

SUMMARY
Spotted fever group (SFG) rickettsiae are human pathogens that infect cells in the vasculature. They disseminate through host tissues by a process of cell-to-cell spread that involves protrusion formation, engulfment and vacuolar escape. Other bacterial pathogens rely on actin-based motility to provide a physical force for spread. Here we show that SFG species Rickettsia parkeri typically lack actin tails during spread and instead manipulate host intercellular tension and mechanotransduction to promote spread. Using transposon mutagenesis, we identified surface cell antigen 4 (Sca4) as a secreted effector of spread that specifically promotes protrusion engulfment. Sca4 interacts with the cell adhesion protein vinculin and blocks association with vinculin’s binding partner, α-catenin. Using traction and monolayer stress microscopy, we show that Sca4 reduces vinculin-dependent mechanotransduction at cell-cell junctions. Our results suggest that Sca4 relieves intercellular tension to promote protrusion engulfment, which represents a distinctive strategy for manipulating cytoskeletal force generation to enable spread.
Graphical Abstract
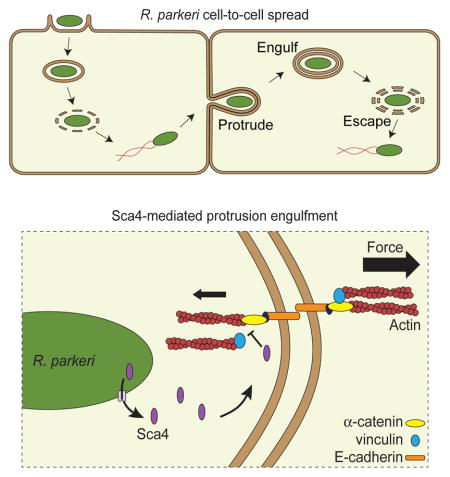
INTRODUCTION
Many intracellular bacterial pathogens that reside in the cytosol have evolved mechanisms to spread through host tissues while remaining within cells, enabling access to cytosolic nutrients and evasion of the immune response (Ray et al., 2009). Cell-to-cell spread has been extensively studied for pathogens such as Listeria monocytogenes and Shigella flexneri. However, less well-studied intracytosolic bacteria also undergo spread, including the spotted fever group (SFG) of Rickettsia spp. These obligate intracellular bacteria are transmitted to humans via arthropod vectors, and spread likely contributes to the vascular and epithelial damage associated with spotted fever disease (Walker and Ismail, 2008). Nonetheless, in comparison with other pathogens, we know very little about the pathways and underlying mechanisms by which SFG rickettsiae spread.
Previous studies of L. monocytogenes and S. flexneri have revealed key steps of cell-to-cell spread. First, intracytosolic bacteria hijack host actin and use the force from actin polymerization to drive movement and form actin comet tails (Bernardini et al., 1989; Tilney and Portnoy, 1989). Next, motile bacteria propel themselves into the donor cell plasma membrane and form long protrusions into the recipient cell (Dragoi and Agaisse, 2015; Kadurugamuwa et al., 1991; Robbins et al., 1999; Tilney and Portnoy, 1989). Eventually, a bacterial protrusion is engulfed into a double-membrane vesicle in the recipient cell, which is followed by lysis of the vesicle and escape of the bacterium into the recipient cell cytosol (Kuehl et al., 2015; Ray et al., 2009). SFG rickettsiae also undergo actin-based motility (Heinzen, 2003) and form protrusions into recipient cells (Gouin et al., 1999). However, the model SFG Rickettsia species R. parkeri uses distinct mechanisms of actin-based motility compared with other pathogens (Haglund et al., 2010; Reed et al., 2014), suggesting their mechanisms of spread may also be different.
During spread, bacteria interact directly with the host plasma membrane at intercellular junctions. These junctions contain protein complexes and signaling platforms that can be usurped by pathogens. For example, S. flexneri target tricellular junctions via the host tight junction protein tricellulin to promote protrusion formation (Fukumatsu et al., 2012). S. flexneri protrusion dynamics are also regulated by localized tyrosine kinase signaling and phospholipid production (Kuehl et al., 2015). In contrast, the L. monocytogenes effector protein InlC promotes protrusion formation by inhibiting the host adaptor protein Tuba and the actin regulatory protein N-WASP (Rajabian et al., 2009). These differences highlight the complexity of this process and suggest that distinct pathways of spread have evolved for different pathogens.
Intercellular junctions also serve as sites for generating cell-cell tension. At adherens junctions, E-cadherin receptors are linked to the underlying actomyosin network through protein complexes that respond to and transmit forces to regulate cell shape, structure and function (Lecuit and Yap, 2015). When tension is applied to E-cadherin, α-catenin is stretched to expose cryptic binding sites for proteins such as vinculin (Buckley et al., 2014; Yonemura et al., 2010). Vinculin recruitment reinforces reciprocal, actin-mediated pulling forces at junctions by facilitating additional connections to the actomyosin network and increasing cell stiffness (Huveneers and de Rooij, 2013). Tension at cell-cell junctions likely represents a key barrier to be overcome by bacteria during spread. Previous work suggested that the forces from actin-based motility were sufficient to overcome cell-cell tension (Monack and Theriot, 2001). Alternatively, bacteria may directly manipulate host subcellular tension forces to promote spread, as has been suggested for L. monocytogenes InlC (Rajabian et al., 2009).
To better understand general and pathogen-specific features of cell-to-cell spread, we investigated spread parameters and molecular mechanisms for R. parkeri. We found that R. parkeri spread occurs with distinct physical and kinetic features when compared with L. monocytogenes. Moreover, most R. parkeri lack actin tails when in protrusions, in contrast with L. monocytogenes, suggesting R. parkeri may not rely on actin-based motility for protrusion dynamics. Instead, we found that the rickettsial protein surface cell antigen 4 (Sca4) promotes efficient protrusion engulfment by reducing vinculin-dependent tension at E-cadherin-mediated adhesion sites. Our results suggest that R. parkeri evolved a strategy to promote spread that may not rely on the force generated by actin-based motility and instead depends on weakening intercellular tension.
RESULTS
R. parkeri utilize a distinctive strategy of cell-to-cell spread
Although previous studies have investigated the parameters of L. monocytogenes and S. flexneri spread (Bernardini et al., 1989; Dragoi and Agaisse, 2015; Gouin et al., 1999; Kadurugamuwa et al., 1991; Robbins et al., 1999; Tilney and Portnoy, 1989), little was known for R. parkeri. Therefore, we compared the spread parameters of GFP-expressing R. parkeri and L. monocytogenes (Rp-GFP and Lm-GFP) using live imaging of infected A549 cells, which form flat, adherent monolayers amenable to live cell imaging. To visualize individual spread events, A549 cells stably expressing a red plasma membrane marker (TagRFP-T-Farnesyl; TRTF) were mixed with unlabeled A549 cells and grown to confluency before infection. We observed each stage of spread, including protrusion formation, protrusion engulfment and bacterial escape into the cytosol (Figure 1A and Movies S1–S2). We discovered that Rp-GFP generated significantly shorter protrusions than Lm-GFP (Figures 1A and 1B; short protrusions were also seen in HMEC-1 cells, data not shown). Moreover, Rp-GFP protrusions resolved into vesicles faster than Lm-GFP protrusions (Figure 1C). Vesicle-escape kinetics were the same for both bacteria (Figure 1D). These morphological and kinetic differences suggested that each pathogen may have evolved different molecular mechanisms for regulating protrusion dynamics.
Figure 1. R. parkeri exhibit a morphologically distinct method of cell-to-cell spread.

(A) Live cell imaging snapshots of the three steps of spread for R. parkeri or L. monocytogenes (Red, TRTF; green, bacterium). (B–D) Quantification of spread parameters from live cell imaging showing (B) maximum protrusion length, (C) time spent in protrusion before engulfment and (D) time spent in vesicle before escape to cytosol. (E) Images of cells infected with Rp-2xTagBFP or Lm-TagBFP. Host membrane stained with anti-β-catenin (green) and F-actin stained with phalloidin (red). Arrow indicates protrusions shown at right. Scale bar for merge image, 5 μm; protrusion image, 1 μm. (F) Percentage of bacteria in protrusions. (G) Percentage of bacterial protrusions with different F-actin phenotypes. Mean ± SEM. (H) Live cell imaging snapshots of Rp-2xTagBFP spread. Arrow indicates a bacterium spreading into unlabeled recipient cell. (Red, TRTF; green, Lifeact-mWasabi; blue, bacterium). (I, J) Infectious life cycles of (I) L. monocytogenes and (J) R. parkeri illustrating differences in the presence or absence of actin tails in protrusions and the organization of actin filaments in tails (actin tail, purple; bacterium, green). For (A) and (H), time shown as min:sec and scale bar, 5 μm. Data from (B–D) and (F) are mean ± SD, unpaired T-test: ** p < 0.01, *** p < 0.001. Rp, R. parkeri; Lm, L. monocytogenes. See Movies S1–S3.
L. monocytogenes relies on the force generated by actin-based motility to initiate and elongate protrusions (Talman et al., 2014). SFG Rickettsia mutants defective in actin-based motility also spread less efficiently (Kleba et al., 2010; Reed et al., 2014), suggesting motility is critical for this process. Thus it was surprising that, in A549 cells infected with R. parkeri (Rp-2xTagBFP) and L. monocytogenes (Lm-TagBFP), only 8% of Rp-2xTagBFP in protrusions had polar actin or actin tails, whereas 82% of Lm-TagBFP had polar actin or actin tails (Figure 1E–G; note, small difference in protrusion frequency (Figure 1F)). The results with R. parkeri were confirmed using live cell imaging of A549 cells co-expressing TRTF and Lifeact-mWasabi (to visualize actin). In these experiments, actin tails were clearly seen associated with motile R. parkeri in the cytosol, but tails were lost once bacteria reached the cell edge and remained absent during protrusion formation and engulfment (Figure 1H and Movie S3). Thus, while actin-based motility may be necessary for R. parkeri to move to the host cell membrane, it may not be needed for initiating or elongating protrusions (Figure 1I and 1J). This highlights important differences in spread mechanisms between these bacterial species and suggests other rickettsial factors could be required for protrusion dynamics.
A mutation in R. parkeri sca4 causes a defect in plaque and infectious focus size
Because no rickettsial genes were known to specifically regulate spread, we sought to better understand the molecular mechanisms of spread by identifying R. parkeri mutants with defects in this process. We used a mariner-based transposon mutagenesis system (Liu et al., 2007) to generate R. parkeri small plaque mutants. One mutant contained a transposon (tn) insertion within the sca4 gene (sca4::tn) (Figure 2A), resulting in a truncation of Sca4 at residue 652 (Figure 2B) and a small plaque phenotype (Figures 2D and S1A). We generated an antibody against Sca4 and confirmed the sca4::tn mutant does not express full-length protein but instead weakly expresses an apparent truncation product (Figure 2C). The Sca4 amino acid sequence includes two antigenic repeat regions of unknown function (Sears et al., 2012) and two vinculin binding sites (VBS) at residues 408–426 (VBS-N) and 813–831 (VBS-C) (Figure 2B). The VBSs are conserved in most Rickettsia spp. (Gillespie et al., 2015) and in R. rickettsii Sca4 were shown to bind to vinculin (Park et al., 2011a). Although Sca4 was hypothesized to be a secreted effector and a major immunogen (Gillespie et al., 2015; Sears et al., 2012; Uchiyama, 1997), its function in infection was unknown.
Figure 2. Transposon mutagenesis of R. parkeri sca4 leads to a defect in cell-to-cell spread.

(A) R. parkeri chromosome region containing the sca4 gene (red arrowhead, tn insertion; bracket, region used for complementation). (B) Sca4 protein schematic indicating the VBSs, predicted antigenic repeats (R) and tn insertion at residue 652 (red arrowhead). (C) Western blot of purified R. parkeri strains, WT (Rp-GFP), sca4::tn and sca4::tn + pSca4 (pRAM18dSGA-Sca4). (OmpA, loading control) (D) Plaque areas in cell monolayers infected with WT and sca4::tn R. parkeri. (E) Images of infectious foci formed by WT (Rp-GFP) and sca4::tn strains. (Red, β-catenin; green, R. parkeri; blue, DAPI). Scale bar, 10 μm. (F) Infectious focus sizes formed by WT (Rp-GFP), sca4::tn and sca4::tn + pSca4 strains. (G) Average time each bacterium spent in a protrusion before resolution into a vesicle (WT data are from Figure 1C). (H) Percentage of bacteria within a protrusion. Data for (D) and (F–H) are mean ± SEM, unpaired T-test: *** p < 0.001 **** p < 0.0001 relative to WT. See Figure S1.
The small plaques formed by the sca4::tn mutant could be caused by defects in the infectious life cycle independent of spread (e.g. growth, motility). However, when compared with WT bacteria, the sca4::tn mutant had similar growth kinetics (Figure S1B), invasion efficiency (Figures S1C and S1D), frequency of actin tails and rate of actin-based motility (Figures S1E and S1F). These data pointed to a role for Sca4 in spread.
To directly measure spread, host cell monolayers were infected at a low multiplicity of infection (MOI), and extracellular bacteria were killed with gentamicin. Foci of infection formed, and the number of infected cells per focus was measured to quantify spread efficiency (Reed et al., 2014). In this infectious focus assay, the sca4::tn mutant generated smaller foci than WT in A549 (Figures 2E and 2F), HMEC-1 and U2OS cells (Figures S1G and S1H). To confirm that this phenotype is caused by sca4 disruption, we complemented the sca4::tn mutant by transforming R. parkeri with a plasmid (pSca4) containing the sca4 genomic region (nt 652,261–655,741 of the R. parkeri chromosome) (Figure 2A). This plasmid was sufficient to mediate full-length Sca4 expression in the sca4::tn mutant (Figure 2C). Moreover, mutant bacteria containing this plasmid (sca4::tn + pSca4) exhibited similar infectious focus sizes compared to WT in A549 cells (Figure 2F). Taken together, these results indicate Sca4 specifically enhances R. parkeri spread in a variety of cell types.
Sca4 promotes efficient resolution of protrusions during spread
To determine the step of spread at which Sca4 acts (protrusion formation, protrusion engulfment and/or vesicle escape), live cell imaging was performed as described above (Figure 1A) to compare WT and sca4::tn spread dynamics. Interestingly, the sca4::tn mutant displayed a kinetic defect in protrusion engulfment, spending over twice as much time in a protrusion (~23 min) compared with WT (~10 min), before resolving into a double-membrane vesicle (Figure 2G). No differences between sca4::tn and WT were observed in the other stages of spread (not shown), suggesting Sca4 is specifically required for efficient protrusion engulfment. Given the apparent importance of Sca4 in protrusion engulfment, we also predicted that more sca4::tn mutant bacteria would be found within protrusions at a fixed time point. In agreement with this prediction, the sca4::tn mutant was found in protrusions at twice the frequency of WT (Figure 2H). This phenotype was also rescued in the sca4::tn + pSca4 strain (Figure 2H). These findings indicate Sca4 acts in spread by specifically enhancing protrusion engulfment efficiency.
Sca4 is a secreted effector
Sca4 orthologs are thought to be secreted from bacteria (Gillespie et al., 2015), which would be consistent with a direct role in promoting spread by targeting host proteins. To determine if Sca4 is secreted from R. parkeri, we generated strains of R. parkeri stably expressing TEM1-Sca4 fusions for use in a β-lactamase reporter assay (Figures 3A–C). Secretion is detected after cleavage of the FRET sensor CCF4/AM in the host cytosol, resulting in a shift from green to blue fluorescence (Charpentier and Oswald, 2004). Infection of host cells with R. parkeri expressing full-length TEM1 (positive control) showed robust secretion and CCF4/AM cleavage (Figures 3A and 3C). Deleting the N-terminal secretion signal from TEM1 (TEM1*, negative control) abolished secretion (Figures 3A and 3C). Importantly, a TEM1*-Sca4 fusion was secreted as shown by abundant CCF4/AM cleavage (Figures 3A and 3C). This strongly suggests R. parkeri secrete Sca4 into the host cell.
Figure 3. Sca4 is secreted from R. parkeri.

(A) Merged images of cells infected with R. parkeri expressing TEM1, TEM1* and TEM1*-Sca4 and treated with CCF4/AM. (Blue, cleaved CCF4/AM; green, uncleaved CCF4/AM). Scale bar, 20 μm. (B) Western blot of TEM1 fusions expressed in purified R. parkeri strains. Lane 1, two TEM1 bands represent uncleaved and processed species. (C) TEM1 assay response ratio (blue:green). Data are mean ± SD, one-way ANOVA: *** p < 0.001 relative to uninfected, which is set to 1. (D) Images of individual bacteria after infection of A549 cells, showing non-secreted (top row) or secreted Sca4 (bottom 2 rows). Scale bar, 1 μm. (E) Images of secreted Sca4 in cells infected with WT, sca4::tn or sca2::tn. Maximum intensity projections shown. Scale bar, 10 μm. Arrows indicate Sca4 that is localized near a bacterium.
Analysis with SignalP 4.0 software (Petersen et al., 2011) revealed R. parkeri Sca4 lacks a classical N-terminal Sec or Tat signal sequence. Secretion may thus be directed by an alternative secretion system, such as the Type I (T1SS) or Type IV secretion systems (T4SS) (Gillespie et al., 2015). Although T1SS and T4SS effectors lack strong consensus signal sequences, such sequences are often at the C-terminus (Cambronne and Roy, 2006; Delepelaire, 2004). Consequently, we tested whether the C-terminal 20 amino acids (aa) of Sca4 are necessary and/or sufficient for secretion. TEM1*-Sca4 harboring a deletion of this C-terminal sequence (TEM1*-Sca4Δ20) was not secreted by R. parkeri, despite being expressed at similar levels as TEM1*-Sca4 (Figures 3B and 3C). However, fusing this 20 aa sequence to TEM1* was not sufficient for secretion (Figures 3B and 3C). Thus, the C-terminal sequence is necessary but not sufficient for secretion, and additional sequences also participate in this process.
To confirm whether endogenous Sca4 is secreted and determine where it localizes in the host cell, we used a differential staining protocol to distinguish secreted proteins from those residing in the bacterial cytosol (Campbell-Valois et al.) (Figure 3D). All WT bacteria displayed Sca4 staining in the bacterial cytosol, whereas only 8 ± 4% of WT bacteria were associated with secreted Sca4 (Figure 3D; bottom 2 rows). The sca4::tn mutant displayed a significantly weaker Sca4 signal in the bacterial cytosol (data not shown) and a much lower fraction was associated with secreted Sca4 (0.6 ± 0.5%), confirming the specificity of the Sca4 antibody. Most of the secreted Sca4 signal was not associated with bacteria, but rather appeared as puncta throughout the host cell cytosol, with no clear pattern emerging (Figure 3E). These puncta were absent when cells were infected with the sca4::tn mutant (Figure 3E). These results confirmed that R. parkeri secretes Sca4 and further suggested Sca4 may target a host protein to promote spread.
Sca4 inhibits vinculin to promote spread
The presence of two VBSs in Sca4 and the known role for vinculin at cell-cell junctions suggested that Sca4 might target vinculin to promote spread. The VBSs in R. parkeri Sca4 are nearly identical to the corresponding sequences in R. rickettsii Sca4 and share significant similarity with VBS sequences in mammalian talin-1 or S. flexneri IpaA2 (Figure 4A). Although R. rickettsii Sca4 binds to the vinculin head (Vh1) domain (residues 1–258) (Park et al., 2011a), the role of this interaction during infection was not reported. To test the functional importance of the Sca4:vinculin interaction, we designed point mutations in both VBSs based on the crystal structure of R. rickettsii Sca4 (Park et al., 2011a) and known VBS mutants from talin-1 (Gingras, 2005) or IpaA2 (Izard et al., 2006; Park et al., 2011b). We then tested whether the VBS-NC* mutant (L415A, S416E, Y814I, V820E) disrupted vinculin binding using a co-immunoprecipitation (co-IP) assay with exogenously expressed vinculin head region (1-836aa) in HEK293T cells. We found that WT Sca4 interacted with HA-vinculin 1-836aa, whereas the VBS-NC* mutant did not (Figure 4B). This confirmed that WT R. parkeri Sca4 binds to vinculin, and that binding requires the VBSs.
Figure 4. Sca4 targets vinculin to promote spread.

(A) Sequence alignments of the indicated VBSs (black, identical residues; grey, similar residues). VBS-NC* mutant amino acid substitutions are indicated in red. (B) Co-IP between FLAG-tagged Sca4 (WT and VBS-NC*) and the 1–836aa truncation of vinculin. IP, immunoprecipitate; IB, immunoblot. (C) Western blot showing expression levels of Sca4 in indicated purified R. parkeri strains. (D) Infectious focus size formed by WT (Rp-GFP), sca4::tn, sca4::tn + pFl-Sca4 (pRAM18dSGA-Sca4pr-Flag-Sca4) and sca4::tn + pFl-Sca4 VBS-NC* strains. Mean ± SEM, unpaired T-test: **** p < 0.0001 relative to WT. (E) Infectious focus size after reverse transfection with control siRNA (nontarget, NT) or vinculin-specific siRNAs (siVCL #1, #2) and infection with WT (Rp-GFP) or sca4::tn. Mean ± SEM, unpaired T-test: **** p < 0.0001 relative to WT + NT. (F) Western blot of A549 lysates from (E) (GAPDH, loading control). Space in blots indicates deletion of irrelevant lane.
To test whether the Sca4:vinculin interaction was necessary to promote spread, we expressed FLAG-tagged WT or VBS-NC* Sca4 in the sca4::tn mutant using the pRAM18dSGA plasmid and quantified spread via the infectious focus assay. The VBS-NC* mutant did not rescue the spread defect for the sca4::tn mutant, despite being expressed at equivalent levels to WT FLAG-Sca4 (Figures 4C and 4D). Thus, Sca4 must bind to vinculin to promote spread.
Based on our data, it was unclear whether vinculin was a positive or negative regulator of spread. We reasoned that if vinculin were a positive regulator, Sca4 would likely activate vinculin. In contrast, if vinculin were a negative regulator, Sca4 would inhibit vinculin to promote spread. We tested this by RNAi-mediated silencing of vinculin expression to see whether loss of vinculin inhibited WT spread (supporting an activating role) or rescued the spread defect of the sca4::tn mutant (supporting an inhibitory role). Consistent with the latter pathway, we found that RNAi-mediated silencing of vinculin by two different siRNAs rescued the sca4::tn mutant spread defect, but did not affect WT spread (Figures 4E and 4F). The lack of an effect on WT spread is expected if Sca4 inhibits vinculin activity, as a loss of vinculin expression would not further improve spread. Overall, our findings suggest secreted Sca4 binds to vinculin and inhibits its activity to promote spread.
Sca4 acts in trans to target vinculin in the donor cell
We next examined whether Sca4 targets vinculin in the donor or recipient cell using a mixed cell infectious focus assay (Figures 5A and 5B). In this assay, the levels of vinculin and Sca4 expression were altered independently in donor or recipient A549 cells using RNAi-mediated silencing of vinculin or lentiviral-mediated stable expression of Sca4 (via FCW2-P2AT-Ires-Sca4) (Figure S2E). Spread was quantified as the percentage of bacteria found in recipient cells. Silencing vinculin expression in the donor cells rescued the spread defect of the sca4::tn mutant (Figure 5C), whereas silencing vinculin expression in the recipient cells had no effect on spread of WT or sca4::tn mutant bacteria (Figure 5C). We also observed that exogenous stable expression of Sca4 in the donor cell trans-complemented the spread defect of the sca4::tn mutant, whereas Sca4 expression in the recipient cell did not (Figure 5D). This evidence strongly suggests Sca4 inhibits vinculin activity in the donor cell.
Figure 5. Sca4 acts in trans to target vinculin in the donor cell.

(A) Mixed cell assay schematic, as described in methods. (B) Images of infectious foci detected in the mixed cell assay showing A549-TRT (soluble TagRFP-T, red), R. parkeri (green) and β-catenin (blue). Scale bar, 10 μm. (C) Mixed cell assay results showing percentage of bacteria that spread to recipient cells in a focus. Donor or recipient cells were reverse transfected with control siRNA (NT) or siVCL #1 and infected with WT (Rp-GFP) or sca4::tn. Mean ± SEM, one-way ANOVA: ** p < 0.01 relative to WT + NT in each set. (D) Mixed cell assay results as in (C) except indicated donors or recipients were transduced with FCW2-P2AT (control) or FCW2-P2AT-Ires-Sca4 (Sca4). One-way ANOVA: *** p < 0.001 relative to WT + P2AT in each set. (E and F) Mixed cell assay as in (A, B) except the donor cell was infected with WT (Rp-GFP), sca4::tn or a mixture of sca4::tn and sca2::tn. (E) Image of an infectious focus co-infected with sca2::tn (bright green, open arrow) and sca4::tn (dim green, solid arrow) and counterstained for Rickettsia (blue) and β-catenin (red). Scale bar, 10 μm. Inset, expanded view of donor cell region indicated by solid arrow (inset scale bar, 5 μm). (F) Percentage of bacteria that spread for WT (Rp-GFP), sca4::tn, or sca4::tn in the presence of sca2::tn. No spread was detected for sca2::tn. Mean ± SEM, one-way ANOVA: * p < 0.05 relative to WT. See Figure S2.
Because Sca4 and vinculin act in the donor cell to promote protrusion engulfment, we initially hypothesized Sca4 might be secreted when bacteria are in protrusions. However, we did not observe specific localization of Sca4 in protrusions and instead, saw puncta throughout the cytosol of infected cells (Figure 3E). This suggested secreted Sca4 might diffuse to its site of action. To test this hypothesis, we sought to trans-complement the sca4::tn mutant phenotype by co-infecting cells with a sca2::tn mutant strain, which secretes Sca4 (Figure 3E) but is unable to move to the cell surface and spread due to an inability to form late actin tails (Reed et al., 2014). To distinguish between sca2::tn and sca4::tn strains, the sca2::tn mutant was transformed with pRAM18dSGA-OmpApr-GFPuv, causing it to fluoresce more brightly than untransformed, dimmer sca4::tn bacteria (Figure 5E). Donor A549-TRT cells were infected with WT or sca4::tn strains, or co-infected with sca4::tn and sca2::tn and then used in the mixed cell infectious focus assay. Strikingly, co-infection with the sca2::tn mutant was sufficient to trans-complement the spread defect of the sca4::tn mutant, as dimmer sca4::tn bacteria were seen in recipient cells (Figures 5E and 5F). These data suggest Sca4 is secreted into the donor cell cytosol where it diffuses to inhibit vinculin at cell-cell junctions and promote spread.
Sca4 disrupts donor cell adherens junction complexes to relieve tension and promote spread
We next investigated how Sca4 inhibits vinculin function. At adherens junctions, vinculin is recruited by α-catenin where it participates in actin-mediated reciprocal pulling forces to stabilize junctions (Huveneers and de Rooij, 2013). Notably, α-catenin binds to the Vh1 domain of vinculin (Weiss et al., 1998), which is the same region targeted by Sca4 (Park et al., 2011a). This suggested that Sca4 might inhibit vinculin function by competing with α-catenin for vinculin association. We tested this using a co-IP assay and detected a robust interaction between myc-tagged α-catenin and HA-tagged vinculin 1-836 (Figure 6A). However, when FLAG-tagged Sca4 was co-expressed at increasing levels, there was a dose-dependent inhibition of the vinculin:α-catenin interaction (Figure 6A). Importantly, high concentrations of the Sca4-VBS-NC* mutant did not displace vinculin (Figure 6A), illustrating that this competition required Sca4:vinculin binding. Sca4 was not isolated in co-IPs with myc-α-catenin, suggesting these two proteins do not interact. Our data supports a model wherein Sca4 binds vinculin and competes with vinculin-binding partners such as α-catenin to inhibit vinculin function. This may weaken interactions between adherens junction complexes and the actomyosin network in the donor cell and reduce intercellular tension to make Rickettsia spread more efficient (Figure 6B).
Figure 6. Sca4 disrupts donor cell adherens junction complexes to relieve tension and promote spread.

(A) Competition co-IP assay between vinculin (HA-vinc 1-836aa), myc-α-catenin and FLAG-Sca4. (B) Model of Sca4 inhibition of vinculin function. In uninfected cells, vinculin and α-catenin promote junction stability via reciprocal actin-mediated pulling forces. Sca4 disrupts the vinculin:α-catenin interaction, thus reducing donor cell actomyosin tension and increasing membrane flexibility to improve spread efficiency. (C) Infectious focus size after 20 h blebbistatin (6.25 μm-25 μM) or DMSO (control) treatment. Mean ± SEM, unpaired T-test: ** p < 0.01 relative to WT + DMSO.
If Sca4 reduces tension by interfering with the actomyosin network, then inhibition of myosin-II activity should rescue the sca4::tn mutant spread defect. To test this, we measured spread efficiency with the infectious focus assay in the presence of blebbistatin, a myosin-II inhibitor. Blebbistatin treatment fully rescued the spread defect of the sca4::tn mutant but did not affect spread of WT bacteria (Figure 6C), suggesting that reducing actomyosin contractility is necessary for efficient spread and that Sca4 may function by reducing intercellular tension.
Sca4 reduces intercellular tension in a vinculin-dependent manner
To further test if Sca4 affects intercellular tension, we directly measured the forces at cell-cell contacts mediated by E-cadherin. We stably expressed Sca4 and Sca4-VBS-NC* proteins in U2OS cells (via FCW2-P2AT-Ires-Sca4 or FCW2-P2AT-Ires-Sca4-VBS-NC*; control, FCW2-P2AT) and confirmed Sca4 was expressed and partially colocalized with F-actin and vinculin (Figure S2A–S2D), as was shown for R. rickettsii Sca4 (Park et al., 2011a). We then performed traction force microscopy (TFM) with cells adherent to polyacrylamide gels coated with E-cadherin-Fc chimeras to mimic cell-cell junctions (Barry et al., 2014). In TFM, the gel on which cells adhere is embedded with fluorescent beads. When cells tug on and deform the gel, they cause bead displacements, which are then used to calculate strain energy (mechanical work) generated by the cells using an in-house Fourier traction force microscopy method (Bastounis et al., 2011; del Álamo et al., 2007). Cells overexpressing Sca4 exerted reduced traction stresses compared with control cells, as shown by instantaneous maps illustrating the magnitude of traction stresses (Figure 7A). The time averaged strain energy was then calculated for multiple cells across several experiments. Sca4-expressing cells generated reduced strain energy compared to control cells (Figure 7B), suggesting Sca4 disabled force transduction at E-cadherin sites. Importantly, expression of the VBS-NC* mutant did not reduce traction stresses or strain energy (Figures 7A and 7B). Thus, Sca4 reduces force transduction at E-cadherin sites, and this is dependent on its ability to interact with vinculin.
Figure 7. Sca4 modulates E-cadherin forces in a vinculin-dependent manner.

TFM results for individual cells adherent to polyacrylamide gels coated with E-cadherin-Fc (A, B) or collagen I (C, D). (A, C) Instantaneous maps showing the magnitude of traction stresses (color indicates stress values in Pa). Cell outlines in white. (B, D) Time-averaged strain energy (nN μm) from individual cells. (E) Model depicting how biomechanical crosstalk allows measurements of traction stresses at cell-ECM junctions to be converted to tension. Cells adhere to a gel containing green fluorescent beads in upper layer. Actin cables (red) connect E-cadherin (orange) and integrin (blue) adhesions. Sca4 specifically targets cell-cell junctions. (F) MSM results for cell monolayers adherent to gels coated with collagen I. Images show the distribution of tension (right) and cells as seen by phase contrast (left). (G) Time-averaged monolayer tension (nN μm−1) calculated for multiple regions within the monolayer. Mann-Whitney rank sum T-test: * p < 0.05, ** p < 0.01, **** p < 0.0001. See Figure S2.
Vinculin is also a critical regulator of force transduction at focal adhesions (Atherton et al., 2015; Peng et al., 2011). We therefore tested whether Sca4 could manipulate cell-extracellular matrix (ECM) forces using TFM of single cells adhered to collagen I-coated gels. Surprisingly, overexpression of Sca4 or Sca4-VBS-NC* did not reduce cell-ECM traction stresses or strain energy (Figures 7C and 7D). This suggests Sca4 does not manipulate cell-ECM adhesions but directly targets cell-cell junctions (Figure 7E).
Cells within a tissue are mechanically linked to one another and to the underlying ECM through adhesion complexes and the cytoskeleton (Figure 7E). This creates considerable biomechanical crosstalk that regulates cell shape, structure and function (Weber et al., 2011). We reasoned that the Sca4-mediated force reduction observed for single cells adherent to E-cadherin would translate to reduced tension between cells in a monolayer. To measure this, we performed TFM with monolayers adhered to collagen I-coated gels and calculated tension using monolayer stress microscopy (MSM) (Banerjee et al., 2015; Tambe et al., 2013). MSM assumes a balance of forces mediated by the biomechanical crosstalk in cells and uses traction stresses exerted at cell-ECM contact sites to calculate tension throughout the monolayer. Using this method, a significant reduction in monolayer tension was observed for Sca4-expressing cells compared to controls (Figures 7F and 7G). In contrast, the Sca4-VBS-NC* mutant did not reduce monolayer tension (Figures 7F and 7G), suggesting Sca4’s effect on monolayer tension requires its ability to bind to vinculin. Overall, our data support a model in which Sca4 targets vinculin in the donor cell to directly modulate intercellular tension forces, but not cell-ECM forces, to facilitate efficient spread.
DISCUSSION
Cell-to-cell spread is critical for bacterial dissemination throughout host tissues and avoidance of immune detection. For pathogens that undergo actin-based motility, it was assumed that forces from motility are harnessed to drive spread. We report the unexpected discovery that R. parkeri instead uses the secreted effector Sca4 to modulate protrusion engulfment by manipulating vinculin-dependent intercellular tension. This challenges long-held views of the role of motility in spread and reveals mechanisms for force manipulation to enable cell-to-cell bacterial transfer.
Despite the general importance of spread in pathogenicity, we know little about the comparative parameters and molecular mechanisms of spread for different bacterial species. We discovered that, in comparison with L. monocytogenes, R. parkeri generate shorter protrusions that resolve more quickly into vesicles in recipient cells. Notably, the vast majority of R. parkeri in protrusions were not associated with actin tails. Thus, unlike L. monocytogenes (Kuehl et al., 2015; Talman et al., 2014), R. parkeri may not harness the pushing forces derived from actin assembly to distend the host membrane. This suggests that R. parkeri uses alternate mechanisms to promote protrusion dynamics.
The fact that R. parkeri and L. monocytogenes exhibit distinct spread parameters indicates these pathogens orchestrate the process of spread to a greater extent than previously appreciated, likely through the contribution of bacterial factors. In agreement with this, we identified Sca4 as a protein from SFG rickettsia that promotes spread. The sca4::tn mutant displayed a specific and significant delay in protrusion resolution, which is noteworthy because only one other bacterial factor – the L. monocytogenes metalloprotease Mpl (Alvarez and Agaisse, 2016) – was recently proposed to regulate protrusion resolution. Though a mutant in the S. flexneri T3SS is defective in protrusion resolution, a specific bacterial secreted factor that acts in this process has not been identified (Kuehl et al., 2014).
If Sca4 directly modulates spread, it should be secreted from bacteria to target host cell factors. We provide direct and quantitative evidence that Sca4 is secreted from bacteria and that its function in spread is restricted to the donor cell. Interestingly, Sca4 may diffuse to its site of action, a notion that is supported by the ability of co-infected bacteria to provide Sca4 and promote spread of the sca4::tn mutant. Therefore, we hypothesize Sca4 may exert global effects on the donor cell that are not restricted to the local environment of the protrusion.
R. rickettsii Sca4 was proposed to activate vinculin by disrupting its autoinhibitory conformation (Park et al., 2011a), suggesting that vinculin might promote spread. R. parkeri Sca4 also interacts with vinculin. However, we discovered that vinculin limits spread, and R. parkeri Sca4 is needed to repress this inhibition. This suggests that Sca4 may regulate the conformational switch in vinculin, but this may lead to inhibition rather than activation of vinculin function in cells.
Sca4 binds to the same vinculin site as α-catenin (Park et al., 2011a; Weiss et al., 1998), and Sca4 competes with α-catenin for vinculin binding. Although this competition might displace vinculin from adherens junctions, the localization of vinculin to cell-cell or cell-ECM junctions was not dramatically reduced in the presence of exogenously expressed Sca4. This suggests that Sca4 exerts a subtler effect on vinculin function. In developing focal adhesions, specific protein components and interactions are spatially segregated at the nanoscale level to regulate vinculin function (Case et al., 2015). Although it is unclear if a similar level of regulation exists in adherens junctions, we speculate that Sca4 might fine-tune vinculin activity to promote bacterial spread without displacing vinculin or destroying junction and tissue integrity.
Cadherin junctions under tension recruit vinculin to promote actin remodeling, which leads to junction stabilization and cell stiffening (Huveneers and de Rooij, 2013). We show that reducing actomyosin contractility rescues the spread defect of the sca4::tn mutant and Sca4 expression decreases vinculin-dependent mechanotransduction at E-cadherin junctions. Interestingly, Sca4 does not reduce integrin-mediated traction forces, suggesting Sca4 specifically modulates intercellular tension and could be a valuable tool for dissecting vinculin function at cadherin junctions. Taken together with our data supporting a role for Sca4 and vinculin in the donor cell, we propose that Sca4 relieves intercellular tension by disrupting donor cell vinculin:α-catenin interactions. This ultimately weakens the interactions between cadherin complexes and the actomyosin network in the donor cell. To satisfy equilibrium of forces between donor and recipient cells, the resulting difference in actomyosin tension is compensated for by an increase in membrane curvature that enhances protrusion engulfment efficiency by facilitating closure of the protrusion into a vesicle.
It is interesting that R. parkeri Sca4 is not required for protrusion initiation, but reduces tension to promote protrusion engulfment. This suggests that other rickettsial factors are needed to promote protrusion formation. In contrast, L. monocytogenes InlC is proposed to reduce tension to promote protrusion initiation (Rajabian et al., 2009), and the force from actin-based motility may be sufficient to promote engulfment. Thus, these pathogens have evolved different strategies to manipulate the cellular force generating machinery to form and resolve protrusions during spread.
Our discovery of a rickettsial effector that specifically regulates one step of spread underscores the notion that this multistep process is mediated by the coordinated action of multiple bacterial effectors. Studying how these factors target host pathways will reveal important interactions that regulate pathogenesis. Previous work has highlighted that bacteria engage different classes of host proteins during spread, including cytoskeletal regulators (Tuba, N-WASP, Arp2/3, Ezrin, diaphanous-related formins, myosin II, myosin X and MLCK), signaling (PIK3C2A, STK11) and cell adhesion proteins (E-cadherin, tricellulin, connexin 26) (Kuehl et al., 2015). However, how these are targeted by each pathogen is still poorly understood. Thus, continued investigation of the bacterial effectors and host targets that mediate spread will enhance our understanding of this crucial mechanism of virulence and reveal cellular pathways that are exploited by pathogens during infection.
METHODS and RESOURCES
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents may be directed to and will be fulfilled by the Lead Contact Matthew Welch welch@berkeley.edu (M.D.W.).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
Mammalian cell lines (Vero, HEK293T, A549 and U2OS) were obtained from the University of California, Berkeley tissue culture facility and grown at 37°C in 5% CO2 Vero cells (monkey, kidney epithelial) were maintained in DMEM (Invitrogen) containing 5% fetal bovine serum (FBS). HEK293T (human, embryonic kidney), A549 (human, lung epithelial), and U2OS (human, osteosarcoma) cells were maintained in DMEM containing 10% FBS. HMEC-1 (human, microvascular endothelial) cells were maintained in MCDB 131 (Invitrogen) with 10% FBS (Hyclone), 2 mM L-glutamine (Gibco), 10 ng/ml epidermal growth factor (BD Biosciences), and 1 μg/ml hydrocortisone (Sigma).
A549 cells stably expressing a farnesylated TagRFP-T fluorophore (A549-TRTF) to mark the plasma membrane were generated using retroviral transduction. Briefly, the 20 amino acid (aa) farnesylation signal from c-HA-Ras (from pEGFP-F Clontech vector) was fused to the C-terminus of TagRFP-T, then cloned into pCLIP2B, a murine moloney leukemia virus derived from pCLIP (Pomerantz et al., 2002). pCLIP2B-TagRFP-T-F virus particles were packaged by transfecting HEK293T cells in 24-well plates as previously described (Lamason et al., 2010), using plasmids kindly provided by Dr. Joel Pomerantz (Johns Hopkins University School of Medicine). Cells were selected with 1.5 μg/ml puromycin and sorted for maximum fluorescence using a Cytopeia INFLUX Sorter (UC Berkeley Flow Cytometry Facility). A549 cells stably expressing a soluble TagRFP-T fluorophore (A549-TRT) were generated as above, except the farnesyl sequence was removed resulting in diffuse TagRFP-T localization (pCLIP2B-TagRFP-T).
To visualize F-actin in A549 cells, the lentiviral vector FCW2IB-Lifeact-3xTagBFP was constructed. First, the Ubiquitin C promoter from FUW2 (Lamason et al., 2010) (kindly provided by Dr. Joel Pomerantz) was replaced with the CMV promoter from pEGFPN1 (Clontech), and an IRES-blastS cassette was added. Next, a fusion between Lifeact and 3 copies of TagBFP (Evrogen) were inserted upstream of the IRES. FCW2IB-Lifeact-mWasabi was made similarly except a single copy of mWasabi was fused to Lifeact. Viral particles were packaged by transfecting HEK293Ts plated 24 h previously at 9×104 cells/well (0.5 ml/well, 24-well plate), via calcium phosphate with 95 ng pMDL-RRE, 95 ng pCMV-VSVg, 95 ng RSV-Rev and 95 ng FCW2IB-Lifeact-3xTagBFP (or mWasabi). Approximately 22 h after transfection, the media was replaced with 300 μl fresh media. After an additional 21 h, the supernatant was collected from each well, cell debris was cleared by filtration through a 0.45 μm syringe filter, and the viral supernatant was added to A549 cells (for Lifeact-2xTagBFP) or A549-TRTF cells (for Lifeact-mWasabi). Fresh media was added 24 hpi, and at 48 hpi, cells were resuspended in fresh media containing 12 μg/ml blasticidin.
For stable expression of Sca4 in A549 and U2OS cells, the lentivirus FCW2-P2AT-Ires-Sca4 was constructed. The sca4 sequence was codon-optimized for expression in human cells and synthesized (GeneArt/Life Technologies) before cloning. The VBS-NC* mutations were introduced via site-directed mutagenesis. To minimize excessive overexpression of Sca4, the sca4 sequence was cloned downstream of an IRES, and a fusion between puro, the self-cleaving peptide 2A and TagRFP-T was inserted upstream of the IRES. This allowed the 3 proteins to be expressed as separate polypeptides. For control cells, the FCW2-P2AT vector was made without the IRES-Sca4 region. Viral particles were packaged by transfecting HEK293Ts, plated 24 h previously at 5×105 cells/well (2 ml/well, 6-well plate), via calcium phosphate with 400 ng pMDL-RRE, 400 ng pCMV-VSVg, 400 ng RSV-Rev and 800 ng lentiviral vector. Approximately 22 h after transfection, the media was replaced with 1.5 ml fresh media. After an additional 21 h the supernatant was collected, cell debris was cleared by filtration through a 0.45 μm syringe filter, and viral supernatant was added to A549 or U2OS cells. Fresh media was added 24 hpi, and 48 hpi cells were resuspended in fresh media containing 1.5 μg/ml puromycin. Cells were sorted using a Cytopeia INFLUX Sorter (UC Berkeley Flow Cytometry Facility).
Bacterial plasmids and strains used in this study
R. parkeri Portsmouth strain was a gift from Dr. Chris Paddock (Centers for Disease Control and Prevention). GFP-expressing L. monocytogenes (Shen and Higgins, 2005) and TagBFP-expressing L. monocytogenes (Lm-TagBFP) were kindly provided by Dr. Daniel Portnoy (UC Berkeley). To generate the TagBFP-expressing L. monocytogenes (Lm-TagBFP), codon optimized TagBFP DNA was fused to a constitutive promoter Phyper derived from GFP-pPL3 plasmid (Gründling et al., 2004) using splicing by overlap extension PCR. The PCR product was cloned into pPL2 plasmid to generate Phyper -TagBFP-pPL2. The resulting plasmid was introduced into Listeria monocytogenes 10403s strain to generate constitutive TagBFP expressing L. monocytogenes strain DPL-7742.
To make the sca4 complementation plasmid (pRAM18dSGA-Sca4), nucleotides 652,261 – 655,741 (GenBank/NCBI accession NC_017044.1) were amplified from R. parkeri genomic DNA and cloned into pRAM18dSGA[MCS]. This region includes a predicted promoter (determined using SoftBerry, BPROM prediction of bacterial promoters (Solovyev and Salamov, 2011)) and several predicted transcriptional terminators (determined using WebGeSTer DB (Mitra et al., 2011).
The vinculin binding site mutations of sca4 were introduced via site-directed mutagenesis to produce Sca4 VBS-NC* (L415A, S416E, Y814I, V820E). For Figure 4C and 4D, Flag-tagged versions of WT sca4 and the sca4 VBS-NC* mutant in pRAM18dSGA were made by fusing the Sca4 promoter (nt 652,261 – 652,548), an NdeI site and a Flag sequence to the 5′ end of the sca4 gene (nt 652,555–655,623). The ompA transcriptional terminator from pMW1650 was added to the 3′ end. The Flag sequence was codon optimized for expression in Rickettsia conorii (5′-GAT TAT AAA GAT GAT GAT AAA -3′) using the JCat online codon optimization tool (Grote et al., 2005).
To generate brighter versions of the single GFP copy pRAM18dRGA[MCS] (Burkhardt et al., 2011) and pRAM18dSGA[MCS] plasmids (kindly provided by Dr. Ulrike Munderloh, University of Minnesota), a second copy of the ompA promoter + GFPuv cassette was amplified from pMW1650 and cloned into each respective multi-cloning site (MCS). This modification increased bacteria brightness at least 4 fold (data not shown). Rp-GFP refers to wild-type (WT) R. parkeri transformed with pRAM18dRGA+OmpApr-GFPuv. The pRAM18dSGA+OmpApr-GFPuv plasmid was stably introduced into the sca2::tn strain (Reed et al., 2014), as detailed below.
To make 2xTagBFP-expressing R. parkeri, the TagBFP sequence was first codon-optimized via JCat for R. conorii expression (Grote et al., 2005). The ompA promoter was fused to TagBFP followed by the ompA transcriptional terminator. To increase brightness, two TagBFP cassettes were cloned into the MCS of pRAM18dRA[MCS] (a derivative of pRAM18dRGA lacking the GFP cassette) to make pRAM18dRA-2xTagBFP. Rp-2xTagBFP refers to WT R. parkeri transformed with pRAM18dRA-2xTagBFP.
To generate the beta-lactamase TEM1 fusion constructs for Figure 3, the full-length beta-lactamase (bla) gene sequence was codon optimized via JCat for R. conorii expression (Grote et al., 2005), synthesized by Integrated DNA Technologies and cloned into the MCS of pRAM18dRA. The ompA promoter from pMW1650 was used to drive expression of all TEM1 constructs. To generate the negative control TEM1 construct (TEM1*), the first 23 amino acids (aa) were deleted to prevent constitutive secretion (Charpentier and Oswald, 2004). TEM1* was then fused to the N-terminus of Sca4, Sca4Δ20 (deletion of last 20aa) and Sca4-SS (fusion between TEM1* and last 20aa of Sca4).
All pRAM-containing R. parkeri strains were generated using small-scale electroporations described below, except the final, washed bacterial pellets were resuspended in 100 μl cold 250 mM sucrose, mixed with 10 μg plasmid DNA, and electroporated at 2.5 kV, 200 ohms, 25 μF, 5 ms in a 0.2 cm cuvette, using a Gene Pulser Xcell (Bio-Rad). Transformants were selected with either 200 ng/ml rifampicin or 50 μg/ml spectinomycin and purified via 30% MD-76R solution.
METHOD DETAILS
Purification of R. parkeri strains
Media was aspirated from confluent flasks of Vero cells, and cells were infected with R. parkeri resuspended in serum free DMEM (2 ml per T175 cm2 flask with ~1–5×106 plaque forming units (pfu) per flask). Flasks were infected for 30 min at 37°C with rocking. 33 ml DMEM with 2% FBS was then added to the flask before being moved to a 33°C incubator. Plaques began to form 3–5 days post infection, and infected cells were scraped and collected at 4–7 days post infection when 80–90% of cells showed a rounded up phenotype (indicative of infection). Scraped cells were centrifuged at 12,000 × g for 30 min at 4°C. Pelleted cells were then resuspended in K-36 Buffer (0.05M KH2PO4, 0.05M K2HPO4, 0.1M KCl, 0.015M NaCl, pH 7) and transferred to a glass dounce homogenizer. Rickettsiae were released from infected cells by repeated douncing (40–60 strokes). The dounced solution was then centrifuged at 200 × g for 5 min at 4°C to pellet host cell debris. The supernatant containing R. parkeri was overlaid on a 30% MD-76R (Merry X-Ray) solution. Gradients were then centrifuged at 18,000 rpm in an SW-28 ultracentrifuge swinging bucket rotor for 30 min at 4°C to separate remaining host cells debris from the bacteria. Bacterial pellets were resuspended in brain heart infusion (BHI) media (Difco) and stored at −80°C.
Titers were determined for R. parkeri stocks via plaque assays. The media from confluent Vero cells in 6-well plates was aspirated, and 100 μl/well of R. parkeri diluted in BHI was added. Plates were rocked for 30 min at 37°C, and each well was then overlaid with 4 ml/well DMEM with 2% FBS and 0.5% agarose. Plaques were counted 4–6 dpi to determine pfu/ml. To measure plaque size, an overlay of neutral red solution (final 0.01% per well; Sigma) diluted in PBS + 0.5% agarose was added at about 4 dpi and incubated about 6 h to overnight until plaques were clearly visible. Images were taken on an Alpha Innotech Gel Imager, and plaque area was measured in ImageJ.
Growth curves in Vero cells were done as described previously (Reed et al., 2014). Briefly, QPCR was used to measure Rickettsia genome equivalents using primers to the 17 kDa antigen gene (Grasperge et al., 2012), relative to a standard curve of 17 kDa gene DNA. Results from each strain were normalized to the 1 hpi time point, and fold change calculated.
Transposon mutagenesis screen in R. parkeri
The pMW1650 mariner-based Himar1 transposon mutagenesis system (a gift from Dr. David Wood, University of South Alabama) was used to generate small plaque mutants. The transposon cassette contains the R. prowazekii arr-2 rifampin resistance gene and a gene coding for a green fluorescent protein (GFPuv) (Liu et al., 2007). To isolate small plaque mutants, a small-scale electroporation protocol was designed. First, a T75 cm2 flask of confluent Vero cells was infected with WT R. parkeri. Three days post infection, Vero cells were at least 90% rounded up and were scraped from the flask. Infected cells were spun down for 5 min at 1800 × g at 4°C and resuspended in 3–6 ml K-36 buffer. To mechanically disrupt infected cells and release the bacteria within, cells were either passed through a 27.5 gauge syringe needle 10 times or vortexed (~2900 rpm, Vortex Genie 2) in a 15 ml conical tube containing 2 g of 1 mm glass beads with two 30 s pulses with 30 s incubations in ice after each pulse. This bead disruption procedure was adopted for a majority of the screen. Host cell debris was pelleted for 5 min at 200 × g at 4°C. The supernatant containing R. parkeri was transferred to 1.5 ml Eppendorf microcentrifuge tubes and spun down for 2 min at 9000 × g at 4°C. The bacterial pellets were washed three times in cold 250 mM sucrose. Washed bacterial pellets were resuspended in 50 μl cold 250 mM sucrose, mixed with 1 μg of the pMW1650 plasmid, placed in a 0.1 cm cuvette, and electroporated at 1.8 kV, 200 ohms, 25 μF, 5 ms using a Gene Pulser Xcell (Bio-Rad). Bacteria were immediately recovered in 1.2 ml BHI. To infect Vero cells in 6-well plates, media was removed from each well, and cells were washed with 1x PBS. 100 μl of electroporated bacteria was added per well, and plates were placed in a humidified chamber and rocked for 30 min at 37°C. An overlay of DMEM with 5% FBS and 0.5% agarose was added to each well. Infected cells were incubated at 33°C, 5% CO2 for 24 h at which point a second overlay was added containing rifampicin (final concentration 200 ng/ml; Sigma) to select for transformants. After 5 d, plaques were visible by eye in the cell monolayer, and smaller plaques relative to neighboring plaques were selected for further analysis.
To isolate and amplify the mutants for mapping, small plaques were picked, resuspended in 200 μl of BHI and used to infect Vero cells in 6-well plates. Media was aspirated from Vero cells, and the isolated plaque resuspension was used to infect the cells at 37°C for 30 min with rocking. Then 3 ml DMEM with 2% FBS and 200 ng/ml rifampicin was added to each well, and infections progressed until monolayers were fully infected. Infected cells were isolated as described above using mechanical disruption, except the bacteria were immediately resuspended in BHI without a sucrose wash and stored at −80°C.
To map the transposon insertion sites, semi-random nested PCR was done as previously described (Reed et al., 2014). Genomic locations were determined using BLAST against the R. parkeri strain Portsmouth genome (GenBank/NCBI accession NC_017044.1). Mapped small plaque mutants were further purified via a second round of plaque purification before final expansion and purification via the 30% MD-76R solution protocol described above. Isolation of sca2::tn was previously described (Reed et al., 2014), and the pRAM18dSGA+OmpApr-GFPuv plasmid was stably introduced into this strain, as detailed above.
Sca4 protein purification, Sca4 antibody production, and Sca4 immunoblotting
Full-length sca4 was PCR-amplified from the pRAMdSGA+Sca4 plasmid and cloned into a version of the pSMT3 expression vector (Winger et al., 2008) containing a 6xHis tag upstream of the SUMO tag. 6xHis-SUMO-Sca4 was expressed in E. coli BL21 (DE3) T1R, and protein expression was induced with 0.5 mM IPTG at 30°C overnight. Pelleted cells were resuspended in 20 mM Tris pH 8, 50 mM KCl, 10% Glycerol, and protease inhibitors (PMSF; leupeptin, pepstatin, chymostatin [EMD Millipore]) and stored at −80°C. Cells were thawed, sonicated at 4°C (3 × 30 s pulses, 60% power), and lysates were cleared by centrifugation at 12,000 × g at 4°C for 30 min. Ni-NTA resin (Qiagen) was equilibrated in wash buffer (50 mM NaH2PO4, 300 mM NaCl, 5 mM Imidazole pH 8), and the supernatant was batch bound at room temperature for 5 min. The resin was washed in wash buffer and eluted with 50 mM NaH2PO4, 300 mM NaCl, 500 mM Imidazole pH 8. 1 mM DTT was added to the elution, and the SUMO tag was cleaved via ULP1 overnight at 4°C while dialyzing into 20 mM Tris pH 8, 150 mM NaCl, 1 mM DTT. Dialyzed, cleaved protein was then concentrated and purified via gel filtration chromatography on a Superdex 200 10/300 GL column (GE Healthcare) into 20 mM Tris pH 8, 150 mM NaCl, 1 mM DTT. Pooled peak fractions were diluted for a final buffer composition of 20 mM Tris pH 8, 50 mM NaCl, 1 mM DTT and further purified via anion exchange on a HiTrap Q column (GE Healthcare). Final storage buffer for collected fractions was 20 mM Tris pH 8, 375 mM NaCl, 1 mM DTT, and all proteins were concentrated and stored at −80°C.
To generate antibodies against Sca4, the purified Sca4 protein was sent to Pocono Rabbit Farm and Laboratory (Canadensis, PA) where a 91-day custom antibody protocol was performed. To affinity purify Sca4 specific antibodies from serum, the purified Sca4 protein was first dialyzed into Ligand Coupling Buffer (200 mM NaHCO3 pH 8.3, 500 mM NaCl) and then coupled onto NHS-ester Sepharose 4 Fast Flow resin (GE Healthcare). The resin was incubated with serum for 1 h at room temperature with rotation, and low pH buffer (100 mM Glycine, pH 2.5) was used to elute antibody from the resin. Eluted fractions were neutralized with 1M Tris pH 8.8 and dialyzed into 1x PBS overnight at 4°C. Affinity purified antibodies were then concentrated and stored at −80°C.
For immunoblotting of endogenous Sca4, purified bacteria were boiled in 3 x SDS loading buffer (150 mM Tris pH 6.8, 6% SDS, 0.3% Bromophenol Blue, 30% Glycerol, 15% 2-mercaptoethanol) for 10 min, then resolved on an 8% SDS-PAGE gel, transferred to PVDF and developed with affinity purified rabbit anti-Sca4 and mouse anti-OmpA 13-3 (kindly provided by Dr. Ted Hackstadt).
For immunoblotting of Sca4 expression in the U2OS and A549 lines, cells were harvested in immunoprecipitation (IP) lysis buffer (50 mM HEPES, 150 mM NaCl, 1 mM EDTA, 10% Glycerol, 1% Igepal), incubated 10 min on ice, and lysates cleared of debris by centrifugation at 16,100 × g at 4°C. Lysates were analyzed via Western blot as described above except mouse anti-GAPDH was used to detect endogenous GAPDH (loading control).
Co-immunoprecipitation assays
The sca4 sequence was codon-optimized for human expression, synthesized by GeneArt (Life Technologies) to include an N-terminal FLAG tag and cloned into pcDNA3. The α-catenin sequence was amplified from acat 1–906 (Addgene plasmid #24194 containing the CTNNA1 gene), and an N-terminal myc tag was cloned into pcDNA3 (kindly provided by Dr. Joel Pomerantz, Johns Hopkins University School of Medicine). Vinculin residues 1–836 were amplified from MGC human VCL cDNA (Clone ID 4520338; ThermoScientific) and cloned into pcDNA3 along with a N-terminal HA tag.
Co-immunoprecipitation assays were done as described previously (Lamason et al., 2010). Briefly, 40 h after transfection of HEK293T cells via calcium phosphate, cells were harvested in 500 μl IP lysis buffer and incubated 10 min on ice. Debris was cleared by centrifugation at 16,100 × g at 4°C. Lysates were pre-cleared by incubating with 7 μl bed volume protein G sepharose (Amersham) twice for 30 min at 4°C with rotation. An aliquot was removed for input analysis, and the remaining lysate was incubated with 1 μg IP antibody (mouse anti-myc (Santa Cruz sc-40) or rabbit anti-Flag (Sigma F7425)) for 1.5 h at 4°C with rotation. Protein G sepharose (7 μl bed volume) pre-blocked with 1% human insulin was added to samples and incubated 1 h at 4°C with rotation. The beads were then washed with rotation for 4 × 5 min at 4°C with IP lysis buffer before being boiled in the presence of SDS loading buffer. IPs were analyzed by Western blot with mouse anti-HA (Covance MMS-101P), mouse anti-myc (Santa Cruz sc-40), rabbit anti-Flag (Cell Signaling 2368S), rabbit anti-Sca4 (unpurified serum) and M2 anti-FLAG (Sigma F1804).
Bacterial Infections
Due to the longer doubling time for rickettsiae (~ 8–12 h versus ≤ 40 min for L. monocytogenes) and the longer time required to initiate spread, R. parkeri spread was imaged at least 26 hpi, whereas L. monocytogenes spread was imaged at 4 hpi. Additionally, infections were typically carried out at 33°C (R. parkeri) and 37°C (for L. monocytogenes) to match physiological conditions.
To measure infectious focus size, 2.25×105 (A549/U2OS) or 2×105 (HMEC-1) cells were plated onto 12 mm coverslips in 24-well plates. 24 h later, monolayers were infected at an MOI of 0.005–0.05, plates were centrifuged at 200 × g for 5 min at 25°C and incubated at 33°C for 1 h. Samples were washed 3 times with PBS before adding complete media with 50 μg/ml gentamicin. Infection progressed for 28 h (minimum time needed optimal R. parkeri spread) at 33°C until fixation and staining. To quantify spread, 10–20 individual foci were imaged, and the number of infected cells per focus was calculated.
For invasion assays with R. parkeri, 0.75×105 A549 cells or 2×105 HMEC-1 cells were plated on 12mm coverslips 48 or 36 h prior to infection respectively. Culture medium was removed and replaced with 0.5 ml 4°C medium, and R. parkeri at an MOI of 6 (A549) or MOI of 3 (HMEC-1) was added to each sample. Infected cells were centrifuged at 200 × g for 5 min at 4°C, 0.5 ml of 37°C media was added to each well, and samples were immediately placed in a 37°C incubator. Infected coverslips were fixed at specific time points and then stained.
To measure the percentage of R. parkeri with actin tails, 2.25×105 A549 cells were plated on 12 mm coverslips and infected the next day with Rp-GFP or sca4::tn (MOI 0.5–0.8). Infected cells were incubated at 33°C for 24 h and fixed and stained for F-actin. Five fields of view were collected, each containing 90–200 bacteria, and the average percent of bacteria with long tails (> 1 bacterial length) was determined.
To measure the velocities of motile R. parkeri, 2×105 A549 cells stably expressing Lifeact-3xTagBFP were plated in 20 mm MatTek glass bottom dishes, incubated for 24 h and infected with Rp-GFP or sca4::tn (MOI 3–4). Bacteria were added directly to cells, and dishes were incubated at 33°C for 28 h before live cell imaging was done in a 33°C environmental chamber with a Nikon Ti Eclipse. For imaging purposes cell culture medium was replaced with Ringer’s Buffer (155 mM NaCl, 5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 2 mM NaH2PO4, 50 mM HEPES, 10 mM Glucose) with 10% FBS, 1:100 oxyrase and 10 mM succinate, and samples were imaged at 33°C at 5 s intervals for 5 min. To calculate actin-tail mediated velocities, the manual tracking plugin in ImageJ was used to track movement over 11 consecutive frames (6 tracks were collected for Rp-GFP and 15 tracks for sca4::tn).
Live cell imaging of spread was done by infecting confluent monolayers of mixed A549 cells. For Rp-GFP and sca4::tn infections, a mix of A549-TRTF and A549 cells at a 1:4 ratio was plated in 20mm MatTek dishes (7×105 cells/dish) 24 h before infection. Rp-GFP or sca4::tn were added directly to cells (MOI 1.5–2), and infections were carried out at 33°C for at least 26 h before imaging. For Rp-2xTagBFP imaging, the same procedure was followed except A549 co-expressing TRTF and Lifeact-mWasabi were mixed with unlabeled A549 cells. For experiments using Lm-GFP, the mix of A549-TRTF and A549 cells were plated as above 48 h before infection. Lm-GFP were added directly to cells (~MOI 0.5), and infections were carried out at 37°C for at least 4 h before imaging. Infected cells were prepared as above using Ringer’s buffer and imaged in an environmental chamber at 33°C (R. parkeri) or 37°C (Lm-GFP). Images were captured on a Nikon Ti Eclipse microscope with a Yokogawa CSU-XI spinning disc confocal, 60X (1.4 NA) Plan Apo objective, a Clara Interline CCD Camera and MetaMorph software. Z-stacks were captured at 30 s or 1 min intervals for less than 2 h. Individual spread events were then observed, and their kinetics and morphology were recorded. Maximum protrusion length refers to the longest protrusion length observed before resolution into the double-membrane vesicle. Images were processed using ImageJ and assembled in Adobe Illustrator.
To measure the percent of bacteria with or without actin in protrusions for Figures 1E–G, 2.25×105 A549 cells were plated on 12 mm coverslips and infected 24 h later with an MOI of 3 (Rp-2xTagBFP) or MOI 0.3 (Lm TagBFP). Plates were spun down for 5 min at 200 × g at 25°C and moved to 33°C (Rp-2xTagBFP) or 37°C (Lm-TagBFP) incubators. Infections progressed for 28 h (Rp-2xTagBFP) or 4.5 h (Lm-TagBFP). Samples were fixed with 4% paraformaldehyde in 1x PBS (PFA/1x PBS) and stained for β-catenin and phalloidin. Five to ten fields of view were captured (each containing ~50–300 bacteria), and the average percent of bacteria in protrusions and the percent of bacterial protrusions with associated actin were calculated from two independent assays. Actin tails are defined by an F-actin signal at one bacterial pole >1 bacterial length, while polar actin represents actin associated with one pole of the bacterium that does not form a tail.
To measure the percent of bacteria in protrusions for Figure 2H, 2.25×105 A549+TagRFP-T-F cells were plated on 12 mm coverslips. Cells were infected 24 h later with an MOI of 0.5–0.8. Plates were spun down for 5 min at 200 × g at 25°C and subsequently incubated at 33°C for 33 h before fixation with 4% PFA/1x PBS and immunofluorescence staining of β-catenin to detect protrusions. Ten fields of view were captured (each containing ~150–300 bacteria), and the average percent of bacteria in protrusions was calculated.
For mixed cell spread assays (Figure 5D and 5F), 1.5×104 donor cells were plated in 96-well plates (Falcon), and 2×105 recipient cells were plated in 24-well plates (Falcon). 24 h after plating, donors were infected at an MOI of 4 for Figure 5C and 5D, and for Figure 5F, at an MOI of 10 for sca2::tn and an MOI of 4 for WT and sca4::tn. Plates were centrifuged at 200 × g for 5 min at 25°C and incubated at 33°C for 1 h. All samples were then washed once with PBS, lifted with 37°C 1x citric saline (135 mM KCl, 15 mM sodium citrate) to preserve cell surface receptors, recovered in complete media and washed twice in complete media to remove residual citric saline. Donors were resuspended in 100 μl and recipients resuspended in 400 μl complete media + 10 μg/ml gentamicin. Cells were then mixed at a ratio of 1:120 (1.7 μl donors plus 198 μl recipients) and plated in a square well, 96-well glass bottom MatriPlate (Brooks Life Science Systems). Plates were placed in a humidified secondary container in the incubator to promote even cell distribution, and infection was allowed to progress at 33°C for 31 h until fixation and staining. Stained samples were overlaid with 50% sterile glycerol in 1x PBS and imaged directly in the plates. To quantify spread, 10–20 individual foci were imaged, and the percentage of bacteria per focus that had spread to recipient cells was calculated.
For Figure 5C, the same procedure was followed as described above except 1.5×104 donor cells/well and 9×104 recipient cells per well were reverse transfected with 5 nM siRNA and Lipofectamine RNAiMAX (Thermo Fisher) 2 days before infection.
In assays where host vinculin expression was reduced, the following Silencer Select siRNAs (Ambion) were used: siVCL #1 (s14763; target sequence: UUCGAAUUUUGAUUGAAGCag) and siVCL #2 (s14764; target sequence: UCCUAAGUAAGAUACGAGCag). The non-target siRNA (NTsi) was Silencer Select Negative Control No. 1 (4390843). To determine the extent of knockdown, cells were first lysed in IP lysis buffer on ice for 10 min, then the cell debris was cleared via centrifugation at 16,100 × g 4°C for 10 min. Lysates were analyzed by Western blotting using mouse anti-vinculin (Sigma V4505) and mouse anti-GAPDH (AM4300, Ambion).
For myosin-II inhibition experiments, infectious focus assays were performed as described above except at 7–8 hpi, DMSO only (final 0.9% to match final DMSO percentage in blebbistatin treated samples) or 6.25–25 μM blebbistatin (Calbiochem #203389 InSolution Blebbistatin, Racemic) was added to infected cells and incubated for an additional 20 h at 33°C. To quantify spread, 10 individual foci were imaged, and the number of infected cells/focus was calculated.
Immunofluorescence microscopy
For infectious focus assays, cells were fixed in 4% PFA in 1x PBS for 10 min at room temperature. To quench residual PFA, samples were incubated for 10 min in 0.1 M glycine in 1x PBS and permeabilized in 0.05% Triton X-100 (Tx100) in 1x PBS. Samples were then blocked in 2% BSA in 1x PBS and incubated with primary and secondary antibodies diluted in blocking buffer, each for 1 h at room temperature. The host plasma membrane was detected with mouse anti-β-catenin (BD Biosciences, 610153), R. parkeri were detected using rabbit anti-Rickettsia I7205 (Dr. Ted Hackstadt, NIH/NIAID Rocky Mountain Laboratories) and nuclei were stained with DAPI. Coverslips for this experiment and the assays described below were mounted in Prolong mounting media (Invitrogen). For Figure 2H, the same procedure was followed except DAPI was omitted.
For invasion assays, differential staining was done as previously described to distinguish extracellular and intracellular bacteria (Reed et al., 2012).
To quantify actin tail frequency in Figure S1E, samples were fixed in 4% PFA/1x PBS for 10 min at room temperature. To quench residual PFA, samples were incubated for 5 min in 0.1 M glycine in 1x PBS and permeabilized in 0.05% Tx100 in 1x PBS. Samples were stained with Alexa-568 phalloidin (Life Technologies) and imaged with the Nikon Ti Eclipse and a 100x objective. In Figure 1E–G, samples were fixed and imaged as above, except F-actin and host plasma membrane were labeled with Alexa-488 phalloidin and mouse anti-β-catenin (BD Biosciences, 610153), respectively.
To detect endogenous Sca4 secretion in Figure 3E, cells were fixed in 4% PFA/1x PBS for 10 min at room temperature. To quench residual PFA, samples were incubated for 10 min in 0.1 M glycine in 1x PBS and permeabilized in 0.05% Tx100 in 1x PBS. Samples were blocked in blocking buffer (2% BSA, 10% Normal Goat Serum, and 1x PBS). The primary antibody was diluted in blocking buffer and incubation was carried out in a humidified chamber for 3 h at 37°C. The secondary antibodies were diluted in 10% NGS in 1x PBS and incubated for 1 h at room temperature. Sca4 was detected using the affinity-purified rabbit anti-Sca4, R. parkeri were stained using mouse anti-Rickettsia 14-13 (kindly provided by Dr. Ted Hackstadt), and nuclei were stained with DAPI.
Secreted versus non-secreted Sca4 in Figure 3D was detected using a differential staining protocol adapted from Campbell-Valois et al. (Campbell-Valois et al.). After fixation, quenching, and permeabilizing as above, infected cells were blocked with blocking buffer for 30 min at room temperature, then stained with mouse anti-Rickettsia (14–13) and affinity purified rabbit anti-Sca4 for 3 h at 37°C in a humidified chamber. After secondary antibody staining at room temperature for 1 h, samples were fixed for 5 min, quenched with 0.1 M Glycine in 1x PBS for 5 min, and incubated for 20 min at 37°C in lysozyme reaction buffer (0.8x PBS, 50 mM Glucose, 5 mM EDTA, 5 mg/ml Lysozyme, 0.1% Tx-100). Total Sca4 was then detected by staining again with affinity-purified rabbit anti-Sca4 for 3 h at 37°C in a humidified chamber and a 1 h room temperature secondary antibody incubation.
To detect Sca4 localization in the stable U2OS lines in Figure S2, cells were fixed and stained for Sca4 as in Figure 3E, but Alexa 488-phalloidin (Life Technologies) and mouse anti-vinculin (Sigma V4505) were used to visualize F-Actin and vinculin with Sca4.
Images were captured on a Nikon Ti Eclipse microscope with a Yokogawa CSU-XI spinning disc confocal, 60X and 100X (1.4 NA) Plan Apo objectives, a Clara Interline CCD Camera and MetaMorph software. Images were processed using ImageJ and assembled in Adobe Illustrator.
TEM1 secretion assays
A549 cells were plated in a 96-well plate (Corning CellStar) at 1×104 cells/well 48 h before infection. Cells were infected at an MOI of 2 by adding bacteria to wells and centrifuging plates at 200 × g for 5 min at 25°C. Plates were then incubated at 33°C for 48 h before beta-lactamase activity was detected. To detect beta-lactamase activity with the LiveBLAzer FRET – B/G Loading kit (Invitrogen), infected or uninfected A549 cells were first washed with serum free DMEM after which CCF4/AM substrate was added at a final concentration of 2 μM in serum-free DMEM with 6 mM probenecid and 1x alternate substrate loading solution (Invitrogen). Cells were incubated for 90 min at room temperature in the dark, washed one time in PBS, and then imaged in PBS. Cleaved and uncleaved CCF4/AM fluorescence was detected with a plate reader (TECAN infinite F200 Pro; Blue - ex400/em460 and Green - ex405/em535 filters). Average response ratio was calculated by subtracting the background from both channels. Corrected values were then used to determine the blue to green ratio. Fluorescent images were also captured on an Olympus IX71 microscope equipped with a 20x LUCPlanFLN (0.45 NA) objective, a CoolSNAP HQ camera (Photometrics) and DAPI and FITC filter sets.
For immunoblotting of TEM1-fusions, purified bacteria were boiled in 3 x SDS loading buffer (150 mM Tris pH 6.8, 6% SDS, 0.3% Bromophenol Blue, 30% Glycerol, 15% 2-mercaptoethanol) for 10 min and analyzed via Western blot using mouse anti-beta lactamase (Abcam ab12251) and mouse anti-OmpA 13-3 (kindly provided by Dr. Ted Hackstadt).
Traction Force Microscopy
Single cell TFM assays were performed as previously described (Bastounis et al., 2014). Briefly, two layered polyacrylamide gels, the upper of which contained 0.04% carboxylate-modified yellow latex beads 0.1 μm in diameter (FluoSpheres; Molecular Probes) were prepared and activated via SulfoSanpah (22589; ThermoFischer Scientific) as previously described (Bastounis et al., 2014), except gels were attached to 24-well glass bottom plates (MatTek) and their Young’s modulus was ~ 5 kPa (final concentrations of acrylamide and Bis-acrylamide: 5% and 0.15%, respectively). Activated gels were coated with recombinant human E-cadherin/Fc chimera (Creative BioMart) or collagen I (Sigma-Aldrich, C3867) to allow adherence of the stable U2OS cell lines transduced with FCW2-P2AT (control), FCW2-P2AT-Ires-Sca4, or FWC2-P2AT-Ires-Sca4 VBS-NC*.
To measure traction forces mediated by E-cadherin, activated gels were first coated with 0.2 mg/ml rabbit anti-human IgG (Fcγ-fragment specific; Jackson ImmunoResearch) and incubated overnight at 4°C. Gels were washed with PBS the next day to remove unbound antibody and incubated for 3 h at 4°C with 50 μg/ml recombinant human E-cadherin/Fc chimera (Creative BioMart). Gels were then washed with PBS, blocked for 30 min at room temperature with 1% BSA/PBS, washed again with PBS, and equilibrated in DMEM + 10% FBS for 30 min at 37°C prior to adding cells. Cells were lifted via 1x citric saline to preserve cadherins as above. Final washed cell pellets were resuspended in 1x PBS with 20 μg/ml integrin blocking antibodies (anti-integrin β1 (Millipore MAB1987Z; PC410) and anti-integrin alphaVbeta3 (Millipore MAB1976Z; LM609)) and incubated for 30 min at 37°C before adding cells to E-cadherin coated dishes. Approximately 2.5–5×105 cells were used per dish to achieve single cell attachment, and cells were allowed to adhere for 3 h at 37°C before imaging.
To measure cell-ECM traction forces, 0.25 mg/ml collagen I (Sigma-Aldrich, C3867) was added directly to activated gels and incubated overnight at 4°C. Approximately 0.125–0.25×105 cells were used for single cell measurements while 4×105 cells were used for monolayer studies. Cells were lifted via 1x citric saline as above and seeded 6–8 h prior to imaging.
Multi-channel time-lapse sequences of fluorescence (to image the beads) and phase contrast images (to image the cells) were acquired using an inverted Nikon Diaphot 200 with a CCD camera (Andor Technologies) using a 40X Plan Fluor NA 0.60 objective and the MicroManager software package. The microscope was surrounded by a cage incubator (Haison) maintained at 37°C and 5% CO2. Images were acquired every 5 min for 4 to 12 h. Subsequently, at each time interval we measured the 2D deformation of the substrate at each point using an image correlation technique similar to particle image velocimetry (Gui and Wereley, 2002). We calculate the local deformation vector by performing image correlation between each image and an undeformed reference image which we acquired by adding 10% SDS at the end of each recording to detach the cells from the gels. We used interrogation windows of 32×16 pixels (window size x window spacing) for cells forming a monolayer, 32×8 pixels for single cells on collagen-coated gels, and 16×8 pixels for single cells on E-cadherin-coated gels.
For single cell experiments, we used a custom algorithm using MATLAB (MathWorks) to automatically identify the contour of the cells from the phase contrast images (del Álamo et al., 2007). We calculated the 2D traction stresses single cells exert to the gel as described elsewhere (del Álamo et al., 2007). We calculated the strain energy (Us) as the mechanical work done by the cell to deform its gel: , where u⃗ is the measured displacement vector field on the free surface of the gel, τ⃗ is the traction stress field and ∫S()dS represents a surface integral.
For cell monolayer experiments, traction stresses were measured as described above and monolayer tension was calculated from the traction stresses as previously described (Banerjee et al., 2015). Specifically, we solved the equations of mechanical equilibrium for a thin elastic plate subject to the reaction forces created by the polyacrylamide gel on the monolayer, which are opposite to the measured traction stresses.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical parameters and significance are reported in the Figures and the Figure Legends. Data are determined to be statistically significant when p < 0.05 by an unpaired Student’s T-Test, a one-way ANOVA, or Mann-Whitney rank sum T-test, where indicated. As such, asterisks denote statistical significance as: *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001, compared to indicated controls. For graphs depicted as box plots, boxes outline the 25th and 75th percentiles, midlines denote medians, and whiskers show minimum and maximum values. All other graphical representations are described in the figure legends. Statistical analysis was performed in GraphPad PRISM 6.
Supplementary Material
(A) Images of neutral red-stained plaques in Vero cell monolayers. (B) Growth curve of WT (Rp-GFP) and sca4::tn mutant bacteria in Vero cells as measured by R. parkeri genome equivalents. Data is normalized to the 1 hpi timepoint for each set, and data are shown as the mean ± SD from duplicate samples. (C, D) Invasion efficiency of WT (Rp-GFP) and sca4::tn into (C) HMEC-1 or (D) A549 cells. Data are mean ± SD. (E) Quantification of bacteria with long actin tails (>1 bacterial length) at 28 hpi for WT (Rp-GFP) and sca4::tn mutant R. parkeri in A549 cells. F-actin visualized using phalloidin. Data are mean ± SEM. (F) Velocity of bacterial actin-based motility in A549 cells stably expressing the F-actin marker (Lifeact-3xTagBFP) at 28 hpi for individual WT (Rp-GFP) and sca4::tn mutant bacteria. Data are mean ± SEM. (G, H) Infectious focus size in (G) HMEC-1 and (H) U2OS monolayers infected with WT (Rp-GFP) and sca4::tn. Data are mean ± SEM (unpaired T-test: * p < 0.05, ** p < 0.01, **** p < 0.0001).
U2OS cells were stably transduced with control lentiviral construct P2AT (control; FCW2-P2AT), lentiviral constructs expressing Sca4 (FCW2-P2AT-Ires-Sca4) or the VBS-NC* mutant (FCW2-P2AT-Ires-Sca4-VBS-NC*), stained for (A) Sca4 (purple) and F-actin (green) or (B, C) Sca4 (purple) and vinculin (green), and imaged via confocal microscopy. Similar staining patterns were also seen in A549 cells (not shown). (A) Z-slice showing partial colocalization between F-actin and Sca4. Scale bar, 10 μm. (B) Z-slice corresponding to the apical region of a cell monolayer, with vinculin localized to adherens junctions. Scale bar, 10 μm. (C) Z-slice corresponding to the basal cell surface, highlighting focal adhesions labeled by vinculin. Note partial colocalization between Sca4 and vinculin. Scale bar, 10 μm. (D, E) Western blots of cell lysates collected from indicated stable (D) U2OS or (E) A549 lines. GAPDH used as loading control. Antibodies used shown at left, and arrow indicates predicted Sca4 band at ~112 kDa.
A549 cells stably expressing a plasma membrane marker (TagRFP-T-Farnesyl; TRTF; red) were mixed with unlabeled A549 cells and grown to confluency before infection with WT (Rp-GFP; green). Imaging began at 28 hpi. Frames captured every 30 s. A z slice is shown. Timestamp shows min:sec. Scale bar, 5 μm.
A549 cells stably expressing a plasma membrane marker (TagRFP-T-Farnesyl; TRTF; red) were mixed with unlabeled A549 cells and grown to confluency before infection with Lm-GFP (green). Bacterium is shown spreading from labeled donor to unlabeled recipient cell. Imaging began at 4 hpi. Frames captured every 30 s. Z projection of 3 slices is shown. Timestamp shows min:sec. Scale bar, 5 μm.
A549 cells stably expressing a plasma membrane marker (TagRFP-T-Farnesyl; TRTF; red) and an F-actin marker (Lifeact-mWasabi; green) were mixed with unlabeled A549A549 cells and grown to confluency before infection with Rp-2xTagBFP (blue). Arrow indicates bacterium spreading from a labeled donor to an unlabeled recipient cell. Imaging began at 28 hpi. Frames captured every 30 s. Z projection of 2 slices is shown. Timestamp shows min:sec. Scale bar, 5 μm.
Acknowledgments
We are grateful to Vida Ahyong and Taro Ohkawa for critical reading of the manuscript. We thank Ulrike Munderloh, Ted Hackstadt, David Wood, Joel Pomerantz, Chen Chen, Dan Portnoy and Chris Paddock for reagents, and Joel Lamason, Taro Ohkawa, Erin Benanti, Ismaeel Mohamed and Hervé Agaisse for technical help. We also thank UC Berkeley core facility members: Ann Fisher and Alison Killilea (Cell Culture facility), Hector Nolla (CRL Flow Cytometry Facility), and Holly Aaron and Jen-Yi Lee (CRL Molecular Imaging Center). This work was performed in part at the CRL MIC (NIH S10RR027696-01). Support is as follows: R.L.L. (Helen Hay Whitney Foundation postdoctoral fellowship; NIH K99GM115765), M.D.W. (NIH/NIAID AI109044; AI109270), J.A.T. (NIH R37-AI036929; Stanford Center for Systems Biology (P50-GM107615); HHMI) and J.C.A. (NIH 2R01GM084227; 1R01HL128630; NSF CBET 1055697).
Footnotes
AUTHOR CONTRIBUTIONS
Conceptualization and Methodology, R.L.L, E.B., R.S., J.C.A., J.A.T., M.D.W.; Software, E.B., R.S.; Investigation, R.L.L., E.B., N.M.K., and R.S.; Writing - Original Draft, R.L.L.; Writing - Review & Editing, R.L.L., E.B., N.M.K., R.S., J.C.A., J.A.T., M.D.W.; Supervision, J.C.A., J.A.T., M.D.W.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alvarez DE, Agaisse H. The metallo-protease Mpl supports Listeria monocytogenes dissemination through resolution of membrane protrusions into vacuoles. Infection and Immunity. 2016;84:1806–1814. doi: 10.1128/IAI.00130-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atherton P, Stutchbury B, Jethwa D, Ballestrem C. Mechanosensitive components of integrin adhesions: Role of vinculin. Exp Cell Res. 2016;343:21–27. doi: 10.1016/j.yexcr.2015.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee I, Carrion K, Serrano R, Dyo J, Sasik R, Lund S, Willems E, Aceves S, Meili R, Mercola M, et al. Cyclic stretch of embryonic cardiomyocytes increases proliferation, growth, and expression while repressing Tgf-β signaling. J Mol Cell Cardiol. 2015;79:133–144. doi: 10.1016/j.yjmcc.2014.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barry AK, Tabdili H, Muhamed I, Wu J, Shashikanth N, Gomez GA, Yap AS, Gottardi CJ, de Rooij J, Wang N, et al. α-catenin cytomechanics--role in cadherin-dependent adhesion and mechanotransduction. J Cell Sci. 2014;127:1779–1791. doi: 10.1242/jcs.139014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastounis E, Meili R, Alonso-Latorre B, del Álamo JC, Lasheras JC, Firtel RA. The SCAR/WAVE complex is necessary for proper regulation of traction stresses during amoeboid motility. Mol Biol Cell. 2011;22:3995–4003. doi: 10.1091/mbc.E11-03-0278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastounis E, Meili R, Álvarez-González B, Francois J, del Álamo JC, Firtel RA, Lasheras JC. Both contractile axial and lateral traction force dynamics drive amoeboid cell motility. J Cell Biol. 2014;204:1045–1061. doi: 10.1083/jcb.201307106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardini ML, Mounier J, d’Hauteville H, Coquis-Rondon M, Sansonetti PJ. Identification of icsA, a plasmid locus of Shigella flexneri that governs bacterial intra- and intercellular spread through interaction with F-actin. Proc Natl Acad Sci USA. 1989;86:3867–3871. doi: 10.1073/pnas.86.10.3867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley CD, Tan J, Anderson KL, Hanein D, Volkmann N, Weis WI, Nelson WJ, Dunn AR. Cell adhesion. The minimal cadherin-catenin complex binds to actin filaments under force. Science. 2014;346:1254211–1254211. doi: 10.1126/science.1254211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkhardt NY, Baldridge GD, Williamson PC, Billingsley PM, Heu CC, Felsheim RF, Kurtti TJ, Munderloh UG. Development of Shuttle Vectors for Transformation of Diverse Rickettsia Species. PLoS ONE. 2011;6:e29511. doi: 10.1371/journal.pone.0029511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cambronne ED, Roy CR. Recognition and Delivery of Effector Proteins into Eukaryotic Cells by Bacterial Secretion Systems. Traffic. 2006;7:929–939. doi: 10.1111/j.1600-0854.2006.00446.x. [DOI] [PubMed] [Google Scholar]
- Campbell-Valois FX, Schnupf P, Sansonetti PJ. Detection of the Secreted and Cytoplasmic Fractions of IpaB, IpaC and IpaD by Lysozyme Permeabilization. Bio-Protocol. 2014;4:e1271. [Google Scholar]
- Case LB, Baird MA, Shtengel G, Campbell SL, Hess HF, Davidson MW, Waterman CM. Molecular mechanism of vinculin activation and nanoscale spatial organization in focal adhesions. Nat Cell Biol. 2015;17:880–892. doi: 10.1038/ncb3180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charpentier X, Oswald E. Identification of the Secretion and Translocation Domain of the Enteropathogenic and Enterohemorrhagic Escherichia coli Effector Cif, Using TEM-1 beta-Lactamase as a New Fluorescence-Based Reporter. Journal of Bacteriology. 2004;186:5486–5495. doi: 10.1128/JB.186.16.5486-5495.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Álamo JC, Meili R, Alonso-Latorre B, Rodríguez-Rodríguez J, Aliseda A, Firtel RA, Lasheras JC. Spatio-temporal analysis of eukaryotic cell motility by improved force cytometry. Proc Natl Acad Sci USa. 2007;104:13343–13348. doi: 10.1073/pnas.0705815104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delepelaire P. Type I secretion in gram-negative bacteria. Biochimica Et Biophysica Acta (BBA) - Molecular Cell Research. 2004;1694:149–161. doi: 10.1016/j.bbamcr.2004.05.001. [DOI] [PubMed] [Google Scholar]
- Dragoi AM, Agaisse H. The class II phosphatidylinositol 3-phosphate kinase PIK3C2A promotes Shigella flexneri dissemination through formation of vacuole-like protrusions. Infection and Immunity. 2015;83:1695–1704. doi: 10.1128/IAI.03138-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukumatsu M, Ogawa M, Arakawa S, Suzuki M, Nakayama K, Shimizu S, Kim M, Mimuro H, Sasakawa C. Shigella targets epithelial tricellular junctions and uses a noncanonical clathrin-dependent endocytic pathway to spread between cells. Cell Host Microbe. 2012;11:325–336. doi: 10.1016/j.chom.2012.03.001. [DOI] [PubMed] [Google Scholar]
- Gillespie JJ, Kaur SJ, Rahman MS, Rennoll-Bankert K, Sears KT, Beier-Sexton M, Azad AF, Dehio C. Secretome of obligate intracellular Rickettsia. FEMS Microbiol Rev. 2015;39:47–80. doi: 10.1111/1574-6976.12084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingras AR. Mapping and Consensus Sequence Identification for Multiple Vinculin Binding Sites within the Talin Rod. Journal of Biological Chemistry. 2005;280:37217–37224. doi: 10.1074/jbc.M508060200. [DOI] [PubMed] [Google Scholar]
- Gouin E, Gantelet H, Egile C, Lasa I, Ohayon H, Villiers V, Gounon P, Sansonetti PJ, Cossart P. A comparative study of the actin-based motilities of the pathogenic bacteria Listeria monocytogenes, Shigella flexneri and Rickettsia conorii. J Cell Sci. 1999;112(Pt 11):1697–1708. doi: 10.1242/jcs.112.11.1697. [DOI] [PubMed] [Google Scholar]
- Grasperge BJ, Reif KE, Morgan TD, Sunyakumthorn P, Bynog J, Paddock CD, Macaluso KR. Susceptibility of inbred mice to Rickettsia parkeri. Infection and Immunity. 2012;80:1846–1852. doi: 10.1128/IAI.00109-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grote A, Hiller K, Scheer M, Münch R, Nörtemann B, Hempel DC, Jahn D. JCat: a novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic Acids Research. 2005;33:W526–W531. doi: 10.1093/nar/gki376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gründling A, Burrack LS, Bouwer HGA, Higgins DE. Listeria monocytogenes regulates flagellar motility gene expression through MogR, a transcriptional repressor required for virulence. Proc Natl Acad Sci USa. 2004;101:12318–12323. doi: 10.1073/pnas.0404924101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gui L, Wereley ST. A correlation-based continuous window-shift technique to reduce the peak-locking effect in digital PIV image evaluation. Experiments in Fluids. 2002;32:506–517. [Google Scholar]
- Haglund CM, Choe JE, Skau CT, Kovar DR, Welch MD. Rickettsia Sca2 is a bacterial formin-like mediator of actin-based motility. Nat Cell Biol. 2010;12:1057–1063. doi: 10.1038/ncb2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinzen RA. Rickettsial actin-based motility: behavior and involvement of cytoskeletal regulators. Ann N Y Acad Sci. 2003;990:535–547. doi: 10.1111/j.1749-6632.2003.tb07424.x. [DOI] [PubMed] [Google Scholar]
- Huveneers S, de Rooij J. Mechanosensitive systems at the cadherin-F-actin interface. J Cell Sci. 2013;126:403–413. doi: 10.1242/jcs.109447. [DOI] [PubMed] [Google Scholar]
- Izard T, Tran Van Nhieu G, Bois PRJ. Shigella applies molecular mimicry to subvert vinculin and invade host cells. 2006;175:465–475. doi: 10.1083/jcb.200605091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadurugamuwa JL, Rohde M, Wehland J, Timmis KN. Intercellular spread of Shigella flexneri through a monolayer mediated by membranous protrusions and associated with reorganization of the cytoskeletal protein vinculin. Infection and Immunity. 1991;59:3463–3471. doi: 10.1128/iai.59.10.3463-3471.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleba B, Clark TR, Lutter EI, Ellison DW, Hackstadt T. Disruption of the Rickettsia rickettsii Sca2 Autotransporter Inhibits Actin-Based Motility. Infection and Immunity. 2010;78:2240–2247. doi: 10.1128/IAI.00100-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuehl CJ, Dragoi AM, Agaisse H. The Shigella flexneri type 3 secretion system is required for tyrosine kinase-dependent protrusion resolution, and vacuole escape during bacterial dissemination. PLoS ONE. 2014;9:e112738. doi: 10.1371/journal.pone.0112738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuehl CJ, Dragoi A-M, Talman A, Agaisse H. Bacterial spread from cell to cell: beyond actin-based motility. Trends Microbiol. 2015;23:558–566. doi: 10.1016/j.tim.2015.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamason RL, Kupfer A, Pomerantz JL. The dynamic distribution of CARD11 at the immunological synapse is regulated by the inhibitory kinesin GAKIN. Mol Cell. 2010;40:798–809. doi: 10.1016/j.molcel.2010.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecuit T, Yap AS. E-cadherin junctions as active mechanical integrators in tissue dynamics. Nat Cell Biol. 2015;17:533–539. doi: 10.1038/ncb3136. [DOI] [PubMed] [Google Scholar]
- Liu ZM, Tucker AM, Driskell LO, Wood DO. Mariner-based transposon mutagenesis of Rickettsia prowazekii. Applied and Environmental Microbiology. 2007;73:6644–6649. doi: 10.1128/AEM.01727-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra A, Kesarwani AK, Pal D, Nagaraja V. Nucleic Acids Research (Database Issue) 2011;39:D129–135. doi: 10.1093/nar/gkq971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monack DM, Theriot JA. Actin-based motility is sufficient for bacterial membrane protrusion formation and host cell uptake. Cell Microbiol. 2001;3:633–647. doi: 10.1046/j.1462-5822.2001.00143.x. [DOI] [PubMed] [Google Scholar]
- Park H, Lee JH, Gouin E, Cossart P, Izard T. The Rickettsia Surface Cell Antigen 4 Applies Mimicry to Bind to and Activate Vinculin. Journal of Biological Chemistry. 2011a;286:35096–35103. doi: 10.1074/jbc.M111.263855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H, Valencia-Gallardo C, Sharff A, Tran Van Nhieu G, Izard T. Novel vinculin binding site of the IpaA invasin of Shigella. Journal of Biological Chemistry. 2011b;286:23214–23221. doi: 10.1074/jbc.M110.184283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng X, Nelson ES, Maiers JL, DeMali KA. New insights into vinculin function and regulation. Int Rev Cell Mol Biol. 2011;287:191–231. doi: 10.1016/B978-0-12-386043-9.00005-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen TN, Brunak S, von Heijne G, Nielsen H. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods. 2011;8:785–786. doi: 10.1038/nmeth.1701. [DOI] [PubMed] [Google Scholar]
- Pomerantz JL, Denny EM, Baltimore D. CARD11 mediates factor-specific activation of NF-kappaB by the T cell receptor complex. Embo J. 2002;21:5184–5194. doi: 10.1093/emboj/cdf505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajabian T, Gavicherla B, Heisig M, Müller-Altrock S, Goebel W, Gray-Owen SD, Ireton K. The bacterial virulence factor InlC perturbs apical cell junctions and promotes cell-to-cell spread of Listeria. Nat Cell Biol. 2009;11:1212–1218. doi: 10.1038/ncb1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray K, Marteyn B, Sansonetti PJ, Tang CM. Life on the inside: the intracellular lifestyle of cytosolic bacteria. Nature Reviews Microbiology. 2009;7:333–340. doi: 10.1038/nrmicro2112. [DOI] [PubMed] [Google Scholar]
- Reed S, Serio AW, Welch MD. Rickettsia parkeri invasion of diverse host cells involves an Arp2/3 complex, WAVE complex and Rho-family GTPase-dependent pathway. Cell Microbiol. 2012;14:529–545. doi: 10.1111/j.1462-5822.2011.01739.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed SCO, Lamason RL, Risca VI, Abernathy E, Welch MD. Rickettsia actin-based motility occurs in distinct phases mediated by different actin nucleators. Curr Biol. 2014;24:98–103. doi: 10.1016/j.cub.2013.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins JR, Barth AI, Marquis H, de Hostos EL, Nelson WJ, Theriot JA. Listeria monocytogenes exploits normal host cell processes to spread from cell to cell. 1999;146:1333–1350. doi: 10.1083/jcb.146.6.1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sears KT, Ceraul SM, Gillespie JJ, Allen ED, Jr, Popov VL, Ammerman NC, Rahman MS, Azad AF. Surface Proteome Analysis and Characterization of Surface Cell Antigen (Sca) or Autotransporter Family of Rickettsia typhi. PLoS Pathog. 2012;8:e1002856. doi: 10.1371/journal.ppat.1002856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen A, Higgins DE. The 5′ untranslated region-mediated enhancement of intracellular listeriolysin O production is required for Listeria monocytogenes pathogenicity. Mol Microbiol. 2005;57:1460–1473. doi: 10.1111/j.1365-2958.2005.04780.x. [DOI] [PubMed] [Google Scholar]
- Solovyev V, Salamov A. Automatic Annotation of Microbial Genomes and Metagenomic Sequences. In: Li R, editor. Metagenomics and its Applications in Agriculture, Biomedicine and Environmental Studies. Nova Science Publishers; 2011. pp. 61–78. [Google Scholar]
- Talman AM, Chong R, Chia J, Svitkina T, Agaisse H. Actin network disassembly powers dissemination of Listeria monocytogenes. J Cell Sci. 2014;127:240–249. doi: 10.1242/jcs.140038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tambe DT, Croutelle U, Trepat X, Park CY, Kim JH, Millet E, Butler JP, Fredberg JJ. Monolayer Stress Microscopy: Limitations, Artifacts, and Accuracy of Recovered Intercellular Stresses. PLoS ONE. 2013;8:e55172–13. doi: 10.1371/journal.pone.0055172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilney LG, Portnoy DA. Actin filaments and the growth, movement, and spread of the intracellular bacterial parasite. Listeria monocytogenes. 1989;109:1597–1608. doi: 10.1083/jcb.109.4.1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchiyama T. Intracytoplasmic localization of antigenic heat-stable 120- to 130-kilodalton proteins (PS120) common to spotted fever group rickettsiae demonstrated by immunoelectron microscopy. Microbiol Immunol. 1997;41:815–818. doi: 10.1111/j.1348-0421.1997.tb01933.x. [DOI] [PubMed] [Google Scholar]
- Walker DH, Ismail N. Emerging and re-emerging rickettsioses: endothelial cell infection and early disease events. Nature Reviews Microbiology. 2008;6:375–386. doi: 10.1038/nrmicro1866. [DOI] [PubMed] [Google Scholar]
- Weber GF, Bjerke MA, DeSimone DW. Integrins and cadherins join forces to form adhesive networks. J Cell Sci. 2011;124:1183–1193. doi: 10.1242/jcs.064618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss EE, Kroemker M, Rüdiger AH, Jockusch BM, Rüdiger M. Vinculin is part of the cadherin-catenin junctional complex: complex formation between alpha-catenin and vinculin. J Cell Biol. 1998;141:755–764. doi: 10.1083/jcb.141.3.755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winger JA, Derbyshire ER, Lamers MH. The crystal structure of the catalytic domain of a eukaryotic guanylate cyclase. BMC Structural Biology. 2008;8:42. doi: 10.1186/1472-6807-8-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonemura S, Yonemura S, Wada Y, Wada Y, Watanabe T, Watanabe T, Nagafuchi A, Nagafuchi A, Shibata M, Shibata M. alpha-Catenin as a tension transducer that induces adherens junction development. Nat Cell Biol. 2010;12:533–542. doi: 10.1038/ncb2055. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Images of neutral red-stained plaques in Vero cell monolayers. (B) Growth curve of WT (Rp-GFP) and sca4::tn mutant bacteria in Vero cells as measured by R. parkeri genome equivalents. Data is normalized to the 1 hpi timepoint for each set, and data are shown as the mean ± SD from duplicate samples. (C, D) Invasion efficiency of WT (Rp-GFP) and sca4::tn into (C) HMEC-1 or (D) A549 cells. Data are mean ± SD. (E) Quantification of bacteria with long actin tails (>1 bacterial length) at 28 hpi for WT (Rp-GFP) and sca4::tn mutant R. parkeri in A549 cells. F-actin visualized using phalloidin. Data are mean ± SEM. (F) Velocity of bacterial actin-based motility in A549 cells stably expressing the F-actin marker (Lifeact-3xTagBFP) at 28 hpi for individual WT (Rp-GFP) and sca4::tn mutant bacteria. Data are mean ± SEM. (G, H) Infectious focus size in (G) HMEC-1 and (H) U2OS monolayers infected with WT (Rp-GFP) and sca4::tn. Data are mean ± SEM (unpaired T-test: * p < 0.05, ** p < 0.01, **** p < 0.0001).
U2OS cells were stably transduced with control lentiviral construct P2AT (control; FCW2-P2AT), lentiviral constructs expressing Sca4 (FCW2-P2AT-Ires-Sca4) or the VBS-NC* mutant (FCW2-P2AT-Ires-Sca4-VBS-NC*), stained for (A) Sca4 (purple) and F-actin (green) or (B, C) Sca4 (purple) and vinculin (green), and imaged via confocal microscopy. Similar staining patterns were also seen in A549 cells (not shown). (A) Z-slice showing partial colocalization between F-actin and Sca4. Scale bar, 10 μm. (B) Z-slice corresponding to the apical region of a cell monolayer, with vinculin localized to adherens junctions. Scale bar, 10 μm. (C) Z-slice corresponding to the basal cell surface, highlighting focal adhesions labeled by vinculin. Note partial colocalization between Sca4 and vinculin. Scale bar, 10 μm. (D, E) Western blots of cell lysates collected from indicated stable (D) U2OS or (E) A549 lines. GAPDH used as loading control. Antibodies used shown at left, and arrow indicates predicted Sca4 band at ~112 kDa.
A549 cells stably expressing a plasma membrane marker (TagRFP-T-Farnesyl; TRTF; red) were mixed with unlabeled A549 cells and grown to confluency before infection with WT (Rp-GFP; green). Imaging began at 28 hpi. Frames captured every 30 s. A z slice is shown. Timestamp shows min:sec. Scale bar, 5 μm.
A549 cells stably expressing a plasma membrane marker (TagRFP-T-Farnesyl; TRTF; red) were mixed with unlabeled A549 cells and grown to confluency before infection with Lm-GFP (green). Bacterium is shown spreading from labeled donor to unlabeled recipient cell. Imaging began at 4 hpi. Frames captured every 30 s. Z projection of 3 slices is shown. Timestamp shows min:sec. Scale bar, 5 μm.
A549 cells stably expressing a plasma membrane marker (TagRFP-T-Farnesyl; TRTF; red) and an F-actin marker (Lifeact-mWasabi; green) were mixed with unlabeled A549A549 cells and grown to confluency before infection with Rp-2xTagBFP (blue). Arrow indicates bacterium spreading from a labeled donor to an unlabeled recipient cell. Imaging began at 28 hpi. Frames captured every 30 s. Z projection of 2 slices is shown. Timestamp shows min:sec. Scale bar, 5 μm.