine key aspects of the OEC without the complication of the protein environment.
The prevalent opinion that the highest valent Mn center in the S4 state of the OEC is best formulated as a Mn(IV)–oxyl species rather than the Mn(V)–oxo moiety has been driven by computational studies and has yet to be verified experimentally. The strong influence of these computational studies on the mechanistic aspects of water oxidation prompted us to conduct a more in depth study on the electronic structure within the metal oxido unit in order to provide an experimental perspective. Our ability to observe EPR signals for our series of Mn–oxo complexes gave us the opportunity to experimentally probe the viability of a M–oxyl formulation of our high valent M–oxo species. The degree of spin delocalization was determined by measuring the broadening of the EPR signal of complexes in which the oxido ligand was labeled with oxygen-17 (I = 5/2).31 Both [MnIIIH3buea(17O)]2- (ρπ = 0.30 spins) and [MnVH3buea(17O)] (ππ = 0.45 spins) were determined to have substantial spin density on the oxido ligand, which we have interpreted to indicate strong covalent bonds within the Mn–oxo unit for both complexes rather than an oxyl radical, which would have an oxygen-17 hyperfine interaction of nearly one full spin on the oxido ligand (ρπ ~ 1). Similar measurements on [FeIVH3buea(17O)]- gave a spin density of 0.56 on the oxido ligand, which again supports strong covalency within the Fe–oxo unit.32 These experimental results offer another possible pathway to form an O–O bond within the OEC that utilizes a high spin MnV–oxo center instead of an oxyl radical species.
Redesigning the H-bonding Cavities
The successes of the [H3buea]3- ligand system in stabilizing unusual M–oxo complexes led us to question how altering the type of intramolecular H-bonding interactions would change the types of complexes that we could prepare and stabilize. One recent development from our laboratory is a tripodal ligand that contains H-bond acceptors instead of H-bond donors. This ligand, N,N′,N″-[2,2′,2″’-nitrilotris(ethane-2,1-diyl)]tris-(2,4,6-trimethylbenzenesulfonamido (denoted [MST]3-), maintains the same primary coordination environment and symmetry of the [H3buea]3- ligand but substitutes the H-bond donating urea moieties for H-bond accepting sulfonamido groups. The switch to sulfonamido groups changed the polarity of the cavity from partially positive to partially negative, which altered the types of ligands that could be stabilized within the cavity—while the urea ligand was best suited to oxido and hydroxido ligands, the sulfonamido tripod supports ligands with multiple protons that were not stable in [H3buea]3-, such as water and ammonia. An unexpected consequence of redesigning the cavity in [MST]3- was that the sulfonamido oxygen atoms create an auxiliary binding site for a second metal ion that allows for systematic preparation of heterobimetallic complexes whose chemistry will be described in the next sections.
Heterobimetallic Complexes: Probes for Multimetallic Sites in Proteins
Scheme 3 illustrates the general synthetic route to the bimetallic complexes that have been generated with the [MST]3– ligand system; reaction of the MII salt ([NMe4][MIIMST], M = MnII or FeII) with dioxygen in the presence of a second metal ion generates a MIII–OH complex with the second metal ion coordinated within the secondary coordination sphere. Using this method, we have generated heterobimetallic complexes that contain a transition metal ion and a group II metal ion (CaII, SrII, or BaII).33,34 The molecular structure of each complex, determined by XRD methods, revealed similar coordination properties of the metal ions in the two binding sites throughout the series (Figure 5); the transition metal center resides within the N4 binding pocket that is established by the three anionic sulfonamide nitrogen atoms and the neutral apical nitrogen atom. The hydroxido ligand occupies the fifth coordination site opposite of the apical nitrogen and bridges to the second metal ion, which is also coordinated to two sulfonamido oxygen atoms of the supporting ligand. The third ligand arm participates in a H-bonding interaction with the hydroxido ligand, which was determined to originate from reduction of O2 via labeling with 18O2. Oxygen atoms from crown ethers occupy the remaining coordination sites on the group II metal ion. For the [15-crown-5⊃CaII-(μ-OH)-MnIIIMST]+ complex (denoted [CaII(OH)MnIII]+), this molecular structure roughly mimics one edge of the Mn3CaO4 cube within the OEC in which an oxido or hydroxido ligand bridges between the CaII ion and the Mn ions.
Scheme 3.

Figure 5.

Molecular structure of [CaII(OH)MnIII]+ complex.
Influence of Group II Metal Ions on Electron Transfer Processes
We found that the rate of dioxygen activation by the MnII and FeII complexes was enhanced by the presence of a redox inactive metal ion.34 For Mn, the reaction of the parent [NMe4][MnIIMST] salt with dioxygen was sluggish to the point that the product of the reaction could not be isolated. However, addition of a group II metal ion as a crown ether adduct (MII(crown ether)(OTf)2, MII(crown ether) = CaII(15-crown-5), SrII(15-crown-5), BaII(18-crown-6)) to the reaction mixture produced as much as an 80-fold increase in the initial rate of the reaction, which was determined by monitoring the growth of the absorption spectrum corresponding to the heterobimetallic product (Figure 6A). In the case of Fe, the NMe4+ salt showed greater reactivity than the analogous Mn salt, but the rate was still enhanced over 30-fold by the presence of a group II metal ion. We postulated that these enhancements resulted from improved electron transfer to dioxygen that was facilitated by binding of the second metal ion. We therefore expected that the three group II metal ions would give a trend in rate enhancement that was consistent with their Lewis acidities. Instead, we found that CaII and SrII ions gave nearly identical rate enhancements despite the larger size and therefore weaker Lewis acidity of SrII compared to CaII. For the Mn complex, the rate enhancements differed by less than 10% between CaII and SrII ions, while with the Fe complex, they gave identical enhancements within error. These enhancements were greater than those imposed by BaII for both complexes of 3-fold (Mn) to 10-fold (Fe) over the NMe4+ salts.
Figure 6.

Initial rate data for the reaction of [MnIIMST]– + O2 (A) and cyclic voltammograms (B) for [CaII(OH)MnIII]+ (gray, −0.72 V), [SrII(OH)MnIII]+ (black, −0.70 V), and [BaII(OH)MnIII]+ (dashed, −0.76 V). Potentials are referenced to [Cp2Fe]+/0.
The redox properties of the [MII(OH)MIII]+ complexes containing Group II metal ions followed a similar unexpected trend as the initial rate of dioxygen activation. The one-electron reduction potential corresponding to the MIII/MII process differed by only 10 mV for the [CaII(OH)FeIII]+ and [SrII(OH)FeIII]+ complexes while the potential of the [BaII(OH)FeIII]+ complex was 100 mV more negative.34 Similarly, the MnII/MnIII couples of the CaII- and SrII-containing complexes were separated by only 20 mV and exhibited similar reversibility (Figure 6B). The potential of the [BaII(OH)MnIII]+ complex occurred only 40 mV more negative but was also much less reversible. A similar match in the influence of CaII and SrII ions on redox properties was observed by Agapie in his multimetallic clusters containing a Mn3M(μ4-O)(μ2-O) core, which together indicates that these two ions have similar influences on electron transfer properties of transition metal centers.35 This result has relevance to the OEC, whose function requires the presence of CaII or SrII ions.
Probing the Influence of Redox Inactive Metal Ions on C–H Cleavage
A deviation in the reactivity of [FeIIMST]– in the presence of CaII ions was observed upon oxidation with oxygen atom transfer reagents such as 4-methylmorpholine-N-oxide (NMO).36 When CaII ions are present in this reaction, the same [CaII(OH)FeIII]+ complex that was generated from dioxygen is isolated in high yield. In contrast, in the reaction between [FeIIMST]– and NMO alone, the major product was a new Fe(III)-alkoxide complex in which an ortho methyl group of a ligand arm has been hydroxylated instead of the expected monometallic [FeIIIMST(OH)]– product (Figure 7). We propose that this difference in reactivity results from a structural influence of CaII ions—before the addition of NMO to the reaction, a CaII ion can precoordinate to the sulfonamido oxygen atoms, rigidifying the structure so that the mesityl units and their ortho methyl groups are positioned far from the iron center and preventing intramolecular oxidation.
Figure 7.

Molecular structure of FeIII–alkoxide product from oxidation of [NMe4][FeIIMST] with NMO.
The formation of the Fe(III)-alkoxide product from NMO highlights both the oxidizing power of the intermediate generated in this reaction as well as a limitation in the [MST]3- ligand system for supporting reactive species. We therefore replaced the susceptible ortho methyl groups with tolyl groups and re-examined the oxidation of this modified complex ([FeIITST]–) with NMO.36 This reaction produced an observable intermediate at room temperature that contains a low intensity band at 905 nm in the electronic absorbance spectrum, which suggests the formation of an Fe(IV)–oxo species, as all synthetic non-heme complexes of this type exhibit a similar band.18 The intermediate reacts further to produce [FeIIITST(OH)]–, which we proposed results from H-atom abstraction from the solvent. This premise was substantiated with the aid of the external substrate 9,10-dihydroanthracene (DHA) that served as the source of the H-atom: DHA was converted to anthracene and other oxidized products as [FeIIITST(OH)]– was formed. Note that no oxidized ligand products were isolated from the oxidation of [FeIITST]–.
Outlook
The processes of making and breaking of O–O bonds in biology share a common mechanistic thread of proceeding through high valent intermediates that contain M–oxo or M–OH units. This connection is illustrated in a series of synthetic complexes that we have described here: O–O bond breaking by an FeII complex produced an FeIII–oxo complex that was converted to a high valent FeIV–oxo species, and high valent Mn–oxo complexes were prepared via stepwise oxidation from a MnIII–oxo species that was generated from water—a pathway that parallels the steps leading to O2 evolution in the OEC. In another system, O2 activation by MnII and FeII complexes lead to the isolation of heterobimetallic complexes that contain two metal ions bridged by a hydroxido ligand. All of the complexes described in this Account are high spin, and their stabilization is made possible through the control of their secondary coordination spheres with synthetic ligands that promote intramolecular H-bonds. However the, symmetrical and relatively static secondary coordination spheres within these synthetic systems differ from most active sites in metalloproteins, which incorporate a combination of non-covalent interactions around the metal ion(s). New ligand designs and synthetic methods are therefore needed to better duplicate these architectural features, and progress in clearly being made. We have designed a series of ligands that vary the number of H-bonds within the secondary sphere,37 and have also developed dynamic ligands that aid in the catalytic reduction dioxygen to water.38 Fout has illustrated another approach in her report of a ligand system that can simultaneously provide either H-bond donors or acceptors, which has produced FeIII–oxo complexes similar to [FeIIIH3buea(O)]2.39 In another example by Szymczak, Lewis acid/base pairs within the secondary coordination sphere stabilized the first V-(η2-N2H3) complex.40 These newer systems illustrate the potential for increased molecular complexity to unveil new types of reactivity.
Acknowledgments
We thank the NIH (GM50781) for support of our work and the many fabulous coworkers who have contributed to this research.
Biographies
Sarah A. Cook graduated from Ohio Wesleyan University in 2010 with a B.A. in Chemistry and is currently a 5th year graduate student in the laboratory of A. S. Borovik at the University of California-Irvine.
A. S. Borovik received his B.S. degree in Chemistry from Humboldt State University, his Ph.D. in Chemistry at the University of North Carolina-Chapel Hill under Tom Sorrell, and held postdoctoral positions with Larry Que at the University of Minnesota and Ken Raymond at the University of California-Berkeley. He has been on the faculty at Ithaca College, Kansas State University, the University of Kansas, and the University of California-Irvine, where he is currently a professor.
References
- 1.Marshall NM, Garner DK, Wilson TD, Gao YG, Robinson H, Nilges MJ, Lu Y. Rationally Tuning the Reduction Potential of a Single Cupredoxin Beyond the Natural Range. Nature. 2009;462:113–116. doi: 10.1038/nature08551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Miller AF. Redox Tuning Over Almost 1 V in a Structurally Conserved Active Site: Lessons From Fe-Containing Superoxide Dismutase. Acc Chem Res. 2008;41:501–510. doi: 10.1021/ar700237u. [DOI] [PubMed] [Google Scholar]
- 3.Wikstrom M. Cytochrome C Oxidase: 25 Years of the Elusive Proton Pump. Biochim Biophys Acta. 2004;1655:241–247. doi: 10.1016/j.bbabio.2003.07.013. [DOI] [PubMed] [Google Scholar]
- 4.Helm ML, Stewart MP, Bullock RM, DuBois MR, DuBois DL. A Synthetic Nickel Electrocatalyst with a Turnover Frequency Above 100,000 S−1 for H2 Production. Science. 2011;333:863–866. doi: 10.1126/science.1205864. [DOI] [PubMed] [Google Scholar]
- 5.Collman JP. Synthetic Models for Oxygen-Binding Hemoproteins. Acc Chem Res. 1977;10:265–272. [Google Scholar]
- 6.Kitajima N, Fujisawa K, Morooka Y, Toriumi K. μ-η2:η2-Peroxo Binuclear Copper Complex, [Cu(HB(3,5-(Me2CH)2pz)3)]2(O2) J Am Chem Soc. 1989;111:8975–8976. [Google Scholar]
- 7.MacBeth CE, Golombek AP, Young VG, Yang C, Kuczera K, Hendrich MP, Borovik AS. O2 Activation by Nonheme Iron Complexes: a Monomeric Fe(III)-Oxo Complex Derived From O2. Science. 2000;289:938–941. doi: 10.1126/science.289.5481.938. [DOI] [PubMed] [Google Scholar]
- 8.Umena Y, Kawakami K, Shen JR, Kamiya N. Crystal Structure of Oxygen-Evolving Photosystem II at a Resolution of 1. 93Å. Nature. 2011;473:55–60. doi: 10.1038/nature09913. [DOI] [PubMed] [Google Scholar]
- 9.Borovik AS. Bioinspired Hydrogen Bond Motifs in Ligand Design: the Role of Noncovalent Interactions in Metal Ion Mediated Activation of Dioxygen. Acc Chem Res. 2005;38:54–61. doi: 10.1021/ar030160q. [DOI] [PubMed] [Google Scholar]
- 10.Shook RL, Borovik AS. The Effects of Hydrogen Bonds on Metal-Mediated O2 Activation and Related Processes. Chem Commun. 2008:6095–6107. doi: 10.1039/b810957e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shook RL, Borovik AS. Role of the Secondary Coordination Sphere in Metal-Mediated Dioxygen Activation. Inorg Chem. 2010;49:3646–3660. doi: 10.1021/ic901550k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shirin Z, Hammes BS, Young VG, Borovik AS. Hydrogen Bonding in Metal Oxo Complexes: Synthesis and Structure of a Monomeric Manganese(III)–Oxo Complex and Its Hydroxo Analogue. J Am Chem Soc. 2000;122:1836–1837. [Google Scholar]
- 13.Ballhausen CJ, Gray HB. The Electronic Structure of the Vanadyl Ion. Inorg Chem. 1962;1:111–122. [Google Scholar]
- 14.Dey A, Hocking RK, Larsen P, Borovik AS, Hodgson KO, Hedman B, Solomon EI. X-Ray Absorption Spectroscopy and Density Functional Theory Studies of [(H3buea)FeIII-X]n- (X = S2-, O2-, OH-): Comparison of Bonding and Hydrogen Bonding in Oxo and Sulfido Complexes. J Am Chem Soc. 2006;128:9825–9833. doi: 10.1021/ja061618x. [DOI] [PubMed] [Google Scholar]
- 15.Rittle J, Green MT. Cytochrome P450 Compound I: Capture, Characterization, and C-H Bond Activation Kinetics. Science. 2010;330:933–937. doi: 10.1126/science.1193478. [DOI] [PubMed] [Google Scholar]
- 16.Krebs C, Galonić Fujimori D, Walsh CT, Bollinger JM. Non-Heme Fe(IV)–Oxo Intermediates. Acc Chem Res. 2007;40:484–492. doi: 10.1021/ar700066p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rohde JU, In JH, Lim MH, Brennessel WW, Bukowski MR, Stubna A, Muenck E, Nam W, Que LJ. Crystallographic and Spectroscopic Characterization of a Nonheme Fe(IV)=O Complex. Science. 2003;299:1037–1039. doi: 10.1126/science.299.5609.1037. [DOI] [PubMed] [Google Scholar]
- 18.Que L. The Road to Non-Heme Oxoferryls and Beyond. Acc Chem Res. 2007;40:493–500. doi: 10.1021/ar700024g. [DOI] [PubMed] [Google Scholar]
- 19.Nam W. Guest Editorial: Dioxygen Activation by Metalloenzymes and Models. Acc Chem Res. 2007;40:465. [Google Scholar]
- 20.Lacy DC, Gupta R, Stone KL, Greaves J, Ziller JW, Hendrich MP, Borovik AS. Formation, Structure, and EPR Detection of a High Spin FeIV—Oxo Species Derived From Either an FeIII—Oxo or FeIII—OH Complex. J Am Chem Soc. 2010;132:12188–12190. doi: 10.1021/ja1047818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.England J, Martinho M, Farquhar ER, Frisch Jonathan R, Bominaar EL, Munck E, Que LJ. A Synthetic High-Spin Oxoiron(IV) Complex: Generation, Spectroscopic Characterization, and Reactivity. Angew Chem, Int Ed. 2009;48:3622–3626. doi: 10.1002/anie.200900863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.England J, Guo Y, Farquhar ER, Young VGJ, Munck E, Que LJ. The Crystal Structure of a High-Spin Oxoiron(IV) Complex and Characterization of Its Self-Decay Pathway. J Am Chem Soc. 2010;132:8635–8644. doi: 10.1021/ja100366c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bigi JP, Harman WH, Lassalle-Kaiser B, Robles DM, Stich TA, Yano J, Britt RD, Chang CJ. A High-Spin Iron(IV)–Oxo Complex Supported by a Trigonal Nonheme Pyrrolide Platform. J Am Chem Soc. 2012;134:1536–1542. doi: 10.1021/ja207048h. [DOI] [PubMed] [Google Scholar]
- 24.England J, Guo Y, Van HKM, Cranswick MA, Rohde GT, Bominaar EL, Munck E, Que LJ. A More Reactive Trigonal-Bipyramidal High-Spin Oxoiron(IV) Complex with a Cis-Labile Site. J Am Chem Soc. 2011;133:11880–11883. doi: 10.1021/ja2040909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hess A, Horz MR, Liable-Sands LM, Lindner DC, Rheingold AL, Theopold KH. Insertion of O2 Into a Chromium-Phenyl Bond: Mechanism of Formation of the Paramagnetic d2 Oxo Complex [TptBu,MeCrIV(O)OPh] Angew Chem Int Ed. 1999;38:166–168. [Google Scholar]
- 26.Parsell TH, Behan RK, Green MT, Hendrich MP, Borovik AS. Preparation and Properties of a Monomeric MnIV–Oxo Complex. J Am Chem Soc. 2006;128:8728–8729. doi: 10.1021/ja062332v. [DOI] [PubMed] [Google Scholar]
- 27.Taguchi T, Gupta R, Lassalle-Kaiser B, Boyce DW, Yachandra VK, Tolman WB, Yano J, Hendrich MP, Borovik AS. Preparation and Properties of a Monomeric High-Spin MnV–Oxo Complex. J Am Chem Soc. 2012;134:1996–1999. doi: 10.1021/ja210957u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Suga M, Akita F, Hirata K, Ueno G, Murakami H, Nakajima Y, Shimizu T, Yamashita K, Yamamoto M, Ago H, Shen JR. Native Structure of Photosystem II at 1. 95 a Resolution Viewed by Femtosecond X-Ray Pulses. Nature. 2015;517:99–103. doi: 10.1038/nature13991. [DOI] [PubMed] [Google Scholar]
- 29.Brudvig GW. Water Oxidation Chemistry of Photosystem II. Philos Trans R Soc, B. 2008;363:1211–1219. doi: 10.1098/rstb.2007.2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cox N, Pantazis DA, Neese F, Lubitz W. Biological Water Oxidation. Acc Chem Res. 2013;46:1588–1596. doi: 10.1021/ar3003249. [DOI] [PubMed] [Google Scholar]
- 31.Gupta R, Taguchi T, Lassalle-Kaiser B, Bominaar EL, Yano J, Hendrich MP, Borovik AS. High-Spin Mn–Oxo Complexes and Their Relevance to the Oxygen-Evolving Complex Within Photosystem II. Proc Natl Acad Sci. 2015;112:5319–5324. doi: 10.1073/pnas.1422800112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gupta R, Lacy DC, Bominaar EL, Borovik AS, Hendrich MP. Electron Paramagnetic Resonance and Mössbauer Spectroscopy and Density Functional Theory Analysis of a High-Spin FeIV–Oxo Complex. J Am Chem Soc. 2012;134:9775–9784. doi: 10.1021/ja303224p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Park YJ, Ziller JW, Borovik AS. The Effects of Redox-Inactive Metal Ions on the Activation of Dioxygen: Isolation and Characterization of a Heterobimetallic Complex Containing a MnIII–(μ-OH)–CaII Core. J Am Chem Soc. 2011;133:9258–9261. doi: 10.1021/ja203458d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Park YJ, Cook SA, Sickerman NS, Sano Y, Ziller JW, Borovik AS. Heterobimetallic Complexes with MIII-(μ-OH)-MII Cores (MIII = Fe, Mn, Ga; MII = Ca, Sr, and Ba): Structural, Kinetic, and Redox Properties. Chem Sci. 2013;4:717–726. doi: 10.1039/C2SC21400H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tsui EY, Tran R, Yano J, Agapie T. Redox-Inactive Metals Modulate the Reduction Potential in Heterometallic Manganese–Oxido Clusters. Nat Chem. 2013;5:293–299. doi: 10.1038/nchem.1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cook SA, Ziller JW, Borovik AS. Iron(II) Complexes Supported by Sulfonamido Tripodal Ligands: Endogenous Versus Exogenous Substrate Oxidation. Inorg Chem. 2014;53:11029–11035. doi: 10.1021/ic501531g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lucas RL, Zart MK, Mukherjee J, Murkerjee J, Sorrell TN, Powell DR, Borovik AS. A Modular Approach Toward Regulating the Secondary Coordination Sphere of Metal Ions: Differential Dioxygen Activation Assisted by Intramolecular Hydrogen Bonds. J Am Chem Soc. 2006;128:15476–15489. doi: 10.1021/ja063935+. [DOI] [PubMed] [Google Scholar]
- 38.Shook RL, Peterson SM, Greaves J, Moore C, Rheingold AL, Borovik AS. Catalytic Reduction of Dioxygen to Water with a Monomeric Manganese Complex at Room Temperature. J Am Chem Soc. 2011;133:5810–5817. doi: 10.1021/ja106564a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Matson EM, Park YJ, Fout AR. Facile Nitrite Reduction in a Non-Heme Iron System: Formation of an Iron(III)-Oxo. J Am Chem Soc. 2014;136:17398–17401. doi: 10.1021/ja510615p. [DOI] [PubMed] [Google Scholar]
- 40.Tutusaus O, Ni C, Szymczak NK. A Transition Metal Lewis Acid/Base Triad System for Cooperative Substrate Binding. J Am Chem Soc. 2013;135:3403–3406. doi: 10.1021/ja400962h. [DOI] [PubMed] [Google Scholar]