

Abstract
The present study is the first to report inhibition of breast cancer cell growth in vitro and in vivo and suppression of self-renewal of breast cancer stem cells (bCSC) by a pine bark component (leelamine). Except for a few recent publications in melanoma, anticancer pharmacology of this interesting phytochemical is largely elusive. Leelamine (LLM) dose-dependently inhibited viability of MDA-MB-231 (triple-negative), MCF-7 (estrogen receptor-positive), and SUM159 (triple-negative) human breast cancer cells in association with apoptotic cell death induction. To the contrary, a normal mammary epithelial cell line derived from fibrocystic breast disease and spontaneously immortalized (MCF-10A) was fully resistant to LLM-mediated cell growth inhibition and apoptosis induction. LLM also inhibited self-renewal of breast cancer stem cells. Apoptosis induction by LLM in breast cancer cells was accompanied by a modest increase in reactive oxygen species production, which was not due to inhibition of mitochondrial electron transport chain complexes. Nevertheless, ectopic expression of manganese superoxide dismutase conferred partial protection against LLM-induced cell death but only at a lower yet pharmacologically relevant concentration. Exposure of breast cancer cells to LLM resulted in (a) induction and/or activation of multidomain proapoptotic proteins Bax and Bak, (b) caspase-9 activation, and (c) cytosolic release of cytochrome c. Bax and Bak deficiency in immortalized fibroblasts conferred significant protection against cell death by LLM. Intraperitoneal administration of LLM (7.5 mg/kg; five times/week) suppressed the growth of orthotopic SUM159 xenografts in mice without any toxicity. In conclusion, the present study provides critical preclinical data to warrant further investigation of LLM.
Keywords: leelamine, breast cancer, apoptosis, breast cancer stem cells
INTRODUCTION
The American Cancer Society predicts diagnosis of about 231,840 and 60,290 new cases of invasive breast cancer and carcinoma in situ, respectively in the United States alone in 2015. Despite our increasingly broader understanding of the risk factors, approximately 40,290 American women are expected to die from breast cancer in 2015 [1-5]. Molecular heterogeneity of breast cancer coupled with inability to modify many underlying risk factors (e.g., reproductive risk factors and genetic predisposition) necessitates pursuit for novel interventions to prevent and treat this devastating disease. Breast cancer is broadly classified into five major subtypes based on gene expression profiling, including: luminal A, luminal B, human epidermal growth factor receptor 2 amplified, basal-type, and normal-like breast cancer [6-9]. Notably, the basal-type breast cancer subtype has a poor prognosis due to limited treatment options [10]. Therefore, identification of agents effective against different subtypes of breast cancer is highly desirable to reduce disease-related cost and mortality associated with this disease. Recent progress in our understanding of the breast cancer biology further complicates clinical management of breast cancer due to involvement of tumor initiating cells (commonly known as breast cancer stem cells; bCSC) in cancer development and progression [11-13]. Consequently, it is only logical to speculate that eradication of proliferating tumor cells constituting bulk of the tumor mass as well as bCSC may be necessary for effective treatment and prevention of breast cancer.
Phytochemicals derived from edible or medicinal plants appear assuring for cancer chemoprevention and possibly treatment [14-16]. Examples of promising phytochemicals proven effective in preclinical models of breast cancer and primed for clinical translation in the near future include isothiocyanates (phenethyl isothiocyanate and benzyl isothiocyanate) abundant in commonly consumed cruciferous vegetables [15-18]. The present study reports in vitro and/or in vivo effects of leelamine (LLM) derived from the bark of pine tree on proliferating breast cancer cells and bCSC population. While the present study is the first to document suppression of bCSC self-renewal by LLM, growth inhibitory effects of this agent or its synthetic analogues have been studied previously in melanoma and in a few other cancer types. For example, daily intraperitoneal administration of LLM (5 or 7.5 mg/kg body weight) or daily intravenous injection of a LLM nanoparticle (nanolipolee-007; 30 mg/kg body weight) significantly inhibited in vivo growth of human melanoma cells subcutaneously implanted in athymic mice [19, 20]. A few reports have also shown inhibition of hepatocellular carcinoma cell growth in vitro by LLM analogues, including N-benzoyl-12-nitro dehydroabietyl amine-7-one [21, 22]. A single publication relevant to breast cancer demonstrated in vitro growth inhibition of MDA-MB-231 and MCF-7 cells by sulfonamide derivatives of LLM [23].
MATERIALS AND METHODS
Reagents
LLM was purchased from Cayman Chemical Company (Ann Arbor, MI). Chemicals required for cell culture were purchased from Invitrogen-Life Technologies (Carlsbad, CA). The antibodies were purchased from the following vendors: polyclonal anti-Bak and anti-Bax antibodies (for western blotting) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA); anti-manganese superoxide dismutase (MnSOD) and anti-active Bak (clone-TC-100 for immunocytochemistry) antibodies were purchased from Calbiochem (Billerica, MA); monoclonal 6A7 antibody specific for detection of active Bax was from BD Biosciences (San Diego, CA); and anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH) antibody was from GeneTex (Irvine, CA). Cell Death Detection ELISAPLUS kit was from Roche Applied Sciences (Indianapolis, IN). MitoSOX Red and MitoTRACKER green were purchased from Invitrogen-Life Technologies. Annexin V-FITC/propidium iodide (PI) Apoptosis detection kit was purchased from BD Biosciences. A kit for detection of active Caspase 9 and anti-cytochrome c antibody were from Abcam (Cambridge, MA).
Cell Culture
MDA-MB-231, MCF-7, and MCF-10A cells were obtained from the American Type Culture Collection (Manassas, VA) and last authenticated by us in 2012 and 2015. SUM159 cells were purchased from Asterand and last authenticated in 2015. Cells were cultured as described previously [24] or according to the supplier’s instructions. Wild-type MDA-MB-231 cells and its Rho-0 variant have been described by us previously [25]. MitoGFP-MDA-MB-231 and MitoGFP-MCF-7 cells were cultured in corresponding growth media supplemented with 100 μg/mL of G418. MCF-7 cells stably transfected with pcDNA3.1 vector or MnSOD plasmid and their culture conditions have been described by us previously [26]. Immortalized mouse embryonic fibroblasts derived from wild-type mice (WT/MEF) and Bax and Bak double knockout mice (DKO/MEF) were generously provided by the late Dr. Stanley J. Korsmeyer (Dana-Farber Cancer Institute, Boston, MA) and maintained as described by us previously [27]. MDA-MB-231 cells stably transfected with pIRES empty vector or pIRES vector encoding ERα were cultured as described by us previously [28].
Cell Viability and Apoptosis Assays
The effect of LLM on cell viability was tested by trypan blue dye exclusion assay as previously described [29]. Apoptosis induction was assessed by quantitation of histone-associated DNA fragment release into the cytosol or by flow cytometry using Annexin V/ PI Apoptosis detection kit and Caspase 9 activation detection kit according to the manufacturer's instructions.
Mammosphere Formation Assay
Mammosphere formation assay was performed as described by Kim et al. [30]. Briefly, after 5 days of cell seeding, the first generation mammospheres were scored and disaggregated to make single cells. The single cells were then re-plated without further treatment with ethanol or LLM for the second generation mammosphere formation. After 7 days of cell seeding, the second generation mammosphere were scored under an inverted microscope.
Flow Cytometric Analysis of Aldehyde Dehydrogenase 1 (ALDH1) Activity and CD49f+/CD24− Population
Effect of LLM treatment on ALDH1 activity was determined using ALDEFLUOR kit (STEMCELL Technologies). DEAB (diethylaminobenzaldehyde), a specific ALDH1 inhibitor was used as a control. For analysis of CD49f+/CD24− population, cells were incubated with anti-CD49f (APC-conjugated) and anti-CD24 (PE-conjugated) antibodies in the dark for 30 min at room temperature. Cells were washed with phosphate-buffered saline and analyzed using Accuri ™C6 flow cytometer.
Detection of Reactive Oxygen Species (ROS)
Flow cytometry and confocal microscopy after staining the cells with MitoSOX Red were performed for ROS detection using a Leica TCS SL microscope (Leica Microsystems, Buffalo Grove, IL). Details of MitoSOX Red staining are reported by us previously [31].
Western Blotting
Control and LLM-treated cells were processed for immunoblotting as described by us previously [32]. Densitometric quantitation was performed with the use of UN-SCAN-IT software version 5.1 (Silk Scientific Corporation, Orem, Utah). Blots were probed with anti-GAPDH antibody for normalization.
Determination of Mitochondrial Electron Transport Chain Complex Activities
MDA-MB-231 or MCF-7 cells (1×106) were plated in 100-mm dishes in triplicate. After overnight incubation, the cells were treated with 0.025% ethanol (final concentration) or 5 μM of leelamine for 6 hours. Complex activities were determined as previously reported [25, 31].
Immunocytochemical Analysis
To detect Bax and Bak activation or cytosolic release of cytochrome c, desired cells (2.5×104 cells/mL) were cultured on coverslips, allowed to attach by overnight incubation, and then exposed to 0.025% ethanol (final concentration) or 5 μM LLM for 24 hours. Immunocytochemical analysis was performed as described by us previously [27]. The cells were examined under a Leica DC300F or DFC450C microscope at 100× objective magnification.
Xenograft Study
The use of mice was approved by the Institutional Animal Care and Use Committee, The University of Pittsburgh. Female nude mice (4-5 weeks old) were purchased from Charles River Laboratories, housed in micro-isolator cages, and acclimated for 1 week prior to start of the experiment. Exponentially growing SUM159 cells (about 60% confluence) were suspended in serum free media consisting of Ham’s F12, 1% penicillin/streptomycin antibiotic mixture, 5 μg/mL insulin and 1 μg/mL hydrocortisone. An aliquot (0.02 mL) containing 2×106 cells was injected orthotopically in right inguinal mammary fat pad of each mouse. Mice were randomized into two groups of five mice per group. One week after tumor cell implantation, the mice were treated intraperitoneally with 100 μL vehicle (10% ethanol, 10% dimethyl sulfoxide, 30% Cremophor EL and 50% phosphate-buffered saline) or LLM (0.184 mg/mouse, which equates to about 7.5 mg/kg body weight assuming average mouse weight of 24.5 g) five times/week (Monday through Friday). The experiment was terminated 53 days after tumor cell injection. Two mice from control group were euthanized one week before study termination due to morbidity from large tumor burden. Body weights of the mice of both groups were determined weekly. After sacrifice, tumor tissues were collected, fixed in 10% neutral buffered formalin, paraffin-embedded, and then the sections from both control and LLM groups were stained with hematoxylin and eosin (H&E) for microscopic observation.
Statistical Analysis
Statistical comparisons were performed using one-way analysis of variance (ANOVA) followed by Dunnett’s adjustment (for dose-response comparisons) or Newman-Keuls adjustment (for multiple group comparisons). Student’s t test was used for binary comparisons.
RESULTS
LLM Inhibited Viability of Breast Cancer Cells by Inducing Apoptosis
The effect of LLM (structure shown in Fig. 1A) treatment on viability of MDA-MB-231, MCF-7, SUM159 human breast cancer cells as well as MCF-10A normal mammary epithelial cell line was assessed by trypan blue dye exclusion assay (Fig. 1B). Viability of breast cancer cells was decreased significantly and dose-dependently upon treatment with LLM. The MCF-7 cell line exhibited a relatively greater sensitivity compared with MDA-MB-231 or SUM159 cells especially at higher concentrations after 48 hour treatment. On the other hand, survival of MCF-10A was minimally affected by LLM treatment even at concentrations that were highly cytotoxic to the breast cancer cells (Fig. 1B).
Fig. 1.
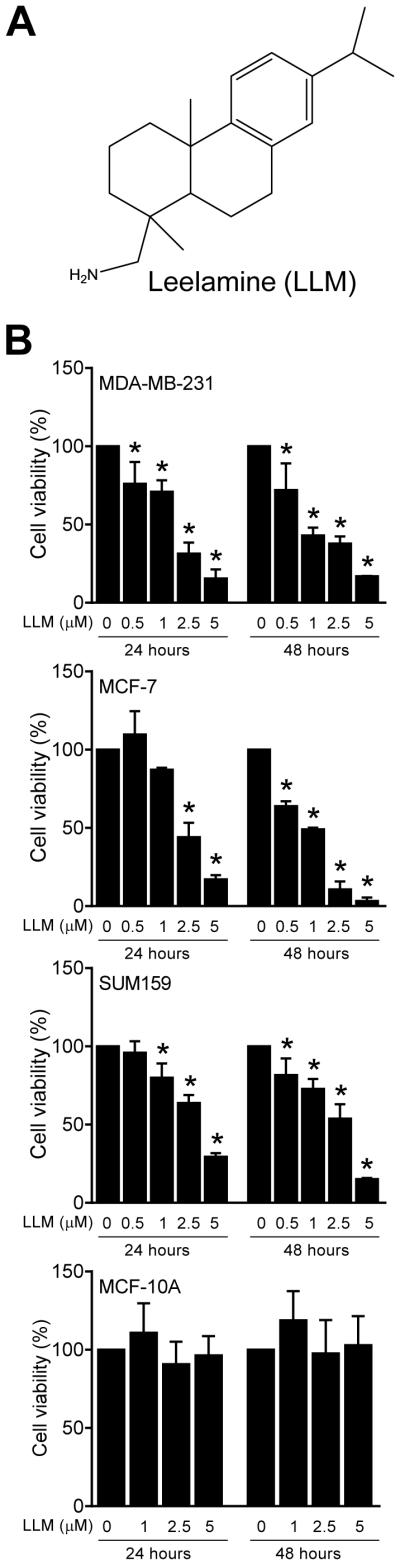
LLM selectively inhibits viability of human breast cancer cells. A Structure of LLM. B Effect of LLM on viability of MDA-MB-231, MCF-7, SUM159, and MCF-10A cells, which was determined by trypan blue dye exclusion assay. Results shown are mean ± SD (n = 3). *Significantly different (P < 0.05) compared with vehicle-treated control by one-way ANOVA followed by Dunnett’s test. The results were consistent in replicate experiments.
Figure 2A exemplifies flow histograms for early + late apoptotic fraction in MDA-MB-231 and MCF-10A cultures after 24-or 48-hour treatment with vehicle or LLM. As can be seen in Fig. 2B, early + late apoptotic fraction was dose-dependently and significantly increased upon 24-and/or 48-hour treatment with LLM in MDA-MB-231, MCF-7 and SUM159 cultures. Likewise, LLM-treated MDA-MB-231, MCF-7 and SUM159 cells (24-or 48-hour treatment) exhibited a dose-dependent and statistically significant increase in histone-associated DNA fragment release into the cytosol, another marker of apoptosis, compared with corresponding ethanol-treated controls (Fig. 2C). In agreement with cell viability data (Fig. 1B), LLM treatment (1-5 μM) failed to increase apoptotic fraction over ethanol-treated control in MCF-10A cells (Fig. 2B, C). Collectively, these results indicated that LLM-mediated inhibition of viability of breast cancer cells was due to apoptosis induction.
Fig. 2.

LLM treatment induces apoptosis in human breast cancer cells. A Representative flow histograms depicting early + late apoptotic fractions in ethanol (control) and LLM-treated MDA-MB-231 or MCF-10A cells. B Quantitation of early + late apoptotic cell enrichment relative to ethanol-treated control in MDA-MB-231, MCF-7, SUM159, and MCF-10A cells after 24- or 48-hour treatment with LLM. C Quantitation of histone-associated DNA fragment release into the cytosol relative to corresponding control in MDA-MB-231, MCF-7, SUM159, and MCF-10A cells after 24- or 48-hour treatment with LLM. Enrichment relative to corresponding control is shown as mean ± SD (n = 3). *Significantly different (P < 0.05) compared with vehicle-treated control by one-way ANOVA followed by Dunnett’s test. Each experiment was done at least twice and representative data from one such experiment are shown. The results in different experiments were consistent.
LLM Inhibited Self-Renewal of bCSC
Because of emerging role of bCSC in breast cancer development and progression [11-13], it was of interest to determine if self-renewal of bCSC was affected by LLM treatment. Fig. 3A shows representative 1st generation mammospheres resulting after 5 days of treatment of cells with ethanol (control) or LLM. The 1st and 2nd generation mammosphere number was dose-dependently decreased following LLM treatment in both cell lines (Fig. 3B). Representative flow histograms for ALDH1 activity in SUM159 cells after 72-hour treatment with ethanol or 1 μM LLM are shown in Fig. 3C. Quantitation of the ALDH1 activity revealed statistically significant decrease in LLM-treated MCF-7 and SUM159 cells when compared with corresponding vehicle-treated controls (Fig. 3D). Inhibition of bCSC fraction after LLM treatment was confirmed by flow cytometric analysis of CD49f+/CD24− fraction (Fig. 3E, F). The CD49f+/CD24− fraction was significantly decreased after treatment of MCF-7 and SUM159 cultures with LLM (Fig. 3 E, F). These results indicated inhibition of bCSC by LLM treatment.
Fig. 3.

LLM inhibits bCSC population. A Representative images of 1st generation mammospheres after 5 days of treatment with ethanol or 1 μM of LLM (100× magnification; scale bar = 200 μm). B Bar graphs show the percentage of 1st generation (after 5 days of cell plating) and 2nd generation (after 7 days of cell plating) mammospheres relative to corresponding ethanol-treated controls. C Representative flow histograms for ALDH1 activity in SUM159 cells after 72-hour treatment with ethanol (control) or 1 μM of LLM. The ALDH1 inhibitor DEAB was used as a control. D Bar graphs show the percentage of ALDH1 activity relative to corresponding controls in MCF-7 and SUM159 cells (72-hour treatment). All data shown are mean ± SD (n = 3). E Representative flow histograms for CD49f+/CD24− fraction in MCF-7 cells after 72-hour treatment with vehicle or LLM. F Bar graphs show quantitation of CD49f+/CD24− fraction in MCF-7 and SUM159 cells relative to corresponding ethanol-treated controls (72-hour treatment). Results shown are mean ± SD (n = 3). *Significantly different (P < 0.05) compared with control by one-way ANOVA with Dunnett’s adjustment. Comparable results were observed in two independent experiments. Representative data from one such experiment are shown.
LLM-Induced Apoptosis Was Associated with ROS Production
Next, we explored the possibility of whether LLM-induced apoptosis was mediated by ROS, which are implicated in cell death induction by structurally diverse phytochemicals [33]. Representative images depicting MitoSOX Red fluorescence in MitoGFP-MDA-MB-231, MitoGFP-MCF-7, and SUM159 cells are shown in Fig. 4A. ROS production by LLM treatment in each cell line was confirmed by flow cytometry (Fig. 4B). Moreover, ectopic expression of MnSOD (Fig. 4C, inset) conferred partial but significant protection against LLM-induced apoptosis at 2.5 and 5 μM concentrations (Fig. 4C). This protection, however, was not observed at the 7.5 μM LLM dose (Fig. 4C)
Fig. 4.

LLM-induced ROS partially contribute to apoptotic cell death in human breast cancer cells. A Representative confocal images for MitoSOX Red fluorescence (63× objective magnification; scale bar = 20 μm) in MitoGFP-MDA-MB-231, MitoGFP-MCF-7, and SUM159 cells treated with ethanol or 5 μM of LLM for 6 hours. B Quantitation of MitoSOX Red fluorescence enrichment relative to corresponding control in MDA-MB-231, MCF-7, and SUM159 cells after 3- or 6-hour treatment with LLM. Results shown are mean ± SD (n = 3). *Statistical significance compared with ethanol-treated control was analyzed by Student’s t test (P < 0.05). C Quantitation of histone-associated DNA fragment release into the cytosol in stably transfected MCF-7 cells after treatment with ethanol (control) or the indicated concentrations of LLM for 24 hours (Inset: Immunoblotting for MnSOD using lysates from pcDNA3.1 vector-transfected control cells or MnSOD-overexpressing MCF-7 cells). Results shown are mean ± SD (n = 3). Statistically significant (P < 0.05) acompared with corresponding control and b between pcDNA3.1 transfected and MnSOD overexpressing cells by one-way ANOVA with Newman-Keuls multiple comparisons test. D Quantitation of histone-associated DNA fragment release into the cytosol in wild-type (WT) MDA-MB-231 cells and its Rho-0 variant after 24-hour treatment with ethanol (control) or the indicated concentrations of LLM. The enrichment results shown are relative to ethanol-treated WT cells. E The effect of LLM treatment (24 hours) on viability in WT and Rho-0 MDA-MB-231 cells. The results shown are relative to ethanol-treated WT MDA-MB-231 cells (mean ± SD; n = 3). Statistically significant (P < 0.05) a compared with corresponding control and b between WT and Rho-0 cells by one-way ANOVA with Newman-Keuls multiple comparisons test (D and E). Each experiment was done at least twice and representative data from one such experiment are shown.
Inhibition of mitochondrial respiratory complexe I or III is one possibility by which LLM may cause ROS production. Measurement of the activities of different complexes in MDA-MB-231 and MCF-7 cells did not show any meaningful inhibition upon 6-hour treatment with LLM (data not shown). We used Rho-0 variant of MDA-MB-231 cells to further rule out inhibition of mitochondrial respiration in LLM-induced apoptosis. The Rho-0 variant of a cell line is generated by culture in the presence of ethidium bromide to deplete mitochondrial DNA. Because mitochondrial DNA encodes some of the respiratory complex subunits, survival of Rho-0 cells is dependent on ATP derived from anaerobic glycolysis [34]. These cells are unable to generate ROS from inhibition of electron transport chain. As can be seen in Fig. 4D, Rho-0 cells were nearly as sensitive to LLM-induced apoptosis as wild-type MDA-MB-231 cells. Results of cell viability assay (Fig. 4E) were consistent with analysis of apoptosis (Fig. 4D) in wild-type MDA-MB-231 cells and its Rho-0 variant. Together, these observations indicated that while ROS production partly contributed to apoptosis induction by LLM, the pro-oxidant effect of LLM was not due to inhibition of mitochondrial electron transport.
LLM-Induced Apoptosis Was Associated With Activation of Bax and Bak
We next studied potential involvement of multidomain proapoptotic proteins Bax and Bak in LLM-induced apoptosis. Many natural chemicals promote ROS-mediated activation of Bax and/or Bak to induce apoptosis in cancer cells [25, 31, 33]. LLM-treated breast cancer cells exhibited sustained (MDA-MB-231 and SUM159) or transient (MCF-7) induction of Bax and/or Bak (Fig. 5A). Activation (conformational change) of Bax and Bak is required for their proapoptotic activity [35]. We performed immunocytochemistry using antibodies specific for detection of active-Bax and active-Bak to test whether LLM treatment caused activation of these proteins. Control (ethanol-treated) MDA-MB-231, MCF-7 and SUM159 cells exhibited very weak green fluorescence (Fig. 5B). Treatment with LLM resulted in enrichment of green fluorescence associated with active-Bax and active-Bak in each cell line (Fig. 5B). These results indicated activation of Bax and Bak post LLM exposure in breast cancer cells, but not in MCF-10A cell line (Fig. 5B). Lack of Bax and Bak activation may partly explain resistance of MCF-10A cells to apoptosis induction by LLM. To further confirm the roles of Bax and Bak in cell death regulation by LLM, we compared sensitivities of WT/MEF and DKO/MEF to LLM-induced apoptosis. Exposure of WT/MEF to LLM resulted in dose-dependent apoptosis that was not evident in DKO/MEF (Fig. 5C). In agreement with these results, DKO/MEF was significantly more resistant to LLM-mediated inhibition of cell viability compared with WT/MEF (Fig. 5D). Overexpression of ERα (Fig. 5E) resulted in a significant decrease in LLM-induced apoptosis compared to empty vector (EV)-transfected MDA-MB-231 cells (Fig. 5E). Together these results indicated that Bax and Bak play an important role in execution of LLM-induced apoptosis. We also conclude that ERα overexpression partly abrogates apoptosis induction by LLM at least in the MDA-MB-231 cell line.
Fig. 5.

The role of Bak and Bax in LLM-induced apoptosis. A Immunoblotting for Bak and Bax using lysates from MDA-MB-231, MCF-7, and SUM159 cells after 6-, 12- or 24-hour treatment with ethanol (control) or the indicated doses of LLM. Numbers above band represent changes in protein levels relative to corresponding control. B Representative microscopic images (100× objective magnification; scale bar = 30 μm) depicting active-Bak and active-Bax in MDA-MB-231, MCF-7, SUM159, and MCF-10A cells after 24-hour treatment with ethanol (control) or 5 μM of LLM. C Quantitation of apoptotic cells (Annexin V-FITC/PI method) normalized to respective control in WT/MEF and DKO/MEF. The cells were treated for 24 hours with ethanol (control) or the indicated concentrations of LLM and processed for flow cytometry. D Percentage of cell viability relative to respective control in WT/MEF and DKO/MEF cells (24-hour treatment). Results shown are mean ± SD (n = 3). Statistically significant (P < 0.05) compared with a respective control and b between WT/MEF and DKO/MEF by one-way ANOVA followed by Newman-Keuls multiple comparisons test. E Percentage of apoptotic fraction (Annexin V-FITC/PI method) in MDA-MB-231 cells stably transfected with empty vector (EV) or ERα plasmid. The inset shows western blot for ERα. The cells were treated for 24 hours with ethanol (control) or 5 μM of LLM and processed for flow cytometry. Results shown are mean ± SD (n = 3). Statistically significant (P < 0.05) compared with a respective control and b between EV and ERα overexpressing cells by one-way ANOVA followed by Newman-Keuls multiple comparisons test. Each experiment was done at least twice and representative data from one such experiment are shown.
LLM Treatment Activated Intrinsic Caspase Pathway
We investigated the role of intrinsic (Caspase 9) and extrinsic (Caspase 8) caspase pathways in cell death induction by LLM. Caspase 9 was activated upon LLM treatment in all three breast cancer cell lines (Fig. 6A). On the other hand, Caspase 8 activation was observed only in SUM159 after 24-hour treatment with 5 μM LLM (data not shown). In vehicle-treated control MitoGFP-MCF-7 cells, cytochrome c was mainly localized in mitochondria as reflected by yellow-orange color due to merging of the red (cytochrome c) and green (GFP) fluorescence. The mitochondrial yellow-orange signal was greatly diminished in a majority of LLM-treated cells (Fig. 6B). Collectively, these results suggested that activation of intrinsic caspase pathway might be the major route of LLM-mediated apoptosis in breast cancer cells.
Fig. 6.

LLM treatment induces Caspase 9 activation and cytochrome c release into the cytosol. A Quantitation of Caspase 9 activation in control and LLM-treated cells. The cells were treated with ethanol (control) or the indicated doses of LLM for 6 or 24 hours and processed for flow cytometry. Results shown are mean ± SD (n = 3). *Significantly different (P < 0.05) compared with control by one-way ANOVA with Dunnett’s adjustment. B Representative microscopic images (100× objective magnification; scale bar = 12.5 μm) depicting cytosolic release of cytochrome c in MitoGFP-MCF-7 cells after 24-hour treatment with ethanol (control) or 5 μM of LLM. Note yellow-orange color in control cells indicating mitochondrial localization of cytochrome c that was decreased in LLM-treated cells. Each experiment was done at least twice and representative data from one such experiment are shown.
In Vivo Effect of LLM Administration on SUM159 Xenograft Growth
Next, we determined the effect of intraperitoneal LLM administration on SUM159 xenograft growth using female nude mice. The body weights of the control and LLM-treated mice were comparable (Fig. 7A). In addition, the average weights of vital organs (spleen, heart, kidney, liver, and lungs) did not differ significantly between control and LLM treatment groups (results not shown). Even though tumor cells were injected into the mammary fat pad, tumor growth also occurred in the peritoneal cavity (abdomen) likely due to more than desired penetration of the injection needle. Because of a large size difference between orthotopic and abdominal tumors, the results were computed as total wet tumor weight and orthotopic tumor weight. The total wet tumor weight in the LLM treatment group was lower by 46% compared with that of control but the difference was not significant (Fig. 7B). However, 3 of the 5 mice of the control group had tumors >1g as opposed to only 1 mouse of the LLM treatment with tumor weight exceeding 1g. The average wet weight of the orthotopic tumors was lower by 69% in the LLM-treated mice compared with control (P = 0.04 (Fig. 7C). Representative H&E-stained sections from orthotopic tumors of control and LLM treatment groups are shown in Fig. 7D. These results indicated that the LLM regimen used in the present study was safe and effective in reducing the growth of at least orthotopic SUM159 xenografts.
Fig. 7.

LLM administration retards orthotopic tumor growth of SUM159 xenograft. A Average body weights over time for mice of control and LLM treatment groups. The results shown are mean ± SD [n = 5 for both groups except at week 7 where n = 3 for control group because two mice from this group were sacrificed one week prior to study termination due to morbidity]. Initial body weight of control group of mice was significantly lower than that of LLM-treated mice by unpaired Student’s t test (P < 0.05). B Total wet tumor weight. Results shown are mean ± SD. C Orthotopic mammary tumor burden in control and LLM-treated mice. Results shown are mean ± SD. * Statistically significant by unpaired Student’s t test. D Representative H&E-stained sections from control and LLM treatment groups (200× magnification; scale bar = 100 μm).
DISCUSSION
The present study achieves critical milestones in the overall process of developing LLM as a preventive or therapeutic intervention against breast cancer. First, LLM exhibits remarkable selectivity towards cancer cells as a normal mammary epithelial cell line is resistant to growth inhibition and apoptosis induction by this agent. Second, LLM is highly effective in suppressing self-renewal of bCSC. Third, we demonstrate that LLM is safe and inhibits growth of orthotopic SUM159 xenografts in immune compromised mice. Finally, inhibitory effect of LLM on proliferating cancer cells and bCSC is not influenced by the p53 status as these effects are observed in both MCF-7 (wild-type p53) and basal-like cells (mutant p53). On the other hand, cell death induction by LLM is partly attenuated by overexpression of ERα. It would be important to determine if LLM suppresses ERα expression. Together, these findings suggest that LLM may be effective against different subtypes of breast cancer warranting further preclinical investigations of this agent. For example, we show in vivo efficacy of the agent against SUM159 cells but its activity against MCF-7 and MDA-MB-231 cells is yet to be determined.
Evidence continues to accumulate to indicate a crucial role for bCSC in mammary cancer development and progression [11-13]. LLM is a potent inhibitor of bCSC as evidenced by a decrease in mammosphere frequency, ALDH1 activity, and CD49f+/CD24− fraction. LLM-mediated inhibition of bCSC is evident in both MCF-7 and SUM159 cells with nearly equal efficacy. It is also interesting to note that bCSC inhibition occurs at concentrations that are not cytotoxic to cancer cells. It is reasonable to propose that bCSC inhibition by LLM may contribute to its anticancer effect.
In vitro inhibition of breast cancer cell growth and bCSC self-renewal by LLM is evident at pharmacologically-relevant doses. Peak-plasma concentration of LLM in mice is about 2 μM after oral administration at 10 mg/kg body weight [36]. Growth of proliferating breast cancer cells and self-renewal of bCSC by LLM is evident 0.5-2.5 μM concentrations. The in vivo growth of orthotopic SUM159 xenografts is decreased following intraperitoneal treatment with 7.5 mg LLM/kg body weight without any overt toxicity. This dose represents 30% of the maximally-tolerated dose of LLM by intraperitoneal route [19].
Mechanistic understanding of LLM-induced apoptosis is limited with only a few publications addressing this question using melanoma cell lines [19, 37]. LLM-mediated apoptotic cell death induction in melanoma cell lines in vitro and in vivo was associated with inhibition of Akt, Stat3, and Erk1/2 activation (reduced phosphorylation). In vitro inhibition of these prosurvival and oncogenic pathways was evident as early as 3 hours after LLM treatment at 3-5 μM concentrations. However, the precise role and contribution of these pathways in cell death induction by LLM is still unclear [19]. In a follow-up study, the same group of investigators reported inhibition of cholesterol transport for killing of melanoma cells by LLM [37]. The present study reveals a critical role for Bax and Bak in LLM-mediated apoptosis. LLM causes induction and/or activation of Bax and Bak in human breast cancer cells but not in the MCF-10A cell line. Our data are in agreement with previous studies showing partial resistance of Bax−/−HCT116 human colon cancer cells to cell viability inhibition by LLM in comparison with WT HCT116 cells [37]; partial resistance in Bax−/−cells is not surprising due to the presence of Bak. The present study also reveals that ROS generation only partially accounts for apoptotic cell death by LLM. It is possible that LLM-mediated inhibition of prosurvival pathways like Akt, Stat3 and/or Erk1/2 may be responsible for the ROS-independent component of the cell death induction. Further research is needed to explore this possibility.
In conclusion, the present study provides in vitro and in vivo evidence for anticancer effect of LLM in a panel of cell lines differing in ERα and p53 status. The anticancer effect of LLM is mediated by ROS generation and Bax/Bak-dependent apoptosis regulated by the intrinsic caspase pathway. Inhibition of bCSC by LLM likely contributes to its anticancer effect. However, additional work is necessary for clinical translation of our findings. First, the in vivo efficacy of LLM for growth inhibition of breast cancer cell lines other than SUM159 is yet to be determined. Second, it is important to determine the oral bioavailability of the agent. Third, the pharmacokinetics of LLM in humans is not yet known.
ACKNOWLEDGMENTS
This work was supported by the grant RO1 CA142604 and RO1 CA129347 awarded by the National Cancer Institute (S.V. Singh). This research used the Animal Facility and Flow Cytometry Facility supported in part by a grant from the National Cancer Institute at the National Institutes of Health (P30 CA047904).
Abbreviations
- ALDH1
aldehyde dehydrogenase 1
- ANOVA
analysis of variance
- BAAA
BODIPY™-aminoacetaldehyde
- DEAB
diethylaminobenzaldehyde
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- PBS
phosphate-buffered saline
- bCSC
breast cancer stem-like cells
- LLM
leelamine
- PI
propidium iodide
Footnotes
Conflict of Interest: The authors declare that they have no conflict of interest.
REFERENCES
- 1.Kelsey JL, Gammon MD, John EM. Reproductive factors and breast cancer. Epidemiol Rev. 1993;15:36–47. doi: 10.1093/oxfordjournals.epirev.a036115. [DOI] [PubMed] [Google Scholar]
- 2.Hulka BS, Stark AT. Breast cancer: cause and prevention. Lancet. 1995;346:883–887. doi: 10.1016/s0140-6736(95)92713-1. [DOI] [PubMed] [Google Scholar]
- 3.Vargo-Gogola Gogola, Rosen JM. Modelling breast cancer: one size does not fit all. Nat Rev Cancer. 2007;7:659–672. doi: 10.1038/nrc2193. [DOI] [PubMed] [Google Scholar]
- 4.Van Zitteren M, van der Net JB, Kundu S, Freedman AN, van Duijn CM, Janssens AC. Genome-based prediction of breast cancer risk in the general population: a modeling study based on meta-analyses of genetic associations. Cancer Epidemiol Biomarkers Prev. 2011;20:9–22. doi: 10.1158/1055-9965.EPI-10-0329. [DOI] [PubMed] [Google Scholar]
- 5.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65:5–29. doi: 10.3322/caac.21254. [DOI] [PubMed] [Google Scholar]
- 6.Perou CM, Sørlie T, Eisen MB, et al. Molecular portraits of human breast tumours. Nature. 2000;406:747–752. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- 7.Sørlie T, Perou CM, Tibshirani R, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci USA. 2001;98:10869–10874. doi: 10.1073/pnas.191367098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sørlie T, Tibshirani R, Parker J, et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc Natl Acad Sci USA. 2003;100:8418–8423. doi: 10.1073/pnas.0932692100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hu Z, Fan C, Oh DS, et al. The molecular portraits of breast tumors are conserved across microarray platforms. BMC Genomics. 2006;71:96. doi: 10.1186/1471-2164-7-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cleator S, Heller W, Coombes RC. Triple-negative breast cancer: therapeutic options. Lancet Oncol. 2007;8:235–244. doi: 10.1016/S1470-2045(07)70074-8. [DOI] [PubMed] [Google Scholar]
- 11.Al-Hajj Hajj, Wicha MS, Benito-Hernandez Hernandez, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA. 2003;100:3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Velasco-Velázquez MA, Popov VM, Lisanti MP, Pestell RG. The role of breast cancer stem cells in metastasis and therapeutic implications. Am J Pathol. 2011;179:2–11. doi: 10.1016/j.ajpath.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McDermott SP, Wicha MS. Targeting breast cancer stem cells. Mol Oncol. 2010;4:404–419. doi: 10.1016/j.molonc.2010.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Surh YJ. Cancer chemoprevention with dietary phytochemicals. Nat Rev Cancer. 2003;3:768–780. doi: 10.1038/nrc1189. [DOI] [PubMed] [Google Scholar]
- 15.Singh SV, Singh K. Cancer chemoprevention with dietary isothiocyanates mature for clinical translational research. Carcinogenesis. 2012;33:1833–1842. doi: 10.1093/carcin/bgs216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stan SD, Kar S, Stoner GD, Singh SV. Bioactive food components and cancer risk reduction. J Cell Biochem. 2008;104:339–356. doi: 10.1002/jcb.21623. [DOI] [PubMed] [Google Scholar]
- 17.Warin R, Chambers WH, Potter DM, Singh SV. Prevention of mammary carcinogenesis in MMTV-neu mice by cruciferous vegetable constituent benzyl isothiocyanate. Cancer Res. 2009;69:9473–9480. doi: 10.1158/0008-5472.CAN-09-2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Singh SV, Kim SH, Sehrawat A, et al. Biomarkers of phenethyl isothiocyanate-mediated mammary cancer chemoprevention in a clinically relevant mouse model. J Natl Cancer Inst. 2012;104:1228–1239. doi: 10.1093/jnci/djs321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gowda R, Madhunapantula SV, Kuzu OF, Sharma A, Robertson GP. Targeting multiple key signaling pathways in melanoma using leelamine. Mol Cancer Ther. 2014;13:1679–1689. doi: 10.1158/1535-7163.MCT-13-0867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gowda R, Madhunapantula SV, Sharma A, Kuzu OF, Robertson GP. Nanolipolee-007, a novel nanoparticle-based drug containing leelamine for the treatment of melanoma. Mol Cancer Ther. 2014;13:2328–2340. doi: 10.1158/1535-7163.MCT-14-0357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lin LY, Bao YL, Chen Y, et al. N-Benzoyl-12-nitrodehydroabietylamine-7-one, a novel dehydroabietylamine derivative, induces apoptosis and inhibits proliferation in HepG2 cells. Chem Biol Interact. 2012;199:63–73. doi: 10.1016/j.cbi.2012.06.002. [DOI] [PubMed] [Google Scholar]
- 22.Li F, He L, Song ZQ, Yao JC, Rao XP, Li HT. Cytotoxic effects and pro-apoptotic mechanism of TBIDOM, a novel dehydroabietylamine derivative, on human hepatocellular carcinoma SMMC-7721 cells. J Pharm Pharmacol. 2008;60:205–211. doi: 10.1211/jpp.60.2.0009. [DOI] [PubMed] [Google Scholar]
- 23.Ling T, Tran M, González MA, et al. (+)-Dehydroabietylamine derivatives target triple-negative breast cancer. Eur J Med Chem. 2015;102:9–13. doi: 10.1016/j.ejmech.2015.07.034. [DOI] [PubMed] [Google Scholar]
- 24.Xiao D, Vogel V, Singh SV. Benzyl isothiocyanate-induced apoptosis in human breast cancer cells is initiated by reactive oxygen species and regulated by Bax and Bak. Mol Cancer Ther. 2006;5:2931–2945. doi: 10.1158/1535-7163.MCT-06-0396. [DOI] [PubMed] [Google Scholar]
- 25.Xiao D, Powolny AA, Singh SV. Benzyl isothiocyanate targets mitochondrial respiratory chain to trigger reactive oxygen species-dependent apoptosis in human breast cancer cells. J Biol Chem. 2008;283:30151–30163. doi: 10.1074/jbc.M802529200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chandra-Kuntal Kuntal, Lee J, Singh SV. Critical role for reactive oxygen species in apoptosis induction and cell migration inhibition by diallyl trisulfide, a cancer chemopreventive component of garlic. Breast Cancer Res Treat. 2013;138:69–79. doi: 10.1007/s10549-013-2440-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Choi S, Singh SV. Bax and Bak are required for apoptosis induction by sulforaphane, a cruciferous vegetable-derived cancer chemopreventive agent. Cancer Res. 2005;65:2035–2043. doi: 10.1158/0008-5472.CAN-04-3616. [DOI] [PubMed] [Google Scholar]
- 28.Hahm ER, Lee J, Huang Y, Singh SV. Withaferin A suppresses estrogen receptor-α expression in human breast cancer cells. Mol Carcinog. 2011;50:614–624. doi: 10.1002/mc.20760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xiao D, Choi S, Johnson DE, et al. Diallyl trisulfide-induced apoptosis in human prostate cancer cells involves c-Jun N-terminal kinase and extracellular-signal regulated kinase-mediated phosphorylation of Bcl-2. Oncogene. 2004;23:5594–5606. doi: 10.1038/sj.onc.1207747. [DOI] [PubMed] [Google Scholar]
- 30.Kim SH, Sehrawat A, Singh SV. Dietary chemopreventative benzyl isothiocyanate inhibits breast cancer stem cells in vitroand in vivo. Cancer Prev Res (Phila.) 2013;6:782–790. doi: 10.1158/1940-6207.CAPR-13-0100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hahm ER, Moura MB, Kelley EE, Van Houten B, Shiva S, Singh SV. Withaferin A-induced apoptosis in human breast cancer cells is mediated by reactive oxygen species. PLoS One. 2011;6:e23354. doi: 10.1371/journal.pone.0023354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xiao D, Srivastava SK, Lew KL, et al. Allyl isothiocyanate, a constituent of cruciferous vegetables, inhibits proliferation of human prostate cancer cells by causing G2/M arrest and inducing apoptosis. Carcinogenesis. 2003;24:891–897. doi: 10.1093/carcin/bgg023. [DOI] [PubMed] [Google Scholar]
- 33.Antosiewicz J, Ziolkowski W, Kar S, Powolny AA, Singh SV. Role of reactive oxygen intermediates in cellular responses to dietary cancer chemopreventive agents. Planta Med. 2008;74:1570–1579. doi: 10.1055/s-2008-1081307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Buchet K, Godinot C. Functional F1-ATPase essential in maintaining growth and membrane potential of human mitochondrial DNA-depleted ρ° cells. J Biol Chem. 1998;273:22983–22989. doi: 10.1074/jbc.273.36.22983. [DOI] [PubMed] [Google Scholar]
- 35.Renault TT, Chipuk JE. Death upon a kiss: Mitochondrial outer membrane composition and organelle communication govern sensitivity to BAK/BAX-dependent apoptosis. Chem Biol. 2014;21:114–123. doi: 10.1016/j.chembiol.2013.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Song M, Lee D, Lee T, Lee S. Determination of leelamine in mouse plasma by LC-MS/MS and its pharmacokinetics. J Chromatography B. 2013;931:170–173. doi: 10.1016/j.jchromb.2013.05.018. [DOI] [PubMed] [Google Scholar]
- 37.Kuzu OF, Gowda R, Sharma A, Robertson GP. Leelamine mediates cancer cell death through inhibition of intracellular cholesterol transport. Mol Cancer Ther. 2014;13:1690–1703. doi: 10.1158/1535-7163.MCT-13-0868. [DOI] [PMC free article] [PubMed] [Google Scholar]