

Abstract
Background
Based on their distinctive histologic and genetic features, the latest WHO classification of soft tissue tumors includes four pathologic variants of rhabdomyosarcoma (RMS): embryonal (ERMS), alveolar (ARMS), spindle cell-sclerosing (SRMS-ScRMS) and pleomorphic RMS. The aim of this study focused on a detailed clinicopathologic and survival analysis of head and neck RMS (HNRMS) using the latest pathologic and molecular criteria reflecting this new subclassification in a large cohort.
Patients and Methods
Patients managed for HNRMS in our institution (1996 - 2015) were analyzed. The presence of a FOXO1 fusion was required for the classification of ARMS. MYOD1 mutations in SRMS-ScRMS were tested when material available. Univariate and multivariate analyses were performed to evaluate variables related to overall survival (OS).
Results
Ninety-nine HNRMS patients (52 males and 47 females, mean of 16 years) were included in the study after pathologic re-review. The most common location was parameningeal (PM) (n=64), followed by non-orbital/non-PM (n=25) and orbital (n=10). There were 53 ERMS, 33 fusion-positive ARMS and 13 SRMS-ScRMS [SRMS (8); ScRMS (5)]. The 5-year OS rate for ERMS patients was significantly higher (82%) compared to ARMS (53%) and SRMS-ScRMS (50%) [SRMS (75%); ScRMS (30%)]. Univariate analysis showed that survival was dependent on histology (P=0.012), tumor size > 5 cm (P<0.001), regional lymph node involvement (P=0.002), metastasis at initial presentation (P<0.001), stage (P<0.001), and recurrence (P=0.002). Multivariate analysis confirmed histologic subtype to be significant (P=0.043).
Conclusion
Our findings reinforce that HNRMS is a heterogenous disease with ARMS and SRMS-ScRMS having an equally unfavorable outcome.
Keywords: Spindle cell rhabdomyosarcoma, Sclerosing rhabdomyosarcoma, Alveolar rhabdomyosarcoma, Embryonal rhabdomyosarcoma, MYOD1 mutations, PAX3/7-FOXO1 fusion
INTRODUCTION
Head and neck soft tissue sarcomas are rare, accounting for 1% of all head and neck neoplasms [1, 2]. Rhabdomyosarcomas are the most common soft tissue sarcomas in children and adolescents, accounting for 5-8% of all childhood malignancies [3]. The head and neck is the most common anatomic site for rhabdomyosarcoma [4, 5]. The incidence of head and neck rhabdomyosarcoma (HNRMS) is currently placed at 0.104 cases per 100,000 [6]. The clinical presentation of HNRMS is divided into 3 sub-sites based on its anatomic location and local relapse: parameningeal (PM), including the paranasal sinuses, nasopharynx, nasal cavity, middle ear, mastoid, parapharyngeal region, pterygopalatine and infratemporal fossa; orbital; and non-PM / non-orbital site, encompassing the neck, face, oral cavity, cheek, external ear, scalp and larynx [7]. PM subsite is the most common presentation and is associated with the least favorable outcome compared to other locations [4-6, 8].
Recently, the WHO classification divided RMS into 4 clinicopathologic variants: embryonal (ERMS), alveolar (ARMS), spindle cell-sclerosing (SRMS-ScRMS) and pleomorphic RMS [9]. ERMS is the most common variant, being associated with the most favorable outcome compared to other variants [6, 8]. ERMS occurs in younger patients and shows a morphologic resemblance to fetal skeletal muscle. Although no genetic abnormality prevails, small subsets of ERMS harbor LOH at 11p15, as well as FGFR4, P53, BCOR, ARID1A and RAS mutations, as shown in recent large genomic studies [10-13]. ARMS is the second most common variant, being associated with a poor prognosis [6, 8]. ARMS has a predilection for older children and young adults, having a histologic appearance of undifferentiated small blue round cells, arranged in a variable alveolar or solid pattern. The genetic hallmark of ARMS is either the more common t(2;13)(q35;q14) translocation, or the t(1;13)(q36;q14), resulting in the PAX3-FOXO1 or PAX7-FOXO1 fusion, respectively [14-16]. SRMS-ScRMS represent a rare and recently recognized stand-alone pathologic entity, separated from the broad umbrella of ERMS. Spindle cell RMS (SRMS) is composed of monomorphic spindle cells arranged in intersecting fascicles, lacking overt rhabdomyoblastic differentiation. A subset of SRMS display areas of hyaline sclerosis suggesting a morphologic overlap with the even less common sclerosing RMS (ScRMS) [17, 18]. ScRMS may show in addition an undifferentiated round cell component arranged in a pseudovascular or pseudoalveolar pattern in a prominent hyalinized stroma [19, 20]. SRMS-ScRMS share genetic alterations, although these vary depending on the clinical presentation: recurrent NCOA2 and VGLL2 related fusions in congenital/infantile setting, which are associated with a favorable outcome [21, 22], or MYOD1-mutations in older children and adults are associated with a poor prognosis [23-26], Pleomorphic RMS is a rare variant, usually diagnosed in patients over the age of 45 years, with a morphologic appearance of large pleomorphic cells [9, 27].
Although most RMS have a sporadic presentation, in a small subset of patients, RMS is part of a genetic syndrome, such as Beckwith-Wiedemann, Von Recklinghausen disease, Costello, Noonan, Gorlin, Rubinstein-Taybi and Li-Fraumeni syndromes [28-38]. The management of HNRMS remains challenging due to an increased failure of local control as well as a high rate of early metastases [6, 39]. In the last three decades, RMS outcomes have improved significantly due to evolving multidisciplinary therapy paradigms, such as improved surgical techniques, aggressive chemotherapy regimen and more effective conformal intensity-modulated radiotherapy [40, 41]. The 5-year overall survival rate of childhood RMS has risen to 62% and the 5-year relative survival rate of HNRMS is estimated at 63% [6, 42]. The prognostic factors in HNRMS have been multifactorial, such as age, tumor site, overall stage, distant metastases, histologic variant, nodal status and status of primary surgical site [6, 8, 43-46]. The aim of this study focused on a detailed clinicopathologic analysis, assessing the oncologic outcome and prognostic factors of HNRMS patients managed at our institution.
PATIENTS AND METHODS
The study was approved by the Institutional Review Board of Memorial Sloan Kettering Cancer Center (MSKCC), New York. The electronic records of the Department of Pathology at MSKCC were searched for RMS cases presenting in the head and neck anatomic location from 1996 to 2015. All cases were re-reviewed and reclassified based on the 2013 WHO classification. The following clinical data was recorded: age at diagnosis, gender, anatomic site, overall stage, presence of distant metastasis, regional lymph node involvement, immunohistochemical results, molecular results (presence of PAX3/7-FOXO1 fusion or rearrangement), modality of therapy (chemotherapy, radiation therapy, and/or surgical resection), outcome, recurrence, and survival time. The pre-treatment staging was based on the Intergroup Rhabdomyosarcoma Study Group (IRS) staging system. Only FOXO1-fusion positive ARMS cases were included in this study. If the molecular diagnosis was not previously done, attempts were made when tissue available to perform FISH studies for the presence of FOXO1 gene rearrangements, as previously reported [21]. Cases with no available molecular information or negative molecular results were excluded. The clinical features and molecular findings (NCOA2 fusion or MYOD1 mutation) of SRMS-ScRMS cases included in this study have been recently reported by Owosho et al.[26].
The staging work-up for evaluating metastatic disease included: bone marrow biopsy, imaging studies such as CT scan, MRI, PET scan and whole body technetium bone scan. The institution's guideline for the treatment of rhabdomyosarcoma includes multiagent induction chemotherapy, such as vincristine, doxorubicin, and cyclophosphamide followed, by ifosfamide and etoposide. In few patients, surgical removal of the entire tumor was achievable and performed after chemotherapy. Typically, radiation therapy is delivered months after chemotherapy, in the form of intensity-modulated radiation therapy/proton beam radiation therapy [39, 47]. Patients with intermediate and high risk RMS are offered additional chemotherapy, such as irinotecan and carboplatin [48].
Statistical analysis
Statistical analysis was performed on an SPSS platform (version 20.0; IBM Corp., Armonk, NY). The associations between the clinical characteristics and the histologic variants were evaluated by ANOVA, Kruskal-Wallis test and Fisher's exact test according to the variable type. The overall survival was measured from the date of diagnosis to the date of death. Kaplan-Meier estimate was used to calculate the overall survival, and the statistical significance of different variables was examined by the log-rank test. The clinicopathologic variables with p<0.05 in the log-rank analysis were subjected to the Cox proportional hazard regression model for determining the independent prognostic factors. Two-sided p values were calculated and, p<0.05 was considered to be significant for all statistical analyses.
RESULTS
Clinicopathologic analysis
The study comprised 99 HNRMS patients managed at a single tertiary center (MSKCC) spanning two decades, between 1996 and 2015. A summary of the clinical characteristics of HNRMS patients is included in Table 1. There were 52 (52%) males and 47 (48%) females, with ages ranging from 1 month to 72 years old, with a mean of 16 years. Almost two-thirds (63%) of patients were of pediatric age (<18 years), with 40 (40%) being younger than age of 10. The most common anatomic location was the parameningeal (PM) site, accounting for 64 (65%) patients, followed by the non-orbital, non-PM subsite, present in 25 (25%) patients. The orbital subsite was the least common location noted in 10 (10%) cases. Imaging demonstrated large lesions associated with mass-effect, erosion and destruction of bony structures (Figures 1). Histopathologically, 3 variants were represented with ERMS > ARMS > SRMS-ScRMS, in that order of frequency (Table 1). There were too few pleomorphic RMS cases identified to include as a separate group for statistical analysis. By immunohistochemistry (IHC), all cases were positive for desmin (typically diffuse and strong) and myogenin. Myogenin was focal in ERMS and SRMS-ScRMS, but was diffuse in ARMS. SRMS-ScRMS typically showed diffuse MyoD1 nuclear expression. However, aberrant IHC staining for CD99, FLI1, CD56, Cytokeratin, epithelial membrane antigen (EMA), WT1 or S100 were observed in 26 cases, typically with a focal and weak pattern. Regional lymph nodes were involved in 40/97 (41%). Twenty-nine (29%) patients were stage I, 10 (10%) patients stage II, 41 (41%) patients stage III and 19 (19%) patients stage IV. All except two HNRMS patients had follow-up information.
Table 1.
Clinical characteristics of head and neck rhabdomyosarcoma patients
| Clinical Characteristics | (%) | Total number | ||
|---|---|---|---|---|
| Gender | 99 (100%) | |||
| Male | 52 | 52.5 | ||
| Female | 47 | 47.5 | ||
| Age (years) | 99 (100%) | |||
| Mean | 16 | |||
| Median | 12 | |||
| Max. | 72 | |||
| Min. | 0.08 | |||
| Age group | 99 (100%) | |||
| Adult | 37 | 37.4 | ||
| Pediatric | 62 | 62.6 | ||
| Tumor site | 99 (100%) | |||
| PM | 64 | 64.6 | ||
| S | 25 | 25.3 | ||
| O | 10 | 10.1 | ||
| Histopathology | 99 (100%) | |||
| ERMS | 33 | 33.3 | ||
| ARMS | 53 | 53.5 | ||
| SRMS-ScRMS | 13 | 13.1 | ||
| Tumor size | 68 (69%) | |||
| Mean | 4.3 | |||
| Median | 4.0 | |||
| Max. | 12 | |||
| Min. | 0.8 | |||
| Regional lymph node | 97 (98%) | |||
| Positive | 40 | 40.8 | ||
| Negative | 57 | 58.2 | ||
| Initial metastasis | 99 (100%) | |||
| Positive | 19 | 19.2 | ||
| Negative | 80 | 80.8 | ||
| Stage | 99 (100%) | |||
| 1 | 29 | 29.3 | ||
| 2 | 10 | 10.1 | ||
| 3 | 41 | 41.4 | ||
| 4 | 19 | 19.2 | ||
| Recurrence | 97 (98%) | |||
| Local | 9 | 9.3 | ||
| Distant | 8 | 8.2 | ||
| Local & distant | 5 | 5.2 | ||
| None | 75 | 77.3 | ||
| Vital status | 97 (98%) | |||
| Alive | 69 | 71.1 | ||
| Deceased | 28 | 28.9 | ||
| Follow-up duration (months) | 97 (98%) | |||
| Mean | 83.6 | |||
| Median | 65 | |||
| Max. | 232 | |||
| Min. | 4 | |||
Figure 1. MRI findings of HNRMS based on anatomic location and histology.
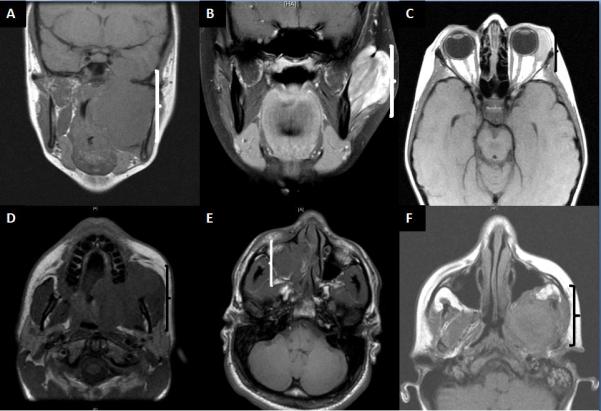
Images illustrate large soft tissue mass associated with mass effect, erosion or destruction of bony structures. (A) Infratemporal fossa region ScRMS (parameningeal site) occuring in a 15-year-old female; deceased at 26 months; (B) Buccal mucosa ScRMS negative for MYOD1 mutation (non-parameningeal/ non-orbital site) in a 17-year-old female; alive at 31 months; (C) Orbital ERMS arising in a 7-year-old female; alive at 95 months; (D) ERMS occuring in the soft palate of a 20-year-old male; alive at 132 months. (E) Paranasal ARMS airsing in a 21-year-old male; alive at 103 months; (F) ScRMS of the infratemporal fossa with MYOD1 mutation, in a 15-year-old female; deceased at 26 months.
ERMS is the most common subtype, presenting in young children with a low to intermediate stage at diagnosis
A summary of the characteristics of ERMS patients is presented in Table 2. There were 53 (53%) patients with ERMS, showing an equal gender distribution and an age at presentation ranging from 4 months - 38 years, with a mean of 8.7 years. Among them, 46 (87%) of them were of pediatric age, with 37 being younger than age of 10. Most of the ERMS were located in PM subsite (n=32, 62%), followed by non-orbital/ non-PM site (11) and orbital site (9). Sixteen patients were stage I, 8 patients staged II, 23 patients staged III and 6 patients staged IV. Six patients presented with metastases at diagnosis, to the bone (n = 3) and lung (n = 4). One patient presented with multiple metastatic sites involving both lung and bone. All patients had follow-up information.
Table 2.
Statistical Correlation of Clinical Variables to Histologic Subtypes in HNRMS
| Clinical variables | Histologic subtype |
P value (Fisher's exact)* | ||||||
|---|---|---|---|---|---|---|---|---|
| ARMS (n = 33) | ERMS (n = 53) | SRMS-ScRMS (n = 13) | ||||||
| Gender | 0.541 | |||||||
| Male | 19 | 58% | 25 | 47% | 8 | 61% | ||
| Female | 14 | 42% | 28 | 53% | 5 | 39% | ||
| Age (years) | Mean | 23 | 8.7 | 29 | <0.001 (ANOVA) | |||
| Median | 23 | 6 | 28 | <0.001 Kruskal-Wallis | ||||
| Max. | 57 | 38 | 72 | |||||
| Min. | 0.08 | 0.3 | 0.7 | |||||
| Age group | <0.001 | |||||||
| Adult | 22 | 67% | 7 | 13% | 8 | 61% | ||
| Pediatric | 11 | 33% | 46 | 87% | 5 | 39% | ||
| Tumor site | <0.001 | |||||||
| PM | 28 | 85% | 33 | 62% | 3 | 23% | ||
| S | 4 | 12% | 11 | 21% | 10 | 77% | ||
| O | 1 | 3% | 9 | 17% | 0 | 0% | ||
| Tumor size | Mean | 4 | 4 | 5 | 0.344 (ANOVA) | |||
| Median | 4 | 4 | 5 | 0.517 Kruskal-Wallis | ||||
| Max. | 9 | 12 | 10 | |||||
| Min. | 1 | 1.3 | 0.8 | |||||
| Regional lymph node | <0.001 | |||||||
| Positive | 24 | 72% | 14 | 27% | 2 | 17% | ||
| Negative | 9 | 28% | 38 | 73% | 10 | 83% | ||
| Initial metastasis | 0.078 | |||||||
| Positive | 10 | 30% | 6 | 11% | 3 | 23% | ||
| Negative | 23 | 70% | 47 | 89% | 10 | 77% | ||
| Stage | 0.001 | |||||||
| 1 | 4 | 12% | 16 | 30% | 9 | 69% | ||
| 2 | 2 | 6% | 8 | 15% | 0 | 0% | ||
| 3 | 17 | 52% | 23 | 44% | 1 | 8% | ||
| 4 | 10 | 30% | 6 | 11% | 3 | 23% | ||
| Recurrence | 0.001 | |||||||
| LR | 6 | 18% | 3 | 6% | 2 | 18% | ||
| DR, (LR & DR) | 8 | 24% | 1 | 2% | 2 | 18% | ||
| NR | 19 | 58% | 49 | 92% | 7 | 64% | ||
| Vital status | 0.05 | |||||||
| Alive | 20 | 61% | 43 | 81% | 6 | 54% | ||
| Deceased | 13 | 39% | 10 | 19% | 5 | 46% | ||
| Follow-up duration (months) | Mean | 63 | 106 | 35 | <0.001 (ANOVA) | |||
| Median | 32 | 104 | 29 | <0.001 Kruskal-Wallis | ||||
| Max. | 232 | 232 | 94 | |||||
| Min. | 4 | 7 | 4 | |||||
PM – Parameningeal, S – Non parameningeal non orbital, O – Orbital, LR – Local recurrence, DR – Distant recurrence, NR – No recurrence
P values calculated with Fisher's exact if not otherwise specified
Most ARMS occur at parameningeal sites with an advanced stage at diagnosis
The characteristics of ARMS patients are summarized in Table 2. There were 33 (33%) ARMS patients, with age at presentation ranging from birth - 57 years, with a mean of 23 years. Two-thirds of cases (n=22) were adult patients, with a male: female ratio of 19:14. The overwhelming majority of cases (n=28, 85%) involved the PM site, while only a minority occurred in the non-orbital/ non-PM site (4) and orbital site (1). Most ARMS patients (n=27, 82%) were classified as intermediate (17 patients stage III) to high stage (10 patients stage IV), with only 4 patients being stage I and 2 patients stage II. Ten patients presented with metastases to the bone (n =10), and one each to the lung and pancreas. One patient had metastases to bone and pancreas, and another patient had metastases to both bone and lung. All 33 cases of ARMS included in this study were confirmed to harbor either a PAX3/7-FOXO1 fusion by RT-PCR or FOXO1 gene rearrangement by FISH. Follow-up information was available in all patients.
MYOD1 mutations occur preferentially in ScRMS
The clinical characteristics of SRMS-ScRMS patients are presented in Table 2. There were 13 (13%) patients with ages ranging 0.67 – 72, mean of 29 years. Most of the patients were adults (n=8, 62%). The male:female was 8:5. Ten (77%) cases involved non-orbital/ non-PM site and PM site (n=3). Nine patients were stage I, one patient stage III and three patients stage IV. Three patients had metastases: bones (n=2) and lung (n=1). Eight cases were classified as SRMS and five cases classified as ScRMS. Six cases were positive for SMA (n=5). Three of the 9 cases tested for MYOD1 mutation were positive, all showing ScRMS subtype morphology. The only infant patient was an 8 month-old baby-girl with a neck mass, which was positive for SRF-NCOA2 fusion, and remained NED at 60 months follow-up. Eleven of thirteen patients had follow-up information.
ARMS and SRMS-ScRMS have an equally unfavorable outcome
The mean survival duration for HNRMS in this study was 164 months (95% CI, 143-185). The 5-year overall survival (OS) rate for the entire cohort of HNRMS patients was 70% (Figure 2A), while the 5-year OS rate related to histologic type was 82% for ERMS, 53% for ARMS and 50% for SRMS-ScRMS (Figure 2B). The 5-year OS rate for patients with localized HNRMS was 77%, while the 5-year OS rate for localized ERMS was 84%, and for both localized ARMS and SRMS-ScRMS was 64%. Based on age, the 5-year OS rate for children with HNRMS pediatric was 71% while for adults 67%. If divided by ≤10 years of age, the 5-year OS was 73% versus 67% for patients older than 10 years. In the pediatric cohort, the 5-year OS rate divided by histology was 81% for ERMS, 36% for ARMS and 67% for SRMS-ScRMS, while, in the adult cohort it was 86%, 66%, and 44%, respectively.
Figure 2. Survival of HNRMS patients based on histology, tumor size and regional lymph node involvment.

(A) Overall survival of the entire patient cohort (5-yr OS: 70%). (B) Patient with ERMS have a favorable outcome compared to other histologies; ARMS and SRMS-ScRMS having an equally unfavorable outcome (P=0.012, log-rank test; *P=0.043, cox regression). HNRMS patients with tumor sizes >5 cm (C, P<0.001) and with positive regional lymph node follow an unfavorable clinical course (D, P=0.002).
Most ERMS patients followed a favorable outcome and a low recurrence rate. About a fifth (19%, 10/53) of ERMS patients died of disease (DOD) (P=0.05) and 8% developed recurrence at a median follow-up of 104 months (P=0.001). In contrast, ARMS was associated with an unfavorable outcome and high recurrence rate, with 39% of patients DOD and 42% developed recurrence at a median follow-up of 32 months. Similarly, SRMS-ScRMS was associated with a poor survival and a high recurrence rate. Almost half (46%, 5/11) of SRMS-ScRMS patients DOD and 36% of patients diagnosed with SRMS-ScRMS developed recurrence at a median follow-up of 29 months. Among the SRMS-ScRMS patients who succumbed to disease, all except one were of adult age and showed sclerosing morphology. Three of the 4 ScRMS patients who succumbed of disease showed the presence of a MYOD1 mutation. By survival analysis, the 5-year OS rate for ScRMS was 30%, compared to 75% for SRMS.
Parameningeal (PM) subsite is associated with an unfavorable outcome and a high recurrence rate
One third (35%) of patients with PM-site DOD, while the non-PM / non-orbital subsite had a 20% mortality and the orbital sub-site 10%. Similarly, the PM sub-site had a higher recurrence rate (30%), compared to 12% in non-PM / non-orbital subsite and 0% in orbital location.
By multivariate analysis histologic type is the only statistically significant factor
On univariate analysis, the following factors were found to be statistically significant variables for survival: histologic subtype, tumor size, regional lymph node involvement, metastasis at initial presentation, stage and recurrence. Other variables such as gender, age group and subsites were not found to be statistically significant (Table 3). On multivariate analysis, only histology was found to be statistically significant (Table 3). Patients diagnosed with SRMS-ScRMS were associated with the lowest mean survival time followed by ARMS and ERMS (P=0.012). Patients with large tumors (> 5 cm) had a lower mean survival time compared to patients with smaller tumor sizes (P<0.001) (Figure 2C). Patients with regional lymph node involvement had a lower mean survival time compared to patients without lymph node metastasis (P=0.002) (Figure 2D). Patients with stage 4 and presenting with metastases had a lower mean survival time (P<0.001) (Figure 3A). Also, patients with distant recurrence had a lower mean survival time (P=0.002) (Figure 3B). Patients with tumors in the PM subsite had a lower mean survival time but not statistically significant (P=0.119). Adult patients had a lower mean survival time compared to pediatric patients, but was not statistically significant (P=0.302) (online supplemental Fig 1). On multivariate analysis, only histology was found to be statistically significant (P=0.043).
Table 3.
Survival Analysis of HNRMS Patients Based on Clinical Variables and Histologic Subtype
| Clinical & pathologic variables | Number | Survival duration (months) | Univariate analysis (log-rank test) | Multivariate analysis (Cox regression) | ||||
|---|---|---|---|---|---|---|---|---|
| Mean | 95% CI | P value | Hazard ratio | 95% CI | P value | |||
| Gender | 0.711 | |||||||
| Female | 46 | 161 | 133.8-189.0 | |||||
| Male | 51 | 159 | 129.97-187.8 | |||||
| Age group | 0.302 | |||||||
| Adult | 36 | 134 | 101.1-167.6 | |||||
| Pediatric | 61 | 173 | 147.8-197.3 | |||||
| Tumor site | 0.119 | |||||||
| O | 10 | 210 | 169.95-250.7 | |||||
| PM | 63 | 139 | 113.2-165.2 | |||||
| S | 24 | 172 | 137.6-208.3 | |||||
| Histology | 0.012 | 0.043 | ||||||
| ARMS | 33 | 123 | 87.9-158.2 | 1 | ||||
| ERMS | 53 | 190 | 166.5-213.3 | 0.8 | 0.3-2.6 | 0.822 | ||
| SRMS-ScRMS | 11 | 53 | 30.8-74.6 | 4.8 | 1.2-20.5 | 0.031 | ||
| Tumor size (CM) | <0.001 | |||||||
| ≤5 | 47 | 206 | 185.4-227.5 | |||||
| >5 | 19 | 68 | 40.9-201.9 | |||||
| Regional lymph node | 0.002 | 0.143 | ||||||
| Negative | 56 | 191 | 168.1-213.6 | 1 | ||||
| Positive | 40 | 119 | 87.1-150.5 | 2.3 | 0.7-7.6 | |||
| Initial metastasis | <0.001 | |||||||
| Negative | 79 | 179 | 158.4-200.4 | |||||
| Positive | 18 | 46 | 24.97-67.5 | |||||
| Stage | <0.001 | 0.193 | ||||||
| 1 | 28 | 189 | 155.95-222.6 | 1 | ||||
| 2 | 10 | 202 | 169.3-233.95 | 1.1 | 0.1-11.6 | 0.912 | ||
| 3 | 41 | 141 | 114.4-167.5 | 2.3 | 0.6-8.5 | 0.213 | ||
| 4 | 18 | 46 | 24.97-67.5 | 4.6 | 1.1-19.5 | 0.036 | ||
| Recurrence | 0.002 | 0.15 | ||||||
| NR | 75 | 184 | 162.8-205.6 | 1 | ||||
| LR | 9 | 89 | 31.97-145.2 | 1.8 | 0.5-6.5 | 0.326 | ||
| DR | 13 | 66 | 29.8-101.5 | 2.5 | 0.98-6.6 | 0.055 | ||
PM – Parameningeal, S – Non parameningeal non orbital, O – Orbital, LR – Local recurrence, DR – Distant recurrence, NR – No recurrence
Figure 3. Survival of HNRMS patients is also influenced by stage at diagnosis and tumor recurrence.

Additional adverse factors found to be statistically significant by univariate analyses included stage IV patients presenting with metastases at diagnosis (A, P<0.001) and patients who developed local or distant recurrence (B, P=0.002).
DISCUSSION
Although head and neck RMS (HNRMS) is rare with an incidence of 0.104 cases per 100,000, it is the most common clinical presentation of RMS [6]. In contrast with other anatomic locations, HNRMS encompass certain particularities such as an increased rate of spindle/sclerosing (SRMS/ScRMS) histology, high stage and recurrence rates, with often limited benefit from surgical management due to its proximity to vital structures. Prior studies of HNRMS have focused mainly on specific subsites and a recent study used the National Cancer Institute's (NCI) SEER database of multiple geographic regions and different therapy regimen [6, 45, 46]. In this study, we performed a detailed clinicopathologic and survival analysis of HNRMS managed at a single tertiary center during a 20-year period (1996-2015). All cases included were subjected to strict pathologic and molecular criteria using the last 2013 WHO classification [9]. As the SRMS-ScRMS histologic type was only recently introduced as a fourth clinicopathologic variant of RMS in the latest WHO, most prior clinical studies do not take into account this new variant characterized by different biology and clinical outcome, which is included under the large umbrella of ERMS. Furthermore, according to recent Children's Oncology Group (COG) studies, a subset of ScRMS displays histologically a dense proliferation of round cells in a dense collagenized background, which have been previously misclassified as fusion-negative ARMS [49]. As a result of all these recent changes in RMS classification as well as to the new developments in the molecular characterization of the SRMS/ScRMS subset, we undertook a thorough re-evaluation of the HNRMS cohort managed at our Institution in order to establish prognostic factors and oncologic outcome.
Based on the NCI SEER database, the 5-year survival rate for patients with childhood RMS is reported as 62%. Similarly, the 5-year relative survival rate for patients with HNRMS was 63%, with a better outcome being noted in patients younger than age of 10, 80%, compared to older than 10, 46% [6, 42]. This is in keeping with the 70% 5-year OS rate of the entire cohort of HNRMS from our study. Studies on localized parameningeal RMS in children have reported a 5-year OS rate as 70-73% [45, 46]. In contrast, the estimated 5-year OS in adults with HNRMS was only 36% [8]. Although the mean survival time for adults in our study was poorer (67%) compared to pediatric patients (71%), there was no statistical difference noted with age.
As recent studies from COG found that the fusion negative ARMS have overlapping gene expression prolife with ERMS and most likely behave like ERMS [50, 51], we included only FOXO1 fusion positive cases in our ARMS subgroup. Cases deemed as having alveolar morphology that were either not tested or negative for FOXO1 gene rearrangements/fusion were excluded from the study. The presence of PAX3/7-FOXO1 gene fusion is a crucial prognostic indicator in RMS associated with poor prognosis [14, 52]. Head and neck ARMS usually occurs in older children or young adults, involves the PM subsite, and is associated with local and distant failures. The 5-year survival rate for childhood ARMS from the NCI SEER database is estimated as 48%, while the 5-year relative survival rate for head and neck ARMS was 44% [6, 42]. In our study, ARMS was also associated with poor outcome, with a 53% 5-year OS and 64% 5-year OS for localized cases with no significant difference between age groups.
ERMS is associated with the most favorable clinical outcome compared to other variants. According to the NCI SEER database, the 5-year survival rate for patients with childhood ERMS is estimated to 73%. In the head and neck location the reported 5-year relative survival rate is 72% [6, 42], while in our study the 5-year OS was 82%. In contrast to earlier literature of ERMS in children having a better outcome compared to ERMS in adults, our results showed a lower 5-year OS of 81% in children with ERMS compared to 86% in adults ERMS, although this could be related to the small number of adult patients (13%) in our ERMS cohort [53]. Some of the adverse factors associated with a poor outcome in ERMS includes the presence of nuclear anaplasia[54] and p53 mutations [11].
Recent studies focusing on SRMS-ScRMS have reported a variable prognosis based on their age at diagnosis and genetic abnormalities. Most SRMS occurring in infants are characterized by recurrent NCOA2 and VGLL2 related fusions and are associated overwhelmingly with a favorable outcome and long-term survival [22]. In contrast, SRMS-ScRMS harboring MYOD1 mutations follow an aggressive clinical behavior and poor prognosis, irrespective of the patient's age [23, 25, 26]. In contrast to a recent COG study reporting a 5-year OS of 86% in a cohort of 22 patients with ScRMS, 41% arising in the head and neck, our results showed a significantly lower 5-year OS of 30% in this histologic subtype [49]. The explanation for this clinical outcome discrepancy might be related to the mixed pediatric and adult patient population in our ScRMS cohort, as well as the fact that their study did not investigate for MYOD1 mutations, to identify the high-risk MYOD1-mutant tumors. In the present study, the poor outcome in head and neck SRMS-ScRMS was associated with adult age, sclerosing morphology and the presence of MYOD1 mutations [26]. All cases of sclerosing morphology in this study were tested for PAX3/7-FOXO1 fusion or rearrangement to evaluate for ARMS and were all found to be negative. The only survivor with ScRMS in this study was found to be negative for MYOD1 mutation. Molecular evaluation of MYOD1 mutations in patients with SRMS-ScRMS for risk stratification has been suggested [26, 49].
In accordance with prior data from the NCI SEER, IRS-III and IRS-IV data, we found that orbital RMS had a better survival compared to other head and neck locations [6], while the parameningeal (PM) subsite was associated with the poorest outcome. As previously reported, our results also showed that the presence of recurrence was associated with ARMS histology [6]. Other variables identified in this study in keeping with prior reports included large tumor size (> 5 cm), the presence of regional lymph node involvement and/or metastasis at diagnosis, advanced stage and recurrence [6, 8, 43-46].
In summary, HNRMS is an uncommon disease composed of a heterogenous group with variable prognosis depending on the histologic subtype, and genetic abnormalities. Our results showed that patients with ARMS and SRMS-ScRMS have an equally unfavorable outcome. In particular patients with ScRMS and MYOD1 mutations followed a highly aggressive clinical course. These findings suggest that molecular evaluation should be performed on RMS for risk stratification, including testing for FOXO1 gene fusion in ARMS, MYOD1 mutations in SRMS-ScRMS, particularly in older children and adults, and NCOA2 and VGLL2 related fusions in congenital/infantile SRMS. Genetic testing not only provides valuable prognostic information but also can select patients who can benefit for treatment intensification.
Supplementary Material
HIGHLIGHTS.
ARMS and SRMS-ScRMS have an equally unfavorable outcome
MYOD1 mutations occur preferentially in ScRMS with an unfavorable outcome
By multivariate analysis histologic type is the only significant prognostic factor
FOXO1 gene fusion and MYOD1 mutations should be performed for risk stratification
Acknowledgments
Supported in part by: P50CA140146-01 (CRA), Cycle for Survival (LW, CRA) and Kristen Ann Carr Foundation (CRA)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure: All authors declare that there are no financial conflicts associated with this study and that the funding source has no role in conceiving and performing the study.
REFERENCES
- 1.Potter BO, Sturgis EM. Sarcomas of the head and neck. Surgical oncology clinics of North America. 2003;12:379–417. doi: 10.1016/s1055-3207(03)00005-x. [DOI] [PubMed] [Google Scholar]
- 2.de Bree R, van der Valk P, Kuik DJ, van Diest PJ, Doornaert P, Buter J, et al. Prognostic factors in adult soft tissue sarcomas of the head and neck: a single-centre experience. Oral Oncol. 2006;42:703–9. doi: 10.1016/j.oraloncology.2005.11.009. [DOI] [PubMed] [Google Scholar]
- 3.Gurney JG, Young JL, Roffers SD, Smith MA, Bunin GR. Soft Tissue Sarcomas. In: Ries LAG, Smith MA, Gurney JG, Linet M, Tamra T, Young JL, Bunin GR, editors. Cancer Incidence and Survival among Children and Adolescents: United States SEER Program 1975-1995. Vol. 1999. National Cancer Institute; SEER Program Bethesda: pp. 111–24. [Google Scholar]
- 4.Pappo AS, Shapiro DN, Crist WM, Maurer HM. Biology and therapy of pediatric rhabdomyosarcoma. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 1995;13:2123–39. doi: 10.1200/JCO.1995.13.8.2123. [DOI] [PubMed] [Google Scholar]
- 5.Hicks J, Flaitz C. Rhabdomyosarcoma of the head and neck in children. Oral oncology. 2002;38:450–9. doi: 10.1016/s1368-8375(01)00105-1. [DOI] [PubMed] [Google Scholar]
- 6.Turner JH, Richmon JD. Head and neck rhabdomyosarcoma: a critical analysis of population-based incidence and survival data. Otolaryngology--head and neck surgery : official journal of American Academy of Otolaryngology-Head and Neck Surgery. 2011;145:967–73. doi: 10.1177/0194599811417063. [DOI] [PubMed] [Google Scholar]
- 7.Wharam MD., Jr Rhabdomyosarcoma of Parameningeal Sites. Seminars in radiation oncology. 1997;7:212–6. doi: 10.1053/SRAO00700212. [DOI] [PubMed] [Google Scholar]
- 8.Wu Y, Li C, Zhong Y, Guo W, Ren G. Head and neck rhabdomyosarcoma in adults. The Journal of craniofacial surgery. 2014;25:922–5. doi: 10.1097/SCS.0000000000000704. [DOI] [PubMed] [Google Scholar]
- 9.Parham DM, Montgomery E, Nascimento AF, Barr FG. Skeletal-muscle tumours. In: Fletcher CDM, Bridge JA, Hogendoorn P, Mertens F, editors. WHO Classification of Tumours of Soft Tissue and Bone. ed 4 International Agency for Research on Cancer; Lyon, France: 2013. pp. 127–35. [Google Scholar]
- 10.Shern JF, Chen L, Chmielecki J, Wei JS, Patidar R, Rosenberg M, et al. Comprehensive genomic analysis of rhabdomyosarcoma reveals a landscape of alterations affecting a common genetic axis in fusion-positive and fusion-negative tumors. Cancer discovery. 2014;4:216–31. doi: 10.1158/2159-8290.CD-13-0639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Seki M, Nishimura R, Yoshida K, Shimamura T, Shiraishi Y, Sato Y, et al. Integrated genetic and epigenetic analysis defines novel molecular subgroups in rhabdomyosarcoma. Nature communications. 2015;6:7557. doi: 10.1038/ncomms8557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shukla N, Ameur N, Yilmaz I, Nafa K, Lau CY, Marchetti A, et al. Oncogene mutation profiling of pediatric solid tumors reveals significant subsets of embryonal rhabdomyosarcoma and neuroblastoma with mutated genes in growth signaling pathways. Clinical cancer research : an official journal of the American Association for Cancer Research. 2012;18:748–57. doi: 10.1158/1078-0432.CCR-11-2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Martinelli S, McDowell HP, Vigne SD, Kokai G, Uccini S, Tartaglia M, et al. RAS signaling dysregulation in human embryonal Rhabdomyosarcoma. Genes, chromosomes & cancer. 2009;48:975–82. doi: 10.1002/gcc.20702. [DOI] [PubMed] [Google Scholar]
- 14.Missiaglia E, Williamson D, Chisholm J, Wirapati P, Pierron G, Petel F, et al. PAX3/FOXO1 fusion gene status is the key prognostic molecular marker in rhabdomyosarcoma and significantly improves current risk stratification. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2012;30:1670–7. doi: 10.1200/JCO.2011.38.5591. [DOI] [PubMed] [Google Scholar]
- 15.Davis RJ, D'Cruz CM, Lovell MA, Biegel JA, Barr FG. Fusion of PAX7 to FKHR by the variant t(1;13)(p36;q14) translocation in alveolar rhabdomyosarcoma. Cancer research. 1994;54:2869–72. [PubMed] [Google Scholar]
- 16.Galili N, Davis RJ, Fredericks WJ, Mukhopadhyay S, Rauscher FJ, 3rd, Emanuel BS, et al. Fusion of a fork head domain gene to PAX3 in the solid tumour alveolar rhabdomyosarcoma. Nature genetics. 1993;5:230–5. doi: 10.1038/ng1193-230. [DOI] [PubMed] [Google Scholar]
- 17.Mentzel T, Kuhnen C. Spindle cell rhabdomyosarcoma in adults: clinicopathological and immunohistochemical analysis of seven new cases. Virchows Archiv : an international journal of pathology. 2006;449:554–60. doi: 10.1007/s00428-006-0284-4. [DOI] [PubMed] [Google Scholar]
- 18.Mentzel T. [Spindle cell rhabdomyosarcoma in adults: a new entity in the spectrum of malignant mesenchymal tumors of soft tissues] Der Pathologe. 2010;31:91–6. doi: 10.1007/s00292-009-1249-6. [DOI] [PubMed] [Google Scholar]
- 19.Mentzel T, Katenkamp D. Sclerosing, pseudovascular rhabdomyosarcoma in adults. Clinicopathological and immunohistochemical analysis of three cases. Virchows Archiv : an international journal of pathology. 2000;436:305–11. doi: 10.1007/s004280050451. [DOI] [PubMed] [Google Scholar]
- 20.Folpe AL, McKenney JK, Bridge JA, Weiss SW. Sclerosing rhabdomyosarcoma in adults: report of four cases of a hyalinizing, matrix-rich variant of rhabdomyosarcoma that may be confused with osteosarcoma, chondrosarcoma, or angiosarcoma. The American journal of surgical pathology. 2002;26:1175–83. doi: 10.1097/00000478-200209000-00008. [DOI] [PubMed] [Google Scholar]
- 21.Mosquera JM, Sboner A, Zhang L, Kitabayashi N, Chen CL, Sung YS, et al. Recurrent NCOA2 gene rearrangements in congenital/infantile spindle cell rhabdomyosarcoma. Genes, chromosomes & cancer. 2013;52:538–50. doi: 10.1002/gcc.22050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Alaggio R, Zhang L, Sung YS, Huang SC, Chen CL, Bisogno G, et al. A Molecular Study of Pediatric Spindle and Sclerosing Rhabdomyosarcoma: Identification of Novel and Recurrent VGLL2-related Fusions in Infantile Cases. The American journal of surgical pathology. 2016;40:224–35. doi: 10.1097/PAS.0000000000000538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kohsaka S, Shukla N, Ameur N, Ito T, Ng CK, Wang L, et al. A recurrent neomorphic mutation in MYOD1 defines a clinically aggressive subset of embryonal rhabdomyosarcoma associated with PI3K AKT pathway mutations. Nature genetics. 2014;46:595–600. doi: 10.1038/ng.2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Szuhai K, de Jong D, Leung WY, Fletcher CD, Hogendoorn PC. Transactivating mutation of the MYOD1 gene is a frequent event in adult spindle cell rhabdomyosarcoma. The Journal of pathology. 2014;232:300–7. doi: 10.1002/path.4307. [DOI] [PubMed] [Google Scholar]
- 25.Agaram NP, Chen CL, Zhang L, LaQuaglia MP, Wexler L, Antonescu CR. Recurrent MYOD1 mutations in pediatric and adult sclerosing and spindle cell rhabdomyosarcomas: evidence for a common pathogenesis. Genes, chromosomes & cancer. 2014;53:779–87. doi: 10.1002/gcc.22187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Owosho AA, Chen S, Kashikar S, Zhang L, Chen CL, Wexler LH, et al. Clinical and molecular heterogeneity of head and neck spindle cell and sclerosing rhabdomyosarcoma. Oral oncology. 2016 doi: 10.1016/j.oraloncology.2016.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Furlong MA, Mentzel T, Fanburg-Smith JC. Pleomorphic rhabdomyosarcoma in adults: a clinicopathologic study of 38 cases with emphasis on morphologic variants and recent skeletal muscle-specific markers. Modern pathology : an official journal of the United States and Canadian Academy of Pathology, Inc. 2001;14:595–603. doi: 10.1038/modpathol.3880357. [DOI] [PubMed] [Google Scholar]
- 28.Smith AC, Squire JA, Thorner P, Zielenska M, Shuman C, Grant R, et al. Association of alveolar rhabdomyosarcoma with the Beckwith-Wiedemann syndrome. Pediatric and developmental pathology : the official journal of the Society for Pediatric Pathology and the Paediatric Pathology Society. 2001;4:550–8. doi: 10.1007/s10024001-0110-6. [DOI] [PubMed] [Google Scholar]
- 29.Sung L, Anderson JR, Arndt C, Raney RB, Meyer WH, Pappo AS. Neurofibromatosis in children with Rhabdomyosarcoma: a report from the Intergroup Rhabdomyosarcoma study IV. The Journal of pediatrics. 2004;144:666–8. doi: 10.1016/j.jpeds.2004.02.026. [DOI] [PubMed] [Google Scholar]
- 30.O'Neal JP, Ramdas J, Wood WE, Pellitteri PK. Parameningeal rhabdomyosarcoma in a patient with Costello syndrome. Journal of pediatric hematology/oncology. 2004;26:389–92. doi: 10.1097/00043426-200406000-00012. [DOI] [PubMed] [Google Scholar]
- 31.Jung A, Bechthold S, Pfluger T, Renner C, Ehrt O. Orbital rhabdomyosarcoma in Noonan syndrome. Journal of pediatric hematology/oncology. 2003;25:330–2. doi: 10.1097/00043426-200304000-00014. [DOI] [PubMed] [Google Scholar]
- 32.Carnevale A, Lieberman E, Cardenas R. Li-Fraumeni syndrome in pediatric patients with soft tissue sarcoma or osteosarcoma. Archives of medical research. 1997;28:383–6. [PubMed] [Google Scholar]
- 33.Sobel RA, Woerner S. Rubinstein-Taybi syndrome and nasopharyngeal rhabdomyosarcoma. The Journal of pediatrics. 1981;99:1000–1. doi: 10.1016/s0022-3476(81)80043-1. [DOI] [PubMed] [Google Scholar]
- 34.Lo Muzio L. Nevoid basal cell carcinoma syndrome (Gorlin syndrome). Orphanet journal of rare diseases. 2008;3:32. doi: 10.1186/1750-1172-3-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gripp KW, Scott CI, Jr., Nicholson L, McDonald-McGinn DM, Ozeran JD, Jones MC, et al. Five additional Costello syndrome patients with rhabdomyosarcoma: proposal for a tumor screening protocol. American journal of medical genetics. 2002;108:80–7. doi: 10.1002/ajmg.10241. [DOI] [PubMed] [Google Scholar]
- 36.Cajaiba MM, Bale AE, Alvarez-Franco M, McNamara J, Reyes-Mugica M. Rhabdomyosarcoma, Wilms tumor, and deletion of the patched gene in Gorlin syndrome. Nature clinical practice Oncology. 2006;3:575–80. doi: 10.1038/ncponc0608. [DOI] [PubMed] [Google Scholar]
- 37.Jongmans MC, Hoogerbrugge PM, Hilkens L, Flucke U, van der Burgt I, Noordam K, et al. Noonan syndrome, the SOS1 gene and embryonal rhabdomyosarcoma. Genes, chromosomes & cancer. 2010;49:635–41. doi: 10.1002/gcc.20773. [DOI] [PubMed] [Google Scholar]
- 38.Crucis A, Richer W, Brugieres L, Bergeron C, Marie-Cardine A, Stephan JL, et al. Rhabdomyosarcomas in children with neurofibromatosis type I: A national historical cohort. Pediatric blood & cancer. 2015;62:1733–8. doi: 10.1002/pbc.25556. [DOI] [PubMed] [Google Scholar]
- 39.Yang JC, Wexler LH, Meyers PA, Wolden SL. Parameningeal rhabdomyosarcoma: outcomes and opportunities. International journal of radiation oncology, biology, physics. 2013;85:e61–6. doi: 10.1016/j.ijrobp.2012.08.019. [DOI] [PubMed] [Google Scholar]
- 40.Zevallos JP, Jain K, Roberts D, Santillan AA, Huh W, Hanna EY, et al. Modern multimodality therapy for pediatric nonorbital parameningeal sarcomas. Head & neck. 2010;32:1501–5. doi: 10.1002/hed.21353. [DOI] [PubMed] [Google Scholar]
- 41.Malempati S, Hawkins DS. Rhabdomyosarcoma: review of the Children's Oncology Group (COG) Soft-Tissue Sarcoma Committee experience and rationale for current COG studies. Pediatric blood & cancer. 2012;59:5–10. doi: 10.1002/pbc.24118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ognjanovic S, Linabery AM, Charbonneau B, Ross JA. Trends in childhood rhabdomyosarcoma incidence and survival in the United States, 1975-2005. Cancer. 2009;115:4218–26. doi: 10.1002/cncr.24465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Meza JL, Anderson J, Pappo AS, Meyer WH, Children's Oncology G. Analysis of prognostic factors in patients with nonmetastatic rhabdomyosarcoma treated on intergroup rhabdomyosarcoma studies III and IV: the Children's Oncology Group. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2006;24:3844–51. doi: 10.1200/JCO.2005.05.3801. [DOI] [PubMed] [Google Scholar]
- 44.Rodeberg DA, Garcia-Henriquez N, Lyden ER, Davicioni E, Parham DM, Skapek SX, et al. Prognostic significance and tumor biology of regional lymph node disease in patients with rhabdomyosarcoma: a report from the Children's Oncology Group. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2011;29:1304–11. doi: 10.1200/JCO.2010.29.4611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Merks JH, De Salvo GL, Bergeron C, Bisogno G, De Paoli A, Ferrari A, et al. Parameningeal rhabdomyosarcoma in pediatric age: results of a pooled analysis from North American and European cooperative groups. Annals of oncology : official journal of the European Society for Medical Oncology / ESMO. 2014;25:231–6. doi: 10.1093/annonc/mdt426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Raney RB, Meza J, Anderson JR, Fryer CJ, Donaldson SS, Breneman JC, et al. Treatment of children and adolescents with localized parameningeal sarcoma: experience of the Intergroup Rhabdomyosarcoma Study Group protocols IRS-II through -IV, 1978-1997. Medical and pediatric oncology. 2002;38:22–32. doi: 10.1002/mpo.1259. [DOI] [PubMed] [Google Scholar]
- 47.Wolden SL, Wexler LH, Kraus DH, Laquaglia MP, Lis E, Meyers PA. Intensity-modulated radiotherapy for head-and-neck rhabdomyosarcoma. International journal of radiation oncology, biology, physics. 2005;61:1432–8. doi: 10.1016/j.ijrobp.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 48.Dharmarajan KV, Wexler LH, Wolden SL. Concurrent radiation with irinotecan and carboplatin in intermediate- and high-risk rhabdomyosarcoma: a report on toxicity and efficacy from a prospective pilot phase II study. Pediatric blood & cancer. 2013;60:242–7. doi: 10.1002/pbc.24205. [DOI] [PubMed] [Google Scholar]
- 49.Rudzinski ER, Anderson JR, Hawkins DS, Skapek SX, Parham DM, Teot LA. The World Health Organization Classification of Skeletal Muscle Tumors in Pediatric Rhabdomyosarcoma: A Report From the Children's Oncology Group. Archives of pathology & laboratory medicine. 2015;139:1281–7. doi: 10.5858/arpa.2014-0475-OA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rudzinski ER, Teot LA, Anderson JR, Moore J, Bridge JA, Barr FG, et al. Dense pattern of embryonal rhabdomyosarcoma, a lesion easily confused with alveolar rhabdomyosarcoma: a report from the Soft Tissue Sarcoma Committee of the Children's Oncology Group. American journal of clinical pathology. 2013;140:82–90. doi: 10.1309/AJCPA1WN7ARPCMKQ. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Arnold MA, Anderson JR, Gastier-Foster JM, Barr FG, Skapek SX, Hawkins DS, et al. Histology, Fusion Status, and Outcome in Alveolar Rhabdomyosarcoma With Low-Risk Clinical Features: A Report From the Children's Oncology Group. Pediatric blood & cancer. 2016;63:634–9. doi: 10.1002/pbc.25862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Skapek SX, Anderson J, Barr FG, Bridge JA, Gastier-Foster JM, Parham DM, et al. PAX-FOXO1 fusion status drives unfavorable outcome for children with rhabdomyosarcoma: a children's oncology group report. Pediatric blood & cancer. 2013;60:1411–7. doi: 10.1002/pbc.24532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sultan I, Qaddoumi I, Yaser S, Rodriguez-Galindo C, Ferrari A. Comparing adult and pediatric rhabdomyosarcoma in the surveillance, epidemiology and end results program, 1973 to 2005: an analysis of 2,600 patients. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2009;27:3391–7. doi: 10.1200/JCO.2008.19.7483. [DOI] [PubMed] [Google Scholar]
- 54.Qualman S, Lynch J, Bridge J, Parham D, Teot L, Meyer W, et al. Prevalence and clinical impact of anaplasia in childhood rhabdomyosarcoma : a report from the Soft Tissue Sarcoma Committee of the Children's Oncology Group. Cancer. 2008;113:3242–7. doi: 10.1002/cncr.23929. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.